

Cell division (cytokinesis) is a fundamental biological process that is incompletely understood for any organism. Division of bacterial cells relies on a ring-like machinery called the septal ring or divisome that assembles along the circumference of the mother cell at the site where constriction eventually occurs. In the well-studied bacterium Escherichia coli, this machinery contains over 30 distinct proteins. We identify functionally important parts of one of these proteins, DedD, and present evidence supporting a role for DedD in helping to induce and/or sustain a self-enhancing cycle of processes that are executed by fellow septal-ring proteins and that drive the active constriction phase of the cell division cycle.
KEYWORDS: FtsA, FtsB, FtsL, FtsQ, cell division
ABSTRACT
Two key tasks of the bacterial septal-ring (SR) machinery during cell constriction are the generation of an inward-growing annulus of septal peptidoglycan (sPG) and the concomitant splitting of its outer edge into two layers of polar PG that will be inherited by the two new cell ends. FtsN is an essential SR protein that helps trigger the active constriction phase in Escherichia coli by inducing a self-enhancing cycle of processes that includes both sPG synthesis and splitting and that we refer to as the sPG loop. DedD is an SR protein that resembles FtsN in several ways. Both are bitopic inner membrane proteins with small N-terminal cytoplasmic parts and larger periplasmic parts that terminate with a SPOR domain. Though absence of DedD normally causes a mild cell-chaining phenotype, the protein is essential for division and survival of cells with limited FtsN activity. Here, we find that a small N-terminal portion of DedD (NDedD; DedD1–54) is required and sufficient to suppress ΔdedD-associated division phenotypes, and we identify residues within its transmembrane domain that are particularly critical to DedD function. Further analyses indicate that DedD and FtsN act in parallel to promote sPG synthesis, possibly by engaging different parts of the FtsBLQ subcomplex to induce a conformation that permits and/or stimulates the activity of sPG synthase complexes composed of FtsW, FtsI (PBP3), and associated proteins. We propose that, like FtsN, DedD promotes cell fission by stimulating sPG synthesis, as well as by providing positive feedback to the sPG loop.
IMPORTANCE Cell division (cytokinesis) is a fundamental biological process that is incompletely understood for any organism. Division of bacterial cells relies on a ring-like machinery called the septal ring or divisome that assembles along the circumference of the mother cell at the site where constriction eventually occurs. In the well-studied bacterium Escherichia coli, this machinery contains over 30 distinct proteins. We identify functionally important parts of one of these proteins, DedD, and present evidence supporting a role for DedD in helping to induce and/or sustain a self-enhancing cycle of processes that are executed by fellow septal-ring proteins and that drive the active constriction phase of the cell division cycle.
INTRODUCTION
Cytokinesis (cell division, septation, or fission) of bacterial cells is mediated by a complex ring-like apparatus called the divisome or septal ring (SR) (1, 2). The mature SR of the Gram-negative rod Escherichia coli is a transenvelope assembly including over 30 different proteins. Ten of these (FtsA, -B, -I, -K, -L, -N, -Q, -W, and -Z and ZipA) are essential for cell fission and form the core of the apparatus. Cells lacking any one core protein form long, smooth filaments with multiple nucleoids and either no (FtsZ−) or immature (FtsZ+) SRs before eventually dying. FtsA and FtsZ reside on the cytoplasmic side of the inner membrane (IM), while all the other core proteins are integral IM species with polytopic (FtsK and -W) or bitopic topology of the N-in (FtsB, -I, -L, -N, and -Q) or N-out (ZipA) variety. Noncore SR proteins reside in the cytoplasm, IM, periplasm, or outer membrane (OM). Though they are individually dispensable for cell fission or viability per se, many have (overlapping) roles in critical or important aspects of SR assembly and/or functions (1, 2).
The tubulin-like GTPase FtsZ forms linear homopolymers (Z polymers) and plays a key role in cytokinesis of most prokaryotes and many plastids (2–4). SR assembly starts with the coaccumulation of Z polymers and several core (FtsA and ZipA) and noncore (ZapA, -C, and -D) FtsZ-binding proteins in a ring-like arrangement on the inner face of the IM at the prospective site of fission. This intermediate assembly is often referred to as the FtsZ or Z ring (ZR) (1, 2, 5). Z polymers have no inherent affinity for membranes, and at a minimum, either FtsA or ZipA is needed to arrange them into a ZR in vivo (6, 7). Both bind a small conserved C-terminal peptide of FtsZ, and while ZipA is a bitopic IM protein, FtsA binds the IM peripherally via a C-terminal amphipathic peptide (8–13). FtsA is a subdomain variant of actin in which subdomain 1B in the “cloverleaf” structure of typical actins is absent and an unrelated domain (1C or SHS2) resides on the other side of the molecule instead (14, 15). Still, FtsA can readily form actin-like oligomers/polymers (A polymers) on a phospholipid surface (16–20). In turn, these engage Z polymers to form interpolymer complexes (IPCs) in which A polymers are sandwiched between the membrane surface and Z polymers, keeping the latter two ∼16 nm apart (20–23). GTPase-dependent treadmilling of the Z polymer component drives movement of these IPCs on the IM surface, and they can be seen to rotate around the ZR in either direction in vivo (22, 24, 25).
Assembly of the ZR is followed by recruitment of the remaining core division proteins in an ordered manner (ZR < FtsK < [FtsB + -L + -Q] < [FtsW + -I] < FtsN) to form a mature, constriction-competent SR apparatus (1, 2, 26, 27). The core and noncore SR proteins then orchestrate the coordinated invagination of the IM, peptidoglycan (PG), and OM envelope layers and the subsequent separation of daughter cells. PG (murein) is a mesh-like material of linear glycan strands that are cross-linked via short peptides. It surrounds the entire IM, forming a cell-size molecule called the PG sacculus that protects the cell from osmotic lysis, helps maintain cell shape, and anchors other envelope components, including the OM in Gram-negative species (28). The basic building block is lipid II [undecaprenyl-pyrophosphoryl-N-acetylmuramic acid (NAM)(-d-Ala–d-iGlu–m-Dap–d-Ala–d-Ala)–N-acetylglucosamine (NAG)], a lipid-linked disaccharide pentapeptide that is assembled on the inner face of the IM (29). Lipid II is then flipped to the periplasmic face of the IM (30–32), where it is used by PG glycosyltransferases (GTs) to polymerize new glycan strands that are subsequently cross-linked to adjacent strands by PG transpeptidases (TPs) (28).
One key task of the SR during cell fission is the generation of an ingrowing annulus of septal peptidoglycan (sPG) that is oriented perpendicular to the cell’s long axis and that can be viewed as an integral and dynamic component of the actively constricting SR itself (1, 2, 33–36). The outer (OM-proximal) edge of this annulus is initially coincident with the cylindrical part of the sacculus, while new material is added to its inner (IM-proximal) edge by IM-associated sPG synthase complexes. Central to the latter are two of the core SR proteins, FtsW and FtsI, which associate to form an FtsW-FtsI subcomplex (37–41). FtsI is also known as penicillin-binding protein 3 (PBP3), as it is a target of β-lactam antibiotics and possesses dd-transpeptidase activity (42–45). Recent evidence indicates that FtsW, like its paralog, RodA, belongs to a newly recognized class of PG glycosyltransferases (45–49). Hence, the FtsW-FtsI subcomplex likely possesses both enzymatic activities needed to convert lipid II to sPG. However, though purified E. coli FtsW was found to bind lipid II, it has yet to display actual GT activity in vitro (41, 50). Similarly, while purified E. coli FtsI displays TP activity on artificial substrates (44), it so far appears to ignore natural substrates (51). Perhaps E. coli FtsW-FtsI is not a complete PG synthase, but it is more likely that it requires precise conditions and/or accessory factors to stimulate its GT and TP activities with lipid II as the starting substrate.
In fact, a variety of evidence indicates that FtsW-FtsI associates, at least transiently, with a number of other factors to form larger sPG synthase complexes in vivo. These include the other core SR proteins FtsB, -L, -N, and -Q (37, 38, 52–55) and the noncore bifunctional PG synthase PBP1B (39, 41, 56). The latter possesses both GT and TP activities and likely contributes to sPG synthesis and/or to maintaining the integrity of the sacculus as the sPG annulus is being processed into two layers of polar PG (47, 51). FtsB, -L, and -Q form a phylogenetically well-conserved subcomplex (FtsBLQ) that is required to recruit FtsW-FtsI during SR maturation (57–60) and likely regulates its sPG synthase activities as well (61, 62).
Soon after synthesis of the sPG annulus starts, and while sPG synthases continue to add new material to its inner edge, its outer edge begins to split into two layers that remain continuous with the mother cell sacculus and become part of the two new nascent cell poles. This splitting of the sPG annulus from the OM-proximal side is accomplished by murein hydrolases, most prominently by the periplasmic N-acetylmuramyl-l-alanine amidases AmiA, -B, and -C (34, 35, 63–66). AmiA and AmiB activities are tightly controlled via interactions with the periplasmic protein EnvC, while that of AmiC is regulated by the OM lipoprotein NlpD (66–69). The amidases cleave the peptide-glycan bond, yielding denuded glycan strands that gradually detach from the sPG mesh and are eventually degraded by lytic tranglycosylases (28, 36). Though transient, denuded glycan strands accumulate sharply at the constricting SR, where they serve to recruit/anchor a set of core (FtsN) and noncore (DamX, DedD, and RlpA) SR proteins that contain a SPOR domain (36, 70, 71).
FtsN is the last core protein to accumulate strongly at the SR and helps trigger the active cell constriction phase of the division process (1, 2). It is a bitopic IM protein with a small cytoplasmic domain (FtsN1–30, or NFtsN) and a large periplasmic domain (FtsN55–319). The former binds the 1C domain of FtsA (61, 72–74). The latter consists of a short linker, an essential peptide of only ∼19 residues (FtsN75–93, or EFtsN), a longer linker, and a C-terminal SPOR domain (FtsN243–319, or SFtsN) that binds denuded PG strands (36, 61, 70, 71, 75). The presence of EFtsN in the periplasm is required and sufficient for cells to divide (61, 71). The peptide fails to accumulate at SRs by itself, however, and needs to be overproduced to rescue ΔftsN cells fully. In contrast, the SFtsN domain can localize sharply to constricting SRs when separated from other parts of the protein but requires EFtsN, FtsW-FtsI, and PG amidase activities to do so (61, 71). Based on these and additional observations, FtsN is proposed to induce a positive-feedback loop consisting of (i) FtsN-stimulated sPG synthesis; (ii) sPG splitting and production of SPOR substrate by Ami proteins; and (iii) enhanced SR recruitment of SPOR domain proteins, including FtsN itself (61, 71). We refer to this self-enhancing cycle of processes as the sPG loop.
