

Abstract
First described as essential to the phagocytic activity of leukocytes, Nox2-derived ROS have emerged as mediators of a range of cellular and tissue responses across species from salubrious to deleterious consequences. Knowledge of their role in inflammation is limited, however. We postulated that TNFα-induced endothelial reactive oxygen species (ROS) generation and pro-inflammatory signaling would be ameliorated by targeting Nox2. Herein, we in silico-modelled two first-in-class Nox2 inhibitors developed in our laboratory, explored their cellular mechanism of action and tested their efficacy in in vitro and mouse in vivo models of inflammation. Our data show that these inhibitors (CPP11G and CPP11H) disrupted canonical Nox2 organizing factor, p47phox, translocation to Nox2 in the plasma membrane; and abolished ROS production, markedly attenuated stress-responsive MAPK signaling and downstream AP-1 and NFκB nuclear translocation in human cells. Consequently, cell adhesion molecule expression and monocyte adherence were significantly inhibited by both inhibitors. In vivo, TNFα-induced ROS and inflammation were ameliorated by targeted Nox2 inhibition, which, in turn, improved hind-limb blood flow. These studies identify a proximal role for Nox2 in propagated inflammatory signaling and support therapeutic value of Nox2 inhibitors in inflammatory disease.
Keywords: NADPH oxidase, Reactive oxygen species, Small-molecule inhibitor, Vascular inflammation, Endothelial dysfunction
Graphical abstract
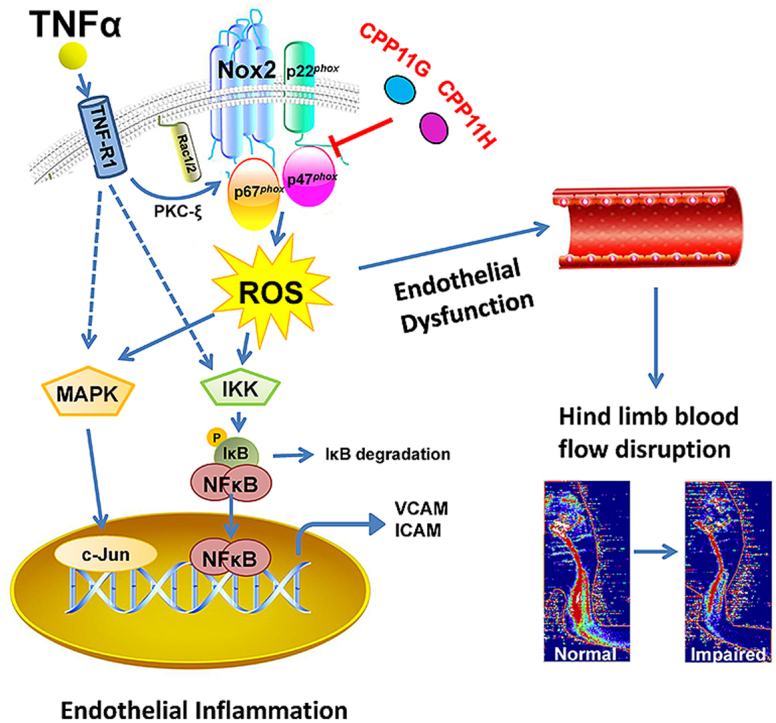
1. Introduction
NADPH oxidases (Noxs) comprise a family of seven isozymes whose only known function is the generation of reactive oxygen species (ROS) [[1], [2], [3], [4], [5], [6], [7]]. Besides their evermore-accepted role as signaling molecules, under pathophysiological conditions, excessive ROS production contributes to cell and tissue damage by oxidative stress and, as a consequence, to the development of myriad diseases. Noxs play a key role in stress and attendant disease, including varied cardiovascular maladies [[7], [8], [9], [10], [11]], cancer [12,13], and neurodegenerative disorders [14,15]. These encompass many of the major leading causes of death in the United States [16] and around the world. As the demand for selective Nox inhibitors for pharmacological and therapeutic purposes has become increasingly clear, drug development both from pharmaceutical industry and academia has intensified in recent years. Still, the need for well-characterized, isoform-specific inhibitors has not been met [7,17,18].
Our laboratory identified two bridged tetrahydroisoquinolines as small molecule inhibitors with high selectivity for the Nox2 isozyme [19]. In that study, the inhibitory potential of these molecules was preliminarily characterized using COS- and HEK- heterologous systems that expressed components of replete Nox isozyme systems and a non-Nox ROS generator. While these inhibitors demonstrated high Nox2 selectivity in a system of Nox subunit overexpression, their inhibitory profile in vivo and in parenchymal primary cells in vitro were neither interrogated nor their mechanism of action in cellular processes explored.
In this study, we beta-tested novel Nox2 inhibitors CPP11G and CPP11H [previously referred to as compounds 11g and 11h [19]] in in vitro and in vivo human and mouse inflammatory models, respectively, and examined the impact of Nox2 on a wide array of oxidant-sensitive signaling pathways leading to adhesion molecule expression, monocyte adhesion, vascular dysfunction and disrupted hind-limb blood flow. Herein, we model, in silico, the interaction of these compounds with 2 crucial binding domains within the canonical Nox2 (comprised of Nox2, p22phox, p47phox, p67phox, and Rac1/2) and report for the first time that they effectively block Nox2 activity by interfering with p47phox cytosol to membrane translocation and its interaction with cytochrome b558 heavy chain (CYBB). Furthermore, our findings reveal that TNFα-stimulated Nox2-derived ROS, MAPK activation, AP-1 and NFκB phosphorylation and nuclear translocation, adhesion molecule expression, endothelial dysfunction and impaired peripheral blood flow are all ameliorated by the Nox2 inhibitors. In aggregate, the findings hold meaningful promise for these agents and their derivatives as viable therapeutics in the treatment of myriad Nox2-actuated disorders.
2. Results
2.1. In silico analysis of Nox2 inhibitors modeling disruption of critical Nox2 oxidase subunit p47phox:p22phox interactions, and in vitro proof of blockade of p47phox binding to cytochrome b558 heavy chain (Nox2)
In silico interaction analysis of CPP11G and CPP11H with atomic structures of key components of the Nox2 oxidase was exploited (Fig. 1A). Modeling predicted that they interfere with a well-defined p47phox-SH3 “super groove” domain key interaction with a proline-rich (PRR) domain on the C-terminus of p22phox that is established as pivotal for the docking of p47phox to the membrane-spanning cytochrome b558 light chain p22phox [6,[20], [21], [22], [23]]. This informed the hypothesis that these compounds would interfere with cytosolic p47phox interactions with membrane-integrated cytochrome components (either p22phox or Nox2) upon Nox activation. To interrogate this potential interference, COS-phox cells overexpressing the Nox2 isozyme subunits were classically stimulated with a phorbol ester. As shown in Fig. 1B, levels of p47phox at the plasma membrane were significantly elevated by phorbol 12-myristate 13-acetate (PMA, 5 μmol/l), a known protein kinase C activator which causes phosphorylation of p47phox and triggers its membrane translocation. This response was inhibited in the presence of CPP11G and CPP11H (Fig. 1B). To further investigate their effects on the interaction between p47phox and Nox2, co-immunoprecipitation was applied on a preparation of COS-phox cells, where PMA (5 μmol/l) stimulation increased Nox2 to p47phox binding (Fig. 1C). Consistent with the results on p47phox membrane translocation, both agents diminished PMA-induced interaction between Nox2 and p47phox. In summary, CPP11G and CPP11H suppressed p47phox cytosol-to-membrane translocation, thus blocking the Nox2 isozyme complex formation and by extension ROS-producing activity.
Fig. 1.

Nox2 inhibitors disrupt p47phox membrane translocation and interaction with the Nox2-p22phox complex. (A) In silico 3D-modeling based on PDB 1OV3 showing (i) both Nox2 inhibitors CPP11G & H interfering with p22phox C-terminal PRR domain interaction with the p47phox Src homology 3 (SH3) domain (super groove) (overlay); (ii) the p22phox C-terminus alone interacting with the p47phox SH3 super groove, (iii, iv) Selective Nox2 inhibitors binding individually to the p47phox super groove. (B) Western blotting of membrane fraction showing the effects of CPP11G (20 μmol/l) and CPP11H (20 μmol/l) on PMA (5 μmol/l)-induced p47phox membrane translocation in COS-phox cells overexpressing Nox2 subunits. The band density of p47phox protein in the membrane fraction was normalized to the density of Nox2 band detected in the same sample, n = 5 (*p < 0.05 vs. control, Ctrl, ##p < 0.01 vs. PMA.) (C) Western blotting showing the effects of CPP11G (20 μmol/l) and CPP11H (20 μmol/l) on PMA (5 μmol/l)-stimulated interaction between p47phox and Nox2 in COS-phox cells overexpressing Nox2 subunits. p47phox was immunoprecipitated (IP) from COS-phox homogenates and detected by Western blot for the presence of Nox2. The densities of Nox2 protein binds were normalized to the levels of p47phox detected in the same samples, n = 6 (*p < 0.05 vs. Ctrl, ###p < 0.001 vs. PMA).
2.2. CPP11G and CPP11H inhibit Nox2-derived ROS production, MAPK/SAPK signaling and AP-1 activation in response to TNFα
To evaluate the effectiveness of these Nox2 inhibitors under more physiological conditions, primary human aortic endothelial cells (HAECs) were employed to establish an acute inflammatory model of TNFα stimulation (10 ng/ml) in which the agents were optimally beta-tested. As expected, HAECs challenged with TNFα yielded increased amounts of O2•‒, which was measured using the fluorescent hydropropidine (HPr+), a cell-impermeant probe for detecting extracellular O2•‒. Importantly, both CPP11G and CPP11H (10 μmol/l) obliterated this response (Fig. 2A). Next, as O2•‒ is rapidly dismuted to H2O2, levels of H2O2 stimulated by TNFα and the effects of the Nox2 inhibitors were assessed using a recently optimized coumarin-7-boronic acid (CBA) as well as the Amplex Red assay on whole cells and cell homogenates, respectively (Figs. 2B and C). Consistent with the O2•‒ results, TNFα-stimulated elevation in H2O2 was effectively blocked by both compounds (each at 10 μmol/l). Moreover, excessive generation of O2•‒ gives rise to peroxynitrite (ONOO−), a highly reactive and often destructive ROS that oxidizes lipoproteins and nitrosates tyrosine residues in many proteins, forming 3-nitrotyrosine (3-NT) [24], a well-established “footprint” biomarker of oxidative damage. Indeed, HAECs treated with TNFα (10 ng/ml) for 24 h exhibited increased (∼2 fold vs. control, Ctrl) 3-NT formation as evidenced by stronger immunofluorescent staining. In comparison, HAECs that were pretreated with CPP11G (10 μmol/l) or CPP11H (10 μmol/l) revealed significantly lower levels of 3-NT than cells with TNFα alone (Fig. 2D), indicating by a fourth independent method that these inhibitors are capable of limiting Nox2-derived ROS in HAECs.
