

Abstract
It has been well documented that neurotrophins, including brain-derived neurotrophic factor (BDNF), are severely affected in Alzheimer's disease (AD), but their administration faces a myriad of technical challenges. Here we took advantage of the early astrogliosis observed in an amyloid mouse model of AD (5xFAD) and used it as an internal sensor to administer BDNF conditionally and locally. We first demonstrate the relevance of BDNF release from astrocytes by evaluating the effects of coculturing WT neurons and BDNF-deficient astrocytes. Next, we crossed 5xFAD mice with pGFAP:BDNF mice (only males were used) to create 5xFAD mice that overexpress BDNF when and where astrogliosis is initiated (5xF:pGB mice). We evaluated the behavioral phenotype of these mice. We first found that BDNF from astrocytes is crucial for dendrite outgrowth and spine number in cultured WT neurons. Double-mutant 5xF:pGB mice displayed improvements in cognitive tasks compared with 5xFAD littermates. In these mice, there was a rescue of BDNF/TrkB downstream signaling activity associated with an improvement of dendritic spine density and morphology. Clusters of synaptic markers, PSD-95 and synaptophysin, were also recovered in 5xF:pGB compared with 5xFAD mice as well as the number of presynaptic vesicles at excitatory synapses. Additionally, experimentally evoked LTP in vivo was increased in 5xF:pGB mice. The beneficial effects of conditional BDNF production and local delivery at the location of active neuropathology highlight the potential to use endogenous biomarkers with early onset, such as astrogliosis, as regulators of neurotrophic therapy in AD.
SIGNIFICANCE STATEMENT Recent evidence places astrocytes as pivotal players during synaptic plasticity and memory processes. In the present work, we first provide evidence that astrocytes are essential for neuronal morphology via BDNF release. We then crossed transgenic mice (5xFAD mice) with the transgenic pGFAP-BDNF mice, which express BDNF under the GFAP promoter. The resultant double-mutant mice 5xF:pGB mice displayed a full rescue of hippocampal BDNF loss and related signaling compared with 5xFAD mice and a significant and specific improvement in all the evaluated cognitive tasks. These improvements did not correlate with amelioration of β amyloid load or hippocampal adult neurogenesis rate but were accompanied by a dramatic recovery of structural and functional synaptic plasticity.
Keywords: Alzheimer's disease, astrocytes, BDNF, long-term potentiation, memory, mice
Introduction
Alzheimer disease (AD) is the most common form of dementia in the aging population, accounting for 60%–80% of the cases. The disease is a progressive neurodegenerative disorder characterized by the presence of extracellular amyloid plaques composed of amyloid-β (Aβ) surrounded by dystrophic neurites and neurofibrillary tangles (NFTs) (Alzheimer's Association, 2012). Further pathological hallmarks of the disease include inflammatory processes, synaptic and neuronal loss, cerebral atrophy, and cerebral amyloid angiopathy (Wirths and Bayer, 2012). The complex progression of neurodegeneration in AD patients results in memory impairment and decline in other cognitive abilities often combined with noncognitive symptoms, such as mood and personality changes (Alzheimer's Association, 2012).
One of the most promising therapies in AD is the use of neurotrophic factors, such as NGF or BDNF (Allen and Dawbarn, 2006). Indeed, BDNF plays important roles in neural survival and synaptic plasticity (Lynch et al., 2008). In line with this, BDNF is reduced early in the course of the disease in transgenic mouse models of AD (Kaminari et al., 2017), and TrkB deletion accelerates and worsens the phenotype of these mice (Devi and Ohno, 2015), whereas TrkB agonists improve it (Devi and Ohno, 2012; Zhang et al., 2014). Alterations in the BDNF-TrkB system could account for memory deficits, neuronal cell death, and synaptic plasticity alterations observed in AD (von Bohlen Und Halbach and von Bohlen Und Halbach, 2018). BDNF-TrkB-associated downstream molecular pathways include PLCγ, ERK, and Akt signaling. Interestingly, dysregulation of these pathways is able to modulate the levels of essential synaptic proteins, such as postsynaptic density-95 (PSD-95) and synaptophysin (Tartaglia et al., 2001; Robinet and Pellerin, 2011; Parsons et al., 2014; Yoshii and Constantine-Paton, 2014; Zhang et al., 2017), which in turn are altered in AD models (Yuki et al., 2014; Dorostkar et al., 2015). Finally, altered BDNF-TrkB pathway could also result in deficient adult neurogenesis in the hippocampus (Waterhouse et al., 2012), which could in turn contribute to the memory loss in AD (Toda and Gage, 2018).
Several strategies have been proposed to produce/deliver BDNF to counteract neurodegeneration. However, all of them are associated with deleterious secondary effects or are not satisfactory enough (Lindvall et al., 2004). Important drawbacks are the risk of tumorigenesis, insufficient cell survival in cell therapy, or invasiveness of some delivery systems, as well as the lack of control on the production and delivery of the neurotrophin because too high levels can be neurotoxic (Martínez-Serrano and Björklund, 1996; Rubio et al., 1999; Pineda et al., 2007; Kells et al., 2008). Furthermore, these strategies do not target specifically the diseased tissue or deliver appropriate levels of the neurotrophin depending on the severity of the symptoms.
Astrocytes in AD are reactive at early stages, have impaired Ca2+ signaling, regulate β-amyloid burden, and influence synaptic plasticity alterations also contributing to excitotoxicity (Acosta et al., 2017). Interestingly, astrocytes normally express the BDNF protein, although at lesser levels than neurons (Saha et al., 2006; Giralt et al., 2010; Fulmer et al., 2014; Hong et al., 2016). Therefore, engineered astrocytes could be good candidates to release neurotrophic factors. Indeed, genetically modified astrocytes have been used with positive results in some models of neurodegeneration (Yoshimoto et al., 1995; Carpenter et al., 1997). This strategy could be relevant for AD as the numbers of reactive astrocytes increases gradually as the disease progress (Song et al., 2015). Moreover, astrogliosis has been shown to be one of the specific hallmarks of disease progression in mouse models of AD (Oakley et al., 2006). Because of this increase in astrocytes, and the fact that astrogliosis leads to increased GFAP promoter activation, one would predict that the use of this promoter as a self-regulator would provide neurotrophic support at the time when it is critically needed. Therefore, here we sought to investigate the role of BDNF from astrocytes in normal neuronal dendritic growth and whether this could be exploited to design advanced and conditional therapies in AD models.
Materials and Methods
Astrocyte/neuron cocultures.
Primary astrocyte cultures were obtained from P1 to P3 BDNF+/+ and BDNF−/− mouse pups (Ernfors et al., 1994) (IMSR catalog #EM:00247, RRID:IMSR_EM:00247) by hippocampal dissections. Extracted tissue was dissociated and placed in 25 cm2 flasks in a MEM 1× conditioned media NM-15 (20% FBS; Invitrogen; d-glucose 90 mm) with l-glutamine and Earle's salts (Invitrogen) and placed in an incubator at 37°C with 5% CO2. A tail biopsy was obtained from each pup for genotyping. After two passages, cultures were purified by agitating in a shaker during 10 min at 400 rpm. Medium with undesired floating cells was replaced, and flasks were placed in an incubator for 2 h at 37°C. Next, flasks were agitated again for 16–18 h at 250 rpm. Finally, medium with floating cells was replaced with new medium. Once astrocytes reached confluence, they were seeded in 24-well plaques and allowed to reach confluence. Then, 1 d before the addition of hippocampal neurons, astrocyte cultures were preincubated with neurobasal medium (Invitrogen) containing 1 ml per 50 ml of B27 supplement (Invitrogen) and 50 ml of GlutaMAX (100×; Invitrogen). Hippocampal neurons were prepared from E17 C57BL/6J mouse embryos (pregnant mice from Charles River). The neuronal cell suspension was low density seeded in the 24-well plaques already containing astrocytes (10,000 cells cm2). Cocultures were fixed at 6 DIV or 20 DIV after neuron seeding to evaluate the number of dendrites and dendritic spines, respectively. Cultures collected at 20 DIV were transfected 48 h before cell fixation with Transfectine (Bio-Rad) following the manufacturer's instructions. Cells were transfected with a previously described construct expressing GFP (Giralt et al., 2017) to allow dendritic spine density counting in isolated hippocampal neurons.
qPCR assay.
Total RNA from astrocyte cultures (see above) and HEK293 cells was extracted using the RNeasy Lipid Tissue Mini Kit (QIAGEN). Total RNA (500 ng) was used to synthesize cDNA using random primers with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA synthesis was performed at 37°C for 120 min in a final volume of 20 μl according to the manufacturer's instructions. The cDNA was then analyzed by qPCR using the following gene expression assays: 18S (NR_003286; Integrated DNA Technologies) and BDNF (NM_001048139; Integrated DNA Technologies). RT-PCR was performed in 12 μl of final volume on 96-well plates using the Premix Ex Taq (Probe qPCR; Takara Biotechnology). Reactions included the following: Segment 1, 1 cycle of 30 s at 95°C; Segment 2, 40 cycles of 5 s at 95°C and 20 s at 60°C. All RT-PCR assays were performed in duplicate and repeated for at least three independent experiments. To provide negative controls and exclude contamination by genomic DNA, the RT was omitted in the cDNA synthesis step, and the samples were subjected to the PCR in the same manner with each TaqMan Gene Expression Assay. The RT-PCR data were analyzed using the MxProTM qPCR analysis software version 3.0 (Stratagene). Quantification was performed with the Comparative Quantitation Analysis program of this software and using the 18S gene expression as internal loading control.
Immunocytochemistry and neuronal morphology assessment.
Fixed cells were permeabilized in Triton X-100 0.5% (v/v) for 10 min, and then blocking was performed with 10 g/L BSA in PBS for 1 h. Cells were incubated with a mouse monoclonal antibody for MAP2 (1:800, Sigma-Aldrich catalog #M1406, RRID:AB_477171) at 4°C overnight. After three washes with PBS, cells were incubated with the corresponding Cy3-coupled fluorescent secondary antibody (1:200; Jackson ImmunoResearch Laboratories catalog #715-165-150, RRID:AB_2340813). After washing twice with PBS, the coverslips were mounted with Vectashield (Vector Laboratories). Hippocampal neuron staining was observed with a confocal SP5-II (see below).
Mouse lines.
For this study, we used the transgenic mouse line 5xFAD (MMRRC catalog #034840-JAX, RRID:MMRRC_034840-JAX). 5xFAD mice overexpress the 695-amino acid isoform of the human amyloid precursor protein (APP695) carrying the Swedish, London, and Florida mutations under the control of the murine Thy-1 promoter. In addition, they express human presenilin-1 (PSEN-1) carrying the M146L/L286V mutation, also under the control of the murine Thy-1 promoter (Oakley et al., 2006). We crossed 5xFAD mice with the previously generated pGFAP-BDNF mice (Giralt et al., 2010) to obtain 5xFAD mice that overexpress BDNF under the GFAP promoter (5xF:pGB mice). For astrocyte cultures, we used P1-P3 BDNF+/+ and BDNF−/− mice (Ernfors et al., 1994) (IMSR catalog #EM:00247, RRID:IMSR_EM:00247). Mouse genotyping for pGFAP-BDNF, 5xFAD, and BDNF−/− mice was performed from a tail biopsy, as previously described (Oakley et al., 2006; Giralt et al., 2009, 2010) by Charles River services. The animals were housed with access to food and water ad libitum in a colony room kept at 19°C–22°C and 40%–60% humidity, under a 12:12 h light/dark cycle. Experimental animals were all males and used at 8 months of age and in accordance with the ethical guidelines (Declaration of Helsinki and NIH Publication no. 85-23, revised 1985, European Community Guidelines, and French Agriculture and Forestry Ministry guidelines for handling animals, decree 87849, license A 75-05-22) and approved by the local ethical committee.
Western blot.
