

Cystic fibrosis (CF) lung infections are increasingly recognized for their polymicrobial nature. These polymicrobial infections may alter the biology of the organisms involved in CF-related infections, leading to changes in growth, virulence, and/or antibiotic tolerance, and could thereby affect patient health and response to treatment. In this study, we demonstrate interactions between P. aeruginosa and streptococci using a coculture model and show that one interaction between these microbes is likely competition for iron. Thus, these data indicate that one CF pathogen may influence the growth of another, and they add to our limited knowledge of polymicrobial interactions in the CF airway.
KEYWORDS: Pseudomonas aeruginosa, Streptococcus, biofilm, cystic fibrosis, polymicrobial
ABSTRACT
Cystic fibrosis (CF) is a genetic disease that causes patients to accumulate thick, dehydrated mucus in the lung and develop chronic, polymicrobial infections due to reduced mucociliary clearance. These chronic polymicrobial infections and subsequent decline in lung function are significant factors in the morbidity and mortality of CF. Pseudomonas aeruginosa and Streptococcus spp. are among the most prevalent organisms in the CF lung; the presence of P. aeruginosa correlates with lung function decline, and the Streptococcus milleri group (SMG), a subgroup of the viridans streptococci, is associated with exacerbations in patients with CF. Here we characterized the interspecies interactions that occur between these two genera. We demonstrated that multiple P. aeruginosa laboratory strains and clinical CF isolates promote the growth of multiple SMG strains and oral streptococci in an in vitro coculture system. We investigated the mechanism by which P. aeruginosa enhances growth of streptococci by screening for mutants of P. aeruginosa PA14 that are unable to enhance Streptococcus growth, and we identified the P. aeruginosa pqsL::TnM mutant, which failed to promote growth of Streptococcus constellatus and S. sanguinis. Characterization of the P. aeruginosa ΔpqsL mutant revealed that this strain cannot promote Streptococcus growth. Our genetic data and growth studies support a model whereby the P. aeruginosa ΔpqsL mutant overproduces siderophores and thus likely outcompetes Streptococcus sanguinis for limited iron. We propose a model whereby competition for iron represents one important means of interaction between P. aeruginosa and Streptococcus spp.
IMPORTANCE Cystic fibrosis (CF) lung infections are increasingly recognized for their polymicrobial nature. These polymicrobial infections may alter the biology of the organisms involved in CF-related infections, leading to changes in growth, virulence, and/or antibiotic tolerance, and could thereby affect patient health and response to treatment. In this study, we demonstrate interactions between P. aeruginosa and streptococci using a coculture model and show that one interaction between these microbes is likely competition for iron. Thus, these data indicate that one CF pathogen may influence the growth of another, and they add to our limited knowledge of polymicrobial interactions in the CF airway.
INTRODUCTION
Cystic fibrosis (CF) is a genetic disease caused by a defect in the cystic fibrosis transmembrane conductance regulator (CFTR) (1), which leads to reduced mucociliary clearance in the lungs of these patients (2). Due to this reduced mucociliary clearance, bacteria colonize the lungs of patients with CF and establish chronic, polymicrobial infections that cause increased inflammation and respiratory function decline (2). Recent studies have demonstrated that the microbiota in the lungs form polymicrobial biofilms and that mixed bacterial biofilm populations can affect antibiotic tolerance and bacterial virulence (3, 65).
The Streptococcus milleri group (SMG), which is composed of three species (S. anginosus, S. constellatus, and S. intermedius), has been isolated from sputum samples of patients with CF. When these microbes are the numerically dominant species in the lung, these organisms correlate with exacerbation in patients with CF (4, 5, 7, 66). In contrast, previous research from our laboratory (8) and two other groups (9, 10) demonstrated that increased relative abundance of Streptococcus spp. within the CF lung microbiome correlates with better lung function and clinical stability. Together these data indicate a possible complex relationship between Streptococcus spp. and the host and between Streptococcus spp. and the other microbes in the CF airway.
Pseudomonas aeruginosa is the dominant microorganism (>50% relative abundance) in the lungs of ∼45% of adults patients with CF (11), is cultured from >80% of these patients (12), and is the predominant microbe in the lung at end-stage disease (13). P. aeruginosa and streptococci have been found to cocolonize CF patients (4, 5, 7, 14, 15), but the polymicrobial interactions that occur between these organisms are not well studied. Previous studies investigating interactions between P. aeruginosa and Streptococcus spp. demonstrated that Streptococcus spp. can influence production of P. aeruginosa virulence factors such as rhamnolipids, elastase, and phenazines (6, 16–19) and can suppress P. aeruginosa growth through hydrogen peroxide production (16) and production of reactive nitrogenous intermediates (20, 21). Conversely, P. aeruginosa was found to influence the growth (16–19, 22, 23) and biofilm formation (23, 24) of Streptococcus spp. Work from our lab demonstrated that P. aeruginosa PA14 produces the surfactants β-hydroxyalkanoyl-β-hydroxyalkanoic acids (HAAs) and monorhamnolipids, which caused a 6-fold reduction in S. constellatus 7155 biofilm formation in coculture (23). The surfactant-induced biofilm suppression was relieved when P. aeruginosa and S. constellatus 7155 were cocultured in the presence of tobramycin, an antibiotic used for maintenance therapy by patients with CF. We determined that tobramycin suppressed P. aeruginosa production of HAAs and monorhamnolipids and that in the presence of tobramycin, P. aeruginosa can enhance S. constellatus 7155 growth on a CF-derived bronchial epithelial cell (CFBE) monolayer (23). These data indicate that P. aeruginosa can both positively and negatively impact cocultured microbes, including Streptococcus spp., and that the interaction between the microbes can be influenced by environmental and/or clinical context.
In this study, we investigated the ability of P. aeruginosa to influence Streptococcus growth in our in vitro coculture system. We demonstrated that multiple P. aeruginosa strains and clinical isolates can enhance the growth of multiple Streptococcus spp. We used a candidate gene approach and a genetic screen to identify P. aeruginosa mutants that were unable to support Streptococcus growth, and we found a single mutant of P. aeruginosa that no longer enhances growth of streptococci. We found that the P. aeruginosa ΔpqsL mutant suppressed S. sanguinis growth, likely via a mechanism that involves siderophore overproduction and thus iron sequestration. These data indicate that competition for iron can impact this polymicrobial interaction.