Fig. 2.

Nox2 inhibitors attenuate TNFα-induced reactive oxygen species (ROS) production in human aortic endothelial cells (HAECs). (A) Effects of CPP11G (10 μmol/l) and CPP11H (10 μmol/l) on TNFα (10 ng/ml)-stimulated extracellular superoxide () measured by hydropropidine (HPr+) Assay, n = 4. (**p < 0.01 vs. Ctrl, ####p < 0.0001 vs. TNFα). (B) Effects of CPP11G (10 μmol/l) and CPP11H (10 μmol/l) on TNFα (10 ng/ml)-stimulated hydrogen peroxide (H2O2) production measured by coumarin-7-boronic acid (CBA) Assay, n = 6. (*p < 0.05 vs. Ctrl, ##p < 0.01 vs. TNFα, ####p < 0.0001 vs. TNFα). (C) Effects of CPP11G (10 μmol/l) and CPP11H (10 μmol/l) on TNFα (10 ng/ml, 10 min)-stimulated H2O2 production examined by Amplex Red Assay, n = 6–7. (*p < 0.05 vs. Ctrl, ##p < 0.01 vs. TNFα, ###p < 0.001 vs. TNFα). (D) Immunofluorescence microscopy (20× magnification) detection of 3-nitrotyrosine (red) formation upon 24h TNFα (10 ng/ml) stimulation in the presence or absence of CPP11G or CPP11H (10 μmol/l). Nuclei were labelled with DAPI (blue). Fluorescence intensity was quantified from 3 images/group, n = 3 independent experiments. (****p < 0.0001 vs. Ctrl, ##p < 0.01 vs. TNFα, ####p < 0.0001 vs. TNFα).
Next, we examined their effects on TNFα-induced Nox2-mediated mitogen-activated protein kinase (MAPK) signaling. TNFα (10 ng/ml) triggered a rapid and transient phosphorylation of p38 MAPK, which peaked at 10 min (6.9 ± 0.6 fold vs. Ctrl) and dropped below baseline by 30 min (Supplementary Fig. 1A). Similarly, SAPK/JNK phosphorylation in response to TNFα (10 ng/ml) was also transiently time-dependent, which achieved a maximum but transitory stimulation at 20 min with TNFα stimulation (10.3 ± 0.6 fold vs. Ctrl) (Supplementary Fig. 1B). Unexpectedly, no stimulations in ERK1/2 (p44/42) MAPK phosphorylation were observed within the first hour or even after prolonged TNFα (10 ng/ml) treatment (up to 24 h) (Supplementary Figs. 1C and D), suggesting that TNFα and subsequent Nox-derived ROS differentially regulate downstream MAPK signaling. As TNFα-induced intracellular events appeared rapid and transient, Nox2 inhibitor effects were evaluated at the time of peak activation. Phosphorylation of p38 by TNFα (10 ng/ml, 10 min) was markedly suppressed by CPP11G and CPP11H by ≥ 50% in a concentration-dependent manner (Fig. 3A). Consistently, activation of SAPK/JNK was also robustly and concentration-dependently inhibited by CPP11G and CPP11H (Fig. 3B).
Fig. 3.

Nox2 inhibitors attenuate TNFα-induced mitogen-activated protein kinase (MAPK) activation in HAECs. (A) Representative Western blots (left) and cumulative data (right) showing concentration dependent inhibition by CPP11G and CPP11H of TNFα (10 ng/ml, 10 min)-stimulated p38 MAPK activation. The densities of phospho-p38 were normalized to the levels of total p38 detected in the same samples, n = 6–8. (****p < 0.0001 vs. unstimulated, #p < 0.05 vs. TNFα, ##p < 0.01 vs. TNFα, ####p < 0.0001 vs. TNFα). (B) Representative Western blots (left) and cumulative data (right) showing concentration dependent inhibition by CPP11G and CPP11H of TNFα (10 ng/ml, 20 min)-stimulated JNK phosphorylation. The densities of phospho-JNK were normalized to the levels of total JNK detected in the same samples, n = 7. (****p < 0.0001 vs. unstimulated, #p < 0.05 vs. TNFα, ##p < 0.01 vs. TNFα). (C) Representative Western blots (left) and cumulative data (right) showing concentration dependent attenuation by CPP11G and CPP11H of TNFα (10 ng/ml, 1h)-stimulated cJun phosphorylation. The densities of phospho-cJun were normalized to the levels of total cJun detected in the same samples, n = 6. (****p < 0.0001 vs. unstimulated, #p < 0.05 vs. TNFα, ####p < 0.0001 vs. TNFα). (D) Representative Western blots (left) and cumulative data (right) showing the effects of CPP11G (10 μmol/l) and CPP11H (10 μmol/l) on TNFα (10 ng/mL, 1h)-modified phospho-cJun levels on the nuclear fraction. The densities of phospho-cJun were normalized to the levels of histone detected in the same samples, n = 3. (***p < 0.001 vs. control, ###p < 0.001 vs. TNFα).
Activated MAPK, particularly SAPK/JNK, phosphorylates the transactivation domain of c-Jun, a key component of the transcription factor AP-1, thereby facilitating AP-1 to initiate inflammatory gene expression [25,26]. Thus, we next investigated the effects of our small molecule Nox2 inhibitors on c-Jun phosphorylation and nuclear translocation. With 1 h of TNFα (10 ng/ml) challenge, the ratio of active (phosphorylated) c-Jun to total c-Jun in whole cell lysates was enhanced (2.7 ± 0.2 fold vs. Ctrl). Both CPP11G and CPP11H significantly inhibited c-Jun phosphorylation concentration-dependently (Fig. 3C). Moreover, we were able to show that c-Jun translocated to the nucleus, where it positively regulates genes involved in endothelial activation [25,26]. Consistently, we found that TNFα (10 ng/ml, 1 h) substantially elevated the levels of phospho-c-Jun in the nuclear fraction of endothelial cells (~5-fold) wherein CPP11H exhibited a notably higher efficacy in diminishing c-Jun nuclear translocation by 60.7 ± 8.0% (p-cJun/Histone); CPP11G displayed a tendency toward an inhibition (not significant; Fig. 3D). Collectively, these results indicate that both CPP11G and CPP11H are broadly effective in suppressing TNFα-induced Nox2-mediated MAPK/SAPK and AP-1 signaling.
2.3. Nox2 inhibitors suppress IKK-NFκB pathway induced by TNFα
In addition to the MAPK and AP-1 signaling cascades, TNFα is implicated in Nox2-dependent NFĸB signaling to endothelial inflammation [27,28]. Hence, we characterized the effects of CPP11G and CPP11H on the NFκB signaling cascade. Mirroring the mode of MAPK activation, IκB kinases α & ß (IKKα/ß) transiently reached a ∼20-fold increase in phosphorylation (represented as p-IKK) within 10 min of TNFα stimulation (Supplementary Fig. 2A). Diminution in endothelial cell IκB (NFĸB attenuator) levels was also observed with TNFα (10 ng/mL) treatment in whole cell lysates. This effect, in keeping with IκB ubiquitination and degradation, is widely reported [[29], [30], [31]]. Within 20 min, IκB levels reached its nadir, and appeared to partially recover at later time points (Supplementary Fig. 2B). These results illustrate that TNFα triggers the canonical NFκB pathway in HAECs in a time-dependent fashion. More importantly, administration of the novel small molecule Nox2 inhibitors revealed that TNFα (10 ng/ml, 10 min)-activated IKKα/ß was concentration-dependently attenuated by both CPP11G and CPP11H (Fig. 4A). Characteristically, TNFα-ablated IκB levels were partially but significantly rescued by both inhibitors at a concentration as low as 5 μmol/l (Fig. 4B). Consistent with these observations, NFκB p65 activation/phosphorylation in whole cell lysates was highly elevated by TNFα (10 ng/ml, 1 h) (4.3 ± 0.2 fold vs. Ctrl). Both agents suppressed this response (Fig. 4C). Upon interrogation of NFκB p65 nuclear translocation using fluorescence microscopy (Fig. 4D), TNFα (10 ng/ml, 1 h) discernibly increased the levels of p65 in HAEC nuclei, which were abolished by CPP11H (10 μmol/l), and were trending towards an inhibition by CPP11G (10 μmol/l). To further validate these results, we compared the phospho-p65 levels in the nuclear extract isolated from endothelial cells. Phospho-p65 in the nuclear fraction (p-p65 vs. histone) was substantially potentiated by TNFα (10 ng/ml) (29.1 ± 2.2 fold vs. control). In accordance with our observation on immunofluorescent images, pretreating cells with CPP11H but not CPP11G significantly attenuated phospho-p65 in the nuclear extract (Supplemental Fig. 2E). These findings demonstrate that while both agents elicit inhibitory actions on the proximal NFκB pathway, CPP11H uniquely displays efficacy with respect to NFκB nuclear translocation.
Fig. 4.

Nox2 inhibitors attenuate TNFα-induced HAEC NFκB pathway activation. (A) Representative Western blots (left) and cumulative data (right) showing concentration-dependent inhibition by CPP11G and CPP11H of TNFα (10 ng/ml, 10 min)-stimulated IKK phosphorylation. The phospho-IKK bands were quantified and normalized to the levels of total IKKβ detected in the same samples, n = 8. (****p < 0.0001 vs. unstimulated, ####p < 0.0001 vs. TNFα). (B) Representative Western blots (left) and cumulative data (right) showing concentration-dependent partial reversal of TNFα (10 ng/ml, 20 min)-diminished IκB levels by CPP11G and CPP11H. The IκB bands were quantified and normalized to the levels of ß-actin detected in the same samples, n = 6. (****p < 0.0001 vs. unstimulated, #p < 0.05 vs. TNFα, ####p < 0.0001 vs. TNFα). (C) Representative Western blots (left) and cumulative data (right) showing concentration-dependent inhibition of TNFα (10 ng/ml, 1 h)-stimulated NFκB p65 phosphorylation by CPP11G and CPP11H. The phospho-bands were quantified and normalized to the total levels of p65 detected in the same samples, n = 6. (****p < 0.0001 vs. unstimulated, #p < 0.05 vs. TNFα, ###p < 0.001 vs. TNFα, ####p < 0.0001 vs. TNFα). (D) Immunofluorescence microscopy (60 X magnification) detection of NFκB (red) nuclear translocation induced by TNFα (10 ng/ml, 1 h) and inhibited by CPP11G (10 μmol/l) and CPP11H (10 μmol/l). Nuclei were labelled with DAPI (blue) to visualize cells. The fluorescence intensity was quantified from 3 images/group, n = 3 independent experiments. (*p < 0.05 vs. Ctrl, #p < 0.05 vs. TNFα).
