

Some bacterial species exhibit astonishing resistance to ionizing radiation, with Deinococcus radiodurans being the archetype. As natural IR sources rarely exceed mGy levels, the capacity of Deinococcus to survive 5,000 Gy has been attributed to desiccation resistance. To understand the molecular basis of true extreme IR resistance, we are using experimental evolution to generate strains of Escherichia coli with IR resistance levels comparable to Deinococcus. Experimental evolution has previously generated moderate radioresistance for multiple bacterial species. However, these efforts could not take advantage of modern genomic sequencing technologies. In this report, we examine four replicate bacterial populations after 50 selection cycles. Genomic sequencing allows us to follow the genesis of mutations in populations throughout selection. Novel mutations affecting genes encoding DNA repair proteins and RNA polymerase enhance radioresistance. However, more contributors are apparent.
KEYWORDS: DNA repair, Deinococcus, Escherichia coli, ionizing radiation, RecD, RecN, RpoB, RpoC, evolution
ABSTRACT
In previous work (D. R. Harris et al., J Bacteriol 191:5240–5252, 2009, https://doi.org/10.1128/JB.00502-09; B. T. Byrne et al., Elife 3:e01322, 2014, https://doi.org/10.7554/eLife.01322), we demonstrated that Escherichia coli could acquire substantial levels of resistance to ionizing radiation (IR) via directed evolution. Major phenotypic contributions involved adaptation of organic systems for DNA repair. We have now undertaken an extended effort to generate E. coli populations that are as resistant to IR as Deinococcus radiodurans. After an initial 50 cycles of selection using high-energy electron beam IR, four replicate populations exhibit major increases in IR resistance but have not yet reached IR resistance equivalent to D. radiodurans. Regular deep sequencing reveals complex evolutionary patterns with abundant clonal interference. Prominent IR resistance mechanisms involve novel adaptations to DNA repair systems and alterations in RNA polymerase. Adaptation is highly specialized to resist IR exposure, since isolates from the evolved populations exhibit highly variable patterns of resistance to other forms of DNA damage. Sequenced isolates from the populations possess between 184 and 280 mutations. IR resistance in one isolate, IR9-50-1, is derived largely from four novel mutations affecting DNA and RNA metabolism: RecD A90E, RecN K429Q, and RpoB S72N/RpoC K1172I. Additional mechanisms of IR resistance are evident.
IMPORTANCE Some bacterial species exhibit astonishing resistance to ionizing radiation, with Deinococcus radiodurans being the archetype. As natural IR sources rarely exceed mGy levels, the capacity of Deinococcus to survive 5,000 Gy has been attributed to desiccation resistance. To understand the molecular basis of true extreme IR resistance, we are using experimental evolution to generate strains of Escherichia coli with IR resistance levels comparable to Deinococcus. Experimental evolution has previously generated moderate radioresistance for multiple bacterial species. However, these efforts could not take advantage of modern genomic sequencing technologies. In this report, we examine four replicate bacterial populations after 50 selection cycles. Genomic sequencing allows us to follow the genesis of mutations in populations throughout selection. Novel mutations affecting genes encoding DNA repair proteins and RNA polymerase enhance radioresistance. However, more contributors are apparent.
INTRODUCTION
Ionizing radiation (IR) can damage any cellular component, either through direct ionization by high energy photons and electrons, or indirect ionization from reactive oxygen species (ROS) produced by radiolysis of water molecules. Fortunately, levels of IR sufficient to cause widespread oxidation of proteins or extensive DNA damage (including double-strand breaks) are rarely encountered in the environment. Surprisingly, a number of microbial species and a few multicellular organisms have evolved extreme resistance to IR (1, 2). The bacterium Deinococcus radiodurans is the first-discovered and remains the best-studied example of this extremophile phenotype. D. radiodurans survives exposure to 5,000 Gy of IR with no lethality, 1,000 times the lethal dose for a human. In the case of this bacterium, extreme IR resistance appears to be a by-product of natural selection for desiccation tolerance (3–5). D. radiodurans is not alone. Organisms with high levels of IR resistance can be found readily, particularly in arid environments (6, 7).
IR resistance in D. radiodurans has been attributed in large measure to an enhanced capacity to ameliorate ROS produced by IR using a variety small molecules and metabolites (8–10). D. radiodurans possesses a fairly standard complement of DNA repair systems. However, specialized adaptations of those systems and the evolution of a few novel DNA metabolic functions may contribute substantially to the extraordinary capacity of D. radiodurans to repair an extensively damaged genome (11–13).
Substantial increases to bacterial IR resistance can be generated in the laboratory by directed evolution (14–17). The advent of advanced DNA sequencing technologies that reveal genomic changes in populations has revolutionized studies of molecular evolution. Mutational trajectories associated with adaptation have been defined in bacteria (18–24), viruses (25–29), yeast (30, 31), and mammalian tumors (32–43). These studies have greatly enhanced the experimental foundation of many phenomena predicted by evolutionary biology, including clonal interference (20, 22, 31), diminishing returns epistasis (18, 44), and genetic parallelism (19–22, 24, 27).
We have utilized directed evolution as a tool providing a facile path to a better understanding of mechanisms underlying the extreme IR resistance phenotype in bacteria. We previously exposed four replicate populations of Escherichia coli to 20 iterative rounds of gamma ray IR and observed significant increases in IR resistance (45). Focusing on isolates from each population, we determined that IR resistance arose from enhanced DNA repair (reflected in mutant alleles of the recombinase RecA, replicative helicase DnaB, putative helicase YfjK, and replication restart machinery), and changes in the regulation of central metabolism and cellular responses to oxidative damage (through an allele of the anaerobic metabolism transcription factor FNR) (45–47).
The overall IR resistance of these populations, while increased substantially, did not reach a level comparable to D. radiodurans. Efforts to further enhance the IR resistance phenotype have been constrained both by source decay (altering irradiation dose rate parameters and increasing times required to impose a particular dose on a sample) and by new governmental policies mandating a general decommissioning of radioactive sources used for research. To generate D. radiodurans-like levels of IR resistance in evolved E. coli, we embarked on a new and more ambitious effort with four new populations. This new effort employs a clinical linear accelerator (Linac) of a type utilized for radiation treatment of cancer patients. Using this device to produce a high energy electron beam, we can achieve a dose rate of 72 Gy/min, nearly 4-fold higher than that used in the previous directed evolution study (45, 46). Use of this instrument eliminates the problem of radioactive source decay and greatly reduces the amount of time required for exposure to kGy levels of IR. The higher dose rate also allows us to explore adaptation to a potentially much more challenging IR environment than that utilized in earlier trials. We can also begin to assess the effect of dose rate on an evolution trial.
In this report, we characterize the four new E. coli lineages (IR9, IR10, IR11, and IR12) after the first 50 rounds of selection (IR9-50, IR10-50, IR11-50, and IR12-50). The dose required to kill 99% of the cells in each population has increased from approximately 750 Gy to >2,000 Gy. These populations are highly resistant compared to the previously evolved isolates CB2000 and CB3000 (45, 46), which appear to be poorly adapted to the higher dose rates applied with the electron beam IR. Deep genomic sequencing of populations after every other selection cycle allow us to explore the full breadth of mutations and provides a revealing window on the evolution of IR resistance. To unveil IR resistance mechanisms, we focus on the genetic parallelism between the four lineages, as reflected in a representative isolate from population IR9-50, IR9-50-1. The results both reinforce some earlier conclusions and offer new insights. The phenotype in IR9-50-1 is largely explained by four mutations, none of which appeared in our previously evolved E. coli populations. Adaptations in DNA repair machinery are again prominent but now feature novel mutations in recD and recN which enhance IR resistance. Furthermore, alterations in RNA polymerase (RNAP; primarily variants in genes rpoB and rpoC) also contribute substantially to an IR-resistant phenotype. The other three evolved populations exhibit patterns that both reinforce those seen in IR9-50 but also suggest direct competition of two distinct pathways of evolved IR resistance (through either paired mutations of the recA and recD genes or a grouping of rpoB/C, recD, and recN). Further adaptation to IR (beyond the mutations described here) has occurred in each of the four populations and remains to be explored.
RESULTS
High-energy electron beam IR kills E. coli at a rate similar to that of high-energy photon IR.
In this new set of evolution trials, we utilized a Linac to deliver doses with a high energy electron beam. Our previous evolution trials to generate IR-resistant E. coli utilized a 60Co source with a dose rate of approximately 19 Gy/min (45). Using the Linac in electron mode, we were able to deliver the same doses at 72 Gy/min. Since there is no source decay with this device, the dose rate will remain constant over the years required for an extended evolution trial. The dose delivered by the Linac was verified using thermoluminescent dosimeters (TLDs). The delivered dose varied by <5% of the calculated dose (see Table S1 in the supplemental material).
Although the Linac electron beam and photon sources are different, the dose delivery mechanisms are very similar. In order to rule out any differences between electron and photon modes, we sought to determine whether high-energy electron beam ionizing radiation (IR) kills E. coli similarly to high-energy photon IR. We note that the 60Co and 137Cs sources used in our previous directed evolution studies (45–47) produced high-energy photons. The Linac used in this study has both an electron and photon mode. By changing the distance of the E. coli cultures to the source of the IR beam, the gray-per-minute dose rate can be made equivalent between these modes. At a dose rate of 17 Gy/min, high energy electrons and photons killed nearly identical percentages of E. coli MG1655 culture at 1,000 Gy, with the electron beam being slightly more potent (Fig. 1). Therefore, the two IR modes appear to have comparable effects at the level of bacterial survival.
FIG 1.
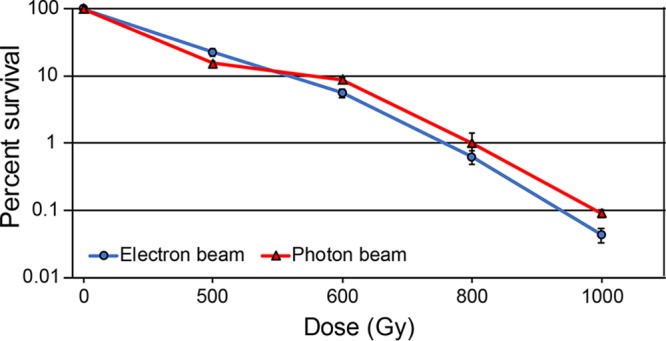
Cell killing with electron beam versus photon beam ionizing radiation. Early-exponential-phase cultures of MG1655 were exposed to electron beam or photon beam IR as described in Materials and Methods. Percent survival was determined via CFU/milliliter counts pre- and postirradiation. The results are representative of a single experiment performed in biological triplicate.
Directed evolution of extreme IR resistance over 50 rounds of selection.
Beginning with a single culture of the E. coli strain MG1655 split into four, we generated four IR resistant populations using a modified version of a previously established directed evolution protocol (45), as described in Materials and Methods and as depicted in Fig. 2A.
FIG 2.

Directed evolution scheme and lineage nomenclature. (A) Scheme. Briefly, overnight cultures were grown from a freezer stock of the parent strain/the evolved population from the previous round of selection. This overnight culture was used to inoculate fresh LB rich medium, and cultures were grown to early exponential phase. These cultures were then separated into multiple aliquots and washed three times in 1× PBS to remove LB medium. One aliquot of each population was irradiated with the same dose that killed 99% of the population in the previous round of selection, and the remaining aliquots were irradiated with higher doses. A portion of the irradiated aliquots were plated to determine percent survival, while the remaining culture was resuspended in fresh LB medium. These were then grown overnight, and the culture that was nearest to 1% survival (as determined by CFU counts from pre- and postirradiation cultures) was stored at −80°C. One round of selection was conducted weekly due to limited access to the Linac. (B) Nomenclature. We have generated four lineages of highly IR-resistant E. coli, designated IR9, IR10, IR11, and IR12. A designated round of selection indicates a population at that round (e.g., IR9-50 is lineage IR9 after 50 rounds of selection). Each population has been stored at –80°C as a “fossil record” of evolution. Clonal isolates generated by streak plating a population then streak plating ten separate isolated colonies are designated by a numeral value added to the parent population nomenclature (e.g., the first isolate from IR9-50 is IR9-50-1).
Using this protocol, we carried out 50 iterative cycles (with selection cycles occurring approximately once per week). The resulting populations were designated IR9-50, IR10-50, IR11-50, and IR12-50 (Fig. 2B). Over these rounds of evolution, the dose required to kill 99% of each culture increased from approximately 750 to 2,300 to 2,500 Gy (Fig. 3A). At round 50, these populations were highly radioresistant compared to the parent MG1655 strain (Fig. 3B). Previously described IR resistant E. coli isolates (45–47), which were evolved to withstand 60Co IR at about 4-fold-lower dose rates, were only moderately more resistant to the beam generated by the Linac compared to the parent strain and were greatly surpassed by the populations generated in this study (Fig. 3B). Populations IR9-50, IR10-50, IR11-50, and IR12-50 have not yet reached the level of IR resistance of the bacterium Deinococcus radiodurans but have made significant progress toward this goal. We note that D. radiodurans was significantly more sensitive to the higher dose rate in the Linac electron source than it was to the lower dose rates in the 60Co photon source (Fig. 3B) (2, 9, 11).