To elucidate how FtsN stimulates cell fission, we and others identified mutant variants of other SR proteins that reduce the need for FtsN activities. Such FtsN−-suppressing versions of SR proteins all appear to promote active cell fission “better” than the native versions and can be described as SF (superfission) variants, encoded by their corresponding sf mutant alleles. SF variants of E. coli FtsA, FtsB, FtsL, and FtsW that, at least partially, bypass the requirement for FtsN have been described so far (61, 62, 76, 77). Their properties support a model wherein FtsN acts allosterically via NFtsN on FtsA in the cytoplasm, and via EFtsN on the FtsBLQ subcomplex in the periplasm, to stimulate sPG synthesis by FtsW-FtsI and to help trigger and sustain the sPG loop (61, 62, 74, 77).
As mentioned above, E. coli also produces three noncore SR proteins with a SPOR domain (71, 78). DamX and DedD have the same bitopic IM topology as FtsN, whereas RlpA is an OM lipoprotein. High levels of DamX cause filamentation in E. coli, and this underlies morphological switching in uropathogenic strains (79, 80). RlpA likely degrades denuded glycan strands, as was demonstrated for the Pseudomonas aeruginosa enzyme (81). Even so, neither ΔdamX nor ΔrlpA single mutants of E. coli show an obvious cell division defect (71, 78).
In contrast, ΔdedD single-mutant cells show a mild cell-chaining phenotype that is exacerbated in ΔdedD ΔdamX double mutants (71, 78) or by stressful growth conditions, such as high hydrostatic pressure (82) or low temperatures (83). Moreover, DedD becomes essential in cells with reduced FtsN function, suggesting it plays an FtsN-like role in stimulating cell constriction (61, 71). Here, we find that an N-terminal portion of DedD (NDedD, or DedD1–54) that includes its transmembrane helix is required and sufficient for its function in cell division. NDedD also contains a weak SR localization determinant that can operate independently of its C-terminal SPOR domain. Further analyses suggest that DedD acts in parallel with FtsN to stimulate FtsW-FtsI activity, possibly via an interaction between DedD and the FtsBLQ subcomplex within, or near, the IM bilayer. We propose that, like FtsN, DedD provides positive feedback to the sPG loop.
RESULTS
An N-terminal portion of DedD (NDedD) is required and sufficient for its function in cell division.
DedD consists of a small cytoplasmic domain (DedD1–8), a transmembrane domain (DedD9–27, or TMDedD), and a large periplasmic domain (DedD28–220) (71, 84). The last contains a C-terminal globular SPOR domain (DedD140–216, or SDedD), and secondary structure predictions (85, 86) suggest that the remainder (DedD28–139) lacks a defined structure save for five possible short α-helices, H1 to H5 (Fig. 1A; see Fig. S1 in the supplemental material).
FIG 1.

Domain analyses of E. coli DedD. (A) Schematic depiction of full-length DedD (DedD1–220). The transmembrane domain (TMDedD) and the C-terminal SPOR domain (SDedD) are indicated. Potential α-helices (H1 to H5), predicted to form in the periplasmic portion between TMDedD and SDedD by the GOR IV secondary-structure prediction method (85), are indicated in black. Also shown are inserts present on plasmids that encode fusions of various portions of DedD to GFP or TTGFP under control of the lac regulatory region. The TTGFP fusion encoded by pMG44 contains the TorA signal peptide (hatched box) that is cleaved upon export to the periplasm via the twin arginine transport (Tat) system. M represents residues 2 to 39 of MalF, which include cytoplasmic residues and the first transmembrane domain (TM1; MalF19–35) of the protein (88). Some fusions end with a nonnative Leu-Glu dipeptide (LE), as indicated. The table on the right shows (left to right) the DedD residues present in each fusion; whether the fusion could fully (++) or partially (+−) or could not (−−) correct the cell-chaining phenotype of BL40 [ΔdamX ΔdedD]; whether the fusion accumulated sharply (+++), weakly (+−−), or not at all (−−−) at division sites in the strain; and the panel with corresponding cell images. (B to I) Images of live BL40 [ΔdamX ΔdedD] cells carrying the vector control pMLB1113ΔH [Plac::] (B), pFB236 [Plac::gfp-dedD] (C), pPC1 [Plac::gfp-dedD1–118] (D), pBL95 [Plac::gfp-dedD1–54] (E), pBL37 [Plac::gfp-dedD1–35] (F), pBL101 [Plac::gfp-dedD1–35-rfp] (G), pBL33 [Plac::gfp-malF2–39-dedD28–220] (H), or pBL27 [Plac::gfp-malF2–39-dedD28–118] (I). Panels C to I comprise a differential interference contrast (DIC) image (left) and a fluorescence (FL) image (right) that corresponds to the boxed area in the DIC image. Bar, 16 μm (all DIC images), 8 μm (FL images in panels H and I), or 4 μm (FL images in panels C to G). Cells were grown for ∼5 mass doublings to an OD600 of 0.5 to 0.6 in LB with 50 μM IPTG. Strong septal localization of TTGFP-DedD140–220 was shown previously (71) and resembled that seen in panel H. Examples of weak (GFP-DedD1–118 and GFP-DedD1–54) or very weak (GFP-DedD1–35-RFP) septal accumulations of some of the SPOR-less fusions are marked by arrowheads in panels D, E, and G.
To identify the part(s) of DedD required for its function in cell division, we created constructs encoding various portions of DedD fused to the C terminus of either cytoplasmic green fluorescent protein (GFP) or Tat-targeted periplasmic GFP (TTGFP) (67) and used them in complementation assays with ΔdedD strains. As noted above, the absence of DedD in otherwise wild-type (wt) cells causes a mild cell-chaining phenotype, but this is significantly exacerbated in ΔdamX cells, even though DedD-replete ΔdamX cells show no obvious cell division phenotype (71, 87) (see Fig. S2 in the supplemental material). Hence, the fusions were produced in strain BL40 [ΔdamX ΔdedD], and cells were observed by microscopy (Fig. 1; see Fig. S2).
Production of a GFP fusion to full-length DedD encoded by pFB236 [Plac::gfp-dedD] corrected the chaining phenotype of BL40 (Fig. 1C; see Fig. S2C), as expected (71). Notably, production of GFP-DedD1–118 (Fig. 1D; see Fig. S2D) or GFP-DedD1–54 (Fig. 1E; see Fig. S2E) did so as well, indicating that the N-terminal 54 residues of DedD are sufficient to prevent cell chaining. In contrast, a GFP-DedD1–35 fusion failed to correct cell chaining in BL40 cells (Fig. 1F; see Fig. S2F and G). Rather than accumulating along the membrane, however, the fluorescent signal in these cells appeared to be mostly cytoplasmic (Fig. 1F). Western analyses indicated that the fusion was not markedly less stable than the functional ones (see Fig. S3A in the supplemental material), suggesting that GFP-DedD1–35 may be poorly inserted or maintained in the membrane due to its small periplasmic domain (∼8 residues). Indeed, appending red fluorescent protein (RFP) to its C terminus yielded a fusion (GFP-DedD1–35-RFP) that more clearly localized to the cell periphery (Fig. 1G). Moreover, the fusion suppressed the chaining phenotype of BL40 to a significant degree but, unlike GFP-DedD, GFP-DedD1–118, or GFP-DedD1–54, did so incompletely even at high levels of expression (Fig. 1G; see Fig. S2H and I). Thus, almost all of the periplasmic residues of DedD, including its SPOR domain and potential α-helices 1 to 5, may be dispensable for its ability to suppress cell chaining in BL40 [ΔdamX ΔdedD] cells, but residues in the 36-to-54 interval clearly contribute to this activity.
A role for residues in the 36-to-54 interval was also evident in a more stringent test for DedD function. The ftsNslm117 allele (ftsN::EZTnKan-2) encodes a truncated SPOR-less version of FtsN, due to a transposon insertion in codon 119 of chromosomal ftsN. In otherwise wt cells, ftsNslm117 causes a cell-chaining phenotype quite similar to that of BL40 [ΔdamX ΔdedD] cells (71). Notably, DedD is essential for viability of ftsNslm117 cells, which cease cell constriction altogether upon depletion of DedD (71).
Hence, pFB236 [Plac::gfp-dedD] or one of its derivatives described above was introduced in the DedD depletion strain MG19/pMG39 [ftsNslm117 ΔdedD/PBAD::dedD], and transformants were tested for the ability to survive in the absence of arabinose. As expected, MG19/pMG39 cells carrying a vector control (pMLB1113ΔH) grew well in the presence of arabinose but formed very long filaments that failed to survive on medium lacking the sugar (Fig. 2 and data not shown). In contrast, cells carrying pFB236 [Plac::gfp-dedD] readily survived the absence of arabinose, even in the absence of added IPTG (isopropyl-β-d-thiogalactopyranoside). Interestingly, the GFP-DedD1–54 fusion rescued cells almost as well as full-length GFP-DedD, but neither GFP-DedD1–35 nor GFP-DedD1–35-RFP did (Fig. 2). The inability of the latter to functionally substitute for DedD in ftsNslm117 ΔdedD cells contrasts with its ability to at least partially correct the chaining phenotype of ΔdamX ΔdedD cells (Fig. 1; see Fig. S2) and supports the notion that DedD residues in the 36-to-54 interval indeed contribute significantly to DedD function.
FIG 2.

Ability of GFP-DedD and variants to rescue DedD-depleted ftsNslm117 cells. Strain MG19/pMG39 [ΔdedD ftsNslm117/PBAD::dedD] carrying one of the indicated plasmids was grown overnight in LB with 0.5% arabinose. Cultures were serially diluted in LB to an OD600 of 4 × 10x, and 5 μl of each dilution was spotted on LB agar containing arabinose, IPTG, or neither, as indicated. The plates were incubated for 16 h. Save for the vector control, pMLB1113ΔH [Plac::], each plasmid encoded GFP-DedD or a mutant derivative under control of the lac regulatory region. The relevant plasmid name and encoded protein are indicated on the left and right of each row, respectively. Plasmids pFB236 and pBL272 are almost identical, except that the GFP-DedD fusion encoded by the latter carries an additional four residues in the linker peptide that connects the GFP and DedD moieties.