Mice were deeply anesthetized in a CO2 chamber, the brains quickly removed, hippocampus dissected out, frozen in dry ice, and stored at −80°C until use. Briefly, tissue was sonicated in 250 ml of lysis buffer (PBS, 1% Nonidet P40 [v/v], 1 g/L SDS, 5 g/L sodium deoxycholate, protease inhibitors mixture 1:1000 [Sigma-Aldrich], and 2 g/L sodium orthovanadate) and centrifuged at 12,000 rpm for 20 min, and the pellet was discarded. Proteins (15 mg) from hippocampal or cortical tissue were analyzed by SDS-PAGE (7.5% acrylamide, w/v) and transferred to nitrocellulose membranes (Millipore). Membranes were blocked in TBS-T (150 mm NaCl, 20 mm Tris-HCl, pH 7.5, 0.05% [v/v] Tween 20) with 50 g/L nonfat dry milk and 50 g/L BSA. Immunoblots were probed with anti-GFAP (1:1000, Agilent Technologies catalog #Z0334, RRID:AB_10013382), anti-BDNF (1:1000, Santa Cruz Biotechnology catalog #sc-546, RRID:AB_630940), anti-Akt (1:1000, Cell Signaling Technology catalog #9272, RRID:AB_329827), anti-ERK (1:1000, Cell Signaling Technology catalog #4695, RRID:AB_390779), anti-PLCγ (1:1000, Cell Signaling Technology catalog #2822, RRID:AB_2163702), anti-phosphor-Akt (1:1000, Cell Signaling Technology catalog #4056, RRID:AB_331163), anti-phosphor-ERK (1:1000, Cell Signaling Technology catalog #9101, RRID:AB_331646), anti-phosphor-PLCγ (1:1000, Cell Signaling Technology catalog #2821, RRID:AB_330855), or anti-α-tubulin (1:10,000, Sigma-Aldrich catalog #T9026, RRID:AB_477593). All blots were incubated overnight at 4°C with shaking, in the presence of the primary antibody in PBS with 0.2 g/L sodium azide. After several washes in TBS-T, blots were incubated with anti-rabbit IgG IRdye800CW-coupled or anti-mouse IgG IRdye700DX-coupled antibodies (1/2000, Rockland Immunochemicals) and signal detected by the Odyssey system (Li-Cor) and analyzed using ImageJ.
Golgi staining, spine counting, and morphological analysis.
Fresh brain hemispheres were processed following the Golgi-Cox method as described previously (Giralt et al., 2017). Essentially, mouse brain hemispheres were incubated in the dark for 21 d in filtered dye solution (10 g L−1 K2Cr2O7, 10 g L−1 HgCl2, and 8 g L−1 K2CrO4). The tissue was then washed 3 × 2 min in water and 30 min in 90% ethanol (EtOH) (v/v); 200 μm sections were cut in 70% EtOH on a vibratome (Leica Microsystems) and washed in water for 5 min. Next, they were reduced in 16% (v/v) ammonia solution for 1 h before washing in water for 2 min and fixation in 10 g l−1 Na2S2O3 for 7 min. After a 2 min final wash in water, sections were mounted on superfrost coverslips, dehydrated for 3 min in 50%, then 70%, 80%, and 100% EtOH, incubated for 2 × 5 min in a 2:1 isopropanol:EtOH mixture, followed by 1 × 5 min in pure isopropanol and 2 × 5 min in xylol. Bright-field images of Golgi-impregnated stratum radiatum dendrites from hippocampal CA1 pyramidal neurons were captured with a Nikon DXM 1200F digital camera attached to a Nikon Eclipse E600 light microscope (100× oil objective). Only fully impregnated pyramidal neurons with their soma found entirely within the thickness of the section were used. Image z stacks were taken every 0.2 mm and at 1024 × 1024 pixel resolution, yielding an image with pixel dimensions of 49.25 × 49.25 mm. Z stacks were deconvolved using the Huygens software (Scientific Volume Imaging) to improve voxel resolution and to reduce optical aberration along the z axis. The total number of spines counting and their morphology categorization were performed by using the NeuronStudio freeware (NeuronStudio, RRID:SCR_013798). At least 60 dendrites per group from at least 5 mice per genotype were counted. For spine morphology analysis, we analyzed between 2000 and 3000 spines (500 spines per mouse from 5 mice per group). Each spine was categorized as having or not having a neck. Spines were defined as stubby if they did not contain a visible neck. Spines with necks were separated into thin and mushroom spines based on head width. Filopodia, defined as protrusions 1.5 μm in length without a head, were excluded from the analysis of spine subtypes. Spines with heads less than the average width were categorized as thin, and those with heads greater than the average width were categorized as mushroom as previously described (Giralt et al., 2017). From these measures, the percentage of the various spine types was obtained. Picture acquisition and subsequent analysis were performed independently by two investigators blind to genotypes and results were then pooled. Overall differences between the results were minor.
Tissue fixation and immunofluorescence.
Animals were deeply anesthetized with pentobarbital (60 mg/kg) and intracardially perfused with a 4% (w/v) PFA solution in 0.12 m sodium phosphate, pH 7.2. Brains were removed and postfixed overnight in the same solution, cryoprotected with 300 g/L sucrose in 20 mm sodium phosphate, pH 7.5, 150 mm NaCl (PBS) with 0.2 g/L sodium azide, and frozen in dry ice-cooled isopentane. Serial coronal sections (30 μm) obtained with a cryostat were processed for immunohistochemistry as free-floating sections. They were washed three times in PBS, permeabilized 15 min by shaking at room temperature with PBS containing (v/v) 0.3% Triton X-100 and 3% normal goat serum (Pierce Biotechnology). After three washes, brain sections were incubated overnight by shaking at 4°C with antibodies for anti-PSD-95 (1:500, Millipore catalog #MAB1596, RRID:AB_2092365), anti-synaptophysin (1:500, Synaptic Systems catalog #101 011, RRID:AB_887824), or anti-phosphor-TrkBY816 (1:350, Abcam catalog #ab75173, RRID:AB_1281172) in PBS with 0.2 g/L sodium azide. After incubation with primary antibody, sections were washed three times and then placed 2 h on a shaking incubator at room temperature with the subtype-specific fluorescent secondary AlexaFluor-488 anti-rabbit (1:250, Thermo Fisher Scientific catalog #A32731, RRID:AB_2633280) or anti-mouse 555 (1:250, Thermo Fisher Scientific catalog #A32727, RRID:AB_2633276). No signal was detected in control sections incubated in the absence of the primary antibody.
Confocal imaging and analysis.
Dorsal hippocampus in fixed tissue and fixed primary cultures were imaged using a Leica Microsystems Confocal SP5-II at the Institut du Fer à Moulin Cell and Tissue Imaging facility, with a 40× or 63× numerical aperture lens with 5× digital zoom and standard (1 Airy disc) pinhole (1 AU) and frame averaging (3 frames per z step) were held constant throughout the study. Confocal z stacks were taken every 0.2 μm for in vitro experiments and every 2 μm for in vivo experiments, and at 1024 × 1024 pixel resolution. The in vitro Sholl analysis, the dendritic spine counting, the in vivo analysis of PSD95- and synaptophysin-positive clusters, and the colocalization of double-labeled PSD-95/phospho-TrkBY816-positive clusters were analyzed with the freeware ImageJ (ImageJ, RRID:SCR_003070). Briefly, for in vivo imaging analysis, for each mouse, at least 3 slices of 30 μm containing dorsal hippocampal tissue were analyzed. Up to 3 representative images, from CA1-stratum radiatum layer, were obtained from each slice. For the in vitro analysis, in the Sholl experiment, we evaluated 45–60 neurons, all of them MAP2-positive from 3 different cultures. To estimate the density of dendritic spines, 31–41 dendrites from MAP2-positive neurons (1 or 2 dendrites/neuron) from 3 different cultures were counted.
Electronic microscopy.
Mice were transcardially perfused with a solution containing 4% PFA and 0.1% glutaraldehyde made up of 0.1 m PB, pH 7.4. Brains were then immersed in the same fixative for 12 h at 4°C. Tissue blocks containing the hippocampus were dissected and washed in 0.1 m PB, cryoprotected in 100 and 200 g/L sucrose in 0.1 m PB, and freeze-thawed in isopentane and liquid nitrogen. Samples were postfixed in 2.5% glutaraldehyde made up of 0.1 m phosphate buffer for 20 min, washed and treated with 2% osmium tetroxide in PB for 20 min. They were dehydrated in a series of ethanol and flat embedded in epoxy resin (EPON 812 Polysciences). After polymerization, blocks from the CA1 region were cut at 70 nm thickness using an ultramicrotome (Ultracut E Leica Microsystems). Sections were cut with a diamond knife, picked up on formvar-coated 200 mesh nickel grids. For etching resin and remove osmium, sections were treated with saturated aqueous sodium periodate (NaIO4). They were then observed with a CM-100 electron microscope (Philips) at the Institut du Fer à Moulin Cell and Tissue Imaging facility. Digital images were obtained with a CCD camera (Gatan Orius). To test method specificity of the immunostaining procedure, the primary antibody was omitted. In ultrathin sections, the density of synaptic vesicles was calculated by counting the number of vesicles within a defined presynaptic area. The area of postsynaptic densities was also evaluated. All these calculations were performed by using the ImageJ.
Behavioral tests.
To analyze mouse anxiety, we used the elevated plus maze paradigm. Briefly, the plus maze was made of plastic and consisted of two opposing 30 × 8 cm open arms, and two opposing 30 × 8 cm arms enclosed by 15-cm-high walls. The maze was raised 50 cm from the floor and lit by dim light. Each mouse was placed in the central square of the raised plus maze, facing an open arm, and its behavior was scored for 5 min. At the end of each trial, any defecation was removed, and the apparatus was wiped with 30% ethanol. We recorded the time spent in the open arms, which normally correlates with low levels of anxiety. Animals were tracked and recorded with SMART junior software (Panlab).
To check spontaneous locomotor activity, we used the open field. Briefly, the apparatus consisted of a white square arena measuring 40 × 40 × 40 cm in length, width, and height, respectively. Dim light intensity was 60 lux throughout the arena. Animals were placed in the arena center and allowed to explore freely for 30 min. Spontaneous locomotor activity was measured. At the end of each trial, any defecation was removed, and the apparatus was wiped with 30% ethanol. Animals were tracked and recorded with SMART junior software (Panlab).
The novelty-suppressed feeding (NSF) measured the mice aversion to eat in a novel environment. This test assesses stress-induced anxiety by measuring the latency of an animal to approach and eat a familiar food in an aversive environment. For this task, mice were food-restricted for a period of 24 h. After food restriction, the animals were placed in a large, brightly lit, open field. A piece of white filter paper was placed in the center of the arena with a small piece of rodent chow. The time to approach and eat a pellet of food located in the center of the arena was measured and used to evaluate anxiety-like behavior.
The forced swimming test was used to evaluate behavioral despair. Animals were subjected to a 6 min trial during which they were forced to swim in an acrylic glass cylinder (35 cm of height × 20 cm of diameter) filled with water, and from which they could not escape. The time that the test animal spent in the cylinder without making any movements beyond those required to keep its head above water was measured.
The spontaneous alternation performance was tested using a symmetrical Y-maze. Each mouse was placed in the center of the Y-maze and could explore freely through the maze during an 8 min session. The sequence and total number of arms entered were recorded. Arm entry was complete when the hindpaws of the mouse had been completely placed in the arm. Percentage alternation is the number of triads containing entries into all three arms divided by the maximum possible alternations (the total number of arms entered − 2) × 100. As the reentry into the same arm was not counted for analysis, the chance performance level in this task was 50% in the choice between the arm mice visited more recently (nonalternation) and the other arm visited less recently (alternation).
The novel object location memory task evaluates spatial memory and is based on the ability of mice to recognize when a familiar object has been relocated. Exploration took place in an open-top arena with quadrangular form (45 × 45 cm). The light intensity was 40 lux throughout the arena. Mice were first habituated to the arena in the absence of objects (1 d, 30 min). Some distal cues were placed throughout the procedure. On the third day during the acquisition phase, mice could explore two duplicate objects (A1 and A2), which were placed close to the far corners of the arena for 10 min. After a delay of 24 h, one object was placed in the diagonally opposite corner. Thus, both objects in the phase were equally familiar, but one was in a new location. The position of the new object was counterbalanced between mice. Animals were tracked and recorded with SMART Junior software (Panlab).