2.4. CPP11G and CPP11H ameliorate endothelial inflammation and monocyte adhesion in response to TNFα
Upon activation of NFκB and AP-1 signaling cascade, inflammatory endothelial activation response ensues with upregulated expression of adhesion molecules [26], including vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1) and E-selectin, all of which facilitate the recruitment of neutrophils and monocytes, and predispose for vascular inflammation and atherosclerosis [32]. We first investigated the ability of the tetrahydroisoquinolines [19] to suppress TNFα-adhesion molecule upregulation at the protein level. Treating HAECs with TNFα for 24 h (a time point selected based on Supplementary Figs. 2C and D illustrating that VCAM-1 and ICAM-1 are stimulated in a time-dependent manner with maximal and robust inductions appearing at 24 h respectively) promoted the levels of VCAM-1 and ICAM-1 by ∼50-fold and ∼20-fold, respectively, which were sharply repressed by CPP11G and CPP11H in a concentration-dependent fashion. Remarkably, VCAM-1 and ICAM-1 expression were virtually abolished at the highest concentrations (10 μmol/l) (Figs. 5A and B). To confirm these observations, a monocyte recruitment assay was performed comparing calcein-labelled monocyte adherence to HAECs in response to different treatments. As is shown in Fig. 5C, TNFα (10 ng/ml, 24 h) stimulation markedly promoted monocyte adhesion to HAECs as indicated by a 2.2 ± 0.2-fold increase in fluorescence intensity of monocytes compared to control. Pretreating HAECs with either inhibitor concentration-dependently diminished the TNFα-induced response, with 10 μmol/l CPP11G or CPP11H by 30.9 ± 1.3% or 29.1 ± 2.4%, respectively. These findings point to the novel Nox2 inhibitors as efficacious agents in ameliorating endothelial activation and chemoattraction of monocytes.
Fig. 5.

Nox2 inhibitors attenuate TNFα-induced endothelial cell activation. (A, B) Representative Western blots (left) and cumulative data (right) showing concentration-dependent inhibition of TNFα (10 ng/ml, 24 h)-stimulated VCAM1 (A) and ICAM (B) expression by CPP11G and CPP11H. VCAM-1 and ICAM-1 bands were quantified and normalized to the levels of ß-actin detected in the same samples, n = 4. (****p < 0.0001 vs. unstimulated, ##p < 0.01 vs. TNFα, ####p < 0.0001 vs. TNFα). (C) Representative data (10 X magnification) (left) of monocyte adhesion assay showing inhibition by CPP11G (10 μmol/l) and CPP11H (10 μmol/l) on TNFα (10 ng/ml, 24 h)-induced monocyte attachment. Summary data (right) showing the concentration-dependent effects of CPP11G and CPP11H on TNFα (10 ng/mL, 24 h)-induced monocyte adhesion. ****p < 0.0001 vs. unstimulated, #p < 0.05 vs. TNFα).
2.5. Nox2 inhibition decreases ROS production and ameliorates endothelial dysfunction in response to TNFα in vivo
To test the clinical relevance of these agents in preventing ROS production and preserving vascular functionality, we employed an acute inflammatory mouse model as per cytokine challenge. Mice were initially injected with either vehicle (30% DMSO in PBS), CPP11G (15 mg/kg) or CPP11H (15 mg/kg) by i.v. bolus. Subsequently, TNFα (20 μg/kg) was administered by i.v. bolus in mice in the TNFα plus/minus CPP11G and CPP11H groups. To evaluate the effectiveness of CPP11G and CPP11H in ROS (hydrogen peroxide, H2O2) production in vivo, we employed Amplex Red to examine levels of H2O2 production in aortic homogenates. We observed that aortas taken from mice treated with TNFα alone generated 69.1 ± 12.5% higher H2O2 in comparison to the control group. Importantly, H2O2 levels were significantly lower in mice pretreated with CPP11H and trending toward a reduction by CPP11G (Fig. 6A). These observations suggest that the inhibitors are efficacious in limiting Nox2-derived ROS generation in vivo. Clinical significance of these novel Nox2 inhibitors was also exemplified by their beneficial effects on endothelial inflammation. In particular, ICAM-1 protein levels on the aortic intima were augmented considerably by TNFα stimulation (Fig. 6B). DAPI (blue) and α-actin (green) staining show the bulk of tissue staining in the media while merged images depict positive ICAM-1 (red) staining along the luminal lining of the vessel. ICAM-1 expression was visibly reduced in mice treated with both inhibitors and this decrease reached significance in the CPP11H group (Fig. 6B; lower magnification images shown in Supplementary Fig. 3).
Fig. 6.

Nox2 inhibitors ameliorate endothelial activation and improve vascular function. (A) Effects of in vivo administered CPP11G (15 mg/kg) and CPP11H (15 mg/kg) on TNFα (10 μg/kg)-stimulated H2O2 in homogenates from aortas taken ex vivo as measured by Amplex Red Assay. n = 6–8 mice/group. (*p < 0.05 vs. Veh, ##p < 0.01 vs. TNFα). (B) Representative images (20 X magnification) (left) and cumulative data (right) showing the immunofluorescence microscopy detection of ICAM-1 (red) on mouse aortic cross sections. ICAM-1 expression is enhanced by TNFα (10 μg/kg) and suppressed by CPP11H (15 mg/kg). Nuclei were labelled with DAPI (blue) to visualize cells. Smooth muscle cells were labelled with α-actin (green). Fluorescence intensity was quantified from 4 to 5 animals/group. (**p < 0.01 vs. Veh, #p < 0.05 vs. TNFα). (C) Laser Doppler flowmetry showing the in vivo effects CPP11G (15 mg/kg) and CPP11H (15 mg/kg) on TNFα (10 μg/kg)-induced reductions in mouse hind-limb blood flow, n = 4–5 mice/group. (*p < 0.05 vs Veh, #p < 0.05 vs. TNFα, ##p < 0.01 vs. TNFα). (D) Two-pin myography showing the effects CPP11G (20 μmol/l, left) and CPP11H (20 μmol/l, right) on TNFα (100 ng/ml)-induced endothelial dysfunction as indicated by diminished acetylcholine-induced vasorelaxation in mouse aortas, n = 5 mice/group. (*p < 0.05 vs. Veh, #p < 0.05 vs. TNFα).
To explore effects on vascular function, we evaluated mouse hind-limb blood flow (Fig. 6C). Compared to mice in the control group, TNFα-stimulated mice exhibited a 25.7 ± 7.3% decrease in basal femoral artery blood flow (highest degree of flux shown in red). However, when mice were pre-injected with either agent, femoral artery blood flow was restored to or even slightly above basal levels, indicating that the Nox2 inhibitors are able to ameliorate TNFα-mediated blood flow disturbance in vivo.
Further, effects on endothelial dilatory function were interrogated using ex vivo two-pin myography. Aortic rings were isolated and mounted on two pins, and stimulated in myograph organ chambers with TNFα (100 ng/ml) for 2 h in the presence or absence of inhibitors. Acetylcholine (ACh: 10−8-10−5 mol/l) evoked a characteristic concentration-dependent vasorelaxation which was substantially impaired in aortic rings incubated with TNFα (Fig. 6D). In contrast, pretreating the aortic rings with either CPP11G (20 μmol/l) or CPP11H (20 μmol/l) prevented TNFα-induced endothelial dysfunction (Fig. 6D), pointing to the capability of both compounds to preserve endothelial cell function. Control experiments using sodium nitroprusside (SNP: 10−9.5-10−6 mol/l) as a direct smooth muscle vasodilator displayed no change among the treatment groups (Supplementary Fig. 4). These results demonstrate that neither TNFα nor Nox2 inhibitors alter smooth muscle cell vasomotor tone.
3. Discussion
Herein, we interrogated the capacity of novel Nox2-selective small molecule inhibitors CPP11G and CPP11H to alter endothelial ROS production, vascular inflammatory responses and dilator function. The major findings of the current study are the ability of these inhibitors to 1) disrupt canonical p47phox-p22phox interaction, p47phox membrane translocation and interaction with the membrane-bound Nox2 subunit in the heterologous Nox2 isozyme; 2) abrogate TNFα-stimulated Nox2-derived ROS, MAPK, AP-1 and NFκB pathway activation in human endothelial cells; 3) limit TNFα-stimulated endothelial adhesion molecules and monocyte adherence; 4) prevent in vivo TNFα-elicited ROS production and vascular endothelial inflammatory response; 5) ameliorate ex vivo endothelial dysfunction; and 6) preserve hind-limb blood flow in mice. Collectively, these findings indicate that the novel inhibitors are highly efficient at inhibiting Nox2-mediated signaling cascades and protecting vascular function against inflammatory insults both in human endothelial cells and in an in vivo mouse model of systemic inflammation.
The small molecule Nox2-selective inhibitors described herein were originally identified by high-throughput screening assay of a subset of approximately 600 small molecules from the University of Pittsburgh Chemical Methodologies and Library Development (UPCMLD) library, followed by structure-activity relationship (SAR) studies of newly synthesized molecules containing a common backbone structure [19]. Among these molecules, CPP11G and CPP11H emerged as highly efficacious and selective inhibitors for the Nox2 isozyme [19] exhibiting no inhibition of Nox1, 4, or 5 isozymes or xanthine oxidase, thus demonstrating no direct ROS scavenging properties. By comparison, the contemporaneous existing tools for Nox2 inhibition, including diphenylene iodonium (DPI), apocynin, VAS2870 and ebselen congeners, presented limitations ranging from non-selective Nox inhibition to formidable off-target effects, prognostic of undesired consequences in vivo and a poor Nox2-selective therapeutic [17,18]. Therefore, with high specificity and no observed off-target effects, CPP11G and CPP11H held significant promise as therapeutic agents for Nox2-mediated pathologies. Their actions, however, in parenchymal cells in vitro and in vivo had not been studied.