FIG 3.
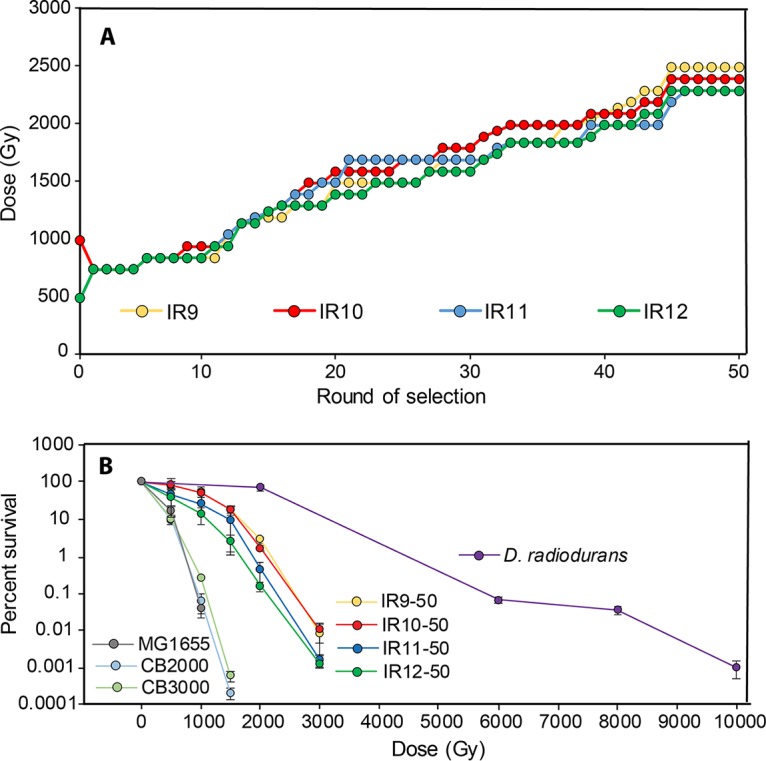
Documentation of increased IR resistance. (A) Increase in dose required to kill 99% of cells. Each data point indicates the dose of IR that each population was given prior to being outgrown overnight (step 6 in Fig. 2) and stored at −80°C (step 7 in Fig. 2). The percent survival was estimated from a single replicate at each round of selection. (B) Survival curves of evolved populations compared to previously evolved Escherichia coli isolates and Deinococcus radiodurans. Early-exponential-phase cultures of the indicated strains were exposed to electron beam IR as described in Materials and Methods. CB2000 and CB3000 are isolates from E. coli populations evolved to withstand gamma ray IR (45, 46). Error bars represent the standard deviation of CFU/milliliter calculations from a single experiment performed in biological triplicate.
Growth phenotypes of evolved population isolates.
The IR resistance phenotype of these experimentally evolved populations was not accompanied by an apparent growth defect in Luria-Bertani (LB) rich medium. An isolate from each population at round 50 of selection (IR9-50-1, IR10-50-1, IR11-50-1, and IR12-50-1) produced growth curves comparable to the parent MG1655 strain in LB (Fig. 4A). However, growth defects of these isolates became apparent when inoculated in other growth media. In a defined rich medium supplemented with glucose, EZ medium, isolate IR12-50-1 failed to grow and isolates IR9-50-1 and IR11-50-1 exhibited growth defects easily observed in a standard growth curve (Fig. 4B). The isolate IR10-50-1 grew similarly to MG1655 in EZ medium and was the only of the four isolates to grow in M9 minimal medium supplemented with glucose (Fig. 4C). Although IR9-50-1, IR11-50-1, and IR12-50-1 have potentially inactivating mutations in key amino acid biosynthetic pathways that could prevent growth in minimal medium (IR10-50-1 does not have a mutation which would clearly affect amino acid biosynthesis), supplementing minimal medium with these amino acids did not rescue the ability of these isolates to grow (Fig. S1).
FIG 4.
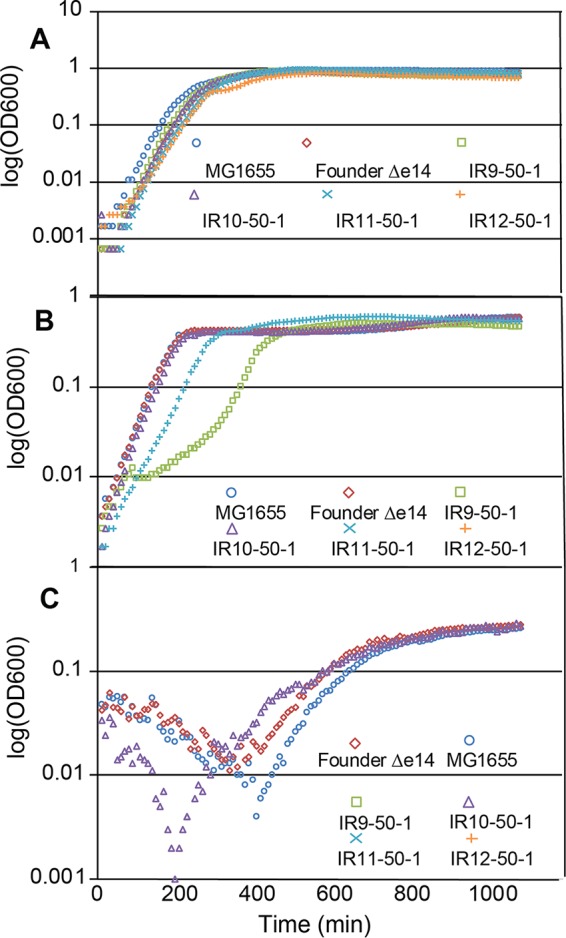
Growth curves of evolved isolates in rich and minimal medium. (A) Growth curves of isolates in LB rich medium. (B) Growth curves of isolates in EZ defined rich medium supplemented with 0.2% glucose. (C) Growth curves of isolates in M9 defined minimal medium supplemented with 0.2% glucose. Cultures of indicated strains were grown in the appropriate medium overnight and then to early exponential phase as described in Materials and Methods. All cultures were incubated at 37°C during growth. Early-exponential-phase cultures were diluted 1:100 in the appropriate medium and then incubated overnight in a BioTek Synergy 2 plate reader, with OD600 measurements taken automatically every 5 min. Each growth curve is a representative replicate from an experiment performed in biological triplicate.
Highly sensitive growth competition assays (48) in LB medium can reveal subtle growth phenotypes of these isolates that are not readily apparent when comparing standard growth curves (Fig. 5). When paired with the parent wild-type E. coli strain in a mixed culture of LB medium, all four evolved isolates were rapidly outcompeted, even with significant dilutions to bias the starting culture ratios toward the evolved strains. These results indicated that 50 rounds of selection in this directed evolution protocol came with an underlying fitness cost, even when selection is not being applied.
FIG 5.
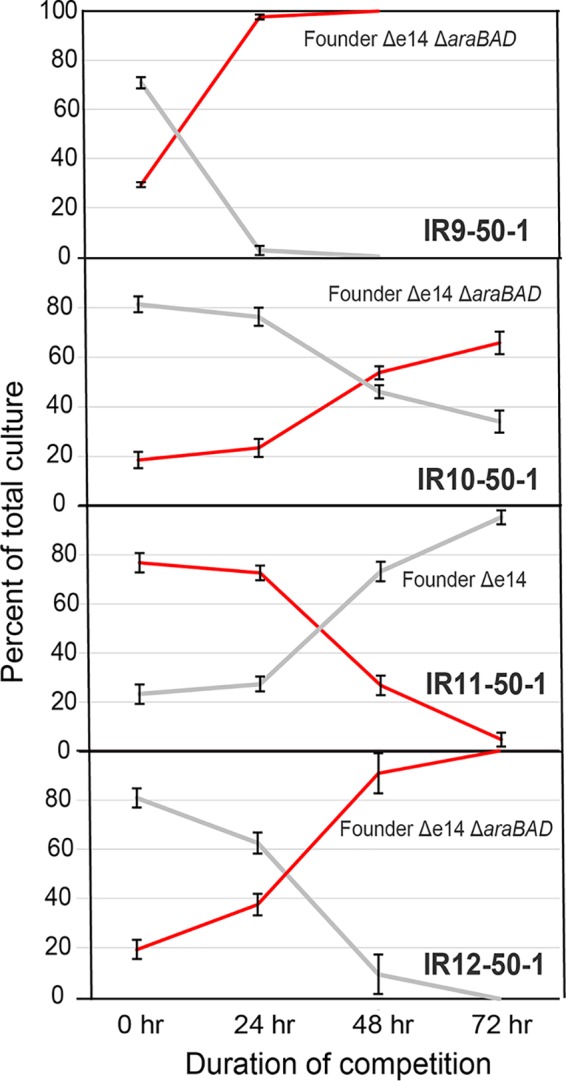
Growth competitions of evolved isolates. (A) Growth competition of IR9-50-1 against Founder Δe14. (B) Growth competition of IR10-50-1 against Founder Δe14. (C) Growth competition of IR11-50-1 against Founder Δe14. (D) Growth competition of IR12-50-1 against Founder Δe14. Growth competitions were performed as described in Materials and Methods. Briefly, competitions were started with an excess of the evolved isolate so that competitions could be carried out to 72 h (at approximately a 9:1 ratio of evolved isolate to Founder Δe14). Even with an excess of IR9-50-1, this isolate was outcompeted by 48 h. Deleting the araBAD operon causes a red colony phenotype with no fitness cost that allows for differentiation of the two strains in competition on TA medium. IR11-50-1 is red on TA medium without alteration of the araBAD operon, so this competition was performed against Founder Δe14 araBAD+. Error bars represent the standard deviations of CFU/milliliter calculations of an experiment performed in biological triplicate. The results shown are representative of two independent experiments.
Evolved isolates exhibit variable resistance to non-IR DNA-damaging agents.
Isolates from the four evolved populations after 50 cycles of selection revealed that resistance to IR does not correlate in any predictable way with resistance to other DNA-damaging agents, even within a single population (Fig. 6 and 7). Multiple isolates from each population exhibited highly variable levels of resistance to UV radiation (Fig. 6), mitomycin C (which causes DNA intrastrand cross-links), ciprofloxacin (which causes DNA double-strand breaks through inhibition of DNA gyrase), bleomycin (which causes DNA double-strand breaks and apurinic/apyrimidinic sites through a ROS mediated mechanism [49, 50]), and hydroxyurea (which has a complex mechanism of action that both reduces dNTP production and causes ROS-mediated DNA damage) (51–54) (Fig. 7). While some isolates exhibited increased resistance to these DNA-damaging agents, others showed no change or increased sensitivity. The results indicate that the directed evolution trials are generating specialists that are uniquely adapted to IR resistance.
FIG 6.
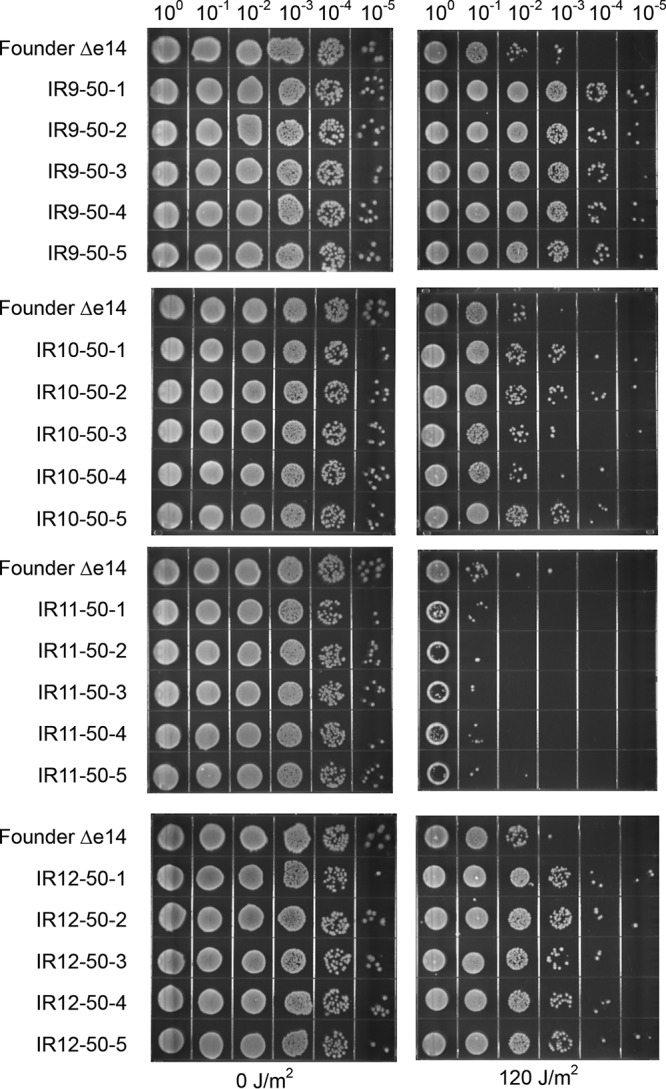
Evolved isolates exhibit variable survival of UV irradiation. Five isolates from each population were grown in LB medium overnight and then to early exponential phase as described in Materials and Methods. Cultures were serial diluted in 1× PBS, and then 10 μl of each dilution was spot plated onto LB agar. Once spots dried, the plates were exposed to UV irradiation and then incubated overnight before imaging. The results shown are representative of two independent experiments performed.