For the passive avoidance (light-dark) paradigm, we conducted the experiments in a 2-compartment box, where 1 compartment was dimly lit (20 lux) and preferable to a rodent and the other compartment was brightly lit (200 lux); both chambers were connected by a door (5 cm × 5 cm). During training, mice were placed into the aversive brightly lit compartment; and upon the entry into the preferred dimly lit compartment (with all 4 paws inside the dark chamber), mice were exposed to a mild foot shock (2 s foot shock, 1 mA intensity). The latency of mice to enter into the dark chamber was recorded. Twenty seconds after receiving the foot shock, mice were returned to the home cage until testing. After 24 h (long-term memory), animals were tested for retention. In the retention test, mice were returned to the brightly lit compartment again, and the latency to enter the shock paired compartment (dark chamber) was measured (retention or recall latency). Ten minutes was used as a time cutoff in the retention test. The animals that learned the task would avoid the location previously paired with the aversive stimulus and showed a greater latency to enter it.
In vivo electrophysiological recordings.
Animals were anesthetized with 0.8%–1.5% isoflurane (Astra Zeneca) delivered via a special mask (Cibertec). Once anesthetized, animals were implanted with bipolar stimulating electrodes aimed at the right Schaffer collateral-commissural pathway of the dorsal hippocampus (2 mm lateral and 1.5 mm posterior to bregma; depth from brain surface, 1.0–1.5 mm) (Paxinos and Franklin, 2013) and with a recording electrode aimed at the ipsilateral stratum radiatum underneath the CA1 area (1.2 mm lateral and 2.2 mm posterior to bregma; depth from brain surface, 1.0–1.5 mm) (Paxinos and Franklin, 2013). Stimulating and recording electrodes were made of 50 μm, Teflon-coated tungsten wire (Advent Research Materials). The final location of the recording electrode in the CA1 area was determined following the field potential depth profile evoked by paired (40 ms interval) pulses presented to the ipsilateral Schaffer collateral pathway (Gruart et al., 2006). Two bare silver wires were affixed to the skull as ground. Electrodes were connected to a 6-pin socket (RS-Amidata) that was latterly fixed with dental cement to the cranial bone. After surgery, each animal was kept in an independent cage with free access to food and water for the rest of the experiment.
Recording sessions were started 1 week after surgery. Electrophysiological recordings were performed using Grass P511 differential amplifiers with a bandwidth of 0.1–10 kHz (Grass-Telefactor). Electrical stimulation was provided by a CS-220 stimulator across CS-220 isolation units (Cibertec). For input/output curves, monosynaptic fEPSPs were evoked in the CA1 area by single (100 μs, square, and negative-positive) pulses applied to Schaffer collaterals. These pulses were presented at increasing intensities ranging from 20 to 400 μA, in steps of 20 μA. To avoid interactions with the preceding stimuli, an interval of 30 s was allowed between each pair of pulses (Madroñal et al., 2007).
For the characterization of the paired-pulse facilitation at the CA3-CA1 synapse, we used the same type of pulses indicated above, but presented in pairs at increasing interpulse intervals (10, 20, 40, 100, 200, and 500 ms). For each animal, the stimulus intensity was set at 30%–40% of the intensity necessary for evoking a maximum fEPSP response (Gureviciene et al., 2004; Gruart et al., 2006). Intervals between pairs of pulses were set at ∼30 s, to avoid unwanted interactions evoked by presynaptic or postsynaptic mechanisms.
LTP experiments were also performed in alert behaving animals. fEPSPs were evoked at the CA3-CA1 synapse for 15 min before LTP induction. For this, single pulses were presented at a rate of 3 per min. The stimulus intensity was set at 30%–40% of the intensity necessary for evoking a maximum fEPSP response, namely, well below the threshold for evoking a population spike (Gruart et al., 2006; Madroñal et al., 2007). For LTP induction, each animal was subjected to a high-frequency stimulation (HFS) protocol consisting of five 200 Hz, 100 ms trains of pulses at a rate of 1 per second. This protocol was presented six times, at intervals of 1 min. The HFS protocol used here (combining low intensity values and a total of 600 electric shocks) allowed us to evoke LTP lasting >2–3 d, without the appearance of abnormal spikes in EEG recordings and/or overt epileptic seizures (Madroñal et al., 2009). Following the HFS session, animals were stimulated again with single pulses applied for 60 min at the same rate of 3 per min. Additional 30 min recordings of fEPSPs were repeated for 3 d following the HFS session.
Experimental design and statistical analysis.
All data are expressed as mean ± SEM. Statistical analysis was performed using the unpaired two-sided Student's t test (95% confidence), one-way ANOVA with the Tukey's as post hoc tests, or two-way ANOVA with the Bonferroni's post hoc test as appropriate and indicated in the figure legends. Values of p < 0.05 were considered statistically significant.
All experiments in this study were blinded and randomized. All mice bred for the experiments were used for preplanned experiments and randomized to experimental groups. Visibly sick animals were excluded before data collection and analysis. Data were collected, processed, and analyzed randomly. The experimental design and handling of mice were identical across experiments. Littermates were used as controls with multiple litters (3–5) examined per experiments. All mice were bred in the Institut du Fer à Moulin Animal Facility.
Results
Astrocytes BDNF production regulates cultured hippocampal neurons morphology and dendritic spine density
Although BDNF is mainly produced in neurons, it is also localized in astrocytes (Saha et al., 2006; Giralt et al., 2010). We verified that this was the case in our experimental conditions using RT-PCR to detect BDNF transcripts (3745 ± 1125% increase, p = 0.0468) (Fig. 1A). However, the precise role of BDNF from astrocytes in neuronal function is still unclear. We evaluated whether BDNF derived from astrocytes was physiologically relevant for neuronal dendrite morphology. We cultured primary astrocytes from mouse pups lacking the BDNF gene (BDNF−/− pups) or WT littermates as a control (BDNF+/+ pups). After astrocyte 100% confluence, we seeded primary WT neurons at E17 on the top of the astrocyte monolayer (Fig. 1B). Then, some astrocyte-neuron cocultures were fixed at 6 DIV to evaluate the morphological characteristics of the imaged neurons stained for MAP2 by using the Sholl analysis (Fig. 1C). The results indicated that the number of intersections in WT neurons grown on BDNF−/− astrocytes were reduced compared with WT neurons on BDNF+/+ astrocytes (Fig. 1D). We fixed other astrocyte-neuron cocultures at 20 DIV, transfected 48 h before fixation with a GFP-expressing construct (see Material and Methods). We thus were able to evaluate spine density in isolated MAP2-positive neurons. We observed that the number of spines in WT neurons cocultured with BDNF−/− astrocytes was decreased compared with neurons cocultured with BDNF+/+ astrocytes (−0.2555 ± 0.046 decrease, p < 0.0001) (Fig. 1E). These results reveal a crucial role of BDNF released from astrocytes in the modulation of neurons, particularly in dendrite and spine development, in coculture conditions. They provide a rationale for the use of astrocytes to overexpress BDNF in brain locations with high levels of pathology in AD.
Figure 1.

BDNF regulates dendrites and spine number in vitro. A, Left, BDNF mRNA levels were determined in WT astrocytes. HEK293 cells were used as negative control (Student's t test, t = 2.496, df = 6; p < 0.05; n = 3–5). Right, Representative agarose gel. Arrow indicates the molecular weight of the BDNF transcript detected at 114 bp. B, The experimental design is depicted illustrating the two in vitro conditions: BDNF+/+ astrocyte monolayer cocultured with WT neurons (i.e., BDNF+/+) and BDNF−/− astrocyte monolayer cocultured with WT neurons (i.e., BDNF−/−). C, Representative images showing the bright-field allowing visualization of the astrocyte monolayer plus some cocultured neurons on the top, hippocampal neurons stained for MAP2 (red), and transfected MAP2-positive neurons transfected with a plasmid coding GFP (green). D, Representative MAP2 images obtained by confocal microscopy from cocultured astrocytes and neurons (top). Scale bar, 40 μm. Bottom, Sholl analysis from MAP2-positive neurons (two-way ANOVA analysis; group effect, F(1,307) = 81.7, p < 0.001). E, Spine density was studied using GFP fluorescence in transfected MAP2-positive neurons (left). Quantification of spine density (right) is shown (Student's t test, t = 5.462, df = 70; p < 0.001). Data are mean ± SEM. D, n = 45 and 60 MAP2-positive neurons/group from 3 different experiments. E, n = 31 and 41 *dendrites/group from 3 different experiments. ***p < 0.001 as compared to BDNF+/+ mice.
Generation and characterization of the 5xFAD mice overexpressing BDNF under the GFAP promoter
To test the potential role of astrocyte-derived BDNF in an AD mouse model, we then took advantage of mice that overexpress BDNF under the control of the GFAP promoter (pGFAP-BDNF mice) (Giralt et al., 2010). In these mice, BDNF production and delivery are increased in neuropathological conditions accompanied by astrogliosis (Giralt et al., 2010, 2011). We crossed pGFAP-BDNF mice with the 5xFAD transgenic mouse model of AD to create 5xF:pGB mice. The 5xFAD mice develop severe amyloid pathology and associated astrogliosis at 1.5–2 months of age, before the onset of the cognitive deficits (observed at 4 months of age) (Oakley et al., 2006). A significant decrease in BDNF levels is also observed in 5xFAD mice (Hongpaisan et al., 2011). We predicted that this decrease would be prevented in the 5xF:pGB double-transgenic mice. To test this hypothesis, we analyzed BDNF hippocampal levels in WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice at 8 months of age, when their phenotype is clear. At this age, astrogliosis was obvious throughout the hippocampus, as evidenced by immunofluorescence (Fig. 2A) and Western blot (146 ± 18 increase, p < 0.001) (Fig. 2B,C). As expected, BDNF levels were reduced (−58 ± 17 decrease, p < 0.05) in the hippocampus of 5xFAD mice compared with WT mice (Fig. 2B,D). Interestingly, BDNF levels in the hippocampus of 5xF:pGB mice were undistinguishable from those in WT mice, indicating a full recovery of BDNF protein levels in this group of mice. We next studied whether such recovery correlated with an improvement in the BNDF-TrkB downstream signaling. We performed Western blot analyses to measure total and phosphorylated levels of ERK, Akt and, PLCγ, which are key proteins in the three major signaling pathways downstream of BDNF-TrkB (Gupta et al., 2013). Total levels of ERK, Akt, and PLCγ were similar in all the four groups of mice (Fig. 2E). Phosphorylation levels of ERK threonine/tyirosine 202/204 and PLCγ tyrosine 783 were decreased in the hippocampus of 5xFAD mice (ERKT202/Y204: −69 ± 8.5 decrease, p < 0.001; PLCγY783: −40 ± 17 decrease, p < 0.05), whereas Akt serine 308 phosphorylation was not significantly affected (Fig. 2E,F). In 5xF:pGB mice, phosphorylation of ERK and PLCγ was like that in WT mice. Interestingly, in 5xF:pGB mice, the phosphorylated levels of ERK and PLCγ were similar to those in WT mice. No changes in these phosphoproteins were observed in the WT mice expressing the pGFAP-BDNF transgene (Fig. 2E,F), supporting the specificity of the astrocyte-dependent BDNF expression. These results indicate that the 5xF:pGB mice develop a significant astrogliosis, which, in turn, induces an overproduction of BDNF from astrocytes preventing the loss of BDNF/TrkB function due to the effects of APP695 and PSEN-1 transgenes. Furthermore, normalization of BDNF levels in the 5xF:pGB mice was accompanied by a complete restoration of BDNF-TrkB downstream signaling.
Figure 2.