Aiming to gain insights into the mechanisms underlying their Nox2 suppression, in silico analyses of various Nox2 subunit interactions were performed. Computational modeling predicts that CPP11G and CPP11H interfere with a critical interaction of a proline-rich binding domain within the C-terminus of p22phox which binds to a well-characterized Src-homology 3 (SH3) “super groove” in the p47phox subunit involved in requisite assembly and canonical Nox2 isozyme activation [[20], [21], [22], [23],33] (Fig. 1A). Consistent with this prediction, we found that both agents: 1) abrogated translocation of p47phox from the cytosol to plasma membrane where Nox2 (aka gp91phox) and p22phox are juxtaposed and reside (Fig. 1B); and 2) disrupted the canonical active enzyme-essential p47phox:Nox2 interaction (Fig. 1C). These findings corroborate that by way of impeded p47phox interaction with the membrane-bound cytochrome (CYBB & CYBA), these agents disrupt the assembly of the Nox2 isozyme complex, thus achieving inhibition. A similar mechanism of action has been reported for the previously described peptidic Nox2 inhibitor, Nox2ds-tat, which blocks the interaction between Nox2 and p47phox by binding to the latter and preventing p47phox membrane translocation [34,35]. The most salient perceived drawback of that agent is a limited oral bioavailability as a peptide in its current form. Another small molecule inhibitor, celastrol, appears to disrupt binding of a p22phox proline-rich peptide to the tandem SH3 domain of both p47phox and NoxO1 (homolog for p47phox in the Nox1 system) [36]. However, celastrol is expected to have multiple off-target effects, as it also inhibits cytosolic factor-independent Nox isoforms such as Noxs 4 and 5 [36]. Furthermore, another group of small molecules, namely ebselen and congeners, appear to also interrupt the binding of the SH3 domain of p47phox to the proline-rich domain of p22phox, and prevent p47phox and p67phox translocation to the plasma membrane in neutrophils [37]. However, ebselen and its analogs exhibited poor selectivity for Nox2 over Nox1 with the exception of one congener, JM-77b, which showed relatively higher selectivity, namely a lower IC50 for Nox2 compared to Nox1 and Nox5 with no detectable inhibition of Nox4 [37]. In addition, ebselen and its analogs have been reported to affect numerous targets, including protein kinase C [38], endothelial nitric oxide synthase [39,40], lipoxygenases [41], c-Jun N-terminal kinase [40] and horseradish peroxidase [42]. Therefore, whether or not they directly prevented p47phox and p67phox membrane translocation or indirectly mitigated Nox activation remains undefined.
An important question that has not been addressed in the current study is whether CPP11G and CPP11H could disrupt Nox1 activity in a hybrid system that employs p47phox as its organizing subunit [43]. That is, the crucial isozyme-activating p47phox: p22phox interaction not only sustains Nox2 isozyme activation, but is also operant in a hybrid Nox1 system wherein, in lieu of NoxO1, p47phox is utilized as a cytosolic organizer. The hybrid Nox1 system has been reported to be functional in vascular smooth muscle cells [43,44] and in a variety of disease settings [17,45,46]. With that said, one limitation of this work is that to-date CPP11G/H mechanism of action studies have only been conducted in a cell system that expresses the canonical Nox2 subunits (Nox2, p22phox, p47phox, p67phox, and Rac 1/2). Thus, whether CPP11G and CPP11H inhibit the hybrid Nox1-p47phox interaction warrants further investigation.
A process wherein Nox2-dependent ROS production has been implicated is inflammation [[47], [48], [49]]. A defense response to injury that involves a cascade of cellular signaling responses, inflammation and its mediators initiate endothelial activation and expression of membrane adhesion molecules, all of which are needed for monocyte recruitment and infiltration [50]. However, when ROS production is sustained, it is noted to give rise to chronic inflammation and endothelial dysfunction, and underlie the pathophysiology of many cardiovascular and non-cardiovascular disorders [49]. One of the most widely studied pro-inflammatory cytokines, tumor necrosis factor alpha (TNFα), has been shown to induce endothelial activation by increasing Nox-derived ROS production and by engaging the NFκB pathway to increase expression of inflammatory genes such as those for cytokines and adhesion molecules [47,51,52].
With respect to the current study, several lines of evidence suggested that the Nox2 isozyme is a source of ROS and a mediator of an inflammatory response in endothelial cells. Specifically, TNFα failed to induce oxidant generation in lung vascular endothelial cells from Nox2 null mice [53], a response normally characterized by NFκB activation and inflammation in wild type mice. Furthermore, TNFα-induced O2•− generation in pulmonary microvascular endothelial cells was shown to be inhibited by selective Nox2 inhibitor, Nox2ds-tat, but not by Nox1-selective inhibitor NoxA1ds, concomitant with a suppressed adhesion molecule expression and monocyte adherence [52]. Still, other lines of evidence show that TNFα also induces ROS production from alternate sources including Nox1 [54] and Nox4 [55]. With that said, further testing of the hypothesis with an additional Nox2-selective agent that is delivered acutely and avoids compensatory roles of other sources spurred the current studies. Moreover, the nature of the current agents as small molecules, their selectivity, their potential for facile oral bioavailability, and alterability for enhanced pharmacokinetic and SAR properties, justified their beta-testing in the current study. On these premises, the characterization of CPP11G and CPP11H on inflammatory pathways was conducted in both human endothelial cells in culture and mouse endothelial cell and vascular tissue both in vivo and ex vivo.
We report that CPP11G and CPP11H are highly effective in reducing TNFα-stimulated ROS production in human aortic endothelial cells (HAECs, Fig. 2). We employed HPr+ (a cell-impermeant O2•−-specific probe [56]) to quantify O2•− generation as the initial product of Nox2 isozyme activity. Our data show that both compounds abrogated the HPr+ signal in these cells, in accordance with a highly effective enzymatic blockade. Coumarin-7-boronic acid (CBA [57]) was used for comprehensive detection of H2O2 (the rapidly dismuted metabolite of O2•−) on the whole cell level and similar results were observed. Employing a third assay of direct ROS detection (Amplex Red), we were able to show that in cell homogenates, the rise in H2O2 was abolished by the novel Nox2 inhibitors. These results corroborate by three independent biochemical assays that ROS generation was maximally inhibited. Furthermore, these findings were confirmed by significant blockade of “footprint” oxidation and nitration marker 3-nitrotyrosine (3-NT) in whole cells in culture (Fig. 2). In aggregate, the data are strongly supportive of the ability of CPP11G and CPP11H to ablate Nox2 activity in these cells.
Our data also show that TNFα triggers activation of inflammation-related MAPK-AP1 signaling and the IKK-NFκB pathway in HAECs. For one, we observed that both compounds inhibited p38 and JNK activation albeit to varying degrees. Subsequent phosphorylation/activation of another key player involved in these pro-inflammatory pathways, c-Jun, that in conjunction with c-Fos, comprises the AP-1 transcription factor complex, was effectively blocked by both CPP11G and CPP11H (Fig. 3). In addition to the MAPK-AP1 signaling cascade, TNFα-triggered IKK-NFκB pathway, characterized by IKKβ activation, IκB degradation and NFκB (p65) phosphorylation, was significantly mitigated by CPP11G and CPP11H (Fig. 4), recapitulating a primary role of Nox2-ROS in mediating TNF-induced endothelial inflammation reported by others [28,52]. Intriguingly, our results on the c-Jun and p65 nuclear translocation consistently show that only CPP11H, but not CPP11G, (Fig. 3, Fig. 4 and Supplementary Fig. 2) was able to significantly reduce their levels in the nucleus. The fact that CPP11G and CPP11H are equally effective preventing ROS generation and enzyme assembly but differ in some downstream endpoints could be explained by the distinct time points at which the measurements are taken, thus reflecting a difference in binding affinity/stability between both compounds. In fact, in Fig. 2D, 3-NT footprint assessed at 24 h supports the idea that CPP11H effects endure more strongly at later time points than those of CPP11G. The reason for this disparity, however, remains unclear but could also suggest a differential penetrability of the two compounds to compartments that connect Nox2 with c-Jun or p65.
Further studies in terms of pharmacokinetic and pharmacodynamic properties of both compounds are required to assess the possibility of unexpected off-target effects or effects on Nox-independent functions of p47phox. It is also worth noting that even relatively high concentrations of CPP11G and CPP11H (10 μmol/l) were not able to completely abolish these TNFα-activated signaling cascades. This may be attributed to a need to test even higher concentrations of the drug that approach or exceed their previously reported IC50 values [19]. Of course, the possibility also exists that TNFα alternatively triggers these pathways independently of Nox2-derived ROS. Several lines of evidence show that TNFα also induces ROS production from other sources in different primary cell types, such as mitochondria [58,59], Nox1 [54] and Nox4 [55]. However, whether they are subjected to TNFα regulation in human aortic endothelial cells, and whether they play a role in endothelial activation and inflammation remains undefined. In addition, though MAPKs and NFκB are generally sensitive to the redox state of their environment, it has also been reported that MAPKs and NFκB can be activated in a ROS-insensitive manner [60,61] as ROS scavengers fail to completely abrogate receptor-induced MAPK activation, and that activation of NFκB signaling can be achieved by direct binding of TNFα receptor with components of the NFκB cascade through TNF receptor‐associated factor 2 (TRAF2) [62]. It is therefore plausible that TNFα-induced endothelial activation is mediated primarily but not exclusively by Nox2/ROS.
Apart from our studies with respect to intracellular signaling pathways, translational relevance was demonstrated by our findings that CPP11G and CPP11H in vitro effectively suppressed upregulation of adhesion molecules in response to TNFα (Figs. 5A and B) on human cells, leading to significantly ameliorated monocyte attachment in the presence of CPP11G or CPP11H (Fig. 5C). Furthermore, in vivo validation in this study of the novel compounds is consistent with our in vitro results. That is, in vivo administration of TNFα prominently enhanced aortic H2O2 production and intimal ICAM-1 expression. Both these responses were more effectively suppressed by CPP11H than CPP11G (Figs. 6A and B). Indeed, it has been well established that the MAPK, AP-1 and NFκB pathways control adhesion molecule expression and monocyte adhesion [26,47,51]. In aggregate, ROS production, adhesion molecule expression and monocyte adhesion in vitro and ROS and adhesion molecule expression in mouse aortas in vivo were ameliorated significantly by CPP11H indicative of its effectiveness in alleviating Nox2-mediated vascular inflammation.
Endothelial activation has been shown to potentiate endothelial dysfunction not only by inhibiting eNOS expression [63], but also by decreasing NO bioavailability through induction of O2•− and generation of ONOO− [64,65]. In line with this notion as well as a number of studies by others [[66], [67], [68], [69]], we also observed that incubation of mouse aortas with TNFα led to a significantly blunted vasodilatory response to the endothelium- and NO-dependent vasodilator acetylcholine. More importantly, the novel small molecule inhibitors were able to largely prevent TNFα-induced endothelial dysfunction (Fig. 6D). The protective role of Nox2 inhibition on endothelial function has been reported by several groups. For instance, in type 2 diabetic mouse models, Nox2ds-tat (aka gp91ds-tat)-treated mesenteric and coronary arteries exhibited improved vasodilation to acetylcholine compared to untreated vessels [70]. Moreover, knocking out p47phox as well as Nox2 prevented disruption of endothelium-dependent vasodilation under pathological conditions [9,70].