FIG 7.

Evolved isolates exhibit variable survival of various DNA-damaging agents. Five isolates from each population were grown in LB medium overnight and then to early exponential phase as described in Materials and Methods. Cultures were serial diluted in 1× PBS, and then 10 μl of each dilution was spot plated onto LB agar with the indicated DNA-damaging agent. Once the spots dried, the plates were incubated overnight before imaging. The results shown are representative of two independent experiments performed.
Deep sequencing reveals unique evolutionary histories of the evolved populations.
We utilized deep-sequencing technology to monitor genomic changes as IR resistance increased with continued selection cycles. At every other round of selection, genomic DNA was prepared from each population and submitted to the Joint Genome Institute (Walnut Creek, CA) for sequencing to determine all mutations present in each population. Stitching together these snapshots revealed the underlying complexity of these evolving populations (Fig. 8A to D). Depicted in Fig. 8 are the allele frequencies of all mutations detected above a 2% frequency at each even round of selection for each lineage. Each line represents a single mutation; groups of mutations that rise and fall in frequency together are inferred to be linked within a subpopulation. The number of subpopulations and total number of mutations increased rapidly (Fig. 8E). The steady accumulation of mutations over time suggests that mutator strains have not yet appeared in these populations. The complete data set upon which Fig. 8 is based is provided in Data Sets S1 through S4.
FIG 8.
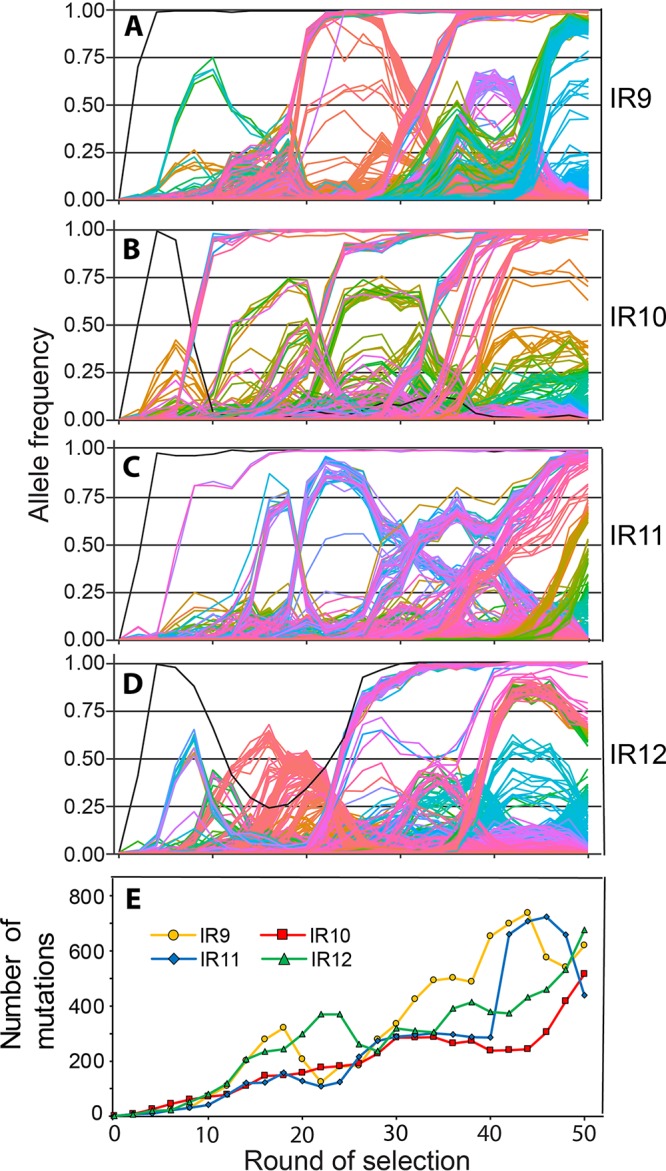
Mutations in evolving populations. (A to D) Allele frequencies as a function of selection round. (A to D) IR9, IR10, IR11, and IR12 populations, respectively. Whole populations at each even round of selection were deep sequenced by the Joint Genome Institute (JGI) as described in Materials and Methods. Every mutation that reached at least 2% frequency at some point in a single population is depicted as a single line. The sole black line in each graph represents the loss of the e14 prophage. Mutations that reach a frequency of “1.00” are fixed, and subsequent mutations occur within that genetic background. The y axis represents the allele frequency of each single mutation within the population (i.e., a mutation at 0.5 is in 50% of the isolates in the population). The x axis represents the round of selection. Allele frequency data were generated from every even cycle of selection. Graphs were generated using the R library “ggplot2.” Line colors are utilized primarily to allow ready distinctions between subpopulations and otherwise have no significance. Data used to generate these plots are contained within Data Sets S1 through S4. (E) Number of mutations over rounds of selection in evolving populations. Each data point represents the total number of detected mutations at or above 2% frequency in each sequenced whole population. The number of mutations generally increases, but experiences dips as major subpopulations are driven to extinction. Data used to generate this plot are contained within Data Sets S1 through S4.
Despite starting from the same parent strain, the evolutionary path of each population was unique. The only event in common to each population was the excision of the e14 prophage, as indicated by four mutations in the icd gene in which the prophage is inserted (as depicted by the initial single black line appearing in each population in Fig. 8A to D). When e14 is excised, the icd gene is restored to the wild-type sequence. Despite excision of e14 occurring quickly in each population, that excision became fixed in only three of them. In the IR10 lineage, the subpopulation that lost the e14 prophage was driven extinct by another subpopulation, and e14 is retained in population IR10-50.
Each lineage experienced significant clonal interference, as depicted by the numerous competing subpopulations in Fig. 8A to D. Competition of subpopulations within lineages IR11 and IR12 significantly reduced the rate of fixation events in each lineage. In IR11 (Fig. 8C), no mutations were able to fix from approximately round 16 to round 50 of selection due to severe clonal interference. In IR12 (Fig. 8D), no subpopulation was able to sweep to fixation until 30 cycles of selection were completed. Although clonal interference is apparent in lineages IR9 and IR10, its effects are far less severe than in IR11 and IR12. In IR9 (Fig. 8A), no subpopulation was able to fix post-e14 phage excision until approximately round 20. After this event, another subpopulation swept to fixation unimpeded (defined as a continuous, linear rate of increasing allele frequency). A final subpopulation was able to eventually reach 100% frequency but was in competition with a separate subpopulation for approximately 12 rounds of selection. Surprisingly, lineage IR10 (Fig. 8B) has approximately six distinct subpopulations which swept to fixation, and each was not noticeably affected by intrapopulation competition. The differing trajectories of evolution in each population may reflect the relative fitness of the mechanisms of IR resistance employed by each eventual successful subpopulation.
At round 50 of selection, there were distinct differences in the amounts and types of mutations between populations. Populations IR9-50 and IR10-50 are the most homogenous, with approximately 16 and 17%, respectively, of the total high confidence (>2% allele frequency) mutations being fixed (detected at >99% frequency). In contrast, only 4 and 11% of high confidence mutations in IR11-50 and IR12-50 are fixed, respectively (Table 1). Although IR9-50 and IR10-50 shared a similar percentage of fixed mutations, IR9-50 and IR12-50 had the highest total number of mutations. Each population had a ratio of nonsynonymous to synonymous mutations greater than 2.
TABLE 1.
Numbers and types of mutations in evolved populations after 50 cycles of selectiona
| Type or frequency | Details | No. of mutations in: |
|||
|---|---|---|---|---|---|
| IR9-50 | IR10-50 | IR11-50 | IR12-50 | ||
| Transition | AT→GC | 91 | 101 | 87 | 109 |
| GC→AT | 179 | 158 | 119 | 218 | |
| Transversion | AT→CG | 26 | 17 | 11 | 25 |
| GC→TA | 154 | 111 | 85 | 137 | |
| AT→TA | 80 | 58 | 59 | 85 | |
| GC→CG | 41 | 24 | 29 | 51 | |
| Total | Transitions | 270 | 259 | 206 | 327 |
| Transversions | 301 | 210 | 184 | 297 | |
| Total | 571 | 469 | 390 | 624 | |
| Coding | Synonymous | 127 | 123 | 88 | 168 |
| Nonsynonymous | 335 | 269 | 234 | 355 | |
| Stop gained | 29 | 15 | 12 | 16 | |
| Stop lost | 1 | 2 | 0 | 1 | |
| Start lost | 0 | 0 | 1 | 2 | |
| Insertions | |||||
| +1 | 1 | 2 | 2 | 3 | |
| +2 | 0 | 0 | 0 | 0 | |
| +3 | 0 | 0 | 1 | 0 | |
| +12 | 0 | 0 | 1 | 0 | |
| Deletions | |||||
| –1 | 40 | 26 | 22 | 24 | |
| –2 | 1 | 2 | 2 | 3 | |
| –3 | 1 | 1 | 0 | 0 | |
| Noncoding | SNPs | 83 | 65 | 55 | 88 |
| Insertions | |||||
| +1 | 0 | 0 | 0 | 0 | |
| +2 | 0 | 0 | 0 | 1 | |
| +3 | 0 | 0 | 0 | 0 | |
| Deletions | |||||
| –1 | 4 | 6 | 3 | 6 | |
| –2 | 0 | 0 | 0 | 1 | |
| –3 | 0 | 0 | 0 | 0 | |
| Allele frequency | Fixed (>99%) | 111 | 83 | 19 | 74 |
| >50% | 241 | 106 | 127 | 148 | |
| >10% | 304 | 192 | 197 | 314 | |
| >2% | 618 | 506 | 420 | 662 | |
Mutations in coding regions that likely cause loss of function of the protein product (single and double base insertions/deletions, gain or loss of stop codons, and loss of start codons) appeared in each population. However, IR9-50 had the most predicted loss-of-function mutations (72) compared to the other three populations (IR10-50, n = 48; IR11-50, n = 40; and IR12-50, n = 49). Of these mutations in all populations, the −1 frameshift mutations were the most common, followed by introduced stop codons.
GC-to-AT and AT-to-GC transitions were detected in high numbers, agreeing with mutational patterns detected in previously evolved IR-resistant E. coli populations (45, 46). Base substitutions are approximately 50% transitions and 50% transversions, whereas E. coli exposed to 60Co gamma ray irradiation exhibit transversions at about four times the frequency of transitions (45, 55). In particular, GC-to-TA transitions appeared at very high levels in each population, indicative of significant template-strand 8-oxodG-mediated mutagenesis (56). These results indicated that the selection utilized in the present study produces a substantially different mutational, and perhaps DNA damage, spectrum relative to the populations evolved to resist 60Co-produced IR.
Mutations which enhance IR resistance in isolate IR9-50-1.
We have begun characterization of evolved IR resistance after 50 cycles of selection. To define mutations that made significant contributions to the phenotype, we counted on a substantial degree of genetic parallelism (19, 20, 24) between the four populations. We focused on mutations meeting the following criteria: (i) the mutations were nonsynonymous, (ii) mutations occurred in a gene that was mutated in at least two additional populations, (iii) mutations in the gene achieved at least 10% abundance at some point during selection in each population in which they appeared, and (iv) the mutation remained in the population at round 50 of selection. These criteria narrowed the search to mutations appearing in six genes, including those involved in recombinational DNA repair (dinI, recD, and recN), transcription (rpoB), regulation of anaerobic metabolism (arcB), and copper ion transport (copA). Two additional gene alterations were analyzed: Nth C203Y and RecA A290S. These did not meet the criteria we set in the present study but were the only two variants, aside from the loss of the e14 prophage, where the exact same mutation appeared in two populations. The RecA A290S variant, which is fixed in population IR10-50 and also appears in the IR11 lineage, is the only variant detected in this present study that was also detected in an earlier study (45). The relevant variants are listed in Table 2.