Validation of the 5xF:pGB model. Crossing 5xFAD mice with pGFAP-BDNF mice resulted in four genotypes, namely, WT, pGFAP-BDNF, 5xFAD, and the double-mutant 5xF:pGB mice. We evaluated these mice at 8 months of age. A, GFAP immunofluorescence microscopy imaging in the dorsal hippocampus of 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Scale bar, 500 μm. B, Immunoblotting for BDNF, GFAP, and tubulin as a loading control in the hippocampus of 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. C, Densitometry quantification of BDNF levels as in B. D, Densitometry quantification of GFAP levels as in B. One-way ANOVA, BDNF genotype effect: F(3,24) = 4.403, p = 0.0103; GFAP genotype effect: F(3,25) = 41.18, p < 0.001. Data were normalized to tubulin for each sample and expressed as a percentage of WT. E, Immunoblotting for pAktser308, pERKT402/Y404, pPLCγY783, ERK, Akt, and PLCγ and tubulin as a loading control in the hippocampus of 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. F, Densitometry quantification of pAktser308, pERKT402/Y404, and pPLCγY783 levels as in E. One-way ANOVA, pERKT402/Y404 genotype effect: F(3,28) = 29.14, p < 0.001; pAktser308 genotype effect: F(3,26) = 0.5666, p < 0.646; pPLCγY783 genotype effect: F(3,28) = 5.251, p < 0.0053. Data were normalized to the corresponding total levels of ERK, Akt, and PLCγ for each sample and expressed as a percentage of WT. Data are mean ± SEM. C–F, Tukey's test as a post hoc analysis was used. *p < 0.05; ***p < 0.001; compared with WT mice. $$p < 0.01; $$$ p < 0.001; compared with 5xFAD mice. B, E, Molecular weight markers positions are indicated in kDa. C–F, n = 6–11/group.
Rescue of memory alterations in 5xF:pGB mice
The 5xFAD transgenic mice display a strong phenotype with a relatively fast time course compared with other AD mouse models. At 8 months of age, they display decreased locomotor activity, altered performance in the elevated plus maze, and memory deficits in hippocampal-related tasks, such as the Y-maze and the novel object location (Oakley et al., 2006; Devi and Ohno, 2015; Schneider et al., 2015; Grinan-Ferre et al., 2016). The passive avoidance, which is also affected in other mouse models of AD (Webster et al., 2014), has not been reported in 5xFAD, to our knowledge. Furthermore, AD patients exhibit high comorbidity with depressive symptoms (Webster et al., 2014) and depressive behaviors have been reported at least in one mouse model of AD (Filali et al., 2009).
To evaluate the potential beneficial effects of reactive astrocytes-targeted BDNF expression in 5XFAD mice, we characterized all these behavioral parameters. We first evaluated the basal locomotor activity and anxiety in the open field in WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. All groups of mice displayed a similar initial habituation and comparable levels of locomotor activity (Fig. 3A) and time spent in the center (Fig. 3B). Next, we investigated anxiety by performing the elevated plus maze and the NSF tests. In the elevated plus maze, both 5xFAD mice (18.45 ± 6.6 increase, p < 0.05) and 5xF:pGB mice (23.50 ± 6.92 increase, p < 0.01) spent more time in the open arms than WT and pGFAP-BDNF mice (Fig. 3C), but there were no differences between 5xFAD and 5xF:pGB mice. In the NSF, the mice from all four groups showed similar latencies to feed in the aggressive environment (Fig. 3D). We also tested the behavioral resignation in the four groups of mice by performing the forced swimming test. The amount of time during which the mice were swimming trying to escape from the water was similar in all mice, independently of their genotype (Fig. 3E). These results indicate that 5XFAD mice displayed only minor anxiety or mood-related alterations, with only an apparent decrease in anxiety in the elevated plus maze, which was not modified by BDNF overexpression.
Figure 3.

Characterization of 5xF:pGB mice. A, WT, pGFAP-BDNF, 5xFAD, and mice at 8 months of age were subjected to a comprehensive behavioral characterization. In the open field, (A) locomotor activity and (B) time spent in the center of the arena were monitored for 30 min in WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Locomotor activity, two-way ANOVA, genotype effect: F(3,116) = 1.2654, p = 0.2899; time in center, one-way ANOVA: F(3,80) = 1.009, p = 0.3934. C, In the plus maze, the time spent in the open arms was monitored for 5 min in the four groups of mice. One-way ANOVA: F(3,77) = 6.907, p = 0.005. D, In the NSF test, the time to reach and eat the pellet in the center of the arena was evaluated in the four genotypes. One-way ANOVA: F(3,76) = 1.467, p = 0.2303. E, In the forced swimming test, the immobility time was evaluated during the last 4 min of the 6 min trial in all groups. One-way ANOVA: F(3,80) = 0.9903, p = 0.417. F, In the novel object location test, spatial long-term memory was evaluated 24 h after a training trial as the percentage of time exploring the object placed in a new location (NL) versus the time exploring the object placed in an old location (OL). Two-way ANOVA, object in a new location effect: F(3,150) = 40.80, p < 0.001. Interaction effect: F(3,150) = 7.14, p = 0.002. G, In the Y-maze, the spontaneous alternation was measured (as triads) in an 8 min trial in all four genotypes. One-way ANOVA: F(3,44) = 7.717, p = 0.003. H, In the passive avoidance paradigm, the latency (seconds) to step-through was evaluated in the training trial and in the testing trial 24 h after receiving an electric shock (2 s/1 mA). One-way ANOVA: F(3,126) = 5.384, p = 0.0016. Data are mean ± SEM. A, B, H, n = 18–25 mice/genotype. G, n = 12–14 mice/genotype. Tukey's post hoc test was used for all behavioral tasks. C, G, H, *p < 0.05; **p < 0.01; ***p < 0.001; compared with WT. $p < 0.05; $$p < 0.01; compared with 5xFAD. F, *p < 0.05; ***p < 0.001; compared with percentage of time exploring the object in an old location (OL).
We next studied memory in these mice. First, we evaluated spatial memory in the novel object location test, which is based on the ability of rodents to recognize when a familiar object has been relocated. After habituation to the open field arena, mice were trained in the presence of 2 identical objects (A1 and A2), and spatial memory was assessed 24 h later by displacing one of the two objects. 5xFAD mice exhibited a significantly lower preference for the displaced object compared (1.877 ± 5.64, not significant between old location and new location) with either WT or pGFAP-BDNF mice (Fig. 3F). The spatial memory deficit was prevented in the 5xF:pGB mice (Fig. 3F). Next, mice were tested in the spontaneous alternation Y-maze paradigm that assesses spatial working memory (Lalonde, 2002). The spontaneous alternation behavior relies on the tendency of mice to enter a less recently visited arm compared with the other one. WT and pGFAP-BDNF mice displayed a spontaneous alternation, whereas the arm choice was decreased to 50% (chance levels) in the 5xFAD mice (−12.70 ± 3.27 decrease, p < 0.01) (Fig. 3G). The spontaneous alternation was restored to WT control levels in 5xF:pGB mice. Finally, we examined associative memory in the passive avoidance task, based on the association formed between an electrical foot shock and a spontaneously preferred specific environmental context (darkness vs light). Latency to step-through during the training session was similar between genotypes (Fig. 3H). However, in the testing session, although all genotypes showed a significant increase in the latency to enter the dark compartment 24 h after receiving an electrical shock (wt: 255 ± 39 increase, p < 0.001; pGFAP-BDNF: 192 ± 32 increase, p < 0.001; 5xFAD 78.8 ± 28 increase, p < 0.05; 5xF:pGB: 145 ± 35 increase p < 0.001), this latency was shorter in mutant 5xFAD mice compared with WT (−178 ± 34 decrease, p < 0.001) or pGFAP-BDNF mice (−117.70 ± 30 decrease, p < 0.01) (Fig. 3H). This alteration was absent in double-mutant 5xF:pGB mice (Fig. 3H). The results of the novel object location, spontaneous alternation, and passive avoidance tests demonstrate that targeted increased expression of BDNF prevented several memory deficits in 5xFAD mice.
Normalization of BDNF levels in double-mutant 5xF:pGB mice does not prevent plaque formation and neurogenesis deficits
To test whether the restoration of BDNF levels modulated β-amyloidosis in 8-month-old 5xFAD mice, toluidine blue staining was performed to determine the number of plaques in the hippocampus and PFC of 5xFAD and 5xF:pGB mice (Fig. 4A). The cortical plaque load in 5xF:pGB mice was indistinguishable from that in age-matched 5xFAD control mice. However, in the hippocampus, we detected an increase in the number of plaques in 5xF:pGB mice compared with 5xFAD mice (0.3507 ± 0.0469 increase, p = 0.017) (Fig. 4B). We hypothesized that the size of the plaques could be different between genotypes, indicating different dynamics in β-amyloidosis. To test this possibility, we characterized the plaques in the hippocampus of 5xFAD and 5xF:pGB mice by using electron microscopy (Fig. 4C). Ultrastructural analysis revealed that hippocampal plaques in 5xF:pGB mice were visually more compact but significantly smaller than the plaques in 5xFAD mice (−131 ± 27 decrease, p = 0.015) (Fig. 4C,D), supporting the hypothesis that β-amyloid accumulation in 5xF:pGB mice is different compared with that in 5xFAD mice. Because findings on hippocampal plaque quantification appeared inconsistent with memory improvements, we then evaluated the rate of newborn cells in the dentate gyrus, whose deficits have been previously reported to be associated with cognitive impairments (Moon et al., 2014). We counted the number of Ki67-positive cells in the dentate gyrus of the four groups of mice. A significant decrease in the number of newborn cells was observed in the dentate gyrus of both 5xFAD (−5 ± 1.69 decrease, p < 0.05) and 5xF:pGB (−5.26 ± 1.84 decrease, p < 0.05) mice compared with WT or pGFAP-BDNF mice (Fig. 4E,F). These data indicate that normalizing BDNF levels in 5xF:pGB mice is not enough to prevent the alterations in neurogenesis observed in 5xFAD mice.
Figure 4.

Analysis of gross neuropathology and neurogenesis in WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. A, Amyloid plaques images stained with blue toluidine obtained in a bright-field microscope in the frontal cortex (top left) and in the hippocampus (bottom left) of 5xFAD and 5xF:pGB mice at 8 months of age. Scale bar, 400 μm. B, The plaque density was determined by manual counting in the PFC and in the hippocampus of all four genotypes (right). Student's unpaired t test: PFC, t = 1.923, df = 4, p = 0.1268; hippocampus, t = 7.446, df = 4, p = 0.0017. C, Amyloid plaque images obtained with electronic microscopy (left). Plaques have been colorized for easy visualization for the reader. Black arrows indicate the plaque boundary. D, The hippocampal plaque area was determined by using the ImageJ software (right). Scale bar, 10 μm. Student's unpaired t test: t = 4.743, df = 8, p = 0.0015. E, Ki67-positive cells obtained by confocal microscopy imaging in the dentate gyrus in 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice (left). Scale bar, 300 μm. F, Quantification of the number of the Ki67-positive cells per field (right). Genotype effect: F(3,20) = 5.323, p < 0.0073. Data are mean ± SEM. B, D, Student's unpaired t test. F, One-way ANOVA and Tukey's post hoc test. B, D, *p < 0.05; **p < 0.01; compared with 5xFAD mice. F, *p < 0.01; compared with WT mice. B, n = 4 mice/genotype. D, n = 4 mice/genotype. F, n = 6 or 7 mice/genotype. Ctx, Frontal cortex; Hipp, hippocampus.