RESULTS
P. aeruginosa promotes streptococcal growth in a coculture system.
We reported previously that P. aeruginosa can enhance viable S. constellatus 7155 cell numbers when grown as a coculture on CF-derived bronchial epithelial (CFBE) cells (23). We first sought to recapitulate the finding that P. aeruginosa promotes the Streptococcus biofilm population by using the model organism S. sanguinis SK36 under coculture conditions in the absence of CFBE cells. We used P. aeruginosa PAO1 and S. sanguinis SK36 for these experiments because both are sequenced strains (25–27) with available genetic mutant libraries (28–30). This simplified coculture system allowed us to test the interaction between P. aeruginosa and streptococci without confounding factors contributed by the CFBE cells.
To test the hypothesis that the S. sanguinis SK36 viable cell number increases in coculture with P. aeruginosa PAO1 in the absence of CFBE cells, we grew P. aeruginosa PAO1 and S. sanguinis SK36 in coculture in the wells of a plastic culture dish in minimal essential medium (MEM) tissue culture medium containing glucose. We observed that the number of viable S. sanguinis SK36 cells in a biofilm was enhanced 100- to 1,055-fold by coculture with P. aeruginosa PAO1 compared to S. sanguinis SK36 grown as a monoculture in MEM (Fig. 1A; see Fig. S1A in the supplemental material). P. aeruginosa PAO1 biofilm growth was not significantly affected by coculture with S. sanguinis SK36 (Fig. S1B). These data also indicate that the enhancement of the S. sanguinis SK36 population in a biofilm by P. aeruginosa PAO1 does not require the CFBE cells.
FIG 1.

P. aeruginosa enhances growth of Streptococcus spp. in coculture. Coculture assays were conducted to investigate streptococcal growth when cocultured with P. aeruginosa. (A) S. sanguinis SK36 (Ss) cocultured with P. aeruginosa PAO1. (B) The coculture growth kinetics of S. sanguinis with P. aeruginosa PA14 were investigated. Each time point represents the average of three biological replicates with three technical replicates. The error bars indicate standard deviation (SD). (C) A representative group of clinical and reference Streptococcus strains were tested in coculture with P. aeruginosa PAO1 (see Fig. S3A in the supplemental material for all Streptococcus species strains tested). Each strain is denoted by its strain number: S. anginosus 5535, S. intermedius 4807, S. constellatus 7155, S. parasanguinis ATCC 15912, S. pneumoniae D39, S. peroris ATCC 700780, S. oralis 7404, and S. salivarius JIM8780. (D) S. sanguinis SK36 was tested in coculture with multiple P. aeruginosa clinical and laboratory strains. (A, C, and D) Each bar represents the average of three biological replicates with three technical replicates. The error bars indicate the standard deviation. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by the paired two-tailed Student t test [A and C], repeated-measures one-way analysis of variance [ANOVA] with Tukey’s multiple-comparison posttest [D], and the paired two-tailed Student t test between Ss + PAO1 DHL08 and Ss + PAO1 mucA22 [D]). The corresponding graphs depicting P. aeruginosa growth in these assays can be found in Fig. S1 to S4 in the supplemental material.
P. aeruginosa enhances growth of S. sanguinis SK36.
We considered two models of polymicrobial interaction that may be enhancing viable S. sanguinis SK36 cells in the biofilm when grown in coculture with P. aeruginosa. P. aeruginosa might promote Streptococcus adhesion and biofilm formation, or P. aeruginosa might promote streptococcal growth. To distinguish between these models, we conducted a time course experiment with P. aeruginosa PA14, P. aeruginosa PAO1, and S. sanguinis SK36. If P. aeruginosa was promoting adhesion of S. sanguinis SK36 cells rather than growth, we predict that we would detect more S. sanguinis SK36 in the biofilm and fewer planktonic cells but that the total cell number would not increase compared to that of S. sanguinis SK36 monoculture. In contrast, if P. aeruginosa was enhancing S. sanguinis SK36 growth, then both total biofilm and planktonic S. sanguinis SK36 populations should increase in coculture compared to that of S. sanguinis SK36 monoculture. As demonstrated by the increased S. sanguinis SK36 biofilm and planktonic cells recovered from coculture compared to monoculture, P. aeruginosa appears to promote the growth of S. sanguinis SK36 (Fig. 1B; see Fig. S2A and B in the supplemental material), thus accounting for the increased population of S. sanguinis SK36 biofilm cells.
Multiple P. aeruginosa strains enhance the growth of multiple streptococci.
Based on current evidence that multiple Streptococcus species inhabit the CF lung (7, 31) and influence patient health (4, 5, 7–10, 14, 15), we sought to determine whether the observed enhancement of Streptococcus viable counts in coculture with P. aeruginosa may be more broadly generalized to other streptococci, including the Streptococcus milleri group (SMG), which has been implicated in CF-related exacerbations (4, 5, 7, 14). To assess the ability of P. aeruginosa to promote multiple Streptococcus spp., we cocultured P. aeruginosa PAO1 with 6 SMG isolates and 8 oral Streptococcus spp. Figure 1C depicts a representative strain of each streptococcal species assayed and shows the biofilm populations obtained from monoculture and coculture with P. aeruginosa PAO1 (see Fig. S3A in the supplemental material for all 14 strains tested). P. aeruginosa PAO1 growth was not significantly affected by coculture with any of the Streptococcus spp. tested (Fig. S3B).
We found that P. aeruginosa PAO1 significantly enhanced the growth of one of the two S. anginosus, two of two S. intermedius, and neither of the two S. constellatus strains tested. Additionally, of the oral Streptococcus spp. tested, P. aeruginosa PAO1 significantly promoted the growth of one of the two S. oralis isolates, the one S. parasanguinis isolate, and one of the three S. salivarius isolates tested but not that of the S. pneumoniae or S. peroris isolates tested (Fig. 1C and S3A). While not every Streptococcus isolate tested demonstrated a significant increase in viable population recovered from the coculture, most species tested exhibited a trend toward increased growth when cocultured with P. aeruginosa PAO1. These data suggest that P. aeruginosa may be promoting Streptococcus growth through a pathway that affects many Streptococcus species.