TABLE 2.
Mutations that are likely candidates for enhancing IR resistancea
| Mutation(s) in: |
|||||
|---|---|---|---|---|---|
| Pathway | Protein | IR9 | IR10 | IR11 | IR12 |
| Anaerobic respiration | ArcB | N405D | D166Y, S280R, L90 fs Y71C | R100H, S74W | S24P, K547 fs |
| Copper metabolism | CopA | V270F | T525A | A812V | |
| DNA metabolism | DinI | R28H, P14A | W71* | I66N | S26Y |
| Nth | C203Y, E160K | C203Y, K85N | |||
| RecA | Y294C | A290S | A290S, E286G | E19K | |
| RecD | A90E, L188*, L223*, P99* | N124D, Q463*, L301M | A550E, A39G, C71 fs, G307W, W534R | S92I, A271E, C103*, T568A | |
| RecN | K429Q, E271G, R118L | F144L | R102P, S382R | A361T, R285C, R368H, S310L | |
| RNA polymerase | RpoB | S72N, S391P | F15L, K1200E | P535L, S574F | T600I |
Proteins listed are encoded by genes that have (i) mutations that were nonsynonymous, (ii) mutations that occurred in a gene that was mutated in at least two additional populations, (iii) mutations that achieved at least 10% abundance at some point during selection in each population in which they appeared, and (iv) mutations that remained in the population at round 50 of selection. The mutations in RecA and Nth were included since these are the only proteins with the same variant present in two populations. The boldface variants are fixed in the indicated population, the variants listed in regular typeface are present at above a 2% frequency at round 50 of selection, and underlined variants were present in the indicated lineage but have been driven extinct by round 50 of selection.
To determine the molecular basis of IR resistance in one of the populations, we more carefully characterized one isolate from population IR9-50: IR9-50-1. IR9-50-1 contained over 300 mutations (Table 3). A complete data set for mutations found within the sequenced isolates is presented in Data Sets S1 through S4, with isolate data in the appropriate lineage data file.
TABLE 3.
Number of mutations in isolates from evolved populations after 50 cycles of selectiona
| Isolate | No. of mutations |
|---|---|
| IR9 | |
| IR9-50-1 | 312 |
| IR9-50-2 | 298 |
| IR9-50-3 | 299 |
| IR9-50-4 | 314 |
| IR9-50-5 | 322 |
| IR10 | |
| IR10-50-1 | 184 |
| IR10-50-2 | 188 |
| IR10-50-3 | 197 |
| IR10-50-4 | 210 |
| IR10-50-5 | 191 |
| IR11 | |
| IR11-50-1 | 200 |
| IR11-50-2 | 192 |
| IR11-50-3 | 194 |
| IR11-50-4 | 182 |
| IR11-50-5 | 197 |
| IR12 | |
| IR12-50-1 | 280 |
| IR12-50-2 | 241 |
| IR12-50-3 | 245 |
| IR12-50-4 | 232 |
| IR12-50-5 | 237 |
Nomenclature of the isolates indicates the lineage (e.g., “IR9”) and round of selection (e.g., “50”) from which the isolate was derived. The remaining numeral distinguishes the individual isolates.
IR9-50-1 is representative of several mutational patterns seen in most of the populations, as it has mutations in dinI, recD, recN, rpoB, rpoC, and copA (which contain the majority of mutations listed in Table 2). This isolate does not have mutations in arcB, nth, and recA. However, a mutation in arcB, leading to the variant gene product ArcB N405D, did appear earlier in the IR9 lineage but was driven to extinction via clonal interference. As such, five protein variants present in IR9-50-1 were investigated for contributions to IR resistance: RecD A90E, RecN K429Q, DinI R28H, RpoB S72N, and CopA V270F. The ArcB N405D mutation was also examined as it represented a mutational pattern in other populations. A mutation in rpoC (which codes for the K1172I variant) was also included in the strains with RpoB S72N, since the genomic proximity of rpoC to rpoB did not allow for ready separation of these two mutations with the methods used here.
To assess the potential contribution of mutations identified in this way to IR resistance, we first isolated each mutation in an otherwise wild-type genetic background. We then took the evolved isolate and converted the mutation being studied to wild type in the otherwise mutant background to determine whether it caused phenotypic loss. We moved these mutations singly and in combination into a derivative of the parent E. coli strain (MG1655) lacking the e14 prophage (as this prophage appears to excise from the chromosome after IR exposure and each subsequent mutation occurred in this genetic background [45, 46]). This strain, Founder Δe14 (45, 46), thus provided the otherwise wild-type genetic background to determine the contribution of each mutation to IR resistance. We determined that the RecD A90E and RpoB S72N/RpoC K1172I variants each provided a significant increase in IR resistance (Fig. 9A) when isolated in this otherwise wild-type background. A combination of these two alleles did not significantly increase IR resistance of the parent strain beyond either individual mutation, indicating that they do not contribute additively. The effects of these two alleles were confirmed by converting each of them singly and in combinations to the wild-type allele in the IR9-50-1 genetic background (Fig. 9B). Interestingly, the RecN K429Q mutation did not significantly alter IR resistance of Founder Δe14. However, when this variant was converted to the wild-type RecN in the IR9-50-1 genetic background, IR resistance was dramatically lowered. When these four mutations were eliminated from a derivative of IR9-50-1 that retained the other 308 mutations, IR resistance was not reduced lower than the wild-type RecN single mutant, suggesting a key role for the RecN K429Q variant.
FIG 9.

Mutations which contribute to IR resistance in the evolved isolate IR9-50-1. (A) IR resistance of Founder Δe14 derivatives containing mutations from lineage IR9. Mutations were moved singly or in combination from IR9-50-1 (in the case of ArcB N405D, from IR9-20-1; the recD deletion was made in the Founder Δe14 parent strain) into the Founder Δe14 strain background as described in Materials and Methods. A “+” symbol indicates that the variant is present in that strain. (B) IR resistance of IR9-50-1 derivatives containing reversions of mutations to the wild-type sequence. Wild-type sequences of the gene encoding the indicated protein variants was moved from Founder Δe14 into IR9-50-1 as described in Materials and Methods (IR9-50-1 does not have a ArcB variant, and therefore no reversion was tested in this strain). A “–” symbol indicates that the variant was reverted to the wild-type allele in that strain. Each strain was assayed for IR resistance alongside biological triplicate of the parent strain (a Founder Δe14 or IR9-50-1 control), and the average percent survival of the experimental strains was compared to that of the parent strain. All strains were exposed to 1,000 Gy of electron beam IR, and the percent survival was determined via calculating CFU/milliliter before and after irradiation. Error bars represent the standard deviations of CFU/milliliter calculations from at least two independent experiments performed in biological triplicate. Statistical significance of percent survival relative to the parent strain was determined using a two-tailed Student t test. * and ***, P < 0.05 and P < 0.001, respectively.
The RecD A90E mutation appeared to be at least a partial loss of function, as deleting the recD gene from Founder Δe14 increased IR resistance just as much as the RecD A90E variant (Fig. 9A). These data agree with the fact that a truncated, and likely inactive, RecD variant appeared earlier in the IR9 lineage and in the IR10 lineage (Table 2). The DinI, CopA, and ArcB variants did not contribute significantly to IR resistance when isolated in the wild-type background; additionally, converting the dinI or copA genes to the wild-type sequence in IR9-50-1 did not increase IR sensitivity of this strain.
Following the allele frequency of mutations in genes that demonstrably contributed to IR resistance (recA, recD, recN, and rpoB), a new pattern emerged (Fig. 10). In the lineages IR9, IR11, and IR12 there was a conserved temporal order in the appearance of mutations. First, the e14 prophage was lost, followed by an rpoB mutation and then a recD mutation along with a recN mutation. In the IR12 lineage, the primary rpoB mutation was a synonymous coding mutation. However, this did not negate a possible effect of this mutation, potentially at the level of codon usage (RpoB D1203, GAC [WT] to GAT [mutant]: 0.37 to 0.63 frequency). The trend was clear in lineages IR9 and IR11, where all the mutations were nonsynonymous. IR10 was the only lineage that did not conform to this trend. Although the e14 prophage did excise in lineage IR10, the subpopulation that lost the prophage was outcompeted by another that maintained the prophage and contained a variant of both RecD (N124D) and RecA (A290S). This RecA variant, A290S, was previously observed in E. coli populations evolved to resist IR (45, 46) and also significantly contributes to electron beam IR resistance (Fig. S2). This is the only mutation observed in these four populations that appeared in the previously evolved E. coli populations, aside from loss of the e14 prophage (45, 46). After the appearance of the RecA A290S and RecD N124D variants, no RpoB variants appeared in the IR10 lineage until nearly round 40 of selection, and those variants (F15L and K1200E) had not yet reached even 50% frequency in IR10 at round 50. It appears that the evolutionary path observed in IR10 nearly occurred in population IR12 as well, where the subpopulation that lost the e14 prophage was almost outcompeted by a different lineage (the competing subpopulation again featuring RecD [T568A] and RecA variants [E19K]), before being carried to fixation with the apparent help of a different RecD variant (S92I). A subpopulation with a RecD (W534R) and RecA variant (E286G) also reached high frequency in IR11 but was slowly outcompeted by a subpopulation with a different RecD variant (A550E) after it gained a second RpoB variant (P535L). In all of these populations, after either RpoB/RecD/RecN or RecA/RecD variants fixed, the opposing grouping of mutations did not appear within the given population. These results imply that these two evolutionary pathways confer competing yet overlapping mechanisms of IR resistance.
FIG 10.

Frequencies of mutations implicated in IR resistance over rounds of selection. Frequencies of mutations in genes implicated in evolved IR resistance (rpoB, recA, recD, and recN) and the loss of the e14 prophage are depicted. Both synonymous and nonsynonymous mutations are included. These data seemingly indicate two separate paths to acquiring IR resistance: loss of the e14 prophage, gain of an rpoB, then recD, then recN mutation, in that order (IR9, IR11, and IR12), or gain of a recA and recD mutation in concert (IR10). In lineages IR11 and IR12, these two pathways to IR resistance appear in direct competition, with the rpoB, recD, and recN path being the apparent “winner.” Data used to generate these plots are contained within Data Sets S1 through S4.
Prominent mutations can contribute to enhanced growth phenotypes.
If variants of proteins such as ArcB, CopA, and DinI do not contribute to IR resistance, do they make some other contribution to fitness that explains their prominence? We have previously observed that high frequency mutations in experimentally evolved populations may contribute to enhanced growth rates, rather than IR resistance (47). Part of the selection protocol involves an outgrowth step, after irradiation, in which mutants that grow slowly could be lost. Consistently, the ArcB N405D mutation significantly enhanced the growth phenotype of Founder Δe14 in a growth competition assay relative to the same strain with a wild-type ArcB (Fig. 11). The DinI R28H and the CopA V270F mutations had no measurable effect on growth (Fig. S3). Mutations that enhance the growth phenotype of these populations are likely important, as evidenced by their inability to grow well in medium aside from LB medium (Fig. 6) and their inability to compete with Founder Δe14 in mixed culture without IR selection (Fig. 5). In effect, certain mutations may arise that compensate for growth defects conferred by mutations that contribute to IR resistance or deleterious, hitchhiking mutations. Founder Δe14 with the RpoB S72N and RpoC K1172I variants is outcompeted quickly by Founder Δe14 with wild-type RNA polymerase (Fig. 11). These results indicate that in order to develop high levels of IR resistance, mutations which buffer against deleterious effects on viability must also rise to prominence in these populations. Although not arising in the same genetic background, the growth phenotype of the ArcB N405D variant indicates that these populations have developed variants which can enhance growth to the degree which mutations such as the RpoB S72N/RpoC K1172I variants impede growth.
FIG 11.
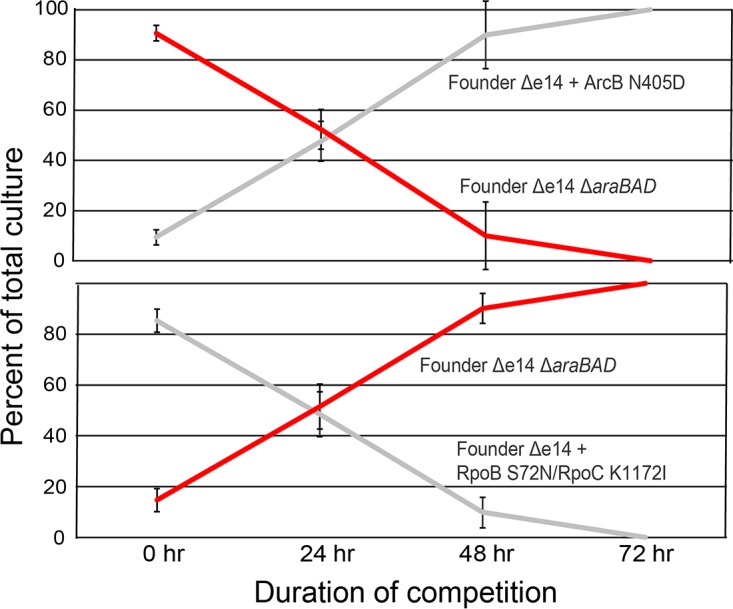
Effects of prevalent mutations on growth. Two mutations from lineage IR9 have opposite effects on growth; ArcB N405D greatly enhances growth of Founder Δe14, whereas the RpoB S72N/RpoC K1172I mutations greatly hinder growth. These results suggest that mutations not seen to affect IR resistance (ArcB N405D) may be selected for in these populations to counteract the deleterious growth effects of mutations which enhance IR resistance. Growth competition assays were performed as described in Materials and Methods. Initial ratios of each strain were purposefully mixed at 9 (loser):1 (winner) so that growth competition assays could be carried out for longer than 24 h. The results shown are representative of two independent experiments performed.