A more surprising result of these experiments was that the same GFP-DedD1–118 fusion that efficiently suppressed chaining of BL40 [ΔdamX ΔdedD] cells (Fig. 1; see Fig. S2), proved to be completely ineffective in rescuing ftsNslm117 ΔdedD cells (Fig. 2 and results not shown). As this contrasted so sharply with the ability of both a larger (full-length GFP-DedD) and a shorter (GFP-DedD1–54) fusion to rescue such cells efficiently, we considered the possibility that GFP-DedD1–118 was not produced well and/or was unstable in this particular strain. However, Western analyses showed that GFP-DedD1–118 was present at about the same level as GFP-DedD or GFP-DedD1–54 in MG19/pMG39 cells, regardless of their DedD depletion state (see Fig. S4 in the supplemental material). Since BL40 is ΔdamX and MG19 is not, we also considered the possibility that the presence of DamX somehow specifically interferes with GFP-DedD1–118 function. However, GFP-DedD1–118 and GFP-DedD1–54 appeared equally capable of suppressing the mild cell-chaining phenotype of MG14 [ΔdedD], the DamX+ parent of BL40 [ΔdamX ΔdedD] (see Fig. S5 in the supplemental material), rendering this possibility unlikely. One remaining possibility is that, in contrast to GFP-DedD or GFP-DedD1–54, proper functioning of GFP-DedD1–118 depends on a property of FtsN that is either lost or gained by its truncation to FtsNslm117. Several scenarios can be imagined, but additional work is required to fully understand why GFP-DedD1–54 readily suppresses the cell division defects associated with ΔdedD in both FtsN+ and FtsNslm117 cells while GFP-DedD1–118 is incapable of doing so in the latter, specifically.
Regardless, these results established that a relatively small N-terminal portion of DedD (NDedD), conservatively corresponding to residues 1 to 54, is sufficient to suppress the cell division phenotypes that are associated with a ΔdedD mutation. Formal evidence that NDedD is also required for this activity of DedD came from the study of fusions in which the cytoplasmic and transmembrane portions of the protein (DedD1–27) are replaced with a peptide (MalF2–39) comprising the cytoplasmic N terminus and first transmembrane helix (TM1) of the MalF protein (88). Neither GFP-MalF2–39-DedD28–118 nor GFP-MalF2–39-DedD28–220 was capable of suppressing chaining of ΔdedD or ΔdamX ΔdedD cells or of rescuing ftsNslm117 cells upon depletion of DedD (Fig. 1I and H and 2 and data not shown). Similarly, the fully periplasmic fusions TTGFP-DedD28–118, TTGFP-DedD140–220, and TTGFP-DedD28–220 also failed to correct division defects associated with ΔdedD (Fig. 1A and data not shown) (71). We conclude that NDedD is both required and sufficient for the ability of DedD to stimulate cell fission in E. coli.
Contribution of SDedD to DedD functionality.
Though the SPOR domain of DedD (SDedD) is not required for its ability to promote normal cell division, it is likely to contribute to DedD functionality, if only by stimulating the accumulation of NDedD at the septal ring (71). To explore this possibility further, we studied BL40 [ΔdamX ΔdedD] cells, which encode either full length GFP-DedD or SPOR-less GFP-DedD1–118 under the control of the lac regulatory region on a CRIM construct that is integrated at the chromosomal attHK022 site (89). Strains BL40(iBL360) [ΔdamX ΔdedD(Plac::gfp-dedD)] and BL40(iBL345) [ΔdamX ΔdedD(Plac::gfp-dedD1–118)] were grown in the presence of different concentrations of inducer (IPTG). Division phenotypes were monitored by microscopy and corresponding levels of GFP fusions by immunoblotting (Fig. 3). Production of GFP-DedD already allowed normal division of BL40(iBL360) cells in the presence of 50 μM IPTG (Fig. 3C). In contrast, at least 250 μM IPTG was required for production of GFP-DedD1–118 to suppress chaining of BL40(iBL345) cells to a comparable extent (Fig. 3J). The accompanying Western analyses indicated that the cellular concentration of SPOR-less GFP-DedD1–118 needed to be elevated 2- to 4-fold relative to full-length GFP-DedD to attain a similar level of function (Fig. 3K). These results are compatible with the idea that SDedD indeed contributes to the efficacy of NDedD action in cells.
FIG 3.

Phenotype of SPOR-less DedD. (A to J) DIC images of live cells of strains BL40(iBL360) [ΔdamX ΔdedD(Plac::gfp-dedD)] (A to E) and BL40(iBL345) [ΔdamX ΔdedD(Plac::gfp-dedD1–118)] (F to J). The cells were grown for ∼3 mass doublings to an OD600 of 0.5 to 0.6 in M9-maltose with 0 μM (A and F), 10 μM (B and G), 50 μM (C and H), 100 μM (D and I), or 250 μM (E and J) IPTG. Bar, 4 μm. (K) Western analysis of full-length GFP-DedD and SPOR-less GFP-DedD1–118. Nonchaining cells of strains BL38 [ΔdamX] (lane 1), BL40(iBL360) [ΔdamX ΔdedD(Plac::gfp-dedD)] (lane 2), and BL40(iBL345) [ΔdamX ΔdedD] [Plac::gfp-dedD1–118] (lane 3) were obtained by growth as described for panel C [50 μM IPTG; BL38 and BL40(iBL360)] or J [250 μM IPTG; BL40(iBL345)] and used to prepare whole-cell extracts. For lanes 4 to 7, the BL40(iBL345) extract (lane 3) was diluted with that of BL38 (lane 1) to yield the fraction of the former indicated above each lane. Each lane contained 40 μg total protein. Fusion proteins were detected using anti-GFP (α-GFP) polyclonal antibodies. Bands corresponding to the fusions of interest are indicated by arrowheads. Migration of molecular mass standards (in kilodaltons) is indicated on the left. Division phenotypes (A to J) and relative band intensities (K) indicated that, relative to GFP-DedD, BL40 [ΔdamX ΔdedD] cells require a 2- to 4-fold higher level of SPOR-less GFP-DedD1–118 to prevent cell chaining.
Weak SDedD-independent accumulation of NDedD at division sites.
Full-length GFP-DedD accumulates strongly at cell constriction sites (Fig. 1C and 4D). This property is maintained in all fusions containing an intact SPOR domain (Fig. 1A and H), consistent with the conclusion that SDedD is a strong septal localization determinant of the protein (71). Even so, several of the fusions lacking SDedD still accumulated at division sites in BL40 cells, albeit significantly more weakly than those containing it. Weak accumulation was especially obvious with GFP-DedD1–118 (Fig. 1D) or GFP-DedD1–54 (Fig. 1E), even though the latter showed a relatively high cytoplasmic signal as well. Even the GFP-DedD1–35-RFP fusion appeared to be enriched at division sites, though its accumulation seemed weaker still (Fig. 1G). In contrast, GFP-MalF2–39-DedD28–118 was distributed evenly along the cell membrane, showing no hint of accumulation at constriction sites (Fig. 1I). These results indicate that, besides its SPOR domain, DedD contains a second SR localization determinant that resides in NDedD and that residues within the 1-to-27 interval are required for the SDedD-independent recruitment of NDedD to division sites.
FIG 4.

Identification of residues important for DedD function. (A) DedD residues 1 to 120 are colored according to phylogenetic conservation (percent identity in the Hogenom gene family HOG000279501 [90]); red, 100 to 98%; blue, 98 to 95%; green, 95 to 90%; brown, 90 to 75%; and black, <75%. DedD is a type II bitopic (N-in) inner membrane protein, and its transmembrane domain is shaded in gray. Pertinent substitutions (orange) are indicated immediately above the affected residues. (B and C) DIC images of live BL38 [ΔdamX] (B) and BL40 [ΔdamX ΔdedD] (C) cells after growth for ∼3 mass doublings to an OD600 of 0.5 to 0.6 in M9-maltose. (D to J) Images of live BL40 [ΔdamX ΔdedD] cells carrying a single copy of iBL360 [Plac::gfp-dedD] (D), iBL360(G11A) [Plac::gfp-dedDG11A] (E), iBL345 [Plac::gfp-dedD1–118] (F), iBL345(G11A) [Plac::gfp-dedD1–118, G11A] (G), iBL345(G11C) [Plac::gfp-dedD1–118, G11C] (H), iBL345(P24A) [Plac::gfp-dedD1–118, P24A] (I), or iBL345(P42G) [Plac::gfp-dedD1–118, P42G] (J) integrated in the chromosome. The cells were grown as for panels B and C, but with 250 μM IPTG included in the medium. The panels comprise a DIC image (top) and an FL image (bottom) that corresponds to the boxed area in the DIC image. Bar, 8 μm (DIC images) or 4 μm (FL images). The arrowheads in panels F, I, and J mark examples of the weak accumulation of GFP-DedD1–118 or mutant derivatives at division sites, which are not seen in panels G and H.
While the weak accumulation of the SPOR-less fusions at division sites could be readily observed in ΔdedD strains MG14 [ΔdedD] and BL40 [ΔdamX ΔdedD], this was very difficult to detect in the dedD+ parent strains TB28 [wt] and BL38 [ΔdamX] (Fig. S6 and data not shown). This suggests that native DedD competes effectively with the SPOR-less fusions for interaction with an SR target that helps recruit the latter in ΔdedD cells and that this presumed binding partner of NDedD is of limiting abundance.
NDedD residues important for DedD function.
Phylogenetic analyses (90) indicate that clear orthologs of dedD are restricted to the gammaproteobacteria and that the best-conserved residues of the protein reside in either NDedD or SDedD (Fig. 4A; see Fig. S1). We replaced three conserved residues in NDedD and assessed their importance to DedD function. G11 and P24 are predicted to reside in TMDedD, while P42 is periplasmic (Fig. 4A; see Fig. S1). To allow detection of potentially subtle defects, the single-residue substitution mutants were studied in the contexts of both SPOR-less GFP-DedD1–118 and full-length GFP-DedD fusion proteins encoded on single- or multicopy constructs.
As described above, when BL40(iBL345) [ΔdamX ΔdedD(Plac::gfp-dedD1–118)] cells were grown in M9-maltose with 250 μM IPTG, production of GFP-DedD1–118 from the integrated CRIM construct was sufficient to suppress the cell-chaining phenotype of the host (Fig. 3J). The fusion accumulated weakly at cell division sites under these conditions (Fig. 4F and Table 1), similar to when it was encoded on a plasmid in cells that were grown in rich medium (Fig. 1D).
TABLE 1.
Septal accumulation of GFP-DedD1–118 variants in BL40 [ΔdedD ΔdamX] cellsa
| CRIM construct name | Variant | % cells withb: |
% rings without const.c | % const. with ringd | |
|---|---|---|---|---|---|
| Const. | Ring | ||||
| iBL345 | WT | 33 | 25 | 16 | 62 |
| iBL345(G11A) | G11A | 80e | 9 | 0 | 6 |
| iBL345(P24A) | P24A | 75e | 35 | 10 | 28 |
| iBL345(P42G) | P42G | 39 | 26 | 28 | 48 |
BL40 [ΔdedD ΔdamX] cells harboring the indicated Plac::gfp-dedD1–118 CRIM construct integrated in the chromosome were grown for 4 mass doublings in M9-maltose with 250 μM IPTG to an OD600 of 0.6. Cells (n = 119 to 168) were imaged live by DIC and fluorescence optics to determine the presence and (co)location of visible cell constrictions (const.) and accumulation of the fusion protein in weakly fluorescent rings.