Next, we assessed whether multiple P. aeruginosa clinical and laboratory strains could promote the growth of S. sanguinis. Additionally, given that S. parasanguinis was found to bind extracellular alginate produced by mucoid P. aeruginosa strains (24), we tested whether mucoid or nonmucoid P. aeruginosa could better promote growth in our coculture system. We cocultured S. sanguinis SK36 with seven nonmucoid P. aeruginosa and four mucoid P. aeruginosa laboratory and clinical strains and observed a significant growth enhancement of S. sanguinis SK36 by 10 out of 11 P. aeruginosa strains tested in coculture biofilms (Fig. 1D) and planktonic growth (see Fig. S4A in the supplemental material). The growth of all tested P. aeruginosa strains was not affected by coculture with S. sanguinis SK36 (Fig. S4B and C).
Additionally, P. aeruginosa PAO1 (parental) and P. aeruginosa PDO300 mucA22 are isogenic nonmucoid and mucoid strains, respectively. We found a significant enhancement in viable S. sanguinis SK36 biofilm cells recovered from coculture with P. aeruginosa PDO300 mucA22 compared to P. aeruginosa PAO1 (Fig. 1D), suggesting that mucoid P. aeruginosa strains may better enhance Streptococcus growth. Additionally, these mucoid P. aeruginosa strains showed among the most robust promotion of viable counts when cocultured with Streptococcus.
To extend our findings here, we next tested the growth-enhancing capability of P. aeruginosa PA14 in rich medium, using both lysogeny broth (LB) and Todd-Hewitt broth supplemented with 0.5% yeast extract (THY). Notably, S. sanguinis SK36 growth in rich medium monoculture reached a level comparable to that for coculture in minimal medium (Fig. S4D). However, there was still a significant increase in S. sanguinis SK36 growth when in coculture with P. aeruginosa PA14 in rich medium compared to S. sanguinis SK36 monoculture under the same conditions.
In summary, we have demonstrated that our minimal medium coculture assay using a plastic substratum can recapitulate our prior observation that P. aeruginosa promotes streptococcal growth on airway cells. This observation extends to coculture assays in rich medium. Furthermore, we were able to determine that P. aeruginosa is likely promoting Streptococcus growth rather than increasing the biofilm population via enhanced adherence. The Streptococcus growth enhancement phenotype occurred among most oral streptococci tested, and the majority of P. aeruginosa clinical and laboratory strains are capable of promoting Streptococcus growth, which lends support to the idea that these interactions are common among these two genera.
Known P. aeruginosa virulence pathways are not involved in the Streptococcus growth-promoting phenotype.
P. aeruginosa has many well-characterized virulence factors that have been demonstrated to impact polymicrobial interactions, including pathways for quorum sensing (32), biofilm formation, and the production of secreted molecules such as phenazines (33, 34), siderophores (35, 65), alginate (67), and rhamnolipids (23). We hypothesized that one or more of these virulence factors might be altering Streptococcus growth in our system. To test this idea, we utilized a candidate genetic approach to assess whether any of these virulence pathways may be involved in the observed growth-enhancing phenotype. We cocultured P. aeruginosa PA14 mutants with mutations in each of the above-mentioned pathways with S. constellatus 7155 as a model streptococcal strain known to positively respond to P. aeruginosa growth enhancement (23) and assessed whether any of these mutants lost the ability to enhance S. constellatus 7155 growth. We found that none of the pathways tested were involved in enhancement of S. constellatus 7155 growth (see Table S1 in the supplemental material).
Given that KatA has been found in the supernatants of P. aeruginosa cultures (36–38), we also constructed P. aeruginosa PA14 ΔkatA, ΔkatB, and ΔkatA ΔkatB mutant strains in order to test the hypothesis that extracellular P. aeruginosa catalase is enhancing S. sanguinis SK36 growth by degrading hydrogen peroxide produced by S. sanguinis SK36. It has previously been reported that S. sanguinis and other oral streptococci can inhibit P. aeruginosa growth through hydrogen peroxide production (16, 20, 21) and that the hydrogen peroxide produced by oral streptococci plays an important role in growth inhibition, extracellular DNA (eDNA) release, and biofilm formation within the oral microbiome (39, 40). We chose to mutate the katA and katB genes and not the katE gene because previous reports indicate that KatA is the major catalase utilized by P. aeruginosa and that KatB can partially recover hydrogen peroxide resistance in the absence of KatA (36, 41). KatE was not demonstrated to play a role in alleviating hydrogen peroxide stress (41). S. sanguinis SK36 did not demonstrate reduced growth in coculture with the P. aeruginosa PA14 ΔkatA, ΔkatB, or ΔkatA ΔkatB mutant strains compared to wild-type P. aeruginosa PA14, indicating that catalase is not playing a role in the Streptococcus growth enhancement phenotype (see Table S1 and Fig. S5A in the supplemental material). The P. aeruginosa ΔkatA mutant displayed a slight but significant, growth defect in the coculture compared to wild-type P. aeruginosa PA14 in coculture, and the ΔkatB mutant displayed a modest but significant, growth defect in monoculture compared to P. aeruginosa PA14 in coculture (Fig. S5B), but these strains still stimulated S. sanguinis SK36 growth to the level observed for wild-type P. aeruginosa. Taken together, these data suggest that known virulence factors, on their own, do not contribute to P. aeruginosa-mediated growth enhancement of Streptococcus spp.
Screening the P. aeruginosa PA14NR set for P. aeruginosa PA14 transposon insertion mutant strains that do not support S. constellatus growth.
The Ausubel lab reported a nonredundant library of P. aeruginosa PA14 transposon insertion mutants (PA14NR set), which contains 5,459 transposon insertion mutant strains with mutations in 4,596 genes (42). Each of these P. aeruginosa PA14 transposon mutant strains was tested in coculture with S. constellatus 7155 (Fig. 2A). Of the 5,459 mutant strains in the library, 48 strains were unable to promote S. constellatus 7155 growth in two replicate experiments (see Table S2 in the supplemental material). Two of these 48 mutants were eliminated when we tested available deletion mutants, as the deletion mutant strains did not recapitulate the phenotype of the transposon mutation (not shown). The remaining 46 transposon mutants (Table S2) were tested in our standard coculture assay with S. sanguinis SK36 to determine which P. aeruginosa PA14 transposon mutants are unable to enhance Streptococcus growth in a second strain. A total of 44 of the 46 P. aeruginosa PA14 transposon mutants were capable of enhancing growth of S. sanguinis SK36, and thus these genes were unlikely to be involved in a general pathway for enhancing growth of Streptococcus. We found that two transposon mutants were unable to promote either S. constellatus 7155 or S. sanguinis SK36 growth: P. aeruginosa pqsL::TnM and P. aeruginosa dbpA::TnM.