In agreement with those findings, novel Nox2 inhibitors CPP11G and CPP11H appear to have conferred protection by abolishing TNFα-induced ROS production and ameliorating endothelial dysfunction. Further, disrupted endothelial function hampers the ability of arteries and arterioles to dilate in response to endogenous vasodilatory agents, and as a result, limits local and systemic blood flow. Indeed, we found that treating mice with TNFα significantly decreased hind limb blood flow, which is consistent with our results regarding endothelial dysfunction in aortas. Though these measurements were employed on different vascular beds (femoral arteries and aortas, respectively), it stands to reason that endothelial function in femoral arteries was also impaired, leading to reduced hind limb blood flow. Surprisingly, though Nox2-selective inhibitor Nox2ds-tat was applied to cerebral circulation to identify the involvement of Nox2 in cerebral blood flow modulation [71,72], there are no other studies, to our knowledge, on Nox2-mediated systemic blood flow regulation under physiological or pathological conditions. Here, for the first time, we show that the novel Nox2 inhibitors CPP11G and CPP11H effectively preserved normal peripheral blood flow against inflammatory insults and protected vascular function. As a decrease in blood flow, particularly in resistance arteries, is associated with increase in blood pressure, our current result is partially supported by one of our previous studies in which Nox2ds-tat attenuated systolic blood pressure elevation in mice infused with AngII [35]. Both studies indicate that targeted Nox2 inhibition promotes vascular protection in various disease models, and that CPP11G and CPP11H hold significant promise clinically for preventing and treating Nox2-associated pathologies.
It is also noteworthy that CPP11H generally exhibits higher efficacy compared to CPP11G. In particular, in vivo results on mouse aortas illustrate that CPP11H reduced ROS production and ICAM expression perhaps to a higher degree than CPP11G. This might be attributed to the different structures of the side chains on the amine group: pentyl and thiophene functional groups for CPP11G and CPP11H, respectively [19]. These functional groups are distinct in their bulkiness, rigidity and hydrophobicity, all of which could influence drug-protein binding affinity. It is plausible that the thiophene functional group on CPP11H presents a better fit for the hydrophobic pocket of the “super groove” on the SH3 domain of p47phox for p22phox docking, thereby yielding higher efficacy. In addition, the distinct functional groups could also affect drug absorption, distribution, metabolism and excretion in vivo, leading to varied efficacy. Interrogation of whether CPP11H possesses higher binding affinity, better tissue penetration, slower metabolism or less excretion than CPP11G will require thorough biochemical and pharmacokinetic studies that are currently beyond the scope this work.
Collectively, these data provide exciting new evidence for the role of Nox2 in vascular inflammation. They also illustrate effectiveness for novel small-molecule Nox2-selective inhibitors CPP11G and CPP11H in ameliorating TNFα-elicited endothelial inflammation and dysfunction in human cells in vitro and in mice in vivo. More development in the way of improving potency will likely be necessary to render more effective and clinically relevant agents. Nevertheless, their manifold and profound effects on key inflammatory pathways as well as cell and tissue function justify their development and appear to hold significant promise for this new class of therapeutics for Nox2-mediated inflammatory vascular diseases, such as atherosclerosis and restenosis.
4. Methods
4.1. Reagents
Superoxide dismutase (SOD) and catalase were purchased from Sigma-Aldrich (St. Louis, MO). Protease inhibitor and phosphatase inhibitor cocktail tablets were purchased from Roche Diagnostics GmbH (Mannheim, Germany). Antibodies for phospho-p38 (#4631), total p38 (#9228), phospho-SAPK/JNK (#9255), total SAPK/JNK (#9252S), phospho-p44/42 (#8544), total p44/42 (#4695), phospho-IKKα/β (#2697), total IKKβ (#8943), total IκB (#4814), phospho-cJun (#3270), total cJun (#9165), phospho-NFκB p65 (#3033), total NFκB p65 (#8242) were purchased from Cell Signaling Technology (Danvers, MA). VCAM-1 (sc-13160), ICAM-1 (sc-19584), and β-Actin (sc-47778) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Nox2 antibody (ab129068) was purchased from Abcam (Cambridge, MA) and p47phox antibody (07-001) was purchased from Millipore (Burlington, MA). Rabbit (925-68070), mouse (925-68071) and goat (925-68074) secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE). The nuclear extract kit (40010) was purchased from Active Motif (Carlsbad, CA). Coumarin 7-boronic acid (CBA) (1357078-03-5) was purchased from Cayman Chemical (Ann Arbor, MI). Hydropropidine (HPr+) was generously provided by Dr. Jacek Zielonka (Medical College of Wisconsin).
4.2. 3-D modeling
Smina [73] was used to dock CPP11G and CPP11H to p40phox (from a p40phox-p47phox complex, PDB 1W70) [74], p47phox (from a p47phox-p22phox complex, PDB 1OV3) [23], and p67phox (from a p67phox-p47phox complex, PDB 1K4U) [75] at the protein-protein interface. The docking box was defined by the corresponding ligand peptide of each structure. Water and cofactors were removed from the receptor protein, and an exhaustiveness level of 50 was used with other parameters kept at their defaults. Poses docked to p47phox exhibited >1 kcal/mol improved binding affinity compared to poses docked to p40phox and p67phox.
4.3. Cell culture
COS-phox cells, kindly provided by Dr. Mary Dinauer, were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 4.5 g/l glucose, l-glutamine and sodium pyruvate containing 10% heat-inactivated fetal bovine serum (FBS), 100 units/ml penicillin and 100 μg/ml streptomycin (complete media) (Gibco, Carlsbad, CA). Human aortic endothelial cells (HAECs) from at least three different donors were obtained from Lonza (CC-2535; Walkersville, MD) and cultured in EBM-2 medium containing EGM-2 bullet kit components (CC-3182, Lonza) at 5% CO2, 37 °C. Passages 3 to 6 were used for subsequent experiments. COS-phox cells and HAECs were grown to 75-80% confluency and subjected to serum deprivation by incubation in Opti-MEM (Gibco, Carlsbad, CA) and endothelial cell media containing 10% of supplied growth factors and 0.2% FBS, respectively. After 16 h, cells were treated with CPP11G, CPP11H or vehicle (DMSO) 20 min prior to the addition of PMA (5 μmol/l) or TNFα (10 ng/ml).
4.4. Measurements of extracellular superoxide production by hydropropidine assay
Hydropropidine (HPr+) probe preparation and methodology were adopted and modified from a publication by Zielonka et al. [56]. HAECs seeded in 96-well, clear-bottom, black-sided plates were serum starved overnight. On the following day, wells were washed with PBS, and incubated in 80 μl assay buffer consisting of Hank's Balanced Salt Solution supplemented with 25 mmol/l HEPES, 1% BSA, 10 μmol/l DTPA, 100 μmol/l l-NAME and 1mmol/l taurine (l-NAME and taurine were added to scavenge peroxynitrite and hypochlorous acid respectively, which react with the probe) for 30 min in an incubator (37 °C, 5% CO2). 250 U/ml superoxide dismutase (SOD) was added to select wells under each treatment as negative controls. After incubation, probe solution was added to each well to reach a final concentration of 100 μmol/l HPr+ and 0.5 mg/ml salmon sperm DNA was added at a final reaction volume of 125 μl. Plates were placed in a Biotek Synergy 4 hybrid multimode microplate reader (preheated to 37 °C), and read kinetically (every minute for 2 h) at excitation 400 nm and emission 574 nm. The average rate of fluorescence generation was determined over the linear portion of the response normalized to SOD negative controls.
4.5. Measurements of hydrogen peroxide (H2O2) production by coumarin boronic acid (CBA) assay
CBA probe preparation was adopted and modified from methods described by Zielonka et al. [57]. HAECs seeded in 96-well, clear-bottom, black-sided plates were serum starved overnight, washed with PBS, and incubated in assay buffer consisting of EC starvation media (phenol red free) in an incubator (37 °C, 5% CO2) for 30 min. Assay buffer was supplemented with 10 μmol/l DTPA, 100 μmol/l l-NAME and 1 μmol/l taurine. 1 KU/ml bovine liver catalase (Sigma-Aldrich, St. Louis, MO) was added to select wells as negative controls. After incubation, probe solution was added to each well to reach a final concentration of 0.5 mmol/l CBA (Cayman Chemicals, Ann Arbor, MI) at a final reaction volume of 125 μl. Plates were placed in a Biotek Synergy 4 hybrid multimode microplate reader (preheated to 37 °C), and read kinetically (every minute for 2 h) at excitation 350 nm and emission 450 nm wavelengths. The average rate of fluorescence generation was determined over the linear portion of the response normalized to catalase negative controls.
4.6. Measurements H2O2 production by Amplex Red Assay
HAECs were collected in lysis buffer (Hank's Balanced Salt Solution with Complete Mini protease inhibitor and PhosStop phosphatase inhibitor from Roche), and were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times to disrupt cells. The cell lysates were centrifuged at 1000 g for 5 min at 4 °C to remove unbroken cells, nuclei, and debris. Throughout all procedures, extreme care was taken to maintain the lysate at a temperature close to 0 °C. Lysates of human aortic endothelial cells or mouse aortic tissue were resuspended in Amplex Red assay mixture (25 mmol/l Hepes, pH 7.4, containing 120 mmol/l NaCl, 3 mmol/l KCl, 1 mmol/l MgCl2, 0.1 mmol/l Amplex red (Invitrogen, Grand Island, NY)), and 0.35 U/ml horseradish peroxidase (HRP) in the presence and absence of catalase (300 U/ml). The reaction was initiated by the addition of 36 μmol/l NADPH (MP Biomedicals, Grand Island, NY). Fluorescence measurements were made using a Biotek Synergy 4 hybrid multimode microplate reader with a 530/25-exitation and a 590/35-emission filter. The reaction was monitored at 25 °C for 1 h.
4.7. Western blotting
Western blot experiments were performed as previously described [76,77]. Tissue or cells were homogenized in ice-cold lysis buffer. Supernatants were collected and lysates quantified using a Bradford assay (Bio-Rad, Hercules, CA). Total protein (30 μg) from cell lysates was added to Tris-glycine SDS sample buffer, boiled, resolved by SDS-PAGE and transferred onto nitrocellulose membrane (Bio-Rad). Blots were blocked with the Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE) and probed using rabbit anti-phospho-p38 (1:500), mouse anti-p38 (1:1000), rabbit anti-phospho-SAPK/JNK (1:500), rabbit anti-SAPK/JNK (1:1000), rabbit anti-phospho-p44/42 (1:500), rabbit anti-44/42 (1:1000), rabbit anti-phospho-IKKα/β (1:500), rabbit anti-IKKβ (1:1000), mouse anti-IκB (1:1000), rabbit anti-phospho-cJun (1:500), rabbit anti-cJun (1:1000), rabbit anti-phospho-NFκB p65 (1:500), rabbit anti-NFκB p65 (1:1000) mouse anti-VCAM-1 (1:1000), mouse anti-ICAM-1 (1:1000), mouse anti-β-Actin (1:2000), rabbit anti-Nox2 (1:1000) or rabbit anti-p47phox (1:1000). Blots were then probed with anti-rabbit or anti-mouse secondary antibodies (1:15000 dilution, LI-COR Biosciences). Bands were visualized on an Odyssey Imaging System (Li-COR) and intensity quantified using ImageJ software.