Percentage of cells in the population with a visible constriction and/or fluorescent ring.
Percentage of all rings at nascent division sites, where constriction was not (yet) obvious.
Percentage of all constrictions with an associated ring.
Due to an uncorrected chaining phenotype, over 26% of cells consisted of more than two cell units and contained more than one constriction. Connected cell units were counted as one cell.
The P42G variant of GFP-DedD1–118 (GFP-DedD1–118, P42G) also suppressed cell chaining and showed similar weak accumulations at division sites (Fig. 4J and Table 1), indicating that the identity of P42 is not critical for function. In contrast, G11A and P24A variants of the fusion failed to suppress cell chaining under these conditions, indicating that G11 and P24 are important for NDedD function (Fig. 4G and I). The possibility that GFP-DedD1–118, G11A or GFP-DedD1–118, P24A was unstable and/or mislocalized to the cytoplasm was refuted by Western analyses (see Fig. S3B) and their accumulation along the peripheries of cell chains (Fig. 4G and I). Notably, while the P24A variant could still be seen to further accumulate weakly at some of the division sites in these chains (Fig. 4I and Table 1), such accumulations were almost undetectable with the G11A variant (Fig. 4G and Table 1). The latter result was strengthened by studies with a G11C variant of GFP-DedD1–118, which similarly failed to localize to division sites or to correct cell chaining (Fig. 4H; see Fig. S3B). These results indicate that G11 is important not only to NDedD function but also for its SDedD-independent recruitment to SRs.
Compared to its importance to the activity of SPOR-less GFP-DedD1–118, the identity of G11 appeared to be less critical to the ability of full-length GFP-DedD to suppress chaining of BL40 cells. Thus, production of CRIM-encoded GFP-DedDG11A in BL40(iBL360, G11A) [ΔdamX ΔdedD(Plac::gfp-dedDG11A)] cells did result in partial suppression of cell chaining in the presence of 250 μM IPTG (Fig. 4E). In comparison, GFP-DedDWT corrected ΔdedD fully (Fig. 4D) and, as discussed above, did so already at a 5-fold lower concentration of inducer (Fig. 3C). Like GFP-DedD, the GFP-DedDG11A fusion accumulated strongly at division sites (Fig. 4D and E), which was expected, as both harbor an intact SDedD domain. Hence, the difference between GFP-DedDG11A and GFP-DedD1–118, G11A activities (Fig. 4E and G) suggests that the functional defect associated with the G11A substitution can, to some degree, be overcome by increasing the local concentration of the mutant variant at the SR. Indeed, when produced from a multicopy plasmid, both GFP-DedDG11A and GFP-DedDP24A were capable of suppressing cell chaining of BL40 cells almost as completely as either GFP-DedDP42G or GFP-DedDWT (see Fig. S7 in the supplemental material). Thus, the G11A and P24A variants of GFP-DedD retain sufficient DedD activity to correct cell chaining of BL40 cells upon overexpression.
In contrast, the GFP-DedDG11A and GFP-DedDP24A variants proved completely ineffective in rescuing ftsNslm117 ΔdedD cells, even when expressed from a multicopy plasmid at high IPTG concentration (Fig. 2). While GFP-DedDWT (encoded by pFB236 or pBL272) readily rescued MG19/pMG39 [ftsNslm117 ΔdedD/PBAD::dedD] cells, even in the absence of added IPTG, these variants failed to do so at any IPTG concentration. In this more stringent test for DedD function, moreover, the P42G variant now also showed a modest defect (Fig. 2).
We conclude that conserved residues G11 and P24 in the TM domain of DedD play an important role in the SDedD-independent recruitment of DedD to SRs (especially G11) and in promoting cell constriction. The identity of neither residue is absolutely critical for DedD function in FtsN+ cells, but their replacement is not tolerated in cells relying on SPOR-less FtsNslm117 for survival.
Absence of DedD is poorly tolerated in cells lacking PBP1B.
While diminished FtsN function in wt or ponA (lacking PBP1A) cells leads to formation of cell chains or filaments, it causes massive cell lysis in ΔponB cells (61). These PBP1B-dependent phenotypes of FtsN− cells parallel those induced by PBP3(FtsI)-specific drugs (91–93) and support the idea that FtsN is needed for full PBP3 activity (61).
Given that the chaining phenotype of ΔdedD cells resembles that of cells with suboptimal FtsN activity (e.g., ftsNslm117 cells) and that DedD becomes essential in the latter (71) (Fig. 2), it seems likely that DedD also contributes to full PBP3 activity. If so, one might expect that depletion or absence of DedD is poorly tolerated in ΔponB cells as well, and we found this to indeed be the case. In contrast to the ftsNslm117 allele, which is lethal in combination with ΔponB (61), the ΔdedD lesion could readily be combined with either ΔponA or ΔponB. However, while ΔponA ΔdedD cells grew as well as wt or single-mutant cells (Fig. 5A), ΔponB ΔdedD cells grew poorly on both solid and liquid (minimal or rich) media (Fig. 5A and data not shown). When grown in liquid LB, for example, overnight cultures of strain BL83 [ΔponB ΔdedD] reached an optical density less than half that of the single-mutant parents or wild-type cells. Upon subculture in fresh medium, BL83 [ΔponB ΔdedD] displayed an extended lag period. This was followed by exponential growth at a rate approaching that of the single-mutant parents in early log phase (optical density at 600 nm [OD600] < 0.08), after which the apparent rate slowed markedly (Fig. 5A, orange curve). Microscopy of cultures in mid-log phase showed the typical ΔdedD-associated chaining phenotype of BL96 [ΔponA ΔdedD] cells (Fig. 5B). In contrast, BL83 [ΔponB ΔdedD] cultures contained a mixture of cell chains, ghosts, and debris. In addition, many of the chains showed signs of septal lysis, such as septal bulges and “rabbit ears” (Fig. 5C), similar to those seen in the ΔponB single-mutant parent strain upon treatment with cephalexin (Fig. 5E). Addition of more NaCl (to 1%) to the medium did not alleviate these phenotypes. As expected, however, the poor growth and lysis of ΔponB ΔdedD cells could be suppressed by ectopic production of full-length DedD or PBP1B (data not shown). Moreover, these ΔponB ΔdedD phenotypes were also suppressed by production of the GFP-DedD1–54 fusion (Fig. 5A, red and green traces, and data not shown), implying that NDedD is sufficient for this function.
FIG 5.
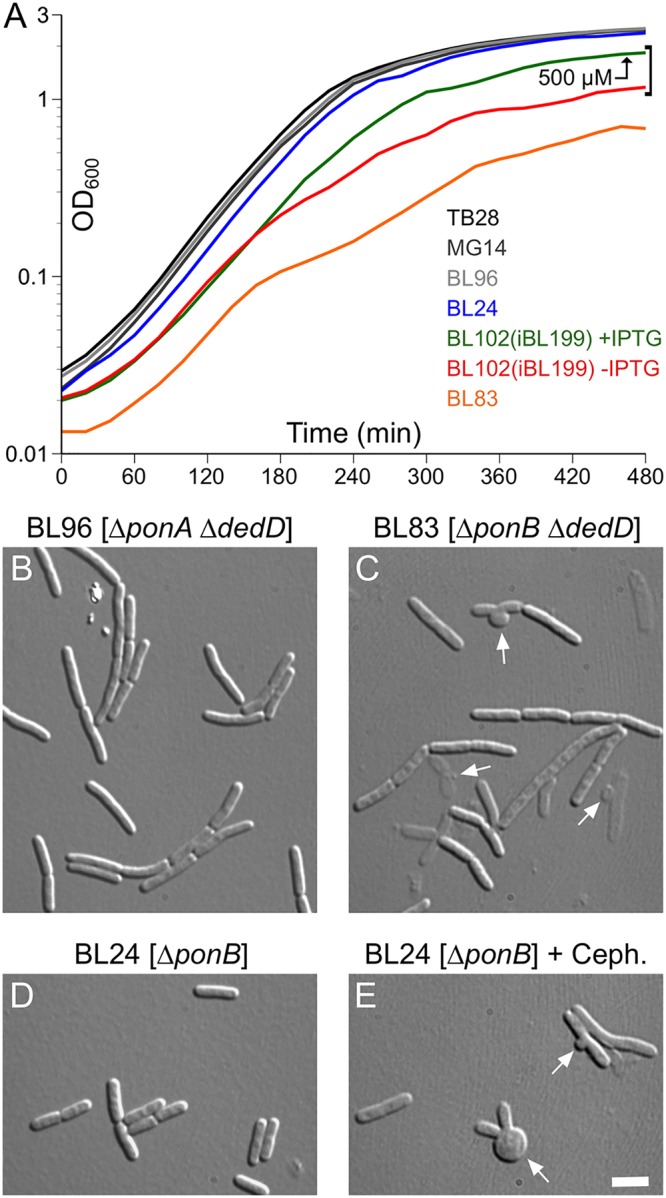
Absence of both DedD and PBP1B causes severe cell lysis. (A) Growth curves of strains TB28 [wt] (black), MG14 [ΔdedD] (dark gray), BL96 [ΔponA ΔdedD] (light gray), BL24 [ΔponB] (blue), BL83 [ΔponB ΔdedD] (orange), and the DedD depletion strain BL102(iBL199) [ΔponB ΔdedD(Plac::gfp-dedD1–54)] (red and green). Cultures were grown overnight in LB with 500 μM IPTG [BL102(iBL199)], or without inducer (all other strains) and diluted 200-fold in LB with 500 μM IPTG (green) or without inducer (all other curves). Growth was continued, and OD600 values were determined every 20 min. Note that the TB28, MG14, BL96, and BL24 curves almost coincide. The shape of the orange curve reflects the relatively low densities attained by overnight cultures of strain BL83 [ΔponB ΔdedD], a relatively long lag period, and a decrease in the rate of optical density increase around 160 min. (B to E) Strains BL96 [ΔponA ΔdedD] (B), BL83 [ΔponB ΔdedD] (C), and BL24 [ΔponB] (D and E) were cultured in parallel as in panel A, and the cells were imaged 3 h (B and D) (OD600, ∼0.6) or 5 h (C) (OD600, ∼0.3) after inoculation. When the culture in panel E reached an OD600 of 0.3, cephalexin was added to 15 μg/ml, and the cells were imaged 30 min later. The arrows in panels C and E indicate examples of septal bulges and rabbit ears, indicative of septal lysis. Bar, 4 μm.