FIG 2.
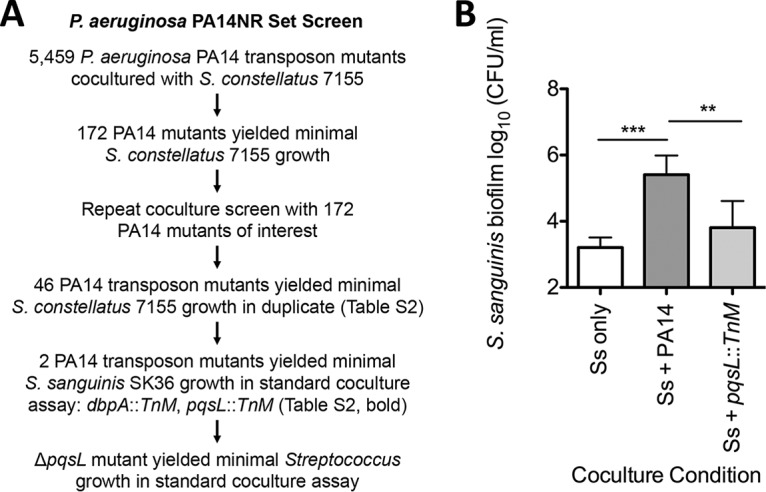
Screening for P. aeruginosa PA14 mutants altered in interaction with Streptococcus. (A) An overview of the P. aeruginosa PA14NR set transposon mutant screen used to identify P. aeruginosa transposon insertion mutants that can no longer enhance S. constellatus 7155 growth and the number of P. aeruginosa PA14 transposon insertion mutants identified in each step. (B) Coculture between a mutant strain identified in the screen, P. aeruginosa pqsL::TnM, and S. sanguinis SK36. Each bar represents the average of three biological replicates, each with three technical replicates. Error bars represent SD. **, P < 0.01; ***, P < 0.001 (by repeated-measures ANOVA with Tukey’s multiple-comparison posttest). P. aeruginosa growth data from this assay can be found in Fig. S7 in the supplemental material.
The dbpA gene codes for the RNA helicase DbpA, which has been demonstrated to play a role in the formation of the 50S ribosomal subunit in Escherichia coli (43). E. coli is able to compensate for a dbpA deletion in forming the 50S ribosomal subunit, as described previously (44); an inability to form the 50S ribosomal subunit would otherwise cause a lethal protein synthesis defect, and a dominant negative dbpA mutation is necessary to observe a defect in DbpA function in E. coli. We built and assayed the P. aeruginosa PA14 ΔdbpA mutant strain and found no significant defect in S. sanguinis SK36 growth enhancement (see Fig. S6A in the supplemental material) or in P. aeruginosa growth (Fig. S6B), and thus we did not pursue further study of this mutant.
We previously studied the effects of the Pseudomonas quinolone signal (PQS) pathway on interactions between P. aeruginosa and Staphylococcus aureus, including the utilization of 2-heptyl-4-hydroxyquinoline-N-oxide (HQNO), a respiratory chain inhibitor, to drive S. aureus to fermentative metabolism (45, 65, 67). As was observed for S. constellatus 7155, there was a significant reduction in the ability of the P. aeruginosa pqsL::TnM mutant to support S. sanguinis SK36 growth compared to the wild-type P. aeruginosa PA14 (Fig. 2B). There was no detectable growth defect of the pqsL::TnM mutant strain compared to wild-type P. aeruginosa PA14 under our assay conditions (see Fig. S7 in the supplemental material). We chose to focus on the P. aeruginosa pqsL::TnM mutant for the remainder of our study.
The P. aeruginosa ΔpqsL mutant has a defect in Streptococcus growth enhancement.
The PQS pathway involves the production of multiple 4-hydroxy-2-alkylquinolones (HAQs) and begins with anthranilic acid, which is converted to intermediates of unknown structure by the enzymes PqsA and PqsD (Fig. 3A). These unknown intermediates can then be converted into HQNO by PqsL, the product of our gene of interest, or 4-hydroxy-2-heptylquinoline (HHQ) by PqsB and PqsC. HHQ can then be converted into 3,4-dihydroxy-2-heptylquinoline (PQS) by PqsH (46–48). MvfR (also known as PqsR) is the transcriptional regulator that is activated by HHQ, PQS, and MvfR (PqsR) positively regulates the transcription of operons involved in PQS production and the LasR and RhlR quorum sensing pathways, as well as the operons required for production of the siderophores pyoverdine and pyochelin (49, 50). MvfR/PqsR and PQS have also been demonstrated to indirectly increase expression of the phenazine pyocyanin (49).
FIG 3.

The P. aeruginosa ΔpqsL mutant inhibits Streptococcus growth. (A) The PQS biosynthetic pathway and the enzymes that catalyze each step. (B) Coculture of S. sanguinis SK36 with wild-type P. aeruginosa PA14 and P. aeruginosa PA14 mutant strains lacking each enzyme in the PQS biosynthetic pathway. (C) Coculture of S. sanguinis SK36 with P. aeruginosa PA14, the ΔpqsL mutant, its complement pMQ72-pqsL, and the vector control pMQ72 in the presence of 0.2% arabinose. (D) Coculture of representative streptococci from Fig. 1 in coculture with the wild-type P. aeruginosa PA14 and the ΔpqsL mutant strain. In each panel, bars represent the average of three biological replicates, each with at least three technical replicates. Error bars indicate SD. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by repeated-measures ANOVA with Dunnett’s multiple-comparison posttest with Streptococcus only as the control condition [B] and repeated-measures ANOVA with Tukey’s multiple-comparison posttest [C and D]). The corresponding P. aeruginosa growth data for these experiments can be found in Fig. S8 to S10 in the supplemental material.