4.8. Co-immunoprecipitation
COS-phox cells were serum starved in Opti-MEM for 16 h, and stimulated with 5 μmol/l PMA for 1 h in the presence or absence of CPP11G, CPP11H or vehicle (DMSO). Cells were washed with ice-cold PBS and collected in lysis buffer (Hank's Balanced Salt Solution with Complete Mini protease inhibitor and PhosStop phosphatase inhibitor from Roche Diagnostics). The cells were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times to disrupt cells. The cell lysate was centrifuged at 1000 g for 5 min at 4 °C to remove unbroken cells, nuclei, and debris. Throughout all procedures, extreme care was taken to maintain the lysate at a temperature close to 0 °C. After pre-washing with protein A/G plus agarose (sc-2003, Santa Cruz Biotechnology), total cell homogenates were immunoprecipitated with anti-p47phox antibody (sc-17845, Santa Cruz Biotechnology) using protein A/G plus agarose slurry. Immunoprecipitates were then subjected to Western blotting using the anti-p47phox antibody (07-001, Millipore) and anti-Nox2 antibody (ab129068, Abcam).
4.9. Measurement of p47phox membrane translocation
COS-phox cells were serum starved in Opti-MEM (Gibco) for 16 h, and stimulated with 5 μmol/l PMA for 1 h in the presence or absence of CPP11G, CPP11H or vehicle (DMSO). Cells were washed with ice-cold PBS and collected in lysis buffer (Hank's Balanced Salt Solution (HBSS) with Complete Mini protease inhibitor and PhosStop phosphatase inhibitor from Roche Diagnostics). The cells were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times to disrupt cells. The cell lysate was centrifuged at 1000 g for 5 min at 4 °C to remove unbroken cells and debris. The fractions of cell nuclei, mitochondria, submitochondria, and small organelles were eliminated by sequential centrifugation at 28,000 g for 20 min at 4 °C and at 100,000 g for 60 min at 4 °C. The final pellet of plasma membrane fraction was resuspended in HBSS quantified using a Bradford assay (Bio-Rad, Hercules, CA). 20 μg of total membrane protein were resolved by SDS-PAGE and transferred onto nitrocellulose membrane (Bio-Rad). Blots were probed using antibodies against p47phox (07-001, Millipore) and Nox2 (ab129068, Abcam).
4.10. Immunofluorescent imaging
Immunofluorescence analysis was performed on HAECs grown on gelatin-coated glass coverslips as well as 5 μm sections of paraffin-embedded mouse aortas. Samples were antigen-retrieved, fixed in 2% paraformaldehyde, permeabilized with 0.1% Triton X-100 and washed using PBS. Sections were blocked in PBS with 2% bovine serum albumin (BSA) for 1 h at room temperature. HAECs on coverslips were then incubated with anti-3-nitrotyrosine (ab61392, Abcam, 1:100) for 1 h at room temperature, or anti-total-NFκB p65 antibody (#8242, Cell Signaling, 1:100) overnight at 4 °C followed by Cy3-conjugated secondary antibody (Life Technologies Inc., 1:1000). Coverslips were then stained for nuclei with Hoechst dye, and cover-slipped using gelvatol mounting media (polyvinylalcohol, glycerol, H2O, sodium azide and Tris pH 8.5). Nonspecific rabbit or goat IgG (5 μg/ml) was used in lieu of primary antibody as a negative control. Confocal images were captured on a Nikon A1 spectral confocal microscope (Nikon Instruments Inc. Melville, NY). For each experiment, 3-4 images per treatment group were captured. Three independent experiments were performed.
4.11. Monocyte adhesion assay
Confluent HAECs grown in black 96-well, clear-bottom plates were pretreated with CPP11G or CPP11H, then stimulated with TNF-α (10 ng/ml) for 24 h. THP-1 monocytes (ATCC, Manassas, VA) were tagged with the compound calcein-AM (Life Technologies, Carlsbad, CA) (10 μmol/L) for 30 min at 37 °C in RPMI-1640 (Sigma-Aldrich, St. Louis, MO) and were washed once with PBS and resuspended in RPMI-1640 at a concentration of 5 × 105 cells/ml. The tagged THP-1 (5 × 104 cells/well) were gently added to the HAEC monolayer and incubated for 1 h at 37 °C. Unbound THP-1 cells were discarded by washing with RPMI-1640 3 times followed by PBS washing once. HAEC-attached THP-1 cells were observed with a Zeiss Axiovert 40 CFL microscope (Carl Zeiss Microscopy, Thornwood, NY) at 10 X magnification. In parallel, fluorescence intensity was determined using a fluorescence microplate reader (BioTek) at 495 nm (excitation wavelength) and 515 nm (emission wavelength). Results were shown as relative monocyte adhesion levels normalized to the control group.
4.12. Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee, University of Pittsburgh and are in accordance with National Institutes of Health guidelines. Mice purchased from Jackson Labs (Bar Harbor, ME, USA) were subjected to i.v. bolus injections of CPP11G or CPP11H (15 mg/kg, 100 μl) or vehicle (30% DMSO in PBS, 100 μl) into the jugular vein 15 min prior to TNF-α treatment (10 μg/kg, 100 μl). Animals were allowed to recover for 4 h, then sacrificed and aortic vessels collected.
4.13. Laser Doppler blood flow analysis
Femoral artery blood flow was assessed 4 h after i.v. bolus injection of TNFα (10 μg/kg) in the presence or absence of CPP11G or CPP11H (15 mg/kg). Briefly, mice were anesthetized with 2.5% isoflurane and placed in a supine position on a heating pad. Core temperatures were maintained at 37 °C and continuously monitored by rectal probe thermometer. In order to reveal the femoral artery and adjacent arterioles and capillaries, the inner hind limb was shaved and the skin removed to better expose subcutaneous tissue. Real-time blood flow was measured using laser Doppler imaging (MoorLDI-2λ; Moor Instruments, Devon, UK).
4.14. Vessel myography
Male C57/B6 mice were anesthetized with CO2 followed by cervical dislocation. Descending thoracic aortas were quickly isolated and cut into 4 rings each 2 mm in length. Aortic rings were placed on myograph stirrups (Danish Myo Technology, Atlanta, GA) in 5 ml physiological saline solution (PSS) buffer (containing 130 mmol/L NaCl, 4.7 mmol/l KCl, 1.17 mmol/l MgSO4, 1.18 mmol/l KH2PO4, 14.9 mmol/l NaHCO3, 5.5 mmol/l d-glucose, 1.6 mmol/l CaCl2, 0.026 mmol/l EDTA) maintained at 37 °C, pH 7.4, gassed with 95% O2 and 5% CO2, and brought to an optimal resting tension of 5000 mg by increasing tone by 500 mg every 5 min. Vessels were then allowed to equilibrate until baseline tension remained constant (30-60 min). Viability of the vessels was ascertained by a series of contractile responses to KCl (25 mmol/l, 50 mmol/l and 100 mmol/l KCl in PSS solution) for 5 min each. Concentration-response curves to phenylephrine were carried out and a concentration that produced 80% maximum contraction (EC80) was chosen for establishing vascular tone prior to additional treatments. In PE-preconstricted vessels, endothelium-dependent dilatation in response to acetylcholine (10−9 to 10−5 mol/l) and endothelium-independent dilatation in response to sodium nitroprusside (SNP) (10−9.5 to 10−6 mol/l) were then tested.
4.15. Statistical analyses
Data are expressed as mean ± SEM, and for comparison of results between two data sets, an unpaired Student's t-test was performed. One-way ANOVA followed by Tukey's multiple comparison test was used for comparison of results among more than two groups. Both analyses were performed using GraphPad Prism software (version 7.1). p < 0.05 was considered statistically significant.
4.16. Study approval
All animal experiments were approved by the Institutional Animal Care and Use Committee, University of Pittsburgh and are in accordance with National Institutes of Health guidelines.
Author contributions
YL, ECP, PJP, DNM conceived and designed the study. YL, ECP, ERD, DdJ, SS collected and analyzed experimental data. YL, MR and CSC obtained imaging data. DK and CC carried out in-silico studies. YL, ECP, PJP wrote and/or edited the manuscript. All authors reviewed and approved the final version.
Conflicts of interest
The authors have declared that no conflict of interest exists.
Acknowledgment
This work was supported by the National Institutes of Health (NIH) Grants R01HL079207 (PJP), P01HL103455-01, and AHA#18TPA34170069 (PJP), NIH Grant 2R01GM097082 (CJC), AHA fellowship 17POST33660330 (YL), and funds from the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101143.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
figs1.

figs2.

figs3.

figs4.