Thus, ΔdedD is synthetically sick with ΔponB but not with ΔponA. In addition, the PBP1B-dependent phenotypes of ΔdedD cells are consistent with a model in which, like EFtsN (61), NDedD function is needed for full PBP3 activity, albeit to a lesser extent than the former.
Parallel roles for FtsN and DedD in cell constriction.
One possibility is that NDedD acts through FtsN to stimulate cell constriction. If so, an increased level of DedD might help alleviate the division defect seen in cells with reduced FtsN activity. As illustrated in Fig. 6B, however, overproduction of GFP-DedD had little to no effect on the chaining phenotype of TB77 [ftsNslm117] cells. Conversely, moreover, overexpression of GFP-FtsN did little to correct the chaining phenotype of BL40 [ΔdamX ΔdedD] cells (Fig. 6F). These results rather favor a model in which FtsN and DedD act independently.
FIG 6.

DedD functions independently of FtsN. (A to J) Cells were grown for ∼5 mass doublings to an OD600 of 0.5 to 0.6 in LB with 50 μM IPTG (A, B, D, and F) or without inducer (C, E, and G to J) and were imaged live (A to F) or after chemical fixation (G to J). Bar, 4 μm. (A to F) Little to no cross-functionality of FtsN and DedD. Cells of strain TB77 [ftsNslm117] (A to C) and BL40 [ΔdamX ΔdedD] (D to F) carrying pMLB1113ΔH [Plac::] (A and D), pFB236 [Plac::gfp-dedD] (B and E), or pCH201 [Plac::gfp-ftsN] (C and F) are shown. (G to J) EFtsN*-suppressing mutations in ftsA, ftsB, or ftsL also suppress the division defect associated with the absence of DedD. Shown are cells of strains BL40 [ΔdamX ΔdedD] (G), CH178 [ΔdamX ΔdedD ftsAE124A] (H), CH177 [ΔdamX ΔdedD ftsBE56A] (I), and CH181 [ΔdamX ΔdedD ftsLD93G] (J). (K) Depletion of DedD from ftsBE56A ΔftsN cells is lethal. Strains BL173 [ftsBE56A ΔftsN] (a and b) and BL188 [ftsBE56A ΔftsN ΔdedD] (c), carrying pMG36 [PBAD::dedD] (b and c) or the vector control pBAD33 [PBAD::] (a), were grown overnight in LB with 0.5% arabinose. The cultures were serially diluted in LB to an OD600 of 1.0 × 10x, and 5 μl of each dilution was spotted on LB agar containing glucose, arabinose, or neither, as indicated. The plates were incubated for 20 h.
We previously showed that the normal requirement for the essential domain of FtsN (EFtsN) can be overcome by EFtsN*-suppressing superfission (sf) mutations in ftsA, ftsB, or ftsL (61). To test if they also alleviate the need for NDedD, we created three BL40 derivatives carrying representative sf alleles (ftsAE124A, ftsBE56A, and ftsLD93G) and observed their division phenotypes. Interestingly, each of the sf alleles indeed suppressed the division defects of ΔdamX ΔdedD cells to a significant degree (Fig. 6G to J). The ftsBE56A allele was recently shown to suppress the filamentation phenotype of ΔdedD cells at very low growth temperatures as well (83). As it allows cells to survive and divide in the complete absence of any part of FtsN, ftsBE56A is considered to be a particularly strong sf allele (61).
Because ftsBE56A suppresses the (lethal) division phenotypes associated with ΔftsN (61) or ΔdedD (Fig. 6I) so efficiently, we tested whether it could compensate for the simultaneous absence of both FtsN and DedD. Attempts to create an ftsBE56A ΔdedD ΔftsN strain were successful only when the strain also harbored a plasmid encoding FtsN or DedD, however, indicating that ftsBE56A cannot overcome the absence of both proteins (data not shown). This was further studied using one of the resulting strains, BL188/pMG39 [ftsBE56A ΔdedD ΔftsN/PBAD::dedD]. This strain lacks chromosomal ftsN and dedD but harbors a plasmid that encodes dedD under control of the ara regulatory region. Cells of the strain survived on media containing arabinose, but they halted cell division and died in the absence of the sugar (Fig. 6K, row c, and results not shown). Thus, the viability of ftsBE56A ΔftsN cells depends on DedD function, and DedD can promote cell constriction and viability in the complete absence of any part of the FtsN protein.
Together, these results support a model in which FtsN and DedD act in parallel to stimulate cell constriction, with their action paths converging at or near the point at which FtsA and the FtsBLQ subcomplex are proposed to undergo conformational changes that promote initiation of sPG synthesis by the FtsW-PBP3 subcomplex (61). Moreover, even if the FtsBE56ALQ subcomplex is inherently more prone to undergo this change than wild-type FtsBLQ (61), the action of either DedD or FtsN is apparently still needed to stimulate the “on” conformation of FtsBE56ALQ to a sufficient degree (see Discussion).
BACTH analyses of DedD and other septal-ring proteins.
To identify potential SR protein partners of DedD, we performed bacterial two-hybrid (BACTH) assays (94, 95). The T18 domain was appended to the cytoplasmic N termini of full-length DedD and of SPOR-less DedD1–118, and each bait fusion was coexpressed with a panel of prey fusions, including all essential SR proteins, bearing the T25 domain at a cytoplasmic terminus of the protein of interest (see Fig. S8 in the supplemental material). The interaction patterns of T18-DedD and T18-DedD1–118 were essentially identical, with strong apparent affinities for FtsQ and the N-terminal domain of FtsK (FtsK1–266); more moderate affinities for DedD itself, FtsA, FtsL, ZipA, FtsI, FtsB, PBP1A, PBP1B, TolA, and FtsN; and weak or no affinities for PBP2, FtsW, FtsEX, ZapC, TolQ, and FtsZ (see Fig. S8A and B). Though relative apparent affinities differed, the patterns were also similar to that seen when T18-FtsN was used as bait, with the notable exception that an interaction between FtsN and FtsL was not obvious in these assays (see Fig. S8C).
As substitution of G11 or P24 is detrimental to DedD function (Fig. 2 and 4), we assessed if such substitutions had a significant impact on the interaction pattern of T18-DedD1–118, using a slightly different panel of prey fusions (see Fig. S9). While the interaction pattern of T18-DedD1–118, P24A did not differ greatly from that of T18-DedD1–118, that of T18-DedD1–118, G11A consistently showed a reduced signal with T25-FtsL (Fig. 7; see Fig. S9). Accordingly, the G11C substitution resulted in a similar reduction in the apparent interaction with FtsL (Fig. 7). These results suggest that an interaction between NDedD and FtsL is important for the ability of DedD to stimulate cell constriction.
FIG 7.

Interactions of DedD1–118 and mutant variants with other division proteins in BACTH assays. Strain BTH101 [cya] was cotransformed with the plasmid pairs indicated at the right and bottom and plated on LB agar containing Amp, Kan, and 0.2% glucose. Purified colonies were subsequently striped on M9-glucose agar supplemented with Amp, Kan, 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), and 250 μM IPTG. The plate was incubated for 48 h before imaging.
DISCUSSION
Activation of FtsW-FtsI-based sPG synthases within the septal ring to start production of an sPG annulus is likely a determining step in the initiation of cell constriction in walled bacteria (96, 97). Several other SR proteins have been implicated in regulation of FtsW-FtsI activities in E. coli. In the current model (Fig. 8), both FtsA and the FtsBLQ subcomplex can exist in an “off” or “on” state/conformation that suppresses or allows/promotes sPG synthesis by FtsW-FtsI, respectively. In turn, the off/on state of FtsA or FtsBLQ is regulated by additional SR proteins, in particular by FtsN (61, 62, 74, 77).
FIG 8.

Roles of DedD in stimulating E. coli cell constriction. The models are based on models proposed previously (61, 62, 71, 77, 102) and incorporate the data on DedD presented here. (A and B) Like EFtsN, NDedD stimulates FtsW-FtsI indirectly via the FtsBLQ subcomplex. For clarity, only the IM (gray line) and a relevant subset of SR proteins are depicted. FtsW (W) and PBP3 (FtsI) (I) form the core of sPG synthases (boxed) within the SR. FtsA (A) helps tether FtsZ (Z) polymers to the cytoplasmic face of the IM. FtsB (purple), FtsL (yellow), and FtsQ (brown) form the transmembrane FtsBLQ subcomplex (BLQ). FtsX (X) forms a transmembrane subcomplex with the cytoplasmic ATPase FtsE (E). EnvC and the murein amidases AmiA and AmiB (Ami) reside in the periplasm. Both FtsN (N) and DedD are bitopic IM proteins with a periplasmic C-terminal SPOR domain. Initiation of cell constriction requires inactive sPG synthases to become active. This switch is controlled by FtsA and the FtsBLQ subcomplex, each of which can adopt conformational states that either suppress (off in panel A), or allow/stimulate (on in panel B) FtsW-FtsI activity. The FtsA off (more polymeric) and FtsA on (less polymeric) conformations likely correspond to the oligomeric state of the protein. The state of FtsBLQ is assumed to be communicated to FtsW-FtsI via direct interactions between the two subcomplexes. Either state of FtsA (off or on) may help to stabilize the corresponding state of FtsBLQ, and vice versa, via direct or indirect interactions (61). The state of FtsA could be communicated to FtsW-FtsI indirectly via such effects on FtsBLQ (double-headed dashed lines). Alternatively, FtsW-FtsI senses the state of FtsA directly or via some other route (single-headed dashed lines). FtsX interacts with both FtsA and EnvC and promotes either the on or off conformations of both proteins, depending on whether FtsEX is actively hydrolyzing ATP or not, respectively. The state of EnvC, in turn, directly regulates whether the murein amidases AmiA and AmiB (Ami) are active (EnvC on) or not (EnvC off). (A) Prior to the initiation of active cell fission, both FtsA and FtsBLQ exist mostly in their off conformations. FtsEX ATPase activity is low and/or uncoupled from conformational changes in its binding partners, favoring the off states of both FtsA and EnvC. No sPG is produced, and AmiA and AmiB are inactive. (B) Both FtsN and DedD allosterically promote FtsBLQ to switch to its on conformation. While FtsN does so in the periplasm via its essential EFtsN peptide (blue), DedD does so within or very near the membrane via NDedD (orange). In the cytoplasm, meanwhile, NFtsN (magenta) directly binds FtsA and stimulates the FtsA on state. FtsEX ATPase activity leads to further stimulation of this state and also promotes the EnvC on conformation. Synthesis of the sPG annulus (sPG) is initiated, and AmiA and -B become active. As FtsW-FtsI adds new material to the inner edge of the sPG annulus, the amidases split its outer edge to generate the polar PG that will shape the two nascent cell poles. Amidase action also results in a high local concentration of denuded glycan strands (G), which are the preferred binding substrate of the SPOR domains of both FtsN and DedD. Hence, additional FtsN and DedD molecules accumulate at the SR, and a positive-feedback loop in the cell constriction process (sPG loop) is established. (C) Proposed roles of DedD within the sPG loop in schematic format. Factors that promote or inhibit progress through the loop are indicated by green or red lines, respectively. FtsEX either inhibits (ATPase inactive) or promotes (ATPase active) progress. See above and the text for further explanation.