We considered two different mechanisms that could explain why the growth of S. sanguinis SK36 is no longer promoted by coculture with the P. aeruginosa PA14 pqsL::TnM. We hypothesized that either the pqsL::TnM strain may no longer be able to promote S. sanguinis SK36 growth or the loss of PqsL function resulted in a P. aeruginosa strain that reduced S. sanguinis SK36 viability. To distinguish between these hypotheses, we assessed the Streptococcus growth enhancement capabilities of P. aeruginosa PA14 mutants with deletions in the pqs pathway when grown in coculture with S. sanguinis SK36. We found that the ΔpqsL mutant was the only pqs pathway mutant that was unable to promote S. sanguinis SK36 growth (Fig. 3B; see Fig. S8A in the supplemental material for S. sanguinis SK36 planktonic growth and Fig. S8B and C for P. aeruginosa biofilm and planktonic growth).
We complemented the ΔpqsL strain with an arabinose-inducible pMQ72-pqsL construct and demonstrated a significant increase in viable S. sanguinis SK36 biofilm cells recovered when the complemented strain was induced with 0.2% arabinose (Fig. 3C; see Fig. S9A in the supplemental material for planktonic growth); there was no significant difference between wild-type P. aeruginosa PA14 and the complemented ΔpqsL/pMQ72-pqsL strain. Additionally, there was no significant difference in P. aeruginosa biofilm and planktonic growth in medium amended with 0.2% arabinose, the inducer of the expression for the PBAD promoter on the pMQ72 plasmid (Fig. S9B and C).
Additionally, we assayed the P. aeruginosa ΔpqsL mutant strain in coculture with a few representative Streptococcus spp. from Fig. 1B to determine if the ΔpqsL mutant strain has a broad defect in Streptococcus growth enhancement (Fig. 3D). We found that for S. intermedius 4807, there was a slight but nonsignificant growth decrease during coculture with P. aeruginosa ΔpqsL, indicating that the mutant strain is unable to enhance Streptococcus growth. Similarly, we saw a nonsignificant decrease in S. parasanguinis 5357 growth in coculture with the ΔpqsL mutant compared to wild-type P. aeruginosa PA14. We did observe a significant decrease in S. anginosus 5535 cells recovered from the coculture with the ΔpqsL mutant strain compared to monoculture and coculture conditions with P. aeruginosa PA14, indicating that the ΔpqsL mutation is contributing to the repression of the growth of S. anginosus 5535. We found that both wild-type P. aeruginosa PA14 and the ΔpqsL mutant strain caused a nonsignificant reduction in S. intermedius 7155 cells recovered from coculture, indicating that both of these P. aeruginosa strains may be able to outcompete S. intermedius 7155. We saw no significant changes to P. aeruginosa growth while in coculture with these representative Streptococcus spp. (see Fig. S10 in the supplemental material). Taking the data together, we can extend our finding to at least one other strain of Streptococcus.
The P. aeruginosa ΔpqsL mutant likely suppresses S. sanguinis SK36 growth by siderophore production via iron sequestration.
It has been demonstrated previously that a pqsL mutant is deficient in HQNO production and overproduces PQS (47). Exogenous PQS has been demonstrated to chelate iron and to increase the expression of the genes coding for siderophore and phenazine biosynthesis enzymes in P. aeruginosa (51–53). Thus, we hypothesized that it was the increased production of PQS and/or increased expression of one or more PQS-regulated genes that caused the observed loss in growth promotion of S. sanguinis SK36.
To test our hypothesis, we assayed S. sanguinis SK36 in coculture with the ΔpqsL mutant strains deficient in production of the virulence factors regulated by PQS: siderophores (ΔpqsL ΔpvdA ΔpchE) and phenazines (ΔpqsL ΔphzA-G1/2) (49, 51–53). We found that the P. aeruginosa ΔpqsL ΔpvdA ΔpchE deletion mutant strain restored S. sanguinis SK36 growth enhancement to levels similar to those with wild-type P. aeruginosa PA14 (Fig. 4A) without affecting P. aeruginosa growth (see Fig. S11 in the supplemental material). In contrast, the P. aeruginosa ΔpqsL ΔphzA-G1/2 mutant did not restore S. sanguinis SK36 growth (Fig. 4A and S11).
FIG 4.

The P. aeruginosa ΔpqsL mutant likely inhibits Streptococcus growth by sequestering iron via siderophore production. (A) Coculture of S. sanguinis SK36 with P. aeruginosa PA14 mutant strains lacking pqsL and siderophore (ΔpqsL ΔpvdA ΔpchE) or phenazine (ΔpqsL ΔphzA-G1/2) genes. (B) Coculture of S. sanguinis SK36 with P. aeruginosa PA14 mutant strains in 5% CO2 (aerobic) or under anaerobic growth conditions. (C) Coculture of S. sanguinis SK36 gene replacement mutants lacking putative iron acquisition genes with P. aeruginosa PA14 and the ΔpqsL mutant. (D) Complementation assays with the rescued Ssx_1742 and Ssx1744 strains. Each bar represents the average of three biological replicates, each with at least three technical replicates. There was no significant difference between the wild-type S. sanguinis SK36 and the two complemented strains. Error bars indicate SD. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by repeated-measures ANOVA with Tukey’s multiple-comparison posttest [A], the paired two-tailed Student t test [B], repeated-measures ANOVA with Dunnett’s multiple-comparisons posttest with S. sanguinis [Ss] as the control condition [C], and the paired two-tailed Student t test comparing each complemented and vector control strain and the paired two-tailed Student t test with a Bonferroni correction with the wild type compared to both complemented strains [D]). The corresponding P. aeruginosa growth data for these experiments can be found in Fig. S11, S13, and S14 in the supplemental material.
We performed a complementation experiment to further confirm a role for siderophores in the ability of the ΔpqsL mutant to reduce the viability of Streptococcus. Specifically, we asked whether complementing the mutation in one of the genes required for siderophore production (pchE) would restore the growth defect of the ΔpqsL ΔpvdA ΔpchE mutant to a phenotype similar to that observed for the ΔpqsL mutant. We cloned pchE into the vector pMQ72 and compared it to the vector control strain (ΔpqsL ΔpvdA ΔpchE/pMQ72). The complemented ΔpqsL ΔpvdA ΔpchE/pMQ72-pchE strain showed a phenotype not significantly different from that of the ΔpqsL mutant (see Fig. S12 in the supplemental material). These data confirmed that it was indeed loss of siderophore production in the ΔpqsL mutant background that allowed the ΔpqsL ΔpvdA ΔpchE strain to enhance the growth of S. sanguinis SK36.