References
- 1.Nauseef W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008 Jun 20;283(25):16961–16965. doi: 10.1074/jbc.R700045200. PubMed PMID: 18420576. Pubmed Central PMCID: PMC2427363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Csanyi G., Taylor W.R., Pagano P.J. NOX and inflammation in the vascular adventitia. Free Radic. Biol. Med. 2009 Nov 1;47(9):1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. PubMed PMID: 19628034. Pubmed Central PMCID: PMC3061339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al Ghouleh I., Khoo N.K., Knaus U.G., Griendling K.K., Touyz R.M., Thannickal V.J. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011 Oct 1;51(7):1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. PubMed PMID: 21722728. Pubmed Central PMCID: PMC3205968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai H., Harrison D.G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 2000 Nov 10;87(10):840–844. doi: 10.1161/01.res.87.10.840. PubMed PMID: 11073878. [DOI] [PubMed] [Google Scholar]
- 5.Frazziano G., Champion H.C., Pagano P.J. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am. J. Physiol. Heart Circ. Physiol. 2012 Jun 1;302(11):H2166–H2177. doi: 10.1152/ajpheart.00780.2011. PubMed PMID: 22427511. Pubmed Central PMCID: PMC3378288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007 Jan;87(1):245–313. doi: 10.1152/physrev.00044.2005. PubMed PMID: 17237347. [DOI] [PubMed] [Google Scholar]
- 7.Li Y., Pagano P.J. Microvascular NADPH oxidase in Health and disease. Free Radic. Biol. Med. 2017 Aug;109 doi: 10.1016/j.freeradbiomed.2017.02.049. PubMed PMID: 28274817, Pubmed Central PMCID: PMC5482368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dourron H.M., Jacobson G.M., Park J.L., Liu J., Reddy D.J., Scheel M.L. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am. J. Physiol. Heart Circ. Physiol. 2005 Feb;288(2):H946–H953. doi: 10.1152/ajpheart.00413.2004. PubMed PMID: 15388496. [DOI] [PubMed] [Google Scholar]
- 9.Jung O., Schreiber J.G., Geiger H., Pedrazzini T., Busse R., Brandes R.P. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004 Apr 13;109(14):1795–1801. doi: 10.1161/01.CIR.0000124223.00113.A4. PubMed PMID: 15037533. [DOI] [PubMed] [Google Scholar]
- 10.Liu J., Yang F., Yang X.P., Jankowski M., Pagano P.J. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2003 May 01;23(5):776–782. doi: 10.1161/01.ATV.0000066684.37829.16. PubMed PMID: 12637340. [DOI] [PubMed] [Google Scholar]
- 11.Ghouleh I.A., Sahoo S., Meijles D.N., Amaral J.H., de Jesus D.S., Sembrat J. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin. Sci. (Lond.) 2017 Aug 1;131(15):2019–2035. doi: 10.1042/CS20160812. PubMed PMID: 28522681. Pubmed Central PMCID: PMC5705051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy K., Wu Y., Meitzler J.L., Juhasz A., Liu H., Jiang G. NADPH oxidases and cancer. Clin. Sci. (Lond.) 2015 Jun;128(12):863–875. doi: 10.1042/CS20140542. PubMed PMID: 25818486. [DOI] [PubMed] [Google Scholar]
- 13.Bonner M.Y., Arbiser J.L. Targeting NADPH oxidases for the treatment of cancer and inflammation. Cell. Mol. Life Sci. 2012 Jul;69(14):2435–2442. doi: 10.1007/s00018-012-1017-2. PubMed PMID: 22581366. Pubmed Central PMCID: PMC4405775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorce S., Stocker R., Seredenina T., Holmdahl R., Aguzzi A., Chio A. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: what is the evidence? Free Radic. Biol. Med. 2017 Nov;112:387–396. doi: 10.1016/j.freeradbiomed.2017.08.006. PubMed PMID: 28811143. [DOI] [PubMed] [Google Scholar]
- 15.Ma M.W., Wang J., Zhang Q., Wang R., Dhandapani K.M., Vadlamudi R.K. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017 Jan 17;12(1):7. doi: 10.1186/s13024-017-0150-7. PubMed PMID: 28095923. Pubmed Central PMCID: PMC5240251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Centers for Disease Control and Prevention . March 17, 2017. Leading Causes of Death.https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm Available from: [Google Scholar]
- 17.Cifuentes-Pagano E., Meijles D.N., Pagano P.J. The quest for selective nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxidants Redox Signal. 2014 Jun 10;20(17):2741–2754. doi: 10.1089/ars.2013.5620. PubMed PMID: 24070014. Pubmed Central PMCID: PMC4026400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cifuentes-Pagano M.E., Meijles D.N., Pagano P.J. Nox inhibitors & therapies: rational design of peptidic and small molecule inhibitors. Curr. Pharmaceut. Des. 2015;21(41):6023–6035. doi: 10.2174/1381612821666151029112013. PubMed PMID: 26510437. Pubmed Central PMCID: PMC4818579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cifuentes-Pagano E., Saha J., Csanyi G., Ghouleh I.A., Sahoo S., Rodriguez A. Bridged tetrahydroisoquinolines as selective NADPH oxidase 2 (Nox2) inhibitors. MedChemComm. 2013 Jul;4(7):1085–1092. doi: 10.1039/C3MD00061C. PubMed PMID: 24466406. Pubmed Central PMCID: 3897123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J., Kleinberg M.E. Activation of the phagocyte NADPH oxidase protein p47(phox). Phosphorylation controls SH3 domain-dependent binding to p22(phox) J. Biol. Chem. 1999 Jul 9;274(28):19731–19737. doi: 10.1074/jbc.274.28.19731. PubMed PMID: 10391914. [DOI] [PubMed] [Google Scholar]
- 21.Leto T.L., Adams A.G., de Mendez I. Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. Proc. Natl. Acad. Sci. U. S. A. 1994 Oct 25;91(22):10650–10654. doi: 10.1073/pnas.91.22.10650. PubMed PMID: 7938008. Pubmed Central PMCID: 45079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumimoto H., Hata K., Mizuki K., Ito T., Kage Y., Sakaki Y. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J. Biol. Chem. 1996 Sep 6;271(36):22152–22158. doi: 10.1074/jbc.271.36.22152. PubMed PMID: 8703027. [DOI] [PubMed] [Google Scholar]
- 23.Groemping Y., Lapouge K., Smerdon S.J., Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003 May 2;113(3):343–355. doi: 10.1016/s0092-8674(03)00314-3. PubMed PMID: 12732142. [DOI] [PubMed] [Google Scholar]
- 24.Halliwell B. What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett. 1997 Jul 14;411(2–3):157–160. doi: 10.1016/s0014-5793(97)00469-9. PubMed PMID: 9271196. [DOI] [PubMed] [Google Scholar]
- 25.Karin M., Liu Z., Zandi E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997 Apr;9(2):240–246. doi: 10.1016/s0955-0674(97)80068-3. PubMed PMID: 9069263. [DOI] [PubMed] [Google Scholar]
- 26.Cabal-Hierro L., Lazo P.S. Signal transduction by tumor necrosis factor receptors. Cell. Signal. 2012 Jun;24(6):1297–1305. doi: 10.1016/j.cellsig.2012.02.006. PubMed PMID: 22374304. [DOI] [PubMed] [Google Scholar]
- 27.Clark R.A., Valente A.J. Nuclear factor kappa B activation by NADPH oxidases. Mech. Ageing Dev. 2004 Oct-Nov;125(10–11):799–810. doi: 10.1016/j.mad.2004.08.009. PubMed PMID: 15541774. [DOI] [PubMed] [Google Scholar]
- 28.Fan J., Frey R.S., Rahman A., Malik A.B. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-alpha -induced NF-kappa B activation and intercellular adhesion molecule-1 expression in endothelial cells. J. Biol. Chem. 2002 Feb 1;277(5):3404–3411. doi: 10.1074/jbc.M110054200. PubMed PMID: 11729200. [DOI] [PubMed] [Google Scholar]
- 29.Martin A.G., Fresno M. Tumor necrosis factor-alpha activation of NF-kappa B requires the phosphorylation of Ser-471 in the transactivation domain of c-Rel. J. Biol. Chem. 2000 Aug 11;275(32):24383–24391. doi: 10.1074/jbc.M909396199. PubMed PMID: 10823840. [DOI] [PubMed] [Google Scholar]
- 30.Steffan N.M., Bren G.D., Frantz B., Tocci M.J., O'Neill E.A., Paya C.V. Regulation of IkB alpha phosphorylation by PKC- and Ca(2+)-dependent signal transduction pathways. J. Immunol. 1995 Nov 15;155(10):4685–4691. PubMed PMID: 7594468. [PubMed] [Google Scholar]
- 31.Kanarek N., Ben-Neriah Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol. Rev. 2012 Mar;246(1):77–94. doi: 10.1111/j.1600-065X.2012.01098.x. PubMed PMID: 22435548. [DOI] [PubMed] [Google Scholar]
- 32.Szmitko P.E., Wang C.H., Weisel R.D., de Almeida J.R., Anderson T.J., Verma S. New markers of inflammation and endothelial cell activation: Part I. Circulation. 2003 Oct 21;108(16):1917–1923. doi: 10.1161/01.CIR.0000089190.95415.9F. PubMed PMID: 14568885. [DOI] [PubMed] [Google Scholar]
- 33.Rastogi R., Geng X., Li F., Ding Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 2016;10:301. doi: 10.3389/fncel.2016.00301. PubMed PMID: 28119569. Pubmed Central PMCID: PMC5222855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csanyi G., Cifuentes-Pagano E., Al Ghouleh I., Ranayhossaini D.J., Egana L., Lopes L.R. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic. Biol. Med. 2011 Sep 15;51(6):1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. PubMed PMID: 21586323. Pubmed Central PMCID: PMC3204933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rey F.E., Cifuentes M.E., Kiarash A., Quinn M.T., Pagano P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ. Res. 2001 Aug 31;89(5):408–414. doi: 10.1161/hh1701.096037. PubMed PMID: 11532901. [DOI] [PubMed] [Google Scholar]
- 36.Jaquet V., Marcoux J., Forest E., Leidal K.G., McCormick S., Westermaier Y. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011 Sep;164(2b):507–520. doi: 10.1111/j.1476-5381.2011.01439.x. PubMed PMID: 21501142. Pubmed Central PMCID: PMC3188888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith S.M., Min J., Ganesh T., Diebold B., Kawahara T., Zhu Y. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem. Biol. 2012 Jun 22;19(6):752–763. doi: 10.1016/j.chembiol.2012.04.015. PubMed PMID: 22726689. Pubmed Central PMCID: 3383625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cotgreave I.A., Duddy S.K., Kass G.E., Thompson D., Moldeus P. Studies on the anti-inflammatory activity of ebselen. Ebselen interferes with granulocyte oxidative burst by dual inhibition of NADPH oxidase and protein kinase C? Biochem. Pharmacol. 1989 Feb 15;38(4):649–656. doi: 10.1016/0006-2952(89)90211-6. PubMed PMID: 2537084. [DOI] [PubMed] [Google Scholar]
- 39.Zembowicz A., Hatchett R.J., Radziszewski W., Gryglewski R.J. Inhibition of endothelial nitric oxide synthase by ebselen. Prevention by thiols suggests the inactivation by ebselen of a critical thiol essential for the catalytic activity of nitric oxide synthase. J. Pharmacol. Exp. Therapeut. 1993 Dec;267(3):1112–1118. PubMed PMID: 7505326. [PubMed] [Google Scholar]
- 40.Shimohashi N., Nakamuta M., Uchimura K., Sugimoto R., Iwamoto H., Enjoji M. Selenoorganic compound, ebselen, inhibits nitric oxide and tumor necrosis factor-alpha production by the modulation of jun-N-terminal kinase and the NF-kappab signaling pathway in rat Kupffer cells. J. Cell. Biochem. 2000 Jun 12;78(4):595–606. PubMed PMID: 10861857. [PubMed] [Google Scholar]