In the case of FtsA, the direct interaction of its 1C domain with NFtsN is proposed to promote the FtsA off to on switch (61, 73, 74). In addition, this switch is likely also controlled by the ATP hydrolysis status of FtsEX, an ABC transporter-like SR component composed of the ATPase FtsE and the polytopic transmembrane protein FtsX (77, 98, 99). FtsEX plays intriguing roles in both assembly and function of the SR. It joins the ZR early and helps recruit subsequent SR proteins, including FtsK and EnvC, during SR maturation (100–102). During active cell fission, FtsEX additionally modulates the progress of the sPG loop by regulating both the sPG synthesis and sPG splitting processes (Fig. 8C). Thus, the FtsX subunit interacts with FtsA and promotes either of its conformations, depending on whether FtsEX ATPase is active (FtsA on) or not (FtsA off) (77). In addition, FtsX interacts with the periplasmic EnvC protein on the other side of the membrane. FtsEX ATPase activity controls the conformation of EnvC as well, in this case to modulate the sPG-splitting activities of AmiA and AmiB (102–104). Given these intricate roles of FtsEX in SR assembly and function, it is remarkable that the division and viability defects of ΔftsEX cells can be largely alleviated by a modest increase in osmolarity of the growth medium (98, 100, 105). As such, FtsEX is only conditionally essential for cell fission and viability in E. coli and, like DedD, belongs to the noncore SR components.
Several mutant superfission variants of FtsA that reduce the need for FtsN, or for FtsEX in low-osmotic medium, have been identified (61, 76, 77, 106). One reasonable interpretation of their phenotype is that these FtsASF variants are inherently more prone to adopt the on conformation than the native protein. Corresponding residue substitutions map to each of the four domains of the protein, and most FtsASF variants display a decreased degree of self-interaction in two-hybrid assays (77, 107). Hence, the off/on states of FtsA appear to be related to its degree of self-interaction, with longer or circular polymers corresponding to off and shorter, noncircular oligomers or monomers to on (20, 74). Accordingly, the interactions of native FtsA with NFtsN or FtsEX are proposed to interfere with FtsA self-interaction, reducing its polymerization state in the SR (Fig. 8) (2, 61, 62, 77).
In the case of FtsBLQ, the off-to-on switch is stimulated by direct or indirect interaction with the EFtsN domain of FtsN in the periplasm (61, 62). So far, SF variants of the subcomplex that bypass the need for EFtsN, or for all of FtsN, have been found to contain residue substitutions in periplasmic CCD (constriction control domain) subdomains of the FtsB or FtsL protein (61, 77). How these residue substitutions, or the action of EFtsN, might affect the FtsBLQ conformation is still unclear. Even so, the prediction is that the FtsBLQ on conformation is communicated to FtsW-FtsI and stimulates (or disinhibits) its sPG synthesis activities (Fig. 8) (61, 62, 77).
DedD resembles FtsN in several ways. It is a bitopic inner membrane protein with a small cytoplasmic domain and a large periplasmic domain that ends with a C-terminal SPOR fold. By binding PG glycan strands that have been at least partially denuded by the action of the PG amidases, the SPOR domain acts as a strong septal targeting domain for both proteins (36, 70, 71). We found here that, like FtsN (61, 72, 74), DedD contains a weak septal localization determinant at its N terminus as well (Fig. 1 and 4 and Table 1; see Fig. S6 in the supplemental material). In the case of FtsN, this weak SPOR-independent accumulation at the SR is driven by its interaction with FtsA, which involves well-conserved residues near the extreme N terminus of NFtsN (61, 72, 74). In comparison, the cytoplasmic domain of DedD is significantly shorter (∼8 residues) than that of FtsN (∼30 residues) and not as highly conserved (Fig. 4A; see Fig. S1). In addition, replacement of residue G11 within the predicted TM domain of DedD abrogated its SPOR-independent accumulation at the SR (Fig. 4 and Table 1), indicating that recruitment of NDedD to the SR is at least in part mediated by interactions of TMDedD within the IM bilayer. The substitutions at G11 also diminished DedD activity (Fig. 2 and 4) and its interaction with FtsL in BACTH assays (Fig. 7; see Fig. S9). Replacement of another conserved residue in TMDedD, P24, similarly diminished DedD function but had less of an effect on SPOR-independent accumulation of NDedD at SRs and on the apparent interaction with FtsL (Fig. 2, 4, and 7 and Table 1; see Fig. S9). One attractive possibility is that the weak accumulation of SPOR-less DedD at the SR reflects TM-TM interactions between NDedD and the FtsBLQ complex and that these interactions are also important for DedD’s function in stimulating cell constriction.
A tentative model for DedD action in stimulating cell fission is added to those proposed for FtsN and FtsEX in Fig. 8. In the model, DedD stimulates FtsW-FtsI activity via NDedD. As NDedD is small (<54 residues) and shows no obvious similarity to known enzymes, the peptide likely acts allosterically on other SR proteins to promote cell constriction, as has also been proposed for NFtsN and EFtsN (61, 71). The proposal that NDedD normally contributes to FtsW-FtsI activity is consistent with the chaining (otherwise wt), lethal filamentous (ftsNslm117), and lysis (ΔponB) phenotypes of DedD-depleted cells. The peptide could communicate directly with the FtsW-FtsI sPG synthase subcomplex. However, the findings that NDedD interacts with all three components of the FtsBLQ subcomplex in BACTH assays and that replacement of the functionally important G11 residue in the transmembrane portion of NDedD reduces the apparent interaction with FtsL most notably favor a more indirect mechanism. Thus, we propose that NDedD interacts with the FtsBLQ complex and promotes its on conformation (Fig. 8B). Similar indirect regulation of FtsW-FtsI activity via the FtsBLQ subcomplex is also proposed for the EFtsN peptide (61, 62). However, one clear distinction between EFtsN and NDedD properties is that the former acts in the periplasmic compartment while action of the latter is likely confined to within, or very near, the IM phospholipid bilayer (Fig. 8B). Another distinction, of course, is that EFtsN activity is essential to cell division and survival in otherwise wild-type cells, while NDedD activity becomes essential for survival only when cells suffer reduced FtsN activity.
Our studies suggest that, as with FtsN (61, 71), DedD supports cell constriction not only by stimulating sPG synthesis, but by providing positive feedback to the sPG loop as well (Fig. 8C). Prior to the start of constriction, the concentrations of FtsN and DedD in the SR are still low, and FtsA and FtsBLQ exist mostly in their off conformations (Fig. 8A). For sPG synthesis to start, a sufficient proportion of FtsA and/or FtsBLQ subcomplexes in the SR must switch to the on state (Fig. 8B). Switching in wild-type cells could be promoted by an increase in the concentration of FtsN and/or DedD at the SR, an increase in FtsEX ATPase activity, stochastic fluctuations in the conformations of FtsA and FtsBLQ themselves, or a combination of these and any number of other factors. The production of some initial sPG, its subsequent processing by amidases, and the SPOR-dependent recruitment of more molecules of both FtsN and DedD to the SR then launch the sPG loop (Fig. 8C), and the cells enter a sustained and self-promoting cell constriction phase.
Though the proposed action of DedD in promoting cell fission is consistent with the available data, additional work is needed to test various aspects of the model more rigorously. The simplest scenario is that both NDedD and EFtsN directly contact different parts of the FtsBLQ subcomplex to modulate its conformation, but biochemical and/or structural evidence will be needed to confirm or refute this. Other pertinent questions include the following. How do FtsBLQ off and FtsBLQ on differ structurally, and how is this sensed by FtsW-FtsI? Does FtsA communicate with FtsW-FtsI directly, via FtsBLQ, or via some other route? How is FtsEX ATPase activity controlled? How are the activities of all these proteins affected by the dynamics of the interpolymer complexes of FtsA and FtsZ? Elucidation of these and related issues will be needed to provide us with a deeper understanding of the bacterial cell constriction process and the regulation of its initiation.
MATERIALS AND METHODS
Strains, genetic constructs, and growth conditions.
The strains and plasmids used in this study are listed in Tables S1 and S2 in the supplemental material, respectively, and their construction is detailed in the supplemental material. Unless stated otherwise, cells were grown at 30°C in either LB (1% tryptone, 0.5% yeast extract, 0.5% NaCl, pH 7.0) or M9 minimal medium (47.7 mM Na2HPO4, 22.0 mM KH2PO4, 8.6 mM NaCl, 18.7 mM NH4Cl, 2.0 mM MgSO4, 0.1 mM CaCl) (108) supplemented with 0.2% Casamino Acids, 50 μg/ml l-tryptophan, 50 μM thiamine, and 0.2% glucose (M9-glucose) or maltose (M9-maltose). When appropriate, the medium was supplemented with 50 μg/ml ampicillin (Amp), 25 μg/ml chloramphenicol (Cam), 25 μg/ml kanamycin (Kan), 50 μg/ml spectinomycin (Spec), or 12.5 μg/ml tetracycline (Tet). Amp or Kan concentrations were reduced to 15 and 10 μg/ml, respectively, when cells carried bla or aph integrated in the chromosome. Arabinose and/or IPTG was included in the media as indicated. For the growth curves in Fig. 5, 0.2-ml cultures were grown with shaking at 30°C in a 96-well Falcon 353072 plate in a BioTek Synergy HT microplate reader, and the OD600 was measured every 20 min. Other details are specified in the text.
Microscopy, immunoblotting, and other methods.
Imaging was performed on a Zeiss Axioplan-2 microscope system as previously described (61, 109). For some experiments, cells were chemically fixed as described previously (110) prior to microscopy. Western analyses with anti-GFP antibodies (Rockland) were done essentially as described previously (63). BACTH assays (94) were performed as described previously (95).
Supplementary Material
ACKNOWLEDGMENTS
We thank Matthew Gerding, Pei-Chung Lee, Kyle Logue, and Thomas Bernhardt for help in plasmid and/or strain construction.
This work was supported by NIH GM57059 (to P.A.J.D.B.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00698-18.