DISCUSSION
It is clear that latent within the E. coli genome is a capacity for resistance to extreme doses of ionizing radiation. After 50 rounds of selection with high-energy electron beam IR, the dose required to kill 99% of each of these four replicate populations increased by >3-fold to approximately 2,500 Gy (Fig. 3A). After a dose of 1,500 Gy, this translates into an increase in survival of 4 to 5 orders of magnitude (Fig. 3B). The dose of electron beam IR that these strains can withstand far exceeds that of previously evolved IR-resistant isolates (45, 46), at least when the new and higher dose rate is applied. These populations vary in their growth phenotypes (Fig. 3B and 4) and resistance to other forms of DNA damage (Fig. 6 and 7), indicating that they are specialized to withstand high doses of IR.
Most laboratory exercises in experimental evolution have been characterized by a rapid decline in the rate of fitness increase, as fitness approaches some biologically determined optimum for a given agent of selection (20, 57, 58). One key difference between the current experiment and many other long-term directed evolution experiments is that the selection agent here is not a constant. The dose of IR inflicted on the populations increases as resistance levels increase. Increased selection pressure to meet a defined, desired phenotype throughout selection is not a novel protocol (58), yet in our evolution experiment there is no evident deceleration in the rate of fitness gains. Genetic patterns (parallelism) have allowed us to identify some key contributors to the growing IR resistance phenotype. However, the continuing advance of IR resistance and allele diversity in all populations also suggests that the pool of potential adaptive mutations is much greater than that now present in any one population. As selection continues, substantial additional gains in unique pathways may be expected. Ionizing radiation is not only a selection agent but also a potent mutagen. Although this study was not initiated with the goal of exploring mechanisms of molecular evolution, we anticipate that the data sets provided in Data Sets S1 through S4 in the supplemental material will be of use to investigators in that field.
Modified DNA repair proteins and RNA polymerase enhance IR resistance.
Each population has traversed unique evolutionary paths to reach the same goal (Fig. 8A to D). Despite varied levels of clonal interference in each population, significant genetic parallelism in these populations allow us to identify at least some of the contributors to extreme IR resistance. In one isolate from population IR9-50, IR9-50-1, mutations in recD, recN, and rpoB/rpoC increase IR resistance of the Founder Δe14 parent derivative. However, these variants—singly or together—do not account entirely for the IR resistance phenotype of IR9-50-1 at a dose of 1,000 Gy (Fig. 9A and B). None of these genes have been previously implicated as mutational targets contributing to IR resistance (45–47).
The effects of mutations in these genes reveal complex interactions. When present in an otherwise wild-type background, the mutations in recD and rpoB exhibit a clear contribution to IR resistance, although results with the recN mutation were variable. The recD and rpoB effects were not additive, and combining them demonstrated diminishing returns epistasis. When the rpoB mutation was reverted to wild type in the IR9-50-1 mutant background, the IR resistance phenotype was reduced little or not at all. However, reverting the recD and recN mutation to wild type resulted in a considerable loss of IR resistance. It appears likely that a complete genetic deciphering of the IR resistance of IR9-50-1 and other isolates will require a more systematic consideration of relationships between mutations and genetic backgrounds.
Although additional contributions are clearly present, the alterations in RecD, RecN, and RpoB/RpoC (Fig. 9) provide the clearest evidence for phenotypic contributions in the current work. RecD is part of the RecBCD heterotrimer responsible for preparing ssDNA required for RecA loading, initiating homologous recombination. The RecD A90E variant is likely a loss of function mutation, as a RecD deletion in the Founder Δe14 background increases IR resistance as much as the A90E variant in this background. RecD inactivation produces a hyperrecombination phenotype (59–62). Increased homologous recombination could be of significant use to repair highly fragmented DNA post-IR, with approximately 15 DSBs generated at 1,000 Gy (1).
RecN is a cohesin-like protein which is involved in RecA-mediated double-strand break repair (63–66). There is little known about the precise function of RecN, though it has been implicated in maintaining proximity of broken dsDNA ends to an active RecA filament. The function of the K429Q variant is unknown, but the K429 residue is positioned in a RecN domain highly conserved among bacteria. The K429Q variant is unlikely to be a loss of function, since recN deletion greatly enhances IR sensitivity (46, 67). Further work on these RecN variants may shed new light on the function of the RecN protein.
RpoB and RpoC are the beta and beta-prime subunits of RNA polymerase, respectively. Stringent mutations of RNAP, which mimic the effects of ppGpp binding and therefore the stringent response, are located primarily in RpoB and RpoC (68). Some of these stringent mutants are capable of rescuing UV sensitivity of ruvABC mutants of E. coli, potentially due to decreased stability of contacts of RNAP with DNA (69, 70). Similar to these previous observations, the mutations in RpoB that are prevalent in our evolved populations do not locate to a single region of RpoB and therefore may affect DNA interaction throughout the DNA channel formed by RpoB/RpoC. Previously described stringent mutations L571Q and H1244Q mutants (69, 70) affect residues near those affected in the present study. These mutations likely decrease stability of RNAP on DNA and may allow for easier removal of RNAP that has stalled at a DNA lesion. Removal of stalled RNAP may be crucial for efficient DNA repair due to RNAP occluding the lesion from repair machinery or providing a major obstacle to DNA replication (71). In addition, many stringent mutants of RNAP also confer resistance to the antibiotic Rifampicin. Mutated variants of RNAP at P535 (72) and S574 (73–75) (which have also been isolated in this study) have previously been isolated using selection for rifampin resistance. Although these are the first RNA polymerase mutations detected in E. coli evolved for IR resistance, similar mutants have been generated during experimental evolution of E. coli for trimethoprim and doxycycline resistance (58), heat tolerance (76, 77), growth in nutrient-limited conditions (78, 79), and acid resistance (80, 81). Modifications in RNA polymerase may confer enhanced fitness throughout serial passaging, a common feature of experimental evolution studies. This advantage, combined with a potential ability to enhance DNA repair, may explain the rapid appearance of rpoB or rpoC alleles in each evolving lineage (except IR10).
Excision of the e14 prophage is induced by selection for IR resistance.
The only event common to each of the four populations is the excision of the e14 prophage, which was evident by round two of selection. However, in lineage IR10 the subpopulation that lost e14 was outcompeted and driven extinct by cycle ten. Despite no apparent mutations within e14 that may prevent its excision, the e14 prophage remains within lineage IR10. It is unclear why e14 excision is the first clear event to happen in each population (an event that also occurred early in evolution of our previously evolved gamma ray IR-resistant E. coli populations (45, 46). Excision of the e14 prophage is predicted to be under regulation of the SOS response, this event may simply be an artifact of the E. coli DNA damage response (82). However, the e14 genome does encode two potentially lethal cell division inhibitor proteins: a homologue of the lambda phage protein Kil and SfiC. Loss of these proteins may itself be selective pressure. Indeed, loss of the e14 has been previously noted during experimental evolution (83) Interestingly, deletion of the e14 prophage has also been observed to cause variable resistance to many antimicrobial agents, including increased resistance to hydrogen peroxide (84).
Direct competition between two pathways to IR resistance dictates adaptation.
Population sequencing also reveals the underlying competition of lineages within each population. Clonal interference is a hallmark of these populations. While much of this study focuses on the current endpoint (each population after 50 cycles of selection), observing evolution over the remaining 49 cycles allows us to understand the genetic context in which new mutations arise and how the successful lineages were able to outcompete other fit, but ultimately unsuccessful, lineages. This competition within populations was almost immediately apparent.
There is an apparent competition between lineages which contain alleles of rpoB and those with recA alleles. In the current protocol, with its much higher IR dose rate, recA alleles are less common than observed in our earlier study, and novel rpoB, recD, and recN alleles predominate. In IR9, a single recA variant appeared (Y294C) but only reached approximately 12% frequency in the population before being driven extinct by round 20 of selection. This population successfully lost the e14 prophage and then gained an rpoB allele followed by recD and recN alleles. The appearance of recD and recN alleles in tandem occurred in three of the four populations, and each contributes similarly to IR resistance in a wild-type background (Fig. S4). In IR10, the RecA/RecD variant pair appears very early, and rapidly outcompetes the lineage that lost the e14 prophage. After this event, the only mutation that became fixed within the population (of those common to all four populations which we are focusing on) is another RecD variant. RpoB variants do not appear until at least round 40 of selection in IR10, and by round 50, these variants do not yet appear to be on a path to fixation. No high-frequency recN allele has appeared in IR10. The IR11 and IR12 lineages followed a path similar to that taken by IR9. In the IR11 lineage, loss of the e14 prophage and subsequent gain of an rpoB allele became fixed. In this genetic background, two separate subpopulations containing RecN/RecD and RecA/RecD variants, respectively, later reached prominence. The RecN/RecD subpopulation gained a variant of RpoB which assisted in driving this subpopulation to fixation and the RecA/RecD to extinction. Finally, IR12 almost began on a similar evolutionary trajectory as IR10. A subpopulation that lost the e14 prophage and gained an rpoB allele (which leads to a synonymous coding mutation) was nearly outcompeted by a RecA/RecD variant subpopulation, before a RecN/RecD variant pair rescued the subpopulation that had lost e14. By round 40 of selection, the winning subpopulation reached fixation.
Despite our focus on the genetic parallelism between these four populations, it is evident that each population has begun to develop unique adaptations. Of the total number of fixation events in each lineage (excluding excision of the e14 prophage, IR9, 2; IR10, 5; IR11, 2; and IR12, 2) tracking mutations in rpoB-recA-recD-recN only fully accounts for such events in IR9 and IR11. In all four lineages, major subpopulations that have arisen after fixation of rpoB-recA-recD-recN combinations are completely unaccounted for by the criteria used to identify the mutations tested in this study. While it is clear that e14 excision and competition between RecA/RecD and RpoB/RecD/RecN provides a path to enhanced IR resistance, this appears to be only the first step on a complicated fitness landscape. New methods must be applied to determine the unique paths taken by each of the four lineages.
Source of IR and dose rate alter the molecular basis for experimentally evolved IR resistance.
Populations IR9-50, IR10-50, IR11-50, and IR12-50 provide us with a platform for understanding the molecular fundamentals of extreme IR resistance. These populations have been evolved using a source of IR with a much higher dose rate than that used previously to evolve IR resistance in E. coli (72 Gy/min versus 19 Gy/min) (45). Isolates from the prior evolved populations, CB2000 and CB3000 (45, 46) are only moderately more resistant to this form of IR compared to the parent strain. The higher dose rate in the current protocol may account for the relative sensitivity of the strains derived from the earlier evolution trial.
There are distinct differences in the mutations which appear in these populations and those that were previously evolved (45, 46). Only a single mutation previously noted to enhance IR resistance, RecA A290S (45), appears in the four new populations. While enhanced DNA repair is a hallmark of the previously evolved populations and the populations presented here, it appears as though the enhancements are indeed different. In addition, the frequency of transversion mutations detected in the new populations (∼50% of all mutations) is far higher than that observed in the earlier studies (∼20% of all mutations) (45, 55). This appears to be due to the extreme amount of detected GC-to-TA transitions, specifically. This transition is a hallmark of A mispairing with template strand 8-oxodG (56), indicating that DNA damage may be more extreme in cells exposed to electron beam IR. However, the high energy electrons and gamma rays produce similar levels of killing (Fig. 1). A more likely explanation lies with the differences in the dose rate applied. In any case, these new E. coli lineages are replete with novel mechanisms of experimentally evolved IR resistance.
We do not yet have experimentally evolved E. coli that can match the natural IR resistance of the bacterium D. radiodurans. However, selection is continuing.
MATERIALS AND METHODS
Growth conditions and bacterial strains used in this study.