Normalization of BDNF levels in double-mutant 5xF:pGB mice restores dendritic spines and synaptic alterations
Because BDNF normalization did not improve neuropathological hallmarks in 5xFAD mice, including astrogliosis (Fig. 2A–C), plaque number (Fig. 4A,B), and neurogenesis deficit (Fig. 4E,F), we assumed that phenotypic improvements observed in 5xF:pGB mice could be related with synaptic changes. To test this hypothesis, we performed Golgi staining in two different brain regions in all 4 genotypes at the age of 8 months to examine dendritic spine density and morphology in pyramidal neurons. We examined the cortical layer V in the PFC and the stratum radiatum of CA1. First, we observed that spine density in secondary dendrites of cortical pyramidal neurons was significantly decreased in 5xFAD mice compared with WT mice (−0.231 ± 0.034 decrease, p < 0.001) (Fig. 5A,B). This decrease was partially rescued in 5xF:pGB mice (−0.089 ± 0.036 decrease, p < 0.05) (Fig. 5A,B). Then, we analyzed spine morphology and the density of the three different spine types (mushroom, thin, and stubby) in the same pyramidal neurons (Fig. 5C). No differences were observed in the density of stubby spines, whereas thin spines were markedly decreased in 5xFAD mice (−0.108 ± 0.021 decrease, p < 0.001) and completely recovered in 5xF:pGB mice (Fig. 5C). In contrast, mushroom spines were decreased equally in 5xFAD (−0.098 ± 0.021 decrease, p < 0.001) and in 5xF:pGB (−0.067 ± 0.023 decrease, p < 0.05) mice compared with WT and pGFAP-BDNF mice. These results revealed that overexpression of astrocytic BDNF in 5xF:pGB mice specifically rescued thin spines but not mushroom spines in the PFC.
Figure 5.
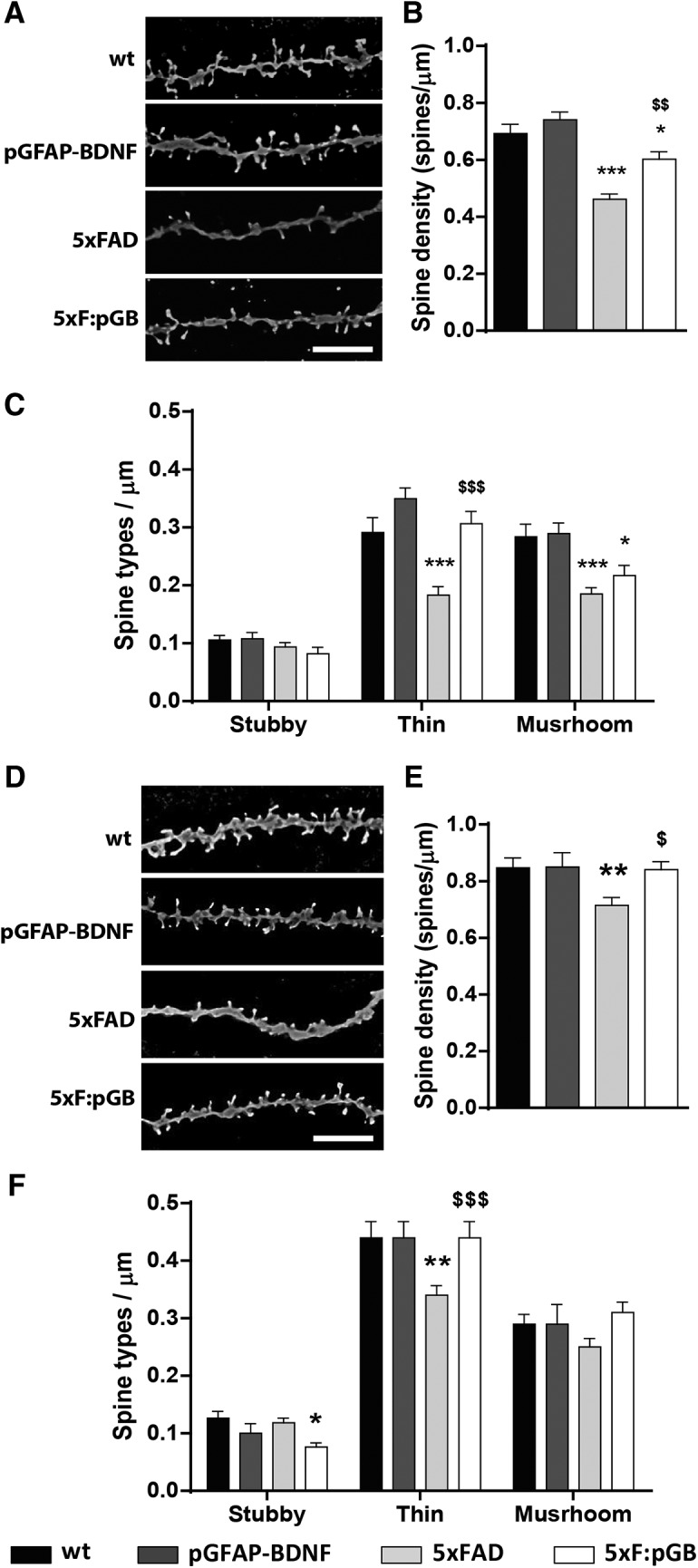
Dendritic spine density and morphology analysis in the PFC and hippocampus of WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. A, Images of apical dendrites from pyramidal neurons of the layer V in PFC stained with Golgi staining obtained in a bright-field microscope in 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Scale bar, 5 μm. B, The dendritic spine density was determined in all four genotypes by using the ImageJ freeware. One-way ANOVA: F(2,100) = 23.09, p < 0.001. C, Density of each type of dendritic spine (stubby, thin, and mushroom) in apical dendrites of pyramidal neurons of the layer V of the PFC in 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Two-way ANOVA, genotype effect: F(3,390) = 21.83, p < 0.001; interaction effect, F(3,390) = 6.036, p < 0.001. D, Images of apical dendrites from pyramidal neurons of the hippocampal CA1 stained with Golgi staining obtained in a bright-field microscope in 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Scale bar, 5 μm. E, The dendritic spine density was determined in all four genotypes by using the ImageJ freeware. One-way ANOVA: F(3,185) = 4.070, p = 0.0079. F, Density of each type of dendritic spine (stubby, thin, and mushroom) in apical dendrites of pyramidal neurons from the hippocampal CA1 in 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Two-way ANOVA, genotype effect: F(3,552) = 3.678, p = 0.0121; interaction effect, F(3,390) = 2.750, p = 0.0122. Data are mean ± SEM. B, E, One-way ANOVA with Tukey's test as a post hoc was used. C, F, Two-way ANOVA and Bonferroni's post hoc test. B, C, E, F, *p < 0.05; **p < 0.01; ***p < 0.001; compared with WT mice. $p < 0.05; $$p < 0.01; $$$p < 0.001; compared with 5xFAD mice. B, C, n = 31–41 dendrites/genotype (from 5 mice/genotype). E, F, n = 35–56 dendrites/genotype (from 5 mice/genotype).
Next, we examined the same parameters in CA1 pyramidal neurons in the four genotypes. Spine density was decreased in 5xFAD mice compared with WT mice (−0.132 ± 0.046 decrease, p < 0.05), and this decrease was rescued in 5xF:pGB mice (Fig. 5D,E). When we analyzed spine morphology, we detected a small decrease in stubby spine density only in 5xF:pGB compared with WT mice (−0.076 ± 0.048 decrease, p < 0.05) whose meaning is uncertain (Fig. 5F). In contrast, a clear decrease in thin spines density was observed in 5xFAD compared with WT mice (−0.100 ± 0.026 decrease, p < 0.01) that was rescued in 5xF:pGB mice (Fig. 5F). Finally, no changes in the density of mushroom spines were detected in any group. These results show that the main alteration in both PFC and hippocampal CA1 region was a decrease in thin spines in 5xFAD mice which was rescued in 5xF:pGB mice.
To explore changes in excitatory synapses likely associated with spine alterations, we evaluated presynaptic and postsynaptic markers. We examined the number of positive clusters for PSD-95 and synaptophysin, which are postsynaptic and presynaptic markers, respectively, previously reported to be decreased in 5xFAD mice, correlating with cognitive decline (Yang et al., 2015; Giralt et al., 2018). We analyzed these two markers in CA1 stratum radiatum of 8-month-old mice. As expected, the number of PSD-95-positive puncta was decreased in 5xFAD compared with WT mice (−754 ± 125 decrease, p < 0.001) (Fig. 6A,B). This decrease was rescued in 5xF:pGB mice (Fig. 6A,B). The number of synaptophysin-positive puncta was also decreased in CA1 stratum radiatum of 5xFAD compared with WT mice (−685 ± 190 decrease, p < 0.05) (Fig. 6C,D). Synaptophysin puncta were recovered in 5xF:pGB mice. Together, these results suggest that the behavioral improvement observed in 5xF:pGB mice compared with 5xFAD mice was accompanied by an improvement in the clusters of presynaptic and postsynaptic markers, indicative of actual synapses. Additionally, we evaluated whether these changes were associated with direct TrkB-mediated signaling in neurons. To do so, we counted the number of double-labeled PSD-95/phospho-TrkBY816-positive puncta in the CA1 stratum radiatum of 8-month-old mice. We observed that 5xFAD mice displayed a reduction of double-labeled PSD-95/phospho-TrkBY816-positive puncta in the stratum radiatum (−65 ± 20 decrease, p < 0.05), whereas such parameter was fully recovered in 5xF:pGB mice (Fig. 6E,F), indicating that 5xF:pGB mice improvements were associated with a TrkB-dependent signaling recovery in neurons. Next, to further examine presynaptic and postsynaptic changes, we performed electronic microscopy experiments and analyzed the PSD area and the number of presynaptic vesicles per synapse, in the CA1 stratum radiatum of the four groups of mice at 8 months. We observed no alteration of the PSD area in 5xFAD mice but an unexpected increase in pGFAP-BDNF controls (0.0059 ± 0.00089 increase, p < 0.001) (Fig. 6G,H). Conversely, the number of presynaptic vesicles per synapse was significantly decreased in 5xFAD mice (−5.088 ± 1.046 decrease, p < 0.001) but was completely rescued in 5xF:pGB mice (Fig. 6G,I). These results on spines, synaptic marker clusters, and synaptic vesicles suggest that the cognitive improvements observed in 5xF:pGB mice resulted from synaptic improvements.
Figure 6.

Hippocampal excitatory synapse characterization of WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. A, Confocal image of PSD-95 immunofluorescence in CA1 stratum radiatum of 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Scale bar, 10 μm. B, Quantification of the number of PSD-95-positive puncta per field. One-way ANOVA: F(3,16) = 14.90, p < 0.001. C, Synaptophysin immunofluorescence. Scale bar, 10 μm. D, Quantification of the number of synaptophysin-positive puncta per field. One-way ANOVA: F(3,16) = 7.194, p < 0.0028. E, Confocal image of a double PSD-95 (green) and phosphoTrkBY816 (red) immunofluorescence in CA1 stratum radiatum of 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Scale bar, 10 μm. F, Quantification of the number of PSD-95/phosphoTrkBY816-positive puncta per field. One-way ANOVA: F(3,26) = 5.303, p < 0.01. G, Electronic microscopy imaging of excitatory synapses in the stratum radiatum of the CA1 in 8-month-old WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. Asterisks indicate the presynaptic component. Black arrows indicate the postsynaptic component. Scale bar, 0.3 μm. H, Quantification of the PSD area. One-way ANOVA: F(3,253) = 17.16, p < 0.001. I, Quantification of the number of presynaptic vesicles per synapse. One-way ANOVA: F(3,201) = 9.18, p < 0.001. One-way ANOVA with Tukey's test as a post hoc was used. Data are mean ± SEM. B, D, F, H, I, *p < 0.05; ***p < 0.001; compared with WT mice. $$p < 0.01; $$$p < 0.001; compared with 5xFAD mice. B, D, F, Two pictures per slice and 3 slices per mouse were taken. B, D, n = 5/genotype. F, n = 7–9/genotype. H, I, 51 ± 3 excitatory synapses from 3 different mice/genotype were evaluated.