REFERENCES
- 1.de Boer PAJ. 2010. Advances in understanding E. coli cell fission. Curr Opin Microbiol 13:730–737. doi: 10.1016/j.mib.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lutkenhaus J, Du S. 2017. E. coli cell cycle machinery. Subcell Biochem 84:27–65. doi: 10.1007/978-3-319-53047-5_2. [DOI] [PubMed] [Google Scholar]
- 3.Erickson HP, Anderson DE, Osawa M. 2010. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev 74:504–528. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Boer PA. 2016. Bacterial physiology: life minus Z. Nat Microbiol 1:16121. doi: 10.1038/nmicrobiol.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bi E, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 6.Hale CA, de Boer PAJ. 1999. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ, and independent of FtsA. J Bacteriol 181:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pichoff S, Lutkenhaus J. 2002. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J 21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Din N, Quardokus EM, Sackett MJ, Brun YV. 1998. Dominant c-terminal deletions of FtsZ that affect its ability to localize in Caulobacter and its interaction with FtsA. Mol Microbiol 27:1051–1063. doi: 10.1046/j.1365-2958.1998.00752.x. [DOI] [PubMed] [Google Scholar]
- 9.Ma X, Margolin W. 1999. Genetic and functional analyses of the conserved C-terminal core domain of Escherichia coli FtsZ. J Bacteriol 181:7531–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haney SA, Glasfeld E, Hale C, Keeney D, He Z, de Boer PA. 2001. Genetic analysis of the E. coli FtsZ-ZipA interaction in the yeast two- hybrid system. Characterization of FtsZ residues essential for the interactions with ZipA and with FtsA. J Biol Chem 276:11980–11987. doi: 10.1074/jbc.M009810200. [DOI] [PubMed] [Google Scholar]
- 11.Hale CA, Rhee AC, de Boer PAJ. 2000. ZipA-induced bundling of FtsZ polymers mediated by an interaction between C-terminal domains. J Bacteriol 182:5153–5166. doi: 10.1128/JB.182.18.5153-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hale CA, de Boer PAJ. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88:175–185. doi: 10.1016/S0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- 13.Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol 55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 14.van Den Ent F, Löwe J. 2000. Crystal structure of the cell division protein FtsA from Thermotoga maritima. EMBO J 19:5300–5307. doi: 10.1093/emboj/19.20.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anantharaman V, Aravind L. 2004. The SHS2 module is a common structural theme in functionally diverse protein groups, like Rpb7p, FtsA, GyrI, and MTH1598/TM1083 superfamilies. Proteins 56:795–807. doi: 10.1002/prot.20140. [DOI] [PubMed] [Google Scholar]
- 16.Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. 2005. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Mol Microbiol 55:699–711. doi: 10.1111/j.1365-2958.2004.04432.x. [DOI] [PubMed] [Google Scholar]
- 17.Pichoff S, Lutkenhaus J. 2007. Identification of a region of FtsA required for interaction with FtsZ. Mol Microbiol 64:1129–1138. doi: 10.1111/j.1365-2958.2007.05735.x. [DOI] [PubMed] [Google Scholar]
- 18.Szwedziak P, Wang Q, Freund SM, Lowe J. 2012. FtsA forms actin-like protofilaments. EMBO J 31:2249–2260. doi: 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krupka M, Cabre EJ, Jimenez M, Rivas G, Rico AI, Vicente M. 2014. Role of the FtsA C terminus as a switch for polymerization and membrane association. mBio 5:e02221. doi: 10.1128/mBio.02221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krupka M, Rowlett VW, Morado D, Vitrac H, Schoenemann K, Liu J, Margolin W. 2017. Escherichia coli FtsA forms lipid-bound minirings that antagonize lateral interactions between FtsZ protofilaments. Nat Commun 8:15957. doi: 10.1038/ncomms15957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osawa M, Erickson HP. 2013. Liposome division by a simple bacterial division machinery. Proc Natl Acad Sci U S A 110:11000–11004. doi: 10.1073/pnas.1222254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loose M, Mitchison TJ. 2014. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat Cell Biol 16:38–46. doi: 10.1038/ncb2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szwedziak P, Wang Q, Bharat TA, Tsim M, Lowe J. 2014. Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. Elife 3:e04601. doi: 10.7554/eLife.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bisson-Filho AW, Hsu YP, Squyres GR, Kuru E, Wu F, Jukes C, Sun Y, Dekker C, Holden S, VanNieuwenhze MS, Brun YV, Garner EC. 2017. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355:739–743. doi: 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, Xiao J. 2017. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355:744–747. doi: 10.1126/science.aak9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goehring NW, Beckwith J. 2005. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr Biol 15:R514–R526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 27.den Blaauwen T, de Pedro MA, Nguyen-Distèche M, Ayala JA. 2008. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev 32:321–344. doi: 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 28.Vollmer W, Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta 1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. 2008. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol Rev 32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 30.Inoue A, Murata Y, Takahashi H, Tsuji N, Fujisaki S, Kato J. 2008. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J Bacteriol 190:7298–7301. doi: 10.1128/JB.00551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruiz N. 2008. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc Natl Acad Sci U S A 105:15553–15557. doi: 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sham LT, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. 2014. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Höltje JV. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev 62:181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Höltje JV. 2001. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol 41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- 35.Priyadarshini R, de Pedro MA, Young KD. 2007. Role of peptidoglycan amidases in the development and morphology of the division septum in Escherichia coli. J Bacteriol 189:5334–5347. doi: 10.1128/JB.00415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yahashiri A, Jorgenson MA, Weiss DS. 2015. Bacterial SPOR domains are recruited to septal peptidoglycan by binding to glycan strands that lack stem peptides. Proc Natl Acad Sci U S A 112:11347–11352. doi: 10.1073/pnas.1508536112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Lallo G, Fagioli M, Barionovi D, Ghelardini P, Paolozzi L. 2003. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology 149:3353–3359. doi: 10.1099/mic.0.26580-0. [DOI] [PubMed] [Google Scholar]
- 38.Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol 187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraipont C, Alexeeva S, Wolf B, van der Ploeg R, Schloesser M, den Blaauwen T, Nguyen-Distèche M. 2011. The integral membrane FtsW protein and peptidoglycan synthase PBP3 form a subcomplex in Escherichia coli. Microbiology 157:251–259. doi: 10.1099/mic.0.040071-0. [DOI] [PubMed] [Google Scholar]
- 40.Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, Kamisetty H, Grishin NV, Baker D. 2015. Large-scale determination of previously unsolved protein structures using evolutionary information. Elife 4:e09248. doi: 10.7554/eLife.09248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leclercq S, Derouaux A, Olatunji S, Fraipont C, Egan AJ, Vollmer W, Breukink E, Terrak M. 2017. Interplay between penicillin-binding proteins and SEDS proteins promotes bacterial cell wall synthesis. Sci Rep 7:43306. doi: 10.1038/srep43306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spratt BG. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A 72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adam M, Damblon C, Jamin M, Zorzi W, Dusart V, Galleni M, el Kharroubi A, Piras G, Spratt BG, Keck W. 1991. Acyltransferase activities of the high-molecular-mass essential penicillin-binding proteins. Biochem J 279:601–604. doi: 10.1042/bj2790601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adam M, Fraipont C, Rhazi N, Nguyen-Disteche M, Lakaye B, Frere JM, Devreese B, Van Beeumen J, van Heijenoort Y, van Heijenoort J, Ghuysen JM. 1997. The bimodular G57-V577 polypeptide chain of the class B penicillin- binding protein 3 of Escherichia coli catalyzes peptide bond formation from thiolesters and does not catalyze glycan chain polymerization from the lipid II intermediate. J Bacteriol 179:6005–6009. doi: 10.1128/jb.179.19.6005-6009.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taguchi A, Welsh MA, Marmont LS, Lee W, Kahne D, Bernhardt TG, Walker S. 2018. FtsW is a peptidoglycan polymerase that is activated by its cognate penicillin-binding protein. bioRxiv doi: 10.1101/358663. [DOI] [PMC free article] [PubMed]
- 46.Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ. 2016. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537:634–638. doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho H, Wivagg CN, Kapoor M, Barry Z, Rohs PD, Suh H, Marto JA, Garner EC, Bernhardt TG. 2016. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat Microbiol 19:16172. doi: 10.1038/nmicrobiol.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emami K, Guyet A, Kawai Y, Devi J, Wu LJ, Allenby N, Daniel RA, Errington J. 2017. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat Microbiol 2:16253. doi: 10.1038/nmicrobiol.2016.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rohs PDA, Buss J, Sim SI, Squyres GR, Srisuknimit V, Smith M, Cho H, Sjodt M, Kruse AC, Garner EC, Walker S, Kahne DE, Bernhardt TG. 2018. A central role for PBP2 in the activation of peptidoglycan polymerization by the bacterial cell elongation machinery. PLoS Genet 14:e1007726. doi: 10.1371/journal.pgen.1007726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohammadi T, Sijbrandi R, Lutters M, Verheul J, Martin NI, den Blaauwen T, de Kruijff B, Breukink E. 2014. Specificity of the transport of lipid II by FtsW in Escherichia coli. J Biol Chem 289:14707–14718. doi: 10.1074/jbc.M114.557371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Egan AJ, Biboy J, van't Veer I, Breukink E, Vollmer W. 2015. Activities and regulation of peptidoglycan synthases. Philos Trans R Soc Lond B Biol Sci 370:20150031. doi: 10.1098/rstb.2015.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corbin BD, Geissler B, Sadasivam M, Margolin W. 2004. Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. J Bacteriol 186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Müller P, Ewers C, Bertsche U, Anstett M, Kallis T, Breukink E, Fraipont C, Terrak M, Nguyen-Distèche M, Vollmer W. 2007. The essential cell division protein FtsN interacts with the murein (peptidoglycan) synthase PBP1B in Escherichia coli. J Biol Chem 282:36394–36402. doi: 10.1074/jbc.M706390200. [DOI] [PubMed] [Google Scholar]
- 54.D’Ulisse V, Fagioli M, Ghelardini P, Paolozzi L. 2007. Three functional subdomains of the Escherichia coli FtsQ protein are involved in its interaction with the other division proteins. Microbiology 153:124–138. doi: 10.1099/mic.0.2006/000265-0. [DOI] [PubMed] [Google Scholar]
- 55.Alexeeva S, Gadella TW Jr, Verheul J, Verhoeven GS, den Blaauwen T. 2010. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol Microbiol 77:384–398. doi: 10.1111/j.1365-2958.2010.07211.x. [DOI] [PubMed] [Google Scholar]
- 56.Bertsche U, Kast T, Wolf B, Fraipont C, Aarsman MEG, Kannenberg K, von Rechenberg M, Nguyen-Distèche M, den Blaauwen T, Höltje J-V, Vollmer W. 2006. Interaction between two murein (peptidoglycan) synthases, PBP3 and PBP1B, in Escherichia coli. Mol Microbiol 61:675–690. doi: 10.1111/j.1365-2958.2006.05280.x. [DOI] [PubMed] [Google Scholar]