Unless otherwise stated, E. coli cultures were grown in LB broth (85) at 37°C with aeration. E. coli were plated on 1.5% LB agar medium (85) and incubated at 37°C. Overnight cultures were grown in a volume of 3 ml for 16 to 18 h. Exponential-phase cultures were routinely diluted 1:100 in 10 ml of LB medium in a 50-ml Erlenmeyer flask and were grown at 37°C with shaking at 200 rpm and were harvested at an optical density at 600 nm (OD600) of 0.2, unless otherwise noted. After growth to an OD600 of 0.2, cultures were placed on ice for 10 min to stop growth before being used for assays.
Cultures were plated on tetrazolium agar (TA) for growth competition assays when noted (48, 86). The defined rich medium EZ was mixed per manufacturer’s specifications and was supplemented with 0.2% glucose or 0.2% glycerol as indicated (Teknova, Hollister, CA). M9 minimal medium (supplemented with 0.2% glucose) was used when indicated (85).
All strains used for in vivo assays in this study are mutants of E. coli K-12 derivative MG1655. Genetic manipulations to transfer mutations or delete genes were performed as previously described (87, 88). Strains used in this study are listed in Table 4.
TABLE 4.
Strains used in this studya
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| MG1655 | YbhJ L54I MntP G25D RIP321 A4296380*ACG GlpR C3560455*CG GatC ACC2173360*A | 91 |
| EAW7704 (founder Δe14) | MG1655 Δe14 RbsR L92R CytR Q110stop fabI/ycjD int (G1351174*A) yifN/ppiC int (T3959934*C) | 45 |
| IR9-50 | MG1655 exposed to 50 iterative rounds of IR; mixed population | This study |
| IR10-50 | MG1655 exposed to 50 iterative rounds of IR; mixed population | This study |
| IR11-50 | MG1655 exposed to 50 iterative rounds of IR; mixed population | This study |
| IR12-50 | MG1655 exposed to 50 iterative rounds of IR; mixed population | This study |
| IR9-50-1 | Isolate from IR9-50 | This study |
| IR9-50-2 | Isolate from IR9-50 | This study |
| IR9-50-3 | Isolate from IR9-50 | This study |
| IR9-50-4 | Isolate from IR9-50 | This study |
| IR9-50-5 | Isolate from IR9-50 | This study |
| IR10-50-1 | Isolate from IR10-50 | This study |
| IR10-50-2 | Isolate from IR10-50 | This study |
| IR10-50-3 | Isolate from IR10-50 | This study |
| IR10-50-4 | Isolate from IR10-50 | This study |
| IR10-50-5 | Isolate from IR10-50 | This study |
| IR11-50-1 | Isolate from IR11-50 | This study |
| IR11-50-2 | Isolate from IR11-50 | This study |
| IR11-50-3 | Isolate from IR11-50 | This study |
| IR11-50-4 | Isolate from IR11-50 | This study |
| IR11-50-5 | Isolate from IR11-50 | This study |
| IR12-50-1 | Isolate from IR12-50 | This study |
| IR12-50-2 | Isolate from IR12-50 | This study |
| IR12-50-3 | Isolate from IR12-50 | This study |
| IR12-50-4 | Isolate from IR12-50 | This study |
| IR12-50-5 | Isolate from IR12-50 | This study |
| CB2000 | Isolate from IR-resistant evolved population IR-2-20 | 45 |
| CB3000 | Isolate from IR-resistant evolved population IR-3-20 | 45 |
| EAW792 | Founder Δe14 ArcB N405D | This study |
| EAW971 | Founder Δe14 CopA V270F | This study |
| EAW766 | Founder Δe14 DinI R28H | This study |
| JDT56 | Founder Δe14 RecA A290S | This study |
| EAW726 | Founder Δe14 RecD A90E | This study |
| EAW725 | Founder Δe14 RecN K429Q | This study |
| EAW756 | Founder Δe14 RpoB S72N/RpoC K1172I | This study |
| EAW748 | Founder Δe14 RecD A90E RecN K428H | This study |
| EAW904 | Founder Δe14 RecD A90E RpoB S72N/RpoC K1172I | This study |
| EAW781 | Founder Δe14 RecD A90E RecN K428H RpoB S72N/RpoC K1172I | This study |
| EAW972 | IR9-50-1 CopA V270 wt | This study |
| EAW1047 | IR9-50-1 DinI R28 wt | This study |
| EAW973 | IR9-50-1 RecD A90 wt | This study |
| EA1024 | IR9-50-1 RecN K429 wt | This study |
| EAW981 | IR9-50-1 RpoB S72/RpoC K1172 wt | This study |
| EAW1021 | IR9-50-1 RecD A90 wt RpoB S72/RpoC K1172 wt | This study |
| EAW1046 | IR9-50-1 RecD A90 wt RpoB S72/RpoC K1172 wt RecN K429 wt | This study |
| STB75 | Founder Δe14 ΔaraBAD | 47 |
Strains generated in this study were constructed as described in Materials and Methods. A “wt” designation indicates that the protein is variant at the indicated allele in the evolved isolate but has been swapped to the wild-type (MG1655) allele. An asterisk indicates that this nucleotide position is in reference to the NCBI GenBank U00096.3 reference sequence.
Serial dilutions and CFU/milliliter determination.
All serial dilutions were performed in 1× phosphate-buffered saline (PBS; for 1 liter: 8 g of NaCl, 0.2 g of KCl, 1.44 g of Na2HPO4, and 0.24 g of KH2PO4 with 800 ml of dH2O, adjusting the pH with HCl to 7.4 and then adding the remaining 200 ml of dH2O). Unless otherwise stated, serial dilutions were performed with serial 1:10 dilutions of 100 μl of culture or previous dilution into 900 μl of 1× PBS. Before transfer to the next dilution tube, samples were vortexed for 2 s and mixed by pipetting to ensure mixing. Appropriate dilutions (100 μl) were aliquoted onto agar plates of the appropriate medium and spread plated utilizing an ethanol-sterilized, bent glass rod. For spot plating, 10 μl of each dilution was aliquoted onto agar plates of the appropriate medium and spots were allowed to dry before plates were incubated as described above.
CFU/milliliter was calculated using the highest CFU count for each strain assayed that remained between 30 and 300 CFU (e.g., 250 CFU on a 10−4 dilution plate would be used for calculation over 40 CFU on a 10−5 dilution plate).
TLD dose validation.
An independent dose verification was performed with thermoluminescent dosimeters (TLDs). TLDs are passive dosimeters that are small, accurate and well suited for dose verification in the routinely used 1.5-ml sample vials. For this project, three TLDs were sealed in a small plastic bag in 1.5-ml tubes containing 900 μl of dH2O, placed horizontally and submerged under 1.3-cm dH2O, and irradiated as described in “Generalized Linac irradiation protocol” below. After the vials were irradiated, the TLDs were read out in the University of Wisconsin Medical Radiation Research Center (UWMRRC) TLD lab and compared to the calculated dose. Nonirradiated control TLDs were read out simultaneously to account for any background radiation. The TLDs were calibrated with a separate 60Co beam in the University of Wisconsin Accredited Dosimetry Calibration Laboratory (UWADCL), which provides independent National Institute of Standards and Technology (NIST) traceability for the measurements. This serves as an independent check of the ion chamber-based dose calculation method used to determine the beam-on time for this project. The estimated overall uncertainty on the TLD measured values is ±5% with a coverage factor of k = 1.
Generalized Linac irradiation protocol.
Samples were maintained at 4°C and transported to the UWMRRC Varian 21EX clinical linear accelerator (Linac) facility for irradiation. The total transport time was approximately 15 min to and from the Linac facility. For each irradiation, the Linac was set to deliver a beam of electrons with 6 MeV of energy to uniformly irradiate all samples (a total of 14) at once. To accomplish this, a special high-dose mode called HDTSē was utilized, which resulted in a dose rate to the samples of approximately 72 Gy/min. The sample tubes were placed horizontally and submerged at a depth of 1.3 cm (measured to the center of the tube’s volume) in an ice-water filled plastic tank and set to a source-to-surface distance (SSD) of 61.7 cm. A 30- by 30-cm2 square field size was set at the Linac console, which gave an effective field size at this SSD of 18.5 by 18.5 cm2. This is ample coverage to provide a uniform dose to all of the sample vials. The monitor unit calculations (determination of the amount of time to leave the Linac on) were based on the American Association of Physicists in Medicine (AAPM) Task Group 51 protocol for reference dosimetry (89). This is the standard method for determining dose per monitor unit in water for radiation therapy calculations. Once the dose was determined in the AAPM Task Group 51 reference protocol conditions (SSD = 100 cm and depth = 10 cm), an ion chamber and water-equivalent plastic slabs were used to translate this dose to the specific conditions used in this project.
Linac photon mode.
The Linac is designed to deliver dose with either electron or photon beams. In photon mode, the electron beam first strikes a flattening filter (not present in electron mode) to produce bremsstrahlung photons and also flatten the beam intensity profile. In order to rule out any effective differences between electron and photon modes during irradiations, the sample vials were irradiated with the same dose rates for both modes by altering the source-to-surface-distance (SSD) for each mode.
Ionizing radiation resistance assay using the Linac.
Strains were grown in biological triplicate overnight and to an OD600 of approximately 0.2 in LB medium as routinely performed. A 1-ml sample for each dose tested (including 0 Gy) was removed and aliquoted into a sterile 1.5-ml microcentrifuge tube. Samples were pelleted by centrifugation at 13 × g for 1 min, and the supernatant was poured off. Samples were resuspended in 1 ml ice-cold 1× PBS, and pelleting was repeated. This process was repeated three more times to wash cells. A 100-μl aliquot of each culture was removed, serial diluted 1:10 in 900 μl of PBS to a final 10,000-fold dilution, and 100 μl was plated on LB agar to determine the CFU/milliliter before irradiation. Samples were maintained at 4°C and irradiated with the appropriate doses as described. A 100-μl aliquot of each culture was removed and plated to determine the CFU/milliliter and percent survival as described.
Directed evolution protocol using Linac.
For each round of directed evolution, separate aliquots of 2 ml of LB medium was inoculated with frozen stock of each population from the previous round of selection. These were incubated overnight with aeration at 37°C and were grown with usual practices in LB medium to an OD600 of 0.2 the next day. Each culture was incubated on ice for 10 min to stop growth. Three 1-ml samples were removed and aliquoted into sterile 1.5-ml microcentrifuge tubes. Samples were washed three times with 1 ml of ice-cold 1× PBS and resuspended in a final volume of 1 ml of 1× PBS. A 100-μl aliquot of each culture was removed, serial diluted 1:10 in 900 μl of PBS to a final 10,000-fold dilution, and 100 μl was plated on LB agar to determine the CFU/milliliter before irradiation. Samples were maintained at 4°C and taken to a Varian 21EX clinical linear accelerator (Linac) for irradiation.
After irradiation, an aliquot of each culture was removed, serial diluted 1:10 in 900 μl of PBS to a final 1,000-fold dilution, and 100 μl of each serial dilution was plated on LB agar for each dose to determine the CFU/milliliter after irradiation. LB agar plates were incubated overnight at 37°C. Remaining irradiated cultures were pelleted by centrifugation at 13 × g, and the supernatant was discarded. These pellets were resuspended in 1 ml of fresh LB medium, and this was added to 1 ml of LB medium in a 5-ml glass culture tube. These resuspensions were incubated overnight with aeration at 37°C. The following day, the percent survival for each dose was calculated using CFU/milliliter calculations before and after irradiation at each dose. The overnight culture of each population replicate showing closest to 1% survival was stored at –80°C and used for the next cycle of selection. One cycle of selection was performed weekly due to limited access to the Linac.
The initiating round of selection was done as described above, except the original culture used was an overnight culture of MG1655 prepared from an isolated colony. This protocol was adapted from a previously used protocol (45).
UV resistance assay.
Cells from a single colony of each strain were cultured overnight and then grown to an OD600 of ∼0.2 as described above in “Growth conditions and bacterial strains used in this study.” Samples were diluted and spotted onto 25 ml of 1.5% LB agar as described above. Spots were dried before the plate lid was removed, and the spots were exposed to the appropriate dose of UV irradiation using a Spectrolinker XL-1000 UV cross-linker (Spectronics Corporation, Westbury, NY). Plates were imaged after incubation for 24 h.
Resistance to DNA-damaging agents.
Cells were grown overnight to an OD600 of approximately 0.2 in LB medium as routinely performed. Samples were mixed by vortexing for 5 s and were serially diluted 1:10 in 900 μl of PBS to a final 100,000-fold dilution. A 10-μl portion was removed from each dilution and spotted onto 30 ml of 1.5% LB agar medium supplemented with 10 or 7.5 ng/ml ciprofloxacin hydrochloride as specified, 0.5 μM bleomycin, 4 μg/ml mitomycin C, or 5 mM hydroxyurea. Spots were dried before being incubated overnight at 37°C. Plates were imaged after 48 h for ciprofloxacin and bleomycin plates.