In vivo study of electrophysiological properties of the CA3-CA1 synapses in 5xF:pGB mice
To determine whether the rescue of histological alterations following BDNF astrocytic expression was associated with functional improvements, we studied the in vivo electrophysiological properties of hippocampal circuits in the four groups of mice. We recorded input/output curves, paired-pulse facilitation, and LTP evoked at the CA3-CA1 synapses (Fig. 7A–C).
Figure 7.

In vivo hippocampal synaptic plasticity in WT, pGFAP-BDNF, 5xFAD, and 5xF:pGB mice. A, Animals were chronically implanted with bipolar stimulating electrodes in CA3 Schaffer (Sch.) collaterals and with a recording electrode in the ipsilateral CA1 area. Two extra wires were attached to the bone as ground. DG, Dentate gyrus. Right, Representative example of fEPSP (averaged 5 times) evoked at the CA3-CA1 synapse in a WT animal. B, Diagram illustrating the location of stimulating and recording electrodes in the intrinsic hippocampal circuit. C, Representative micrographs illustrating the final location of stimulating and recording electrodes. D, L, M, and V, dorsal, lateral, medial, and ventral, respectively. Calibration bar: 0.2 mm. D, Input/output curves of fEPSPs evoked at the CA3-CA1 synapse by single pulses of increasing intensities (0.02–0.4 in mA) in WT (n = 13), pGFAP-BDNF (n = 15), 5xFAD (n = 11), and 5xF:pGB (n = 13) mice. Data are mean ± SEM. No significant differences (F(3,912) = 0.718; p = 0.546) were observed between groups. E, No significant (F(15,240) = 0.464; p = 0.956) differences in paired-pulse facilitation between the four experimental groups were observed (wt = 15; pGFAP-BDNF = 19; 5xFAD = 13; 5xF:pGB = 14). Data are mean ± SEM slopes of the second fEPSP expressed as the percentage of the first for six (10, 20, 40, 100, 200, and 500) interpulse intervals. F, Graphs represent the time course of LTP evoked in the CA3-CA1 synapse following an HFS session presented to mice included in the four experimental groups (wt, n = 16; pGFAP-BDNF, n = 19; 5xFAD, n = 13; 5xF:pGB, n = 14). The HFS was presented after 15 min of baseline recordings, at the time marked by the dashed line. LTP evolution was followed for 3 d. Top, Representative examples of fEPSPs collected at the times indicated in the bottom graphs from a representative animal of each group. fEPSP slopes are given as a percentage of fEPSP values collected during baseline recordings (100%). Although the four groups presented significant (F(114,1976) = 1.863; p < 0.001) increases (ANOVA, two-tailed) in fEPSP slopes following HFS compared with baseline recordings, the 5xF:pGB group did present a larger LTP than that presented by the other three groups: *5xF:pGB versus WT; +5xF:pGB versus pGFAP-BDNF; &5xF:pGB versus 5xFAD; p ≤ 0.045. Top left, inset, The evolution of paired-pulse facilitation (determined at 40 ms of interpulse interval) for fEPSPs recorded during baseline and following the HFS session is illustrated for the four experimental groups. Dotted/dashed line indicates the 5xFAD group.
We first analyzed the response of CA1 pyramidal neurons to single pulses of increasing intensity (0.02–0.4 mA) presented to the ipsilateral Schaffer collaterals. As illustrated in Figure 7D, the four groups (wt, n = 13; pGFAP-BDNF, n = 15; 5xFAD, n = 11; 5xF:pGB, n = 13) presented similar increases (F(57,912) = 1.460; p < 0.017; two-way repeated-measures ANOVA) in the slope of fEPSP evoked in CA1 by stimuli presented to the ipsilateral Schaffer collaterals. No significant differences (F(3,912) = 0.718; p = 0.546) between the four collected curves were observed. These two relationships were best fitted by sigmoid curves (r ≥ 0.99; p < 0.0001; not illustrated), suggesting a normal basal function of the CA3-CA1 synapses in the four mouse genotypes.
Changes in synaptic strength evoked by a pair of pulses are a form of presynaptic short-term plasticity, mostly related to variations in neurotransmitter release (Zucker and Regehr, 2002). In this regard, paired-pulse stimulation is commonly used as an indirect measurement of changes in the probability of neurotransmitter release at presynaptic terminals of hippocampal synapses (Zucker and Regehr, 2002). In addition, it has been shown that these synaptic properties can be studied in alert behaving mice (Madroñal et al., 2009). The paired-pulse facilitation evoked in the four groups (wt, n = 15; pGFAP-BDNF, n = 19; 5xFAD, n = 13; 5xF:pGB, n = 14) was analyzed presenting a fix stimulus intensity (30%–40% of asymptotic values) with increasing interpulse intervals (see Materials and Methods). As illustrated in Figure 7E, the four groups of mice presented a paired-pulse facilitation at short (10, 20, and 40 ms) interpulse intervals (F(5,240) = 21.290; p < 0.001). However, no significant differences were observed between groups (F(15,240) = 0.464; p = 0.956).
Finally, we performed an LTP study in the four groups of behaving mice. It is generally accepted that CA3-CA1 synapses are involved in the acquisition of different types of associative (classical eye blink conditioning) and nonassociative (object recognition, spatial orientation) learning tasks, and it is usually selected for evoking LTP in behaving mice (Gruart et al., 2006). For baseline values, animals were stimulated at the implanted Schaffer collaterals 3 times/min for 15 min (Fig. 7F). Then, animals (wt, n = 16; pGFAP-BDNF, n = 19; 5xFAD, n = 13; 5xF:pGB, n = 14) were presented with the selected HFS protocol (Fig. 7F, dashed line). Following HFS, the same single stimulus used to generate baseline records was presented at the initial rate (3/min) for another 60 min. Recording sessions were repeated for three additional days (30 min each; Fig. 7F). The four groups of mice presented a significant increase in fEPSP slopes following the HFS session (F(114,1976) = 1.863; p < 0.001). Nevertheless, a point-to-point comparison between fEPSPs evoked in the four groups of mice after the HFS protocol indicated that 5xF:pGB animals presented larger LTP values for the first recording session (p ≤ 0.045) than the three other groups.
Figure 7F (inset) illustrates the evolution of paired-pulse facilitation in the four groups of mice. In accordance with previous descriptions (Madroñal et al., 2009), and as observed here for WT, pGFAP-BDNF, and 5xF:pGB mice, paired-pulse facilitation decreased immediately after HFS and was recovered in the following days. Interestingly, the 5xFAD group did not show any sign of recovery of paired-pulse facilitation for the four recording days after the HFS session, indicating an increase in the response to the first pulse in this stressful situation.
In conclusion, the four groups of mice presented similar basal synaptic properties and short-term plasticity. Interestingly, although the four groups of mice presented a significant LTP with respect to baseline values, LTP was significantly larger in the 5xF:pGB group than in the other three groups during the first recording session following the HFS protocol. The 5xFAD group presented LTP values like those reached by WT and pGFAP-BDNF group; this result could be ascribed to an increase in neurotransmitter release evoked by a first pulse because their paired-pulse facilitation was decreased and remained in this situation across the four recording sessions after HFS.
Discussion
In the present study, we tested an approach to deliver BDNF in a conditional targeted fashion in an AD mouse model. We used a transgenic mouse model recently generated and characterized by our team (Giralt et al., 2010, 2011) to overexpress BDNF under the GFAP promoter. Here we show how this method restores the production and delivery of the neurotrophin in the diseased neural tissue when the pathology starts because of the accompanying astrogliosis. We first observed that endogenous BDNF from astrocytes regulates neurite formation and spine density in neurons in vitro. We then used a pGFAP:BDNF transgene to rescue the BDNF loss in 5xFAD mice and observed a significant improvement of learning and memory deficits in the hybrid transgenic mice. These improvements were associated with a restoration of dendritic spines density and morphology and a recovery of clusters of presynaptic and postsynaptic markers, synaptophysin and PSD-95. We also observed a stronger LTP in CA1 in vivo in behaving 5xF:pGB mice, which could be related with the other changes.
We first demonstrated that BDNF produced by astrocytes can play a relevant role in dendrite maturation. Although the present results do not demonstrate a clear in vivo proof that BDNF is produced and released in physiological conditions, they are in line with the idea that astrocytes may play a role in synapse formation and plasticity (Ronzano, 2017) and could play such a role by regulating the BDNF availability to neurons (Vignoli et al., 2016). They also reinforce the idea of astrocytes as potent regulators of neurotrophin availability to neurons. After showing that astrocytes are a potential source of BDNF with morphological effects on neurons delivery, we generated the double-mutant mice 5xF:pGB. We demonstrated that BDNF levels and the activation of TrkB and its downstream pathways (PLCγ and ERK1/2) were fully recovered in these double-mutant 5xF:pGB mice, which are the most important regulating synaptic plasticity pathways upon TrkB activation (Yoshii and Constantine-Paton, 2010). Indeed, PLCγ downstream signaling is essential for hippocampal associative learning and CA3-CA1 LTP (Minichiello et al., 2002; Gruart et al., 2007).
By comparing these double-mutant mice with 5xFAD mice, we observed that the cognitive alterations related to hippocampal dysfunction were rescued. Regarding the neuropathology, genetically recovered BDNF levels did not change astrogliosis in 5xFAD mice. Normalization of BDNF levels increased the number of plaques but reduced their size. These findings contrast with the belief that reduced plaque number associates with an amelioration of the 5xFAD mice phenotype (Murphy and LeVine, 2010). However, the role of plaques is complex, and previous reports have correlated a hyperaggregation of Aβ with an improvement in AD transgenic mice phenotype due to a lower presence of soluble Aβ, which is the most toxic form of the molecule (Castellani et al., 2009; Cohen et al., 2009; Lublin and Gandy, 2010). We also checked for neurogenesis in the dentate gyrus, which has been described to be altered (Moon et al., 2014), as a possible neural correlate of the observed improvements. However, the number of Ki67-positive cells in the dentate gyrus were equally reduced in 5xFAD and 5xF:pGB compared with WT mice. These results revealed that behavioral improvements in 5xF:pGB mice did not result from plaque number reduction or neurogenesis improvement. We therefore hypothesized that the rescued phenotype in 5xF:pGB mice could be rather due to changes in structural and functional synaptic properties. In 5xFAD mice, as well as in AD patients, there is a prominent dendritic spine pathology and loss of synaptic markers, such as synaptophysin and PSD-95 (Hongpaisan et al., 2011; Crowe and Ellis-Davies, 2014; Yuki et al., 2014; Dorostkar et al., 2015; Yang et al., 2015). We found several synaptic changes associated with the cognitive improvements in 5xF:pGB mice. We hypothesize that BDNF delivered from transgenic astrocytes may have induced these changes by reactivating the neuronal TrkB-PLCγ/ERK1/2 pathway. The PLCγ pathway is important for correct PSD-95 location, whereas BDNF-TrkB-ERK is important for PSD-95 expression (Robinet and Pellerin, 2011; Parsons et al., 2014; Yoshii and Constantine-Paton, 2014). BDNF signaling can also regulate synaptophysin levels (Tartaglia et al., 2001; Zhang et al., 2017) and its function/location (Bamji et al., 2006). BDNF could also be directly responsible for the rescue of dendritic spine density and morphology as described previously (Kellner et al., 2014). However, although here we show that BDNF from astrocytes activates TrkB in neurons, we cannot rule out a collateral effect of BDNF on neighboring astrocytes or even microglia as previously described in the literature (Mizoguchi et al., 2011; Sasi et al., 2017), producing a more widespread effect than the one specifically evaluated in the present work. Overall, our results strongly indicate that 5xF:pGB mice improvements could be due to a local and self-regulated delivery of BDNF in the diseased tissue, likely rescuing the ERK1/2 and PLCγ pathways in neurons, which in turn improved dendritic spine pathology.