- 57.Buddelmeijer N, Beckwith J. 2004. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol Microbiol 52:1315–1327. doi: 10.1111/j.1365-2958.2004.04044.x. [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez MD, Akbay EA, Boyd D, Beckwith J. 2010. Multiple interaction domains in FtsL, a protein component of the widely conserved bacterial FtsLBQ cell division complex. J Bacteriol 192:2757–2768. doi: 10.1128/JB.01609-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glas M, van den Berg van Saparoea HB, McLaughlin SH, Roseboom W, Liu F, Koningstein GM, Fish A, den Blaauwen T, Heck AJ, de Jong L, Bitter W, de Esch IJ, Luirink J. 2015. The soluble periplasmic domains of Escherichia coli cell division proteins FtsQ/FtsB/FtsL form a trimeric complex with submicromolar affinity. J Biol Chem 290:21498–21509. doi: 10.1074/jbc.M115.654756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mercer KL, Weiss DS. 2002. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J Bacteriol 184:904–912. doi: 10.1128/jb.184.4.904-912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu B, Persons L, Lee L, de Boer PA. 2015. Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol Microbiol 95:945–970. doi: 10.1111/mmi.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsang MJ, Bernhardt TG. 2015. A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol Microbiol 95:925–944. doi: 10.1111/mmi.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bernhardt TG, de Boer PAJ. 2003. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol 48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Priyadarshini R, Popham DL, Young KD. 2006. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J Bacteriol 188:5345–5355. doi: 10.1128/JB.00476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uehara T, Park JT. 2008. Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and septation. J Bacteriol 190:3914–3922. doi: 10.1128/JB.00207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uehara T, Parzych KR, Dinh T, Bernhardt TG. 2010. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J 29:1412–1422. doi: 10.1038/emboj.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernhardt TG, de Boer PA. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol Microbiol 52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uehara T, Dinh T, Bernhardt TG. 2009. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J Bacteriol 191:5094–5107. doi: 10.1128/JB.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsang MJ, Yakhnina AA, Bernhardt TG. 2017. NlpD links cell wall remodeling and outer membrane invagination during cytokinesis in Escherichia coli. PLoS Genet 13:e1006888. doi: 10.1371/journal.pgen.1006888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ursinus A, van den Ent F, Brechtel S, de Pedro M, Höltje J-V, Löwe J, Vollmer W. 2004. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli. J Bacteriol 186:6728–6737. doi: 10.1128/JB.186.20.6728-6737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerding MA, Liu B, Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. 2009. Self-enhanced accumulation of FtsN at division sites, and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J Bacteriol 191:7383–7401. doi: 10.1128/JB.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Busiek KK, Margolin W. 2014. A role for FtsA in SPOR-independent localization of the essential Escherichia coli cell division protein FtsN. Mol Microbiol 92:1212–1226. doi: 10.1111/mmi.12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Busiek KK, Eraso JM, Wang Y, Margolin W. 2012. The early divisome protein FtsA interacts directly through its 1c subdomain with the cytoplasmic domain of the late divisome protein FtsN. J Bacteriol 194:1989–2000. doi: 10.1128/JB.06683-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pichoff S, Du S, Lutkenhaus J. 2015. The bypass of ZipA by overexpression of FtsN requires a previously unknown conserved FtsN motif essential for FtsA-FtsN interaction supporting a model in which FtsA monomers recruit late cell division proteins to the Z ring. Mol Microbiol 95:971–987. doi: 10.1111/mmi.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J-C, Van Den Ent F, Neuhaus D, Brevier J, Löwe J. 2004. Solution structure and domain architecture of the divisome protein FtsN. Mol Microbiol 52:651–660. doi: 10.1111/j.1365-2958.2004.03991.x. [DOI] [PubMed] [Google Scholar]
- 76.Bernard CS, Sadasivam M, Shiomi D, Margolin W. 2007. An altered FtsA can compensate for the loss of essential cell division protein FtsN in Escherichia coli. Mol Microbiol 64:1289–1305. doi: 10.1111/j.1365-2958.2007.05738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Du S, Pichoff S, Lutkenhaus J. 2016. FtsEX acts on FtsA to regulate divisome assembly and activity. Proc Natl Acad Sci U S A 113:E5052–E5061. doi: 10.1073/pnas.1606656113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arends SJ, Williams K, Scott RJ, Rolong S, Popham DL, Weiss DS. 2010. Discovery and characterization of three new Escherichia coli septal ring proteins that contain a SPOR domain: DamX, DedD, and RlpA. J Bacteriol 192:242–255. doi: 10.1128/JB.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lyngstadaas A, Lobner-Olesen A, Boye E. 1995. Characterization of three genes in the dam-containing operon of Escherichia coli. Mol Gen Genet 247:546–554. doi: 10.1007/BF00290345. [DOI] [PubMed] [Google Scholar]
- 80.Khandige S, Asferg CA, Rasmussen KJ, Larsen MJ, Overgaard M, Andersen TE, Moller-Jensen J. 2016. DamX controls reversible cell morphology switching in uropathogenic Escherichia coli. mBio 7:e00642-16. doi: 10.1128/mBio.00642-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jorgenson MA, Chen Y, Yahashiri A, Popham DL, Weiss DS. 2014. The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa. Mol Microbiol 93:113–128. doi: 10.1111/mmi.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Black SL, Dawson A, Ward FB, Allen RJ. 2013. Genes required for growth at high hydrostatic pressure in Escherichia coli K-12 identified by genome-wide screening. PLoS One 8:e73995. doi: 10.1371/journal.pone.0073995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Porter T, Frederick D, Johnson E, Jones PG. 2016. A requirement for cell elongation protein RodZ and cell division proteins FtsN and DedD to maintain the small rod morphology of Escherichia coli at growth temperatures near 8°C. J Gen Appl Microbiol 62:189–198. doi: 10.2323/jgam.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 84.Kall L, Krogh A, Sonnhammer EL. 2007. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res 35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garnier J, Gibrat JF, Robson B. 1996. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol 266:540–553. doi: 10.1016/S0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 86.Combet C, Blanchet C, Geourjon C, Deleage G. 2000. NPS@: network protein sequence analysis. Trends Biochem Sci 25:147–150. doi: 10.1016/S0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 87.Williams KB, Yahashiri A, Arends SJ, Popham DL, Fowler CA, Weiss DS. 2013. Nuclear magnetic resonance solution structure of the peptidoglycan-binding SPOR domain from Escherichia coli DamX: insights into septal localization. Biochemistry 52:627–639. doi: 10.1021/bi301609e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. 2007. Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 89.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Penel S, Arigon AM, Dufayard JF, Sertier AS, Daubin V, Duret L, Gouy M, Perriere G. 2009. Databases of homologous gene families for comparative genomics. BMC Bioinform 10:S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmidt LS, Botta G, Park JT. 1981. Effects of furazlocillin, a beta-lactam antibiotic which binds selectively to penicillin-binding protein 3, on Escherichia coli mutants deficient in other penicillin-binding proteins. J Bacteriol 145:632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garcia del Portillo F, de Pedro MA. 1990. Differential effect of mutational impairment of penicillin-binding proteins 1A and 1B on Escherichia coli strains harboring thermosensitive mutations in the cell division genes ftsA, ftsQ, ftsZ, and pbpB. J Bacteriol 172:5863–5870. doi: 10.1128/jb.172.10.5863-5870.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol 181:3981–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. 2009. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J 28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Coltharp C, Buss J, Plumer TM, Xiao J. 2016. Defining the rate-limiting processes of bacterial cytokinesis. Proc Natl Acad Sci U S A 113:E1044–E1053. doi: 10.1073/pnas.1514296113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Daley DO, Skoglund U, Soderstrom B. 2016. FtsZ does not initiate membrane constriction at the onset of division. Sci Rep 6:33138. doi: 10.1038/srep33138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Leeuw E, Graham B, Phillips GJ, ten Hagen-Jongman CM, Oudega B, Luirink J. 1999. Molecular characterization of Escherichia coli FtsE and FtsX. Mol Microbiol 31:983–993. doi: 10.1046/j.1365-2958.1999.01245.x. [DOI] [PubMed] [Google Scholar]
- 99.Arends SJ, Kustusch RJ, Weiss DS. 2009. ATP-binding site lesions in FtsE impair cell division. J Bacteriol 191:3772–3784. doi: 10.1128/JB.00179-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmidt KL, Peterson ND, Kustusch RJ, Wissel MC, Graham B, Phillips GJ, Weiss DS. 2004. A predicted ABC transporter, FtsEX, is needed for cell division in Escherichia coli. J Bacteriol 186:785–793. doi: 10.1128/JB.186.3.785-793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Corbin BD, Wang Y, Beuria TK, Margolin W. 2007. Interaction between cell division proteins FtsE and FtsZ. J Bacteriol 189:3026–3035. doi: 10.1128/JB.01581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang DC, Peters NT, Parzych KR, Uehara T, Markovski M, Bernhardt TG. 2011. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc Natl Acad Sci U S A 108:E1052–E1060. doi: 10.1073/pnas.1107780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang DC, Tan K, Joachimiak A, Bernhardt TG. 2012. A conformational switch controls cell wall-remodelling enzymes required for bacterial cell division. Mol Microbiol 85:768–781. doi: 10.1111/j.1365-2958.2012.08138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peters NT, Morlot C, Yang DC, Uehara T, Vernet T, Bernhardt TG. 2013. Structure-function analysis of the LytM domain of EnvC, an activator of cell wall remodelling at the Escherichia coli division site. Mol Microbiol 89:690–701. doi: 10.1111/mmi.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reddy M. 2007. Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional SufI or high osmotic strength. J Bacteriol 189:98–108. doi: 10.1128/JB.01347-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Samaluru H, SaiSree L, Reddy M. 2007. Role of SufI (FtsP) in cell division of Escherichia coli: evidence for its involvement in stabilizing the assembly of the divisome. J Bacteriol 189:8044–8052. doi: 10.1128/JB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pichoff S, Shen B, Sullivan B, Lutkenhaus J. 2012. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA's self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol 83:151–167. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roskams J, Rodgers L. 2002. Lab Ref: a handbook of recipes, reagents, and other reference tools for use at the bench. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. [Google Scholar]
- 109.Johnson JE, Lackner LL, de Boer PAJ. 2002. Targeting of DMinC/MinD and DMinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ-rings. J Bacteriol 184:2951–2962. doi: 10.1128/JB.184.11.2951-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bendezu FO, de Boer PA. 2008. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J Bacteriol 190:1792–1811. doi: 10.1128/JB.01322-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.