Growth competition assay.
This assay was adapted from a previously published protocol (46, 48). To differentiate strains within the competition, a fitness-neutral deletion of the araBAD operon was introduced into one of the two strains. This deletion results in red colonies on tetrazolium arabinose (TA) agar plates (48). Briefly, overnight culture of each strain to be competed were mixed 1:1 or 1:9 (for growth competitions using isolates from the evolved populations after round 50 of selection or RpoB or ArcB mutations, in favor of the losing competitor) in a 1.5-ml tube. Samples were mixed by vortexing for 5 s and were serial diluted 1:10 in 900 μl of PBS to a final dilution of 1:100,000. A 100-μl portion of the final dilution was spread plated onto TA agar plates to assay for CFU. Then, 70 μl of the remaining cell mixture was used to inoculate 5 ml of fresh LB medium for growth overnight. This overnight culture was used to inoculate fresh medium the following day, and 100 μl was serially diluted and plated as noted above. This procedure was repeated twice more over a period of 2 days. The number of white versus red CFU was noted after each day of the competition, and the total percentage of the culture for each competitor was determined.
Deep sequencing.
Genomic DNA was prepped from overnight cultures prepared from frozen stocks of populations from every even round of selection using the Wizard Genomic DNA purification kit (Promega, Madison, WI). DNA samples were submitted to the Department of Energy Joint Genome Institute (Walnut Creek, CA) for sequencing and analysis. DNA was randomly sheared into ∼500-bp fragments, and the resulting fragments were used to create an Illumina library. This library was sequenced on Illumina HiSeq generating 100-bp paired-end reads. Reads were aligned to the reference genome using BWA (90), downsampled to an average depth of 250-fold coverage with Picard (http://broadinstitute.github.io/picard), and putative mutations and small indels were called using callvariants.sh from BBMap (sourceforge.net/projects/bbmap).
Sequencing results are reported in their entirety in Data Sets S1 through S4 in the supplemental material. However, for analysis of numbers and types of mutations in each population (and consequently for generating Fig. 8 and Tables 1 to 3), mutations in genes with high homology throughout the genome (as in rrs, rrl, rrn, rhs, and ins genes) were not considered due to increased likelihood of a false-positive mutation call. In addition, mutations with inconsistent frequency calls (e.g., jumping from 0 to 100 to 0% allele frequency) were also not used. All mutations removed from consideration are also listed in Data Sets S1 through S4.
Growth curves.
Strains were cultured described as above overnight and to an OD600 of 0.2 in LB medium, EZ medium supplemented with 0.2% glucose (Teknova, Hollister, CA) and M9 minimal medium (85) supplemented with 0.2% glucose. Cultures were then diluted 1:100 in the appropriate medium in a clear, flat-bottom 96-well plate (Fisher, product number 07-200-656) and incubated overnight in a Synergy 2 plate reader (BioTek, Winooski, VT) at 37°C with shaking, with OD600 readings taken by the plate reader every 10 min.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grant GM112757 from the National Institute of General Medical Sciences (NIGMS) and by grants 2817 and 502930 from the Joint Genome Institute, U.S. Department of Energy. S.T.B. was supported by a Morgridge Biotechnology Scholarship from the Vice Chancellor’s Office for Research and Graduate Education, University of Wisconsin—Madison. J.D.T. was supported by a Hilldale Undergraduate Fellowship (University of Wisconsin—Madison), an Honors Senior Thesis Summer Research Grant (UW—Madison), and a Biochemistry Undergraduate Summer Research Award (UW—Madison).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00784-18.
REFERENCES
- 1.Daly MJ. 2012. Death by protein damage in irradiated cells. DNA Repair (Amst) 11:12–21. doi: 10.1016/j.dnarep.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 2.Cox MM, Battista JR. 2005. Deinococcus radiodurans: the consummate survivor. Nat Rev Microbiol 3:882–892. doi: 10.1038/nrmicro1264. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka M, Earl AM, Howell HA, Park MJ, Eisen JA, Peterson SN, Battista JR. 2004. Analysis of Deinococcus radiodurans’ transcriptional response to ionizing radiation and desiccation reveals novel proteins that contribute to extreme radioresistance. Genetics 168:21–33. doi: 10.1534/genetics.104.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fredrickson JK, Li SM, Gaidamakova EK, Matrosova VY, Zhai M, Sulloway HM, Scholten JC, Brown MG, Balkwill DL, Daly MJ. 2008. Protein oxidation: key to bacterial desiccation resistance? ISME J 2:393–403. doi: 10.1038/ismej.2007.116. [DOI] [PubMed] [Google Scholar]
- 5.Battista JR, Earl AM, Park MJ. 1999. Why is Deinococcus radiodurans so resistant to ionizing radiation? Trends Microbiol 7:362–365. doi: 10.1016/S0966-842X(99)01566-8. [DOI] [PubMed] [Google Scholar]
- 6.Rainey FA, Ray K, Ferreira M, Gatz BZ, Nobre MF, Bagaley D, Rash BA, Park MJ, Earl AM, Shank NC, Small AM, Henk MC, Battista JR, Kampfer P, da Costa MS. 2005. Extensive diversity of ionizing-radiation-resistant bacteria recovered from Sonoran desert soil and description of nine new species of the genus Deinococcus obtained from a single soil sample. Appl Environ Microbiol 71:5225–5235. doi: 10.1128/AEM.71.9.5225-5235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rainey FA, Nobre MF, Schumann P, Stackebrandt E, da Costa MS. 1997. Phylogenetic diversity of the deinococci as determined by 16s ribosomal DNA sequence comparison. Int J Syst Bacteriol 47:510–514. doi: 10.1099/00207713-47-2-510. [DOI] [PubMed] [Google Scholar]
- 8.Daly MJ, Gaidamakova EK, Matrosova VY, Kiang JG, Fukumoto R, Lee DY, Wehr NB, Viteri GA, Berlett BS, Levine RL. 2010. Small-molecule antioxidant proteome-shields in Deinococcus radiodurans. PLoS One 5:e12570. doi: 10.1371/journal.pone.0012570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Venkateswaran A, Hess M, Omelchenko MV, Kostandarithes HM, Makarova KS, Wackett LP, Fredrickson JK, Ghosal D. 2004. Accumulation of Mn(II) in, Deinococcus radiodurans facilitates gamma-radiation resistance. Science 306:1025–1028. doi: 10.1126/science.1103185. [DOI] [PubMed] [Google Scholar]
- 10.Slade D, Radman M. 2011. Oxidative stress resistance in Deinococcus radiodurans. Microbiol Mol Biol Rev 75:133–191. doi: 10.1128/MMBR.00015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvam K, Duncan JR, Tanaka M, Battista JR. 2013. DdrA, DdrD, and PprA: components of UV and mitomycin C resistance in Deinococcus radiodurans R1. PLoS One 8:e69007. doi: 10.1371/journal.pone.0069007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Earl AM, Mohundro MM, Mian IS, Battista JR. 2002. The IrrE protein of Deinococcus radiodurans R1 is a novel regulator of recA expression. J Bacteriol 184:6216–6224. doi: 10.1128/JB.184.22.6216-6224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris DR, Tanaka M, Saveliev SV, Jolivet E, Earl AM, Cox MM, Battista JR. 2004. Preserving genome integrity: the DdrA protein of Deinococcus radiodurans R1. PLoS Biol 2:e304. doi: 10.1371/journal.pbio.0020304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witkin EM. 1946. Inherited differences in sensitivity to radiation in Escherichia coli. Proc Natl Acad Sci U S A 32:59–68. doi: 10.1073/pnas.32.3.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parisi A, Antoine AD. 1974. Increased radiation resistance of vegetative Bacillus pumilus. Appl Microbiol 28:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies R, Sinskey AJ. 1973. Radiation-resistant mutants of Salmonella typhimurium LT2: development and characterization. J Bacteriol 113:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erdman IE, Thatcher FS, Macqueen KF. 1961. Studies on the irradiation of microorganisms in relation to food preservation. II. Irradiation-resistant mutants. Can J Microbiol 7:207–215. doi: 10.1139/m61-027. [DOI] [PubMed] [Google Scholar]
- 18.Chou HH, Chiu HC, Delaney NF, Segre D, Marx CJ. 2011. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper TF, Rozen DE, Lenski RE. 2003. Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli. Proc Natl Acad Sci U S A 100:1072–1077. doi: 10.1073/pnas.0334340100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. 2017. The dynamics of molecular evolution over 60,000 generations. Nature 551:45–50. doi: 10.1038/nature24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieberman TD, Michel JB, Aingaran M, Potter-Bynoe G, Roux D, Davis MR, Skurnik D, Leiby N, LiPuma JJ, Goldberg JB, McAdam AJ, Priebe GP, Kishony R. 2011. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet 43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Medigue C, Schneider D, Lenski RE. 2016. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536:165–170. doi: 10.1038/nature18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wannier TM, Kunjapur AM, Rice DP, McDonald MJ, Desai MM, Church GM. 2018. Adaptive evolution of genomically recoded Escherichia coli. Proc Natl Acad Sci U S A 115:3090–3095. doi: 10.1073/pnas.1715530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woods R, Schneider D, Winkworth CL, Riley MA, Lenski RE. 2006. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A 103:9107–9112. doi: 10.1073/pnas.0602917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luksza M, Lassig M. 2014. A predictive fitness model for influenza. Nature 507:57–61. doi: 10.1038/nature13087. [DOI] [PubMed] [Google Scholar]
- 26.Miller CR, Joyce P, Wichman HA. 2011. Mutational effects and population dynamics during viral adaptation challenge current models. Genetics 187:185–202. doi: 10.1534/genetics.110.121400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller CR, Nagel AC, Scott L, Settles M, Joyce P, Wichman HA. 2016. Love the one you’re with: replicate viral adaptations converge on the same phenotypic change. PeerJ 4:e2227. doi: 10.7717/peerj.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris DH, Gostic KM, Pompei S, Bedford T, Łuksza M, Neher RA, Grenfell BT, Lässig M, McCauley JW. 2018. Predictive modeling of influenza shows the promise of applied evolutionary biology. Trends Microbiol 26:102–118. doi: 10.1016/j.tim.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zanini F, Brodin J, Thebo L, Lanz C, Bratt G, Albert J, Neher RA. 2015. Population genomics of intrapatient HIV-1 evolution. Elife 4:e11282. doi: 10.7554/eLife.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kvitek DJ, Sherlock G. 2013. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet 9:e1003972. doi: 10.1371/journal.pgen.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM. 2013. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500:571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Andrew Futreal P, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Australian Pancreatic Cancer, Genome, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain . 2013. Signatures of mutational processes in human cancer. Nature 500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. 2015. Clock-like mutational processes in human somatic cells. Nat Genet 47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR. 2016. Mutational signatures associated with tobacco smoking in human cancer. Science 354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, Dawson KJ, Iorio F, Nik-Zainal S, Bignell GR, Hinton JW, Li YL, Tubio JMC, McLaren S, Meara SO, Butler AP, Teague JW, Mudie L, Anderson E, Rashid N, Tai YT, Shammas MA, Sperling AS, Fulciniti M, Richardson PG, Parmigiani G, Magrangeas F, Minvielle S, Moreau P, Attal M, Facon T, Futreal PA, Anderson KC, Campbell PJ, Munshi NC. 2014. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, Brewer DS, Kallio HML, Hoegnas G, Annala M, Kivinummi K, Goody V, Latimer C, O’Meara S, Dawson KJ, Isaacs W, Emmert-Buck MR, Nykter M, Foster C, Kote-Jarai Z, Easton D, Whitaker HC, Neal DE, Cooper CS, Eeles RA, Visakorpi T, Campbell PJ, McDermott U, Wedge DC, Bova GS. 2015. The evolutionary history of lethal metastatic prostate cancer. Nature 520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter KW, Amin R, Deasy S, Ha NH, Wakefield L. 2018. Genetic insights into the morass of metastatic heterogeneity. Nat Rev Cancer 18:211–223. doi: 10.1038/nrc.