A prediction of this iron sequestration model is that iron supplementation should restore the Streptococcus-enhancing activity of the P. aeruginosa ΔpqsL mutant. We added 50 μM FeCl3 to our minimal medium coculture conditions and saw restoration of S. sanguinis SK36 growth enhancement 6 out of the 18 times we performed the experiment. We explored this phenotype using a range of FeCl3 concentrations from 5 μM to 50 μM, making a fresh FeCl3 solution daily and using buffered media in our assay and when making the FeCl3 stock solution, but the phenotype was still variable. We do not fully understand why the iron rescue phenotype was so variable. We measured the iron levels of the medium used in our coculture conditions (MEM) using inductively coupled plasma mass spectrometry (ICP-MS) and showed that the concentration of iron is below the limit of detection (<5 ppb), so it is plausible that the streptococci are iron limited under our coculture conditions.
Next, we tested the idea that coculture under anaerobic conditions would also lead to recovery of the Streptococcus growth promotion phenotype in the ΔpqsL mutant, because P. aeruginosa has been demonstrated to reduce pyoverdine and pyochelin production under anoxic conditions (54). Upon anaerobic coculture in an AnaeroPak-Anaero container with a GasPak sachet, the P. aeruginosa ΔpqsL mutant significantly enhanced S. sanguinis SK36 viability compared to that in coculture under aerobic conditions (Fig. 4B; see Fig. S13A in the supplemental material). The level of S. sanguinis growth enhancement promoted by the P. aeruginosa ΔpqsL mutant under anaerobic conditions was equivalent to that observed for the wild-type P. aeruginosa. Additionally, if P. aeruginosa were enhancing S. sanguinis SK36 growth solely through oxygen consumption, we would expect to see enhanced S. sanguinis SK36 growth in monoculture under anaerobic conditions, which we did not observe here.
We note that P. aeruginosa biofilm and planktonic growth were decreased under the anaerobic growth conditions used in these experiments compared to what we typically observe under aerobic conditions (Fig. S13B and C), which is not surprising given that aerobic respiration is the main means of energy generation for this microbe. Together, these data indicate that wild-type P. aeruginosa contributes to the growth of S. sanguinis SK36 via a mechanism independent of oxygen consumption in our coculture system, and they are consistent with our hypothesis that reduced siderophore production under anaerobic conditions mitigates the phenotype of the ΔpqsL mutant.
An iron ABC transporter of S. sanguinis SK36 participates in competition with P. aeruginosa.
Our data suggest that one component of the interaction between P. aeruginosa and Streptococcus spp. is the competition for iron. The genome of S. sanguinis SK36 has been sequenced and annotated, and using this information, we identified several gene products that, based on their annotation, might be involved in iron uptake. We predicted that if S. sanguinis SK36 is indeed competing with P. aeruginosa for iron, loss of one or more of these iron uptake system would compromise the ability of S. sanguinis SK36 to grow in coculture with P. aeruginosa. Given that the ΔpqsL mutant likely has an enhanced capacity to scavenge iron as indicated by the restoration of S. sanguinis SK36 growth enhancement by the ΔpqsL ΔpvdA ΔpchE mutant, any compromise observed for S. sanguinis SK36 iron acquisition mutants should be exacerbated in coculture with the P. aeruginosa ΔpqsL mutant.
Xu and colleagues reported a mutant library of S. sanguinis SK36 wherein nonessential genes are deleted and replaced with a kanamycin resistance cassette (28, 29). Using this library, we examined whether selected S. sanguinis SK36 mutant strains lacking genes involved in iron uptake (Table 1) have reduced growth in the presence of wild-type P. aeruginosa PA14 or the P. aeruginosa ΔpqsL strain. We tested S. sanguinis SK36 strains carrying mutations in genes coding for iron-regulatory proteins, an iron-binding lipoprotein, a ferrichrome-binding protein, and predicted iron uptake ABC transporters using the coculture assay. We found that all of the mutant strains tested behaved like wild-type S. sanguinis SK36 in coculture, except for the S. sanguinis SK36 Ssx_1742 and Ssx_1744 mutant strains (Fig. 4C; see Fig. S14A in the supplemental material). The SSA_1742 gene codes for a predicted ferrichrome-binding protein, and the SSA_1744 gene codes for a predicted permease protein of an iron compound ABC transporter.
TABLE 1.
S. sanguinis SK36 iron-related gene products
| Gene | Predicted gene product |
|---|---|
| Ssx_0256 | ScaR metalloregulator |
| Ssx_0686 | Fe2+/Zn2+ uptake regulation protein |
| Ssx_1129 | Periplasmic iron transport lipoprotein |
| Ssx_1578 | ABC-type Fe3+-siderophore transport system, permease component |
| Ssx_1581 | FatB, metal-binding ABC transporter |
| Ssx_1741 | ABC-type Fe3+-siderophore transporter, ATPase component |
| Ssx_1742 | Ferrichrome-binding protein |
| Ssx_1743 | ABC-type Fe3+-siderophore transport system, permease component |
| Ssx_1744 | Iron compound ABC transporter, permease protein |
| Ssx_1745 | General stress response protein CsbD |
The S. sanguinis SK36 Ssx_1742 and Ssx_1744 mutant strains demonstrated reduced growth under monoculture conditions (6.5-fold and 6.9-fold, respectively), indicating that they may be iron starved under our minimal medium growth conditions (Fig. 4C). This iron starvation phenotype is exacerbated in coculture with wild-type P. aeruginosa PA14, with a 44.9-fold reduction in Ssx_1742 cells and a 5.5-fold reduction in Ssx_1744 cells obtained from coculture compared to wild-type S. sanguinis SK36 coculture.
To confirm that the observed competition defect was indeed due to the Ssx_1742 and Ssx_1744 mutations, we complemented each of the mutants as described in Materials and Methods. As shown in Fig. 4D, the complemented mutants showed competition phenotypes similar to that observed for wild-type S. sanguinis SK36.