A significant body of evidence indicates that enhancement of the BDNF delivery or function via activation of its high-affinity receptor TrkB could be a promising therapeutic approach in AD (Allen and Dawbarn, 2006; Devi and Ohno, 2012, 2015; Zhang et al., 2014; Kaminari et al., 2017). Based on our results, we propose that the use of engineered astrocytes could be an interesting means to achieve this objective. Astrocytes are currently considered as very promising potential targets for AD treatment, including through genetic manipulation to regulate neurotrophin production (Bronzuoli et al., 2017; Gorshkov et al., 2018). Indeed, astrocytes are uniquely positioned to promote the regeneration of damaged nerve cells or protect existing cells from degeneration and dysfunction in the CNS (Anderson et al., 2016; Blanco-Suárez et al., 2017). At least three main strategies have been proposed: astrocyte transplantation or pharmacological correction of their dysfunction or by its genetic manipulation (Gorshkov et al., 2018). We propose that targeting astrocytes to produce and deliver BDNF could be a potential therapeutic tool for the treatment of AD. Much previous and present evidence supports this idea. First, astrocytes have been recently shown to be crucial for hippocampal-related cognitive function and synaptic plasticity (Adamsky et al., 2018). Second, facilitating BDNF expression from astrocytes has previously been shown to be useful in another neurodegenerative disease, Huntington's disease (Giralt et al., 2011; Corbett et al., 2013; Reick et al., 2016). Furthermore, astrocytes per se not only produce, but also buffer, BDNF in case of necessity (Stary et al., 2015) or recycle it for LTP maintenance and memory retention (Vignoli et al., 2016), thus making them perfect candidates to deliver the neurotrophin in a controlled fashion. Additionally, in the field of cell therapy, astrocytes would be good therapeutic tools because they survive for a long time when grafted (Giralt et al., 2010), and they are specifically reactive where Aβ is accumulated and where neuritic dystrophy is localized (Song et al., 2015). Furthermore, astrocytes are easily manipulated, do not proliferate aberrantly as for example engineered cell lines (Hoffman et al., 1993), and they do not suffer of teratogenic potential, in contrast to stem cells (Martínez-Serrano and Björklund, 1996; Pineda et al., 2007; Rubio et al., 1999). Neural cells may be another candidate source for the overexpression of BDNF. However, as an excessive amount of this neurotrophin is deleterious (Kells et al., 2008), its release must be controlled (Martínez-Serrano and Björklund, 1996; Rubio et al., 1999). Transgenic astrocytes would overcome all these drawbacks. A promising therapeutic strategy could be the use of astrocytes from induced pluripotent stem cells (Gorshkov et al., 2018). Once astrocytes have been obtained and characterized and genetically modified to express BDNF under the GFAP promoter, they could be transplanted to promote the survival and appropriate functioning of existing neurons, such as synaptic plasticity processes.
Although 5xFAD mice presented noticeable losses in the expression of PSD-95 and synaptophysin, they did not present any significant alteration in input/output curves, paired-pulse facilitation, or in LTP evoked in vivo. In this regard, it has been already reported that LTP can be preserved longer than other behavioral functions in aging mice (López-Ramos et al., 2012) and that motor exercise can help to recover some behavioral and associative learning abilities, but not synaptic changes and LTP in 3xTg-AD mice (García-Mesa et al., 2011). In addition, age seems to be a critical factor; for example, in 3-month-old mice, it is almost impossible to distinguish differences in learning abilities and LTP strength and duration between WT versus APP, PS1, and APP-PS1 mice (Gruart et al., 2008). For the present experiments, we can assume that baseline measures of transmission (input/output curves and paired-pulse facilitation) and LTP were sustained in 8-month-old 5xFAD mice by an increase in neurotransmitter release (Fig. 7F), as reported by some of us in TgNTRK3 transgenic mice (Sahún et al., 2007). Indeed, the latter study is an excellent example of dissociation between CA3-CA1 synaptic plasticity and associative learning capabilities in genetically manipulated mice.
The present results have some limitations. First, although the 5xFAD mouse model has been shown to recapitulate several hallmarks of AD pathology, they do not show tauopathy or formation of intraneuronal neurofibrillary tangles. From a therapeutic point of view, the interpretation of our results should be taken with caution, and studies evaluating our approach in models of tauopathy are needed. Second, we observed recovered BDNF levels in 5xF:pGB mice with respect to 5xFAD mice at 8 months of age, but we cannot rule out the possibility that earlier beneficial effects could have taken place in our double-mutant mice. It is known that astrocytes synthesize GFAP from the first days of age (Guo et al., 2013). Thereby, putative early increases on BDNF levels in 5xF:pGB mice could also counteract the deleterious effects of the five transgenic mutations at very early stages.
In conclusion, our study supports the idea that the use of engineered astrocytes to deliver BDNF under the control of the GFAP promoter in AD has a strong potential. It may correspond to the increase of a physiological function; and the delivery is conditionally and locally administered, creating then a customized neurotrophin-based treatment.
Footnotes
A. Giralt is a Ramón y Cajal fellow (RYC-2016-19466). The J.-A.G. laboratory was supported in part by Institut National de la Santé et de la Recherche Médicale, Sorbonne Université, Fondation pour la Recherche Médicale, and Bio-Psy (Biology for Psychiatry) laboratory of excellence. The cell and tissue imaging facility at the IFM benefited from support of Espoir en Tête/Fondation pour la Recherche sur le Cerveau, Région Ile-de-France, and Institut National de la Santé et de la Recherche Médicale. A. Gruart and J.M.D.-G. were supported by MINECO Grant BFU2017-82375-R and Tatiana Pérez de Guzman el Bueno Foundation. J.A. was supported by Grants SAF2017-88076 (Ministerio de Ciencia, Innovación y Universidades) and Marató TV3 Foundation. S.G. was supported by Grant SAF2015-67474-R (Ministerio de Ciencia, Innovación y Universidades). We thank María Sánchez Enciso and José M. González Martín for help in animal handling and care during in vivo experiments.
The authors declare no competing financial interests.
References
- Acosta C, Anderson HD, Anderson CM (2017) Astrocyte dysfunction in Alzheimer disease. J Neurosci Res 95:2430–2447. 10.1002/jnr.24075 [DOI] [PubMed] [Google Scholar]
- Adamsky A, Kol A, Kreisel T, Doron A, Ozeri-Engelhard N, Melcer T, Refaeli R, Horn H, Regev L, Groysman M, London M, Goshen I (2018) Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell 174:59–71.e14 10.1016/j.cell.2018.05.002 [DOI] [PubMed] [Google Scholar]
- Allen SJ, Dawbarn D (2006) Clinical relevance of the neurotrophins and their receptors. Clin Sci (Lond) 110:175–191. 10.1042/CS20050161 [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association (2012) 2012 Alzheimer's disease facts and figures. Alzheimers Dement 8:131–168. 10.1016/j.jalz.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532:195–200. 10.1038/nature17623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Rico B, Kimes N, Reichardt LF (2006) BDNF mobilizes synaptic vesicles and enhances synapse formation by disrupting cadherin-beta-catenin interactions. J Cell Biol 174:289–299. 10.1083/jcb.200601087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Suárez E, Caldwell AL, Allen NJ (2017) Role of astrocyte-synapse interactions in CNS disorders. J Physiol 595:1903–1916. 10.1113/JP270988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronzuoli MR, Facchinetti R, Steardo L, Scuderi C (2017) Astrocyte: an innovative approach for Alzheimer's disease therapy. Curr Pharm Des 23:4979–4989. 10.2174/1381612823666170710163411 [DOI] [PubMed] [Google Scholar]
- Carpenter MK, Winkler C, Fricker R, Emerich DF, Wong SC, Greco C, Chen EY, Chu Y, Kordower JH, Messing A, Björklund A, Hammang JP (1997) Generation and transplantation of EGF-responsive neural stem cells derived from GFAP-hNGF transgenic mice. Exp Neurol 148:187–204. 10.1006/exnr.1997.6657 [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, Zhu X, Perry G, Smith MA (2009) Reexamining Alzheimer's disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. J Alzheimers Dis 18:447–452. 10.3233/JAD-2009-1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A (2009) Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139:1157–1169. 10.1016/j.cell.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett GT, Roy A, Pahan K (2013) Sodium phenylbutyrate enhances astrocytic neurotrophin synthesis via protein kinase C (PKC)-mediated activation of cAMP-response element-binding protein (CREB): implications for Alzheimer disease therapy. J Biol Chem 288:8299–8312. 10.1074/jbc.M112.426536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SE, Ellis-Davies GC (2014) Spine pruning in 5xFAD mice starts on basal dendrites of layer 5 pyramidal neurons. Brain Struct Funct 219:571–580. 10.1007/s00429-013-0518-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M (2012) 7,8-Dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology 37:434–444. 10.1038/npp.2011.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M (2015) TrkB reduction exacerbates Alzheimer's disease-like signaling aberrations and memory deficits without affecting beta-amyloidosis in 5XFAD mice. Transl Psychiatry 5:e562. 10.1038/tp.2015.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar MM, Zou C, Blazquez-Llorca L, Herms J (2015) Analyzing dendritic spine pathology in Alzheimer's disease: problems and opportunities. Acta Neuropathol 130:1–19. 10.1007/s00401-015-1449-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R (1994) Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368:147–150. 10.1038/368147a0 [DOI] [PubMed] [Google Scholar]
- Filali M, Lalonde R, Rivest S (2009) Cognitive and non-cognitive behaviors in an APPswe/PS1 bigenic model of Alzheimer's disease. Genes Brain Behav 8:143–148. 10.1111/j.1601-183X.2008.00453.x [DOI] [PubMed] [Google Scholar]
- Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF (2014) Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci 34:8186–8196. 10.1523/JNEUROSCI.4267-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mesa Y, López-Ramos JC, Giménez-Llort L, Revilla S, Guerra R, Gruart A, Laferla FM, Cristòfol R, Delgado-García JM, Sanfeliu C (2011) Physical exercise protects against Alzheimer's disease in 3xTg-AD mice. J Alzheimers Dis 24:421–454. 10.3233/JAD-2011-101635 [DOI] [PubMed] [Google Scholar]
- Giralt A, Rodrigo T, Martín ED, Gonzalez JR, Milà M, Ceña V, Dierssen M, Canals JM, Alberch J (2009) Brain-derived neurotrophic factor modulates the severity of cognitive alterations induced by mutant huntingtin: involvement of phospholipaseCgamma activity and glutamate receptor expression. Neuroscience 158:1234–1250. 10.1016/j.neuroscience.2008.11.024 [DOI] [PubMed] [Google Scholar]
- Giralt A, Friedman HC, Caneda-Ferrón B, Urbán N, Moreno E, Rubio N, Blanco J, Peterson A, Canals JM, Alberch J (2010) BDNF regulation under GFAP promoter provides engineered astrocytes as a new approach for long-term protection in Huntington's disease. Gene Ther 17:1294–1308. 10.1038/gt.2010.71 [DOI] [PubMed] [Google Scholar]
- Giralt A, Carretón O, Lao-Peregrin C, Martín ED, Alberch J (2011) Conditional BDNF release under pathological conditions improves Huntington's disease pathology by delaying neuronal dysfunction. Mol Neurodegener 6:71. 10.1186/1750-1326-6-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Brito V, Chevy Q, Simonnet C, Otsu Y, Cifuentes-Díaz C, de Pins B, Coura R, Alberch J, Ginés S, Poncer JC, Girault JA (2017) Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington's disease model. Nat Commun 8:15592. 10.1038/ncomms15592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, de Pins B, Cifuentes-Díaz C, López-Molina L, Farah AT, Tible M, Deramecourt V, Arold ST, Ginés S, Hugon J, Girault JA (2018) PTK2B/Pyk2 overexpression improves a mouse model of Alzheimer's disease. Exp Neurol 307:62–73. 10.1016/j.expneurol.2018.05.020 [DOI] [PubMed] [Google Scholar]