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kandoth C, McLellan MD, Vandin F, Ye K, Niu BF, Lu C, Xie MC, Zhang QY, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martincorena I, Campbell PJ. 2015. Somatic mutation in cancer and normal cells. Science 349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 40.Morganella S, Alexandrov LB, Glodzik D, Zou XQ, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, Butler A, Kim HY, Borg A, Sotiriou C, Futreal PA, Campbell PJ, Span PN, Van Laere S, Lakhani SR, Eyfjord JE, Thompson AM, Stunnenberg HG, Vijver Mjv d, Martens JWM, Borresen-Dale AL, Richardson AL, Kong G, Thomas G, Sale J, Rada C, Stratton MR, Birney E, Nik-Zainal S. 2016. The topography of mutational processes in breast cancer genomes. Nat Commun 7:11383. doi: 10.1038/ncomms11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, Menzies A, Stebbings LA, Leroy C, Jia MM, Rance R, Mudie LJ, Gamble SJ, Stephens PJ, McLaren S, Tarpey PS, Papaemmanuil E, Davies HR, Varela I, McBride DJ, Bignell GR, Leung K, Butler AP, Teague JW, Martin S, Jonsson G, Mariani O, Boyault S, Miron P, Fatima A, Langerod A, Aparicio S, Tutt A, Sieuwerts AM, Borg A, Thomas G, Salomon AV, Richardson AL, Borresen-Dale AL, Futreal PA, Stratton MR, Campbell PJ. 2012. The life history of 21 breast cancers. Cell 149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou SB, Diaz LA, Kinzler KW. 2013. Cancer genome landscapes. Science 339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M, Zamora J, Butler A, Whitaker H, Kote-Jarai Z, Alexandrov LB, Van Loo P, Massie CE, Dentro S, Warren AY, Verrill C, Berney DM, Dennis N, Merson S, Hawkins S, Howat W, Lu YJ, Lambert A, Kay J, Kremeyer B, Karaszi K, Luxton H, Camacho N, Marsden L, Edwards S, Matthews L, Bo V, Leongamornlert D, McLaren S, Ng A, Yu YW, Zhang HW, Dadaev T, Thomas S, Easton DF, Ahmed M, Bancroft E, Fisher C, Livni N, Nicol D, Tavare S, Gill P, Greenman C, Khoo V, Van As N, Kumar P, Ogden C, Cahill D, Thompson A, Mayer E, Rowe E, Dudderidge T, Gnanapragasam V, Shah NC, Raine K, Jones D, Menzies A, Stebbings L, Teague J, Hazell S, Corbishley C, de Bono J, Attard G, Isaacs W, Visakorpi T, Fraser M, Boutros PC, Bristow RG, Workman P, Sander C, Hamdy FC, Futreal A, McDermott U, Al-Lazikani B, Lynch AG, Bova GS, Foster CS, Brewer DS, Neal DE, Cooper CS, Eeles RA, Camcap S, TCGA Consortium . 2018. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet 50:682–692. doi: 10.1038/s41588-018-0086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rokyta DR, Joyce P, Caudle SB, Miller C, Beisel CJ, Wichman HA. 2011. Epistasis between beneficial mutations and the phenotype-to-fitness map for a ssDNA virus. PLoS Genet 7:e1002075. doi: 10.1371/journal.pgen.1002075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris DR, Pollock SV, Wood EA, Goiffon RJ, Klingele AJ, Cabot EL, Schackwitz W, Martin J, Eggington J, Durfee TJ, Middle CM, Norton JE, Popelars M, Li H, Klugman SA, Hamilton LL, Bane LB, Pennacchio L, Albert TJ, Perna NT, Cox MM, Battista JR. 2009. Directed evolution of radiation resistance in Escherichia coli. J Bacteriol 191:5240–5252. doi: 10.1128/JB.00502-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Byrne RT, Klingele AJ, Cabot EL, Schackwitz WS, Martin JA, Martin J, Wang Z, Wood EA, Pennacchio C, Pennacchio LA, Perna NT, Battista JR, Cox MM. 2014. Evolution of extreme resistance to ionizing radiation via genetic adaptation of DNA repair. Elife 3:e01322. doi: 10.7554/eLife.01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruckbauer ST, Trimarco JD, Henry C, Wood EA, Battista JR, Cox MM. 2019. A variant of the Escherichia coli anaerobic transcription factor FNR exhibiting diminished promoter activation function enhances ionizing radiation resistance. PLoS One 14:e0199482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lenski RE, Rose MR, Simpson SC, Tadler SC. 1991. Long-term experimental evolution in Escherichia-coli. 1. Adaptation and divergence during 2,000 generations. Am Nat 138:1315–1341. doi: 10.1086/285289. [DOI] [Google Scholar]
- 49.Povirk LF. 1987. Mutational specificity of bleomycin. Proc Am Assoc Cancer Res 28:117–117. [Google Scholar]
- 50.Povirk LF. 1987. Bleomycin-induced base substitutions in the lambda-cI gene. Environ Mutagen 9:86–86. [Google Scholar]
- 51.Hendricks SP, Mathews CK. 1998. Differential effects of hydroxyurea upon deoxyribonucleoside triphosphate pools, analyzed with vaccinia virus ribonucleotide reductase. J Biol Chem 273:29519–29523. doi: 10.1074/jbc.273.45.29519. [DOI] [PubMed] [Google Scholar]
- 52.Sakano K, Oikawa S, Hasegawa K, Kawanishi S. 2001. Hydroxyurea induces site-specific DNA damage via formation of hydrogen peroxide and nitric oxide. Jpn J Cancer Res 92:1166–1174. doi: 10.1111/j.1349-7006.2001.tb02136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh A, Xu YJ. 2016. The cell killing mechanisms of hydroxyurea. Genes 7:99. doi: 10.3390/genes7110099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinberger M, Trabold PA, Lu M, Sharma K, Huberman JA, Burhans WC. 1999. Induction by adozelesin and hydroxyurea of origin recognition complex-dependent DNA damage and DNA replication checkpoints in Saccharomyces cerevisiae. J Biol Chem 274:35975–35984. doi: 10.1074/jbc.274.50.35975. [DOI] [PubMed] [Google Scholar]
- 55.Xie C-X, Xu A, Wu L-J, Yao J-M, Yang J-B, Yu Z-L. 2004. Comparison of base substitutions in response to nitrogen ion implantation and 60Co-gamma ray irradiation in Escherichia coli. Genet Mol Biol 27:284–290. doi: 10.1590/S1415-47572004000200025. [DOI] [Google Scholar]
- 56.Wang D, Kreutzer DA, Essigmann JM. 1998. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res Fund Mol Mech Mutagen 400:99–115. doi: 10.1016/S0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 57.Elena SF, Lenski RE. 2003. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- 58.Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. 2012. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet 44:101–U140. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amundsen SK, Taylor AF, Chaudhury AM, Smith GR. 1986. recD: the gene for an essential third subunit of exonuclease V. Proc Natl Acad Sci U S A 83:5558–5562. doi: 10.1073/pnas.83.15.5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biek DP, Cohen SN. 1986. Identification and characterization of recD, a gene affecting plasmid maintenance and recombination in Escherichia coli. J Bacteriol 167:594–603. doi: 10.1128/jb.167.2.594-603.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaudhury AM, Smith GR. 1984. A new class of Escherichia coli recBC mutants: implications for the role of RecBC enzyme in homologous recombination. Proc Natl Acad Sci U S A 81:7850–7854. doi: 10.1073/pnas.81.24.7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thaler DS, Sampson E, Siddiqi I, Rosenberg SM, Thomason LC, Stahl FW, Stahl MM. 1989. Recombination of bacteriophage lambda in recD mutants of Escherichia coli. Genome 31:53–67. doi: 10.1139/g89-013. [DOI] [PubMed] [Google Scholar]
- 63.Uranga LA, Reyes ED, Patidar PL, Redman LN, Lusetti SL. 2017. The cohesin-like RecN protein stimulates RecA-mediated recombinational repair of DNA double-strand breaks. Nat Comms 8:15282. doi: 10.1038/ncomms15282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang G, Maier RJ. 2008. Critical role of RecN in recombinational DNA repair and survival of Helicobacter pylori. Infect Immun 76:153–160. doi: 10.1128/IAI.00791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Picksley SM, Morton SJ, Lloyd RG. 1985. The recN locus of Escherichia coli K12: molecular analysis and identification of the gene product. Mol Gen Genet 201:301–307. doi: 10.1007/BF00425675. [DOI] [PubMed] [Google Scholar]
- 66.Meddows TR, Savory AP, Grove JI, Moore T, Lloyd RG. 2005. RecN protein and transcription factor DksA combine to promote faithful recombinational repair of DNA double-strand breaks. Mol Microbiol 57:97–110. doi: 10.1111/j.1365-2958.2005.04677.x. [DOI] [PubMed] [Google Scholar]
- 67.Brena-Valle M, Serment-Guerrero J. 1998. SOS induction by gamma-radiation in Escherichia coli strains defective in repair and/or recombination mechanisms. Mutagen 13:637–641. doi: 10.1093/mutage/13.6.637. [DOI] [PubMed] [Google Scholar]
- 68.Cashel M, Gentry DR, Hernandez VJ, Vinella D. 1996. The stringent response, p 1458–1495. In Neidhardt FC, Curtiss RI (ed), Escherichia coli and Salmonella: molecular and cellular biology. ASM Press, Washington, DC. [Google Scholar]
- 69.Trautinger BW, Lloyd RG. 2002. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J 21:6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGlynn P, Lloyd RG. 2000. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 71.Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, Mironov A, Nudler E. 2014. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505:372–377. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Landick R, Stewart J, Lee DN. 1990. Amino acid changes in conserved regions of the beta subunit of Escherichia coli RNA polymerase alter transcription pausing and termination. Genes Dev 4:1623–1636. doi: 10.1101/gad.4.9.1623. [DOI] [PubMed] [Google Scholar]
- 73.Barrick JE, Kauth MR, Strelioff CC, Lenski RE. 2010. Escherichia coli rpoB mutants have increased evolvability in proportion to their fitness defects. Mol Biol Evol 27:1338–1347. doi: 10.1093/molbev/msq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Severinov K, Soushko M, Goldfarb A, Nikiforov V. 1993. Rifampicin region revisited: new rifampicin-resistant and streptolydigin-resistant mutants in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem 268:14820–14825. [PubMed] [Google Scholar]
- 75.Zhou YN, Lubkowska L, Hui M, Court C, Chen S, Court DL, Strathern J, Jin DJ, Kashlev M. 2013. Isolation and characterization of RNA polymerase rpoB mutations that alter transcription slippage during elongation in Escherichia coli. J Biol Chem 288:2700–2710. doi: 10.1074/jbc.M112.429464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deatherage DE, Kepner JL, Bennett AF, Lenski RE, Barrick JE. 2017. Specificity of genome evolution in experimental populations of Escherichia coli evolved at different temperatures. Proc Natl Acad Sci U S A 114:E1904–E1912. doi: 10.1073/pnas.1616132114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tenaillon O, Rodriguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS. 2012. The molecular diversity of adaptive convergence. Science 335:457–461. doi: 10.1126/science.1212986. [DOI] [PubMed] [Google Scholar]
- 78.Avrani S, Bolotin E, Katz S, Hershberg R. 2017. Rapid genetic adaptation during the first four months of survival under resource exhaustion. Mol Biol Evol 34:1758–1769. doi: 10.1093/molbev/msx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Conrad TM, Frazier M, Joyce AR, Cho BK, Knight EM, Lewis NE, Landick R, Palsson BO. 2010. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc Natl Acad Sci U S A 107:20500–20505. doi: 10.1073/pnas.0911253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harden MM, He A, Creamer K, Clark MW, Hamdallah I, Martinez KA, Kresslein RL, Bush SP, Slonczewski JL. 2015. Acid-adapted strains of Escherichia coli K-12 obtained by experimental evolution. Appl Environ Microbiol 81:1932–1941. doi: 10.1128/AEM.03494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He A, Penix SR, Basting PJ, Griffith JM, Creamer KE, Camperchioli D, Clark MW, Gonzales AS, Erazo JSC, George NS, Bhagwat AA, Slonczewski JL. 2017. Acid evolution of Escherichia coli K-12 eliminates amino acid decarboxylases and reregulates catabolism. Appl Environ Microbiol 83:e00442-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mehta P, Casjens S, Krishnaswamy S. 2004. Analysis of the lambdoid prophage element e14 in the Escherichia coli K-12 genome. BMC Microbiol 4:4. doi: 10.1186/1471-2180-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Charusanti P, Conrad TM, Knight EM, Venkataraman K, Fong NL, Xie B, Gao YA, Palsson BO. 2010. Genetic basis of growth adaptation of Escherichia coli after deletion of pgi, a major metabolic gene. PLoS Genet 6:e1001186. doi: 10.1371/journal.pgen.1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, Wood TK. 2010. Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1:147. doi: 10.1038/ncomms1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 86.Lenski RE. 1991. Quantifying fitness and gene stability in microorganisms. Bio/Technology 15:173–192. [DOI] [PubMed] [Google Scholar]
- 87.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using gaIK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Almond PR, Biggs PJ, Coursey BM, Hanson WF, Huq MS, Nath R, Rogers DWO. 1999. AAPM’s TG-51 protocol for clinical reference dosimetry of high-energy photon and electron beams. Med Phys 26:1847–1870. doi: 10.1118/1.598691. [DOI] [PubMed] [Google Scholar]
- 90.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.