We next explored the phenotypes of the Ssx_1742 and Ssx_1744 mutants in coculture with the P. aeruginosa ΔpqsL mutant strain. In coculture with the P. aeruginosa ΔpqsL mutant strain there was a significant, 4.9-fold reduction in the Ssx_1742 mutant compared to wild-type S. sanguinis SK36 in coculture with the ΔpqsL mutant. Importantly, coculture of the S. sanguinis SK36 Ssx_1744 mutant strain showed no additional, significant growth defect when grown in coculture with the P. aeruginosa ΔpqsL mutant compared to the wild-type P. aeruginosa strain. We take this result to mean that the increased iron sequestration by the ΔpqsL mutant is competing for the iron typically transported by the S. sanguinis SK36 Ssx_1744-encoded iron ABC transporter; thus, loss of Ssx_1744 confers no additional phenotype when cocultured with the P. aeruginosa PA14 ΔpqsL mutant. Finally, we observed no significant difference in P. aeruginosa PA14 and the ΔpqsL mutant strain growing in coculture with S. sanguinis SK36 mutant strains (Fig. S14B and C).
DISCUSSION
In this study, we sought to characterize a polymicrobial interaction that occurs between P. aeruginosa and Streptococcus spp. We previously demonstrated that P. aeruginosa can suppress S. constellatus 7155 biofilms through surfactant production and that this suppression can be alleviated through treatment with the CF maintenance antibiotic tobramycin (23). Our current work adds to our understanding of P. aeruginosa-Streptococcus interactions by demonstrating the widespread ability of multiple P. aeruginosa clinical isolates from CF patients and laboratory strains to enhance the growth of multiple species of Streptococcus. To better understand the basis of the ability of P. aeruginosa to promote the growth of Streptococcus spp., we screened P. aeruginosa transposon insertion mutants to identify factors that contribute to the ability of P. aeruginosa to enhance growth of S. constellatus, and we identified 46 candidate mutants. Following up on these mutants, we identified only one strain, carrying a mutation in the pqsL gene, that has a consistent, reduced Streptococcus growth enhancement phenotype versus multiple species of Streptococcus. Upon further investigation, we revealed that this mutant no longer promotes Streptococcus growth because the P. aeruginosa ΔpqsL mutant strain likely actively competes with Streptococcus for iron. Loss of PqsL function has been reported to enhance PQS production (47), excess PQS has been demonstrated to enhance siderophore biosynthesis gene transcription (51–53), and PQS-mediated iron sequestration by P. aeruginosa has been demonstrated to reduce growth of both Gram-positive and Gram-negative soil bacteria (32). This PQS-mediated inhibition of soil bacterial growth can be restored upon addition of 50 μM FeCl3 (32). Similarly, our data show the ability to restore Streptococcus growth by introducing mutations in the siderophore genes to the ΔpqsL mutant or by growing the cocultures anaerobically, a growth condition where P. aeruginosa is known to reduce pyoverdine and pyochelin production (54). Consistent with these data, we observed that S. sanguinis SK36 grew slightly more in the presence of a P. aeruginosa PA14 ΔpqsL ΔpvdA ΔpchE mutant than in the presence of wild-type P. aeruginosa PA14. The increased S. sanguinis SK36 growth indicates that these microbes are competing for iron during coculture and that changes to iron uptake can alter the competition between these two microbes. In addition, PQS has been demonstrated to act as both an anti- and pro-oxidant under different conditions (55), and we cannot rule out that the increased PQS production in the ΔpqsL mutant impacts production of reactive oxygen species that may be toxic to Streptococcus spp.; the fact that growth of the ΔpqsL mutant under anaerobic conditions reverses the growth phenotype is consistent with this idea.
An interesting observation from this study was the demonstration of a significant increase in S. sanguinis SK36 biofilm growth between an isogenic nonmucoid and mucoid P. aeruginosa PAO1 strain. Previous work demonstrated that S. parasanguinis is able to use the streptococcal surface adhesin BapA1 to bind alginate produced by mucoid P. aeruginosa and enhance S. parasanguinis biofilm formation in vitro; however, S. gordonii and S. sanguinis SK36 did not demonstrate enhanced biofilm formation (24). Here we demonstrate a significant growth increase of S. sanguinis SK36 when in coculture with P. aeruginosa PDO300 mucA22 compared to the isogenic nonmucoid strain. It is possible that S. sanguinis SK36 can also bind to alginate. Alternatively, we hypothesize that the growth enhancement induced by the mucoid P. aeruginosa strain may be due to decreased rhamnolipid production, which has been described in mucoid strains (67), and the corresponding relief of rhamnolipid-induced Streptococcus killing (23). Furthermore, mucoid strains were shown to produce lower levels of products of the PQS pathway and reduced levels of siderophores (67). Thus, Streptococcus spp. may more readily coexist, and perhaps grow to larger numbers, in patients with mucoid P. aeruginosa, a question that could be answered by performing a clinical study assessing relative levels of Streptococcus spp. as a function of mucoid P. aeruginosa. Furthermore, these data indicate that the interactions between P. aeruginosa and Streptococcus may change over the lifetime of patients with CF as the colonizing P. aeruginosa converts to mucoidy.
Oral streptococci have been demonstrated to utilize hydrogen peroxide to inhibit the growth and colonization of competing microorganisms (16, 20, 21, 40), and we hypothesized that P. aeruginosa catalase might play a role in enhancing Streptococcus growth, as catalase has been found in the supernatants of P. aeruginosa cultures (37, 38, 41). However, we found no significant defect in S. sanguinis SK36 growth enhancement by the P. aeruginosa ΔkatA, ΔkatB, and ΔkatA ΔkatB mutants compared to wild-type P. aeruginosa PA14, indicating that catalase is not the factor produced by P. aeruginosa that enhances Streptococcus growth. It has been demonstrated that P. aeruginosa does not secrete catalase and that it is found in the supernatant due P. aeruginosa cell lysis (37); it may be that catalase found in the supernatant is too dilute to have a positive influence on Streptococcus growth in coculture or that the hydrogen peroxide is not growth limiting to the streptococci under our coculture conditions. Thus, we conclude that P. aeruginosa catalase is not influencing Streptococcus growth in our model system. It is also worth noting that anaerobic coculture was not sufficient to enhance S. sanguinis SK36 monoculture growth to the same levels achieved during coculture with P. aeruginosa PA14 under aerobic conditions. These data indicate that P. aeruginosa-mediated growth enhancement of streptococci cannot be explained by oxygen consumption via P. aeruginosa.