- 41.Schewe C., Schewe T., Wendel A. Strong inhibition of mammalian lipoxygenases by the antiinflammatory seleno-organic compound ebselen in the absence of glutathione. Biochem. Pharmacol. 1994 Jul 5;48(1):65–74. doi: 10.1016/0006-2952(94)90224-0. PubMed PMID: 8043032. [DOI] [PubMed] [Google Scholar]
- 42.Mishra B., Priyadarsini K.I., Mohan H., Mugesh G. Horseradish peroxidase inhibition and antioxidant activity of ebselen and related organoselenium compounds. Bioorg. Med. Chem. Lett. 2006 Oct 15;16(20):5334–5338. doi: 10.1016/j.bmcl.2006.07.085. PubMed PMID: 16919452. [DOI] [PubMed] [Google Scholar]
- 43.Ambasta R.K., Schreiber J.G., Janiszewski M., Busse R., Brandes R.P. Noxa1 is a central component of the smooth muscle NADPH oxidase in mice. Free Radic. Biol. Med. 2006 Jul 15;41(2):193–201. doi: 10.1016/j.freeradbiomed.2005.12.035. PubMed PMID: 16814099. [DOI] [PubMed] [Google Scholar]
- 44.Al Ghouleh I., Meijles D.N., Mutchler S., Zhang Q., Sahoo S., Gorelova A. Binding of EBP50 to Nox organizing subunit p47phox is pivotal to cellular reactive species generation and altered vascular phenotype. Proc. Natl. Acad. Sci. U. S. A. 2016 Sep 6;113(36):E5308–E5317. doi: 10.1073/pnas.1514161113. PubMed PMID: 27540115. Pubmed Central PMCID: PMC5018796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niu X.L., Madamanchi N.R., Vendrov A.E., Tchivilev I., Rojas M., Madamanchi C. Nox activator 1: a potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation. 2010 Feb 2;121(4):549–559. doi: 10.1161/CIRCULATIONAHA.109.908319. PubMed PMID: 20083677. Pubmed Central PMCID: 2843418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youn J.Y., Gao L., Cai H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia. 2012 Jul;55(7):2069–2079. doi: 10.1007/s00125-012-2557-6. PubMed PMID: 22549734. Pubmed Central PMCID: 3694990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J.M., Fan L.M., Christie M.R., Shah A.M. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol. Cell Biol. 2005 Mar;25(6):2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. PubMed PMID: 15743827. Pubmed Central PMCID: 1061612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al-Shabrawey M., Rojas M., Sanders T., Behzadian A., El-Remessy A., Bartoli M. Role of NADPH oxidase in retinal vascular inflammation. Invest. Ophthalmol. Vis. Sci. 2008 Jul;49(7):3239–3244. doi: 10.1167/iovs.08-1755. PubMed PMID: 18378574. Pubmed Central PMCID: PMC3798055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meijles D.N., Pagano P.J. Nox and inflammation in the vascular adventitia. Hypertension. 2016 Jan;67(1):14–19. doi: 10.1161/HYPERTENSIONAHA.115.03622. PubMed PMID: 26553230. Pubmed Central PMCID: PMC4750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pober J.S. Endothelial activation: intracellular signaling pathways. Arthritis Res. 2002;4(Suppl 3):S109–S116. doi: 10.1186/ar576. PubMed PMID: 12110129. Pubmed Central PMCID: PMC3240152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jamaluddin M., Wang S., Boldogh I., Tian B., Brasier A.R. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal. 2007 Jul;19(7):1419–1433. doi: 10.1016/j.cellsig.2007.01.020. PubMed PMID: 17317104. [DOI] [PubMed] [Google Scholar]
- 52.Meijles D.N., Fan L.M., Ghazaly M.M., Howlin B., Kronke M., Brooks G. p22phox C242T single-nucleotide polymorphism inhibits inflammatory oxidative damage to endothelial cells and vessels. Circulation. 2016 Jun 14;133(24):2391–2403. doi: 10.1161/CIRCULATIONAHA.116.021993. PubMed PMID: 27162237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frey R.S., Rahman A., Kefer J.C., Minshall R.D., Malik A.B. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ. Res. 2002 May 17;90(9):1012–1019. doi: 10.1161/01.res.0000017631.28815.8e. PubMed PMID: 12016268. [DOI] [PubMed] [Google Scholar]
- 54.Kim Y.S., Morgan M.J., Choksi S., Liu Z.G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell. 2007 Jun 8;26(5):675–687. doi: 10.1016/j.molcel.2007.04.021. PubMed PMID: 17560373. [DOI] [PubMed] [Google Scholar]
- 55.Anilkumar N., Weber R., Zhang M., Brewer A., Shah A.M. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler. Thromb. Vasc. Biol. 2008 Jul;28(7):1347–1354. doi: 10.1161/ATVBAHA.108.164277. PubMed PMID: 18467643. [DOI] [PubMed] [Google Scholar]
- 56.Michalski R., Zielonka J., Hardy M., Joseph J., Kalyanaraman B. Hydropropidine: a novel, cell-impermeant fluorogenic probe for detecting extracellular superoxide. Free Radic. Biol. Med. 2013 Jan;54:135–147. doi: 10.1016/j.freeradbiomed.2012.09.018. PubMed PMID: 23051008. Pubmed Central PMCID: PMC3711142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zielonka J., Sikora A., Hardy M., Joseph J., Dranka B.P., Kalyanaraman B. Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem. Res. Toxicol. 2012 Sep 17;25(9):1793–1799. doi: 10.1021/tx300164j. PubMed PMID: 22731669. Pubmed Central PMCID: 3501381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corda S., Laplace C., Vicaut E., Duranteau J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am. J. Respir. Cell Mol. Biol. 2001 Jun;24(6):762–768. doi: 10.1165/ajrcmb.24.6.4228. PubMed PMID: 11415943. [DOI] [PubMed] [Google Scholar]
- 59.Kim J.J., Lee S.B., Park J.K., Yoo Y.D. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L) Cell Death Differ. 2010 Sep;17(9):1420–1434. doi: 10.1038/cdd.2010.19. PubMed PMID: 20203691. [DOI] [PubMed] [Google Scholar]
- 60.Rajesh M., Mukhopadhyay P., Hasko G., Liaudet L., Mackie K., Pacher P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br. J. Pharmacol. 2010 Jun;160(3):688–700. doi: 10.1111/j.1476-5381.2010.00712.x. PubMed PMID: 20590572. Pubmed Central PMCID: PMC2931568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee R.L., Westendorf J., Gold M.R. Differential role of reactive oxygen species in the activation of mitogen-activated protein kinases and Akt by key receptors on B-lymphocytes: CD40, the B cell antigen receptor, and CXCR4. J. Cell Commun. Signal. 2007 Jun;1(1):33–43. doi: 10.1007/s12079-007-0006-y. PubMed PMID: 18481208. Pubmed Central PMCID: PMC2267655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wajant H., Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011 Apr;278(6):862–876. doi: 10.1111/j.1742-4658.2011.08015.x. PubMed PMID: 21232017. [DOI] [PubMed] [Google Scholar]
- 63.Yoshizumi M., Perrella M.A., Burnett J.C., Jr., Lee M.E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 1993 Jul;73(1):205–209. doi: 10.1161/01.res.73.1.205. PubMed PMID: 7685252. [DOI] [PubMed] [Google Scholar]
- 64.Gryglewski R.J., Palmer R.M., Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986 Apr 3-9;320(6061):454–456. doi: 10.1038/320454a0. PubMed PMID: 3007998. [DOI] [PubMed] [Google Scholar]
- 65.Laursen J.B., Somers M., Kurz S., McCann L., Warnholtz A., Freeman B.A. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103(9):1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 66.Greenberg S., Xie J., Wang Y., Cai B., Kolls J., Nelson S. Tumor necrosis factor-alpha inhibits endothelium-dependent relaxation. J. Appl. Physiol. 1993 May;74(5):2394–2403. doi: 10.1152/jappl.1993.74.5.2394. PubMed PMID: 8335573. [DOI] [PubMed] [Google Scholar]
- 67.Wang P., Ba Z.F., Chaudry I.H. Administration of tumor necrosis factor-alpha in vivo depresses endothelium-dependent relaxation. Am. J. Physiol. 1994 Jun;266(6 Pt 2):H2535–H2541. doi: 10.1152/ajpheart.1994.266.6.H2535. PubMed PMID: 8024016. [DOI] [PubMed] [Google Scholar]
- 68.Davis J.R., Giardina J.B., Green G.M., Alexander B.T., Granger J.P., Khalil R.A. Reduced endothelial NO-cGMP vascular relaxation pathway during TNF-alpha-induced hypertension in pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002 Feb;282(2):R390–R399. doi: 10.1152/ajpregu.00270.2001. PubMed PMID: 11792648. [DOI] [PubMed] [Google Scholar]
- 69.Chen X., Andresen B.T., Hill M., Zhang J., Booth F., Zhang C. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr. Hypertens. Rev. 2008 Nov;4(4):245–255. doi: 10.2174/157340208786241336. PubMed PMID: 20559453. Pubmed Central PMCID: PMC2886300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kassan M., Ait-Aissa K., Ali M., Trebak M., Matrougui K. Augmented EGF receptor tyrosine kinase activity impairs vascular function by NADPH oxidase-dependent mechanism in type 2 diabetic mouse. BBA - Mol. Cell Res. 2015;1853(10):2404–2410. doi: 10.1016/j.bbamcr.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kazama K., Anrather J., Zhou P., Girouard H., Frys K., Milner T.A. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase–derived radicals. Circ. Res. 2004;95(10):1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- 72.Park L., Anrather J., Zhou P., Frys K., Pitstick R., Younkin S. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 2005 Feb 16;25(7):1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. PubMed PMID: 15716413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koes D.R., Baumgartner M.P., Camacho C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013 Aug 26;53(8):1893–1904. doi: 10.1021/ci300604z. PubMed PMID: 23379370. Pubmed Central PMCID: 3726561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Massenet C., Chenavas S., Cohen-Addad C., Dagher M.C., Brandolin G., Pebay-Peyroula E. Effects of p47phox C terminus phosphorylations on binding interactions with p40phox and p67phox. Structural and functional comparison of p40phox and p67phox SH3 domains. J. Biol. Chem. 2005 Apr 8;280(14):13752–13761. doi: 10.1074/jbc.M412897200. PubMed PMID: 15657040. [DOI] [PubMed] [Google Scholar]
- 75.Kami K., Takeya R., Sumimoto H., Kohda D. Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67(phox), Grb2 and Pex13p. EMBO J. 2002 Aug 15;21(16):4268–4276. doi: 10.1093/emboj/cdf428. PubMed PMID: 12169629. Pubmed Central PMCID: 126167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Al Ghouleh I., Rodriguez A., Pagano P.J., Csanyi G. Proteomic analysis identifies an NADPH oxidase 1 (Nox1)-mediated role for actin-related protein 2/3 complex subunit 2 (ARPC2) in promoting smooth muscle cell migration. Int. J. Mol. Sci. 2013 Oct 11;14(10):20220–20235. doi: 10.3390/ijms141020220. PubMed PMID: 24152438. Pubmed Central PMCID: PMC3821612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frazziano G., Al Ghouleh I., Baust J., Shiva S., Champion H.C., Pagano P.J. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: indications for a seminal role for mitochondrial Nox4. Am. J. Physiol. Heart Circ. Physiol. 2014 Jan 15;306(2):H197–H205. doi: 10.1152/ajpheart.00977.2012. PubMed PMID: 24213612. Pubmed Central PMCID: PMC3920131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.