- Gorshkov K, Aguisanda F, Thorne N, Zheng W (2018) Astrocytes as targets for drug discovery. Drug Discov Today 23:673–680. 10.1016/j.drudis.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinan-Ferre C, Sarroca S, Ivanova A, Puigoriol-Illamola D, Aguado F, Camins A, Sanfeliu C, Pallas M (2016) Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY) 8:664–684. 10.18632/aging.100906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, Muñoz MD, Delgado-García JM (2006) Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci 26:1077–1087. 10.1523/JNEUROSCI.2834-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, Sciarretta C, Valenzuela-Harrington M, Delgado-García JM, Minichiello L (2007) Mutation at the TrkB PLCγ-docking site affects hippocampal LTP and associative learning in conscious mice. Learn Mem 14:54–62. 10.1101/lm.428307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, López-Ramos JC, Muñoz MD, Delgado-García JM (2008) Aged wild-type and APP, PS1, and APP + PS1 mice present similar deficits in associative learning and synaptic plasticity independent of amyloid load. Neurobiol Dis 30:439–450. 10.1016/j.nbd.2008.03.001 [DOI] [PubMed] [Google Scholar]
- Guo Z, Wang X, Xiao J, Wang Y, Lu H, Teng J, Wang W (2013) Early postnatal GFAP-expressing cells produce multilineage progeny in cerebrum and astrocytes in cerebellum of adult mice. Brain Res 1532:14–20. 10.1016/j.brainres.2013.08.003 [DOI] [PubMed] [Google Scholar]
- Gupta VK, You Y, Gupta VB, Klistorner A, Graham SL (2013) TrkB receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. Int J Mol Sci 14:10122–10142. 10.3390/ijms140510122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureviciene I, Ikonen S, Gurevicius K, Sarkaki A, van Groen T, Pussinen R, Ylinen A, Tanila H (2004) Normal induction but accelerated decay of LTP in APP + PS1 transgenic mice. Neurobiol Dis 15:188–195. 10.1016/j.nbd.2003.11.011 [DOI] [PubMed] [Google Scholar]
- Hoffman D, Breakefield XO, Short MP, Aebischer P (1993) Transplantation of a polymer-encapsulated cell line genetically engineered to release NGF. Exp Neurol 122:100–106. 10.1006/exnr.1993.1111 [DOI] [PubMed] [Google Scholar]
- Hong Y, Zhao T, Li XJ, Li S (2016) Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J Neurosci 36:8790–8801. 10.1523/JNEUROSCI.0168-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongpaisan J, Sun MK, Alkon DL (2011) PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer's disease transgenic mice. J Neurosci 31:630–643. 10.1523/JNEUROSCI.5209-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminari A, Giannakas N, Tzinia A, Tsilibary EC (2017) Overexpression of matrix metalloproteinase-9 (MMP-9) rescues insulin-mediated impairment in the 5XFAD model of Alzheimer's disease. Sci Rep 7:683. 10.1038/s41598-017-00794-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner Y, Gödecke N, Dierkes T, Thieme N, Zagrebelsky M, Korte M (2014) The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front Synaptic Neurosci 6:5. 10.3389/fnsyn.2014.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kells AP, Henry RA, Connor B (2008) AAV-BDNF mediated attenuation of quinolinic acid-induced neuropathology and motor function impairment. Gene Ther 15:966–977. 10.1038/gt.2008.23 [DOI] [PubMed] [Google Scholar]
- Lalonde R. (2002) The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev 26:91–104. 10.1016/S0149-7634(01)00041-0 [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z, Martínez-Serrano A (2004) Stem cell therapy for human neurodegenerative disorders: how to make it work. Nat Med 10 [Suppl]:S42–S50. 10.1038/nm1064 [DOI] [PubMed] [Google Scholar]
- López-Ramos JC, Jurado-Parras MT, Sanfeliu C, Acuña-Castroviejo D, Delgado-García JM (2012) Learning capabilities and CA1-prefrontal synaptic plasticity in a mice model of accelerated senescence. Neurobiol Aging 33:627–726. 10.1016/j.neurobiolaging.2011.04.005 [DOI] [PubMed] [Google Scholar]
- Lublin AL, Gandy S (2010) Amyloid-beta oligomers: possible roles as key neurotoxins in Alzheimer's disease. Mt Sinai J Med 77:43–49. 10.1002/msj.20160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM (2008) The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol 585:2–13. 10.1016/j.ejphar.2007.11.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madroñal N, Delgado-García JM, Gruart A (2007) Differential effects of long-term potentiation evoked at the CA3 CA1 synapse before, during, and after the acquisition of classical eyeblink conditioning in behaving mice. J Neurosci 27:12139–12146. 10.1523/JNEUROSCI.3397-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madroñal N, Gruart A, Delgado-García JM (2009) Differing presynaptic contributions to LTP and associative learning in behaving mice. Front Behav Neurosci 3:7. 10.3389/neuro.08.007.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Serrano A, Björklund A (1996) Protection of the neostriatum against excitotoxic damage by neurotrophin-producing, genetically modified neural stem cells. J Neurosci 16:4604–4616. 10.1523/JNEUROSCI.16-15-04604.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M (2002) Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36:121–137. 10.1016/S0896-6273(02)00942-X [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Monji A, Kato TA, Horikawa H, Seki Y, Kasai M, Kanba S, Yamada S (2011) Possible role of BDNF-induced microglial intracellular Ca(2+) elevation in the pathophysiology of neuropsychiatric disorders. Mini Rev Med Chem 11:575–581. 10.2174/138955711795906932 [DOI] [PubMed] [Google Scholar]
- Moon M, Cha MY, Mook-Jung I (2014) Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J Alzheimers Dis 41:233–241. 10.3233/JAD-132417 [DOI] [PubMed] [Google Scholar]
- Murphy MP, LeVine H 3rd (2010) Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis 19:311–323. 10.3233/JAD-2010-1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140. 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, Kang R, Buren C, Dau A, Southwell AL, Doty CN, Sanders SS, Hayden MR, Raymond LA (2014) Bidirectional control of postsynaptic density-95 (PSD-95) clustering by huntingtin. J Biol Chem 289:3518–3528. 10.1074/jbc.M113.513945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB (2013) The mouse brain in stereotaxic coordinates. San Diego: Academic. [Google Scholar]
- Pineda JR, Rubio N, Akerud P, Urbán N, Badimon L, Arenas E, Alberch J, Blanco J, Canals JM (2007) Neuroprotection by GDNF-secreting stem cells in a Huntington's disease model: optical neuroimage tracking of brain-grafted cells. Gene Ther 14:118–128. 10.1038/sj.gt.3302847 [DOI] [PubMed] [Google Scholar]
- Reick C, Ellrichmann G, Tsai T, Lee DH, Wiese S, Gold R, Saft C, Linker RA (2016) Expression of brain-derived neurotrophic factor in astrocytes: beneficial effects of glatiramer acetate in the R6/2 and YAC128 mouse models of Huntington's disease. Exp Neurol 285:12–23. 10.1016/j.expneurol.2016.08.012 [DOI] [PubMed] [Google Scholar]
- Robinet C, Pellerin L (2011) Brain-derived neurotrophic factor enhances the hippocampal expression of key postsynaptic proteins in vivo including the monocarboxylate transporter MCT2. Neuroscience 192:155–163. 10.1016/j.neuroscience.2011.06.059 [DOI] [PubMed] [Google Scholar]
- Ronzano R. (2017) Astrocytes and microglia: active players in synaptic plasticity. Med Sci (Paris) 33:1071–1078. 10.1051/medsci/20173312014 [DOI] [PubMed] [Google Scholar]
- Rubio FJ, Kokaia Z, del Arco A, García-Simón MI, Snyder EY, Lindvall O, Satrústegui J, Martínez-Serrano A (1999) BDNF gene transfer to the mammalian brain using CNS-derived neural precursors. Gene Ther 6:1851–1866. 10.1038/sj.gt.3301028 [DOI] [PubMed] [Google Scholar]
- Saha RN, Liu X, Pahan K (2006) Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol 1:212–222. 10.1007/s11481-006-9020-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahún I, Delgado-García JM, Amador-Arjona A, Giralt A, Alberch J, Dierssen M, Gruart A (2007) Dissociation between CA3-CA1 synaptic plasticity and associative learning in TgNTRK3 transgenic mice. J Neurosci 27:2253–2260. 10.1523/JNEUROSCI.4055-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasi M, Vignoli B, Canossa M, Blum R (2017) Neurobiology of local and intercellular BDNF signaling. Pflugers Arch 469:593–610. 10.1007/s00424-017-1964-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Baldauf K, Wetzel W, Reymann KG (2015) Effects of methylphenidate on the behavior of male 5xFAD mice. Pharmacol Biochem Behav 128:68–77. 10.1016/j.pbb.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Song MS, Learman CR, Ahn KC, Baker GB, Kippe J, Field EM, Dunbar GL (2015) In vitro validation of effects of BDNF-expressing mesenchymal stem cells on neurodegeneration in primary cultured neurons of APP/PS1 mice. Neuroscience 307:37–50. 10.1016/j.neuroscience.2015.08.011 [DOI] [PubMed] [Google Scholar]
- Stary CM, Sun X, Giffard RG (2015) Astrocytes protect against isoflurane neurotoxicity by buffering pro-brain-derived neurotrophic factor. Anesthesiology 123:810–819. 10.1097/ALN.0000000000000824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B (2001) Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem 276:37585–37593. 10.1074/jbc.M101683200 [DOI] [PubMed] [Google Scholar]
- Toda T, Gage FH (2018) Review: adult neurogenesis contributes to hippocampal plasticity. Cell Tissue Res 373:693–709. 10.1007/s00441-017-2735-4 [DOI] [PubMed] [Google Scholar]
- Vignoli B, Battistini G, Melani R, Blum R, Santi S, Berardi N, Canossa M (2016) Peri-synaptic glia recycles brain-derived neurotrophic factor for LTP stabilization and memory retention. Neuron 92:873–887. 10.1016/j.neuron.2016.09.031 [DOI] [PubMed] [Google Scholar]
- von Bohlen Und Halbach O, von Bohlen Und Halbach V (2018) BDNF effects on dendritic spine morphology and hippocampal function. Cell Tissue Res 373:729–741. 10.1007/s00441-017-2782-x [DOI] [PubMed] [Google Scholar]
- Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao GY, Zheng K, Lu B, Xu B (2012) BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J Neurosci 32:14318–14330. 10.1523/JNEUROSCI.0709-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ (2014) Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet 5:88. 10.3389/fgene.2014.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Bayer TA (2012) Intraneuronal abeta accumulation and neurodegeneration: lessons from transgenic models. Life Sci 91:1148–1152. 10.1016/j.lfs.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Yang EJ, Ahn S, Ryu J, Choi MS, Choi S, Chong YH, Hyun JW, Chang MJ, Kim HS (2015) Phloroglucinol attenuates the cognitive deficits of the 5XFAD mouse model of Alzheimer's disease. PLoS One 10:e0135686. 10.1371/journal.pone.0135686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M (2010) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70:304–322. 10.1002/dneu.20765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M (2014) Postsynaptic localization of PSD-95 is regulated by all three pathways downstream of TrkB signaling. Front Synaptic Neurosci 6:6. 10.3389/fnsyn.2014.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto Y, Lin Q, Collier TJ, Frim DM, Breakefield XO, Bohn MC (1995) Astrocytes retrovirally transduced with BDNF elicit behavioral improvement in a rat model of Parkinson's disease. Brain Res 691:25–36. 10.1016/0006-8993(95)00596-I [DOI] [PubMed] [Google Scholar]
- Yuki D, Sugiura Y, Zaima N, Akatsu H, Takei S, Yao I, Maesako M, Kinoshita A, Yamamoto T, Kon R, Sugiyama K, Setou M (2014) DHA-PC and PSD-95 decrease after loss of synaptophysin and before neuronal loss in patients with Alzheimer's disease. Sci Rep 4:7130. 10.1038/srep07130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Qiu B, Wang J, Yao Y, Wang C, Liu J (2017) Effects of BDNF-transfected BMSCs on neural functional recovery and synaptophysin expression in rats with cerebral infarction. Mol Neurobiol 54:3813–3824. 10.1007/s12035-016-9946-7 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K (2014) 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology 39:638–650. 10.1038/npp.2013.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]