To better understand how S. sanguinis SK36 might compete with P. aeruginosa for iron under iron-limiting conditions, we examined a set of S. sanguinis SK36 mutants lacking putative iron uptake systems or regulatory genes. Of the nine mutants tested, only two showed reduced growth of S. sanguinis SK36 when in coculture with P. aeruginosa PA14: the Ssx_1742 mutant, lacking a ferrichrome-binding protein, and the Ssx_1744 mutant, lacking the permease protein of an iron compound ABC transporter. The Ssx_1742 mutant demonstrated a significant growth defect in monoculture and during coculture with P. aeruginosa PA14. The growth defect of the Ssx_1742 mutant was worsened when it was cocultured with the ΔpqsL mutant. Together, these data indicate that the Ssx_1742 mutant strain is unable to compete with P. aeruginosa for the limited iron under our coculture conditions and that the ferrichrome-binding protein encoded by Ssx_1742 is not involved in the competition for this metal with P. aeruginosa. In contrast, the Ssx_1744 mutant showed no additional defect when cocultured with the ΔpqsL mutant versus the wild-type P. aeruginosa. We take this result to mean that the increased production of the siderophores in the ΔpqsL mutant is competing for the iron typically transported by the S. sanguinis SK36 Ssx_1744-encoded iron ABC transporter; thus, loss of Ssx_1744 confers no additional phenotype when cocultured with the P. aeruginosa PA14 ΔpqsL mutant. These data indicate that the Ssx_1744-encoded iron ABC transporter of S. sanguinis SK36 plays a key role in the competition with P. aeruginosa.
Our data support a second mechanism whereby P. aeruginosa can limit the growth of Streptococcus spp. (Fig. 5), including the SMG, via iron sequestration. We previously reported that P. aeruginosa rhamnolipid surfactants could reduce the viability of S. constellatus. P. aeruginosa can also influence the biofilm formation of S. parasanguinis through alginate production (24) and the growth of Streptococcus spp. via a currently undescribed mechanisms (16, 17, 23). Conversely, previous studies investigating interactions between P. aeruginosa and Streptococcus spp. also showed that Streptococcus spp. influence transcription of P. aeruginosa virulence genes, including rhamnolipid, elastase, and phenazine biosynthesis genes, through AI-2 signaling (18) and an undescribed mechanism (6, 16, 17) and can suppress P. aeruginosa growth when they are a primary colonizer through production of H2O2 (16, 21) and reactive nitrogenous intermediates (20, 21). Thus, this polymicrobial interaction is complex.
FIG 5.

A model for P. aeruginosa-Streptococcus interactions. P. aeruginosa has both positive and negative interactions with Streptococcus spp. (A) It has been demonstrated previously that P. aeruginosa can enhance S. parasanguinis biofilm formation through alginate secretion (24) or inhibit Streptococcus growth via rhamnolipid secretion (23). Here we propose a negative interaction wherein iron sequestration by P. aeruginosa limits Streptococcus sp. growth. Streptococcus-promoting factors produced by P. aeruginosa have not yet been identified (indicated by the question mark). (B) Previous evidence also demonstrates that streptococci can influence P. aeruginosa through AI-2 signaling (18), leading to enhanced lasB, rhlA, exoT, and phzA1/2 gene expression, or can inhibit P. aeruginosa viability through H2O2 production (16, 20, 21) and subsequent generation of reactive nitrogenous intermediates (20, 21).
Our data also indicate that P. aeruginosa can promote the growth of various Streptococcus spp., but we do not understand the basis of this growth promotion. We anticipated that the genetic screen described here would likely identify components of such a growth-promoting pathway in P. aeruginosa; instead, our screen identified only a single locus apparently involved in a competitive interaction. We suggest two possible explanations for our findings. First, perhaps P. aeruginosa determinants that promote Streptococcus growth are essential; we think that this explanation is unlikely, but it is a formal possibility. More likely is that P. aeruginosa has multiple, redundant pathways to boost Streptococcus growth. Thus, our genetic approach would be expected to fail to identify such redundant pathways, and alternative strategies to explore P. aeruginosa- Streptococcus interactions must be employed in future studies.
Finally, the observations we present here may be of relevance in the CF lung, as many patients are cocolonized by P. aeruginosa and Streptococcus spp. (8, 56). Analysis of the average available iron in the airway varies markedly, between ∼0.02 μM in healthy individuals and ∼8 μM in patients with CF, and there is a great deal of variability among patients with CF (57, 58). The increased iron in the CF airway is likely due to the reported enhanced levels of extracellular iron in the apical surface liquid of airway cells with a mutation in CFTR (59) and the bleeding into the airway (hemoptysis) associated with this patient population (60). Thus, in CF patients, iron levels in the airway can range from concentrations where we might expect direct competition between P. aeruginosa and Streptococcus for this limited resource to levels where abundant iron would mitigate such competition. Additional studies are necessary to determine if Streptococcus spp. are iron limited (or not) in the CF airway or in sufficiently close proximity to P. aeruginosa in the airway (i.e., in mixed microcolonies) to expect direct competition for iron in a local niche.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in this study are listed in Table S3 in the supplemental material. P. aeruginosa strains were grown on lysogeny broth (LB) agar or in LB liquid with shaking at 37°C and, where indicated, in the presence of antibiotics at the following concentrations: 25 μg/ml gentamicin, 250 μg/ml kanamycin, and 75 μg/ml tetracycline. Streptococcus spp. were grown as previously described (23) on tryptic soy agar supplemented with 5% defibrinated sheep’s blood (blood agar) or statically in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) and 20 μl/ml Oxyrase (Oxyrase, Inc.) at 37°C with 5% CO2. S. sanguinis SK36 gene replacement mutant strains were grown on blood agar or THY with 500 μg/ml kanamycin (29). For antibiotic selection during construction of the S. sanguinis SK36 complementation strains, spectinomycin (Spc) was used at a concentration of 100 μg/ml in E. coli and 200 μg/ml in S. sanguinis SK36.
At the end of each coculture assay, P. aeruginosa was grown overnight on Pseudomonas isolation agar (PIA) at 37°C, and Streptococcus spp. were grown overnight on blood agar at 37°C anaerobically in AnaeroPak-Anaero containers (Thermo Fisher) or on blood agar supplemented with 10 μg/ml neomycin and 10 μg/ml polymyxin B (Streptococcus selection agar) when specified. Saccharomyces cerevisiae strain InvSc1 (Invitrogen), was used for homologous recombination to build the pMQ30-katA, pMQ-30-katB, and pMQ30-dbpA deletion vectors and pMQ72-pqsL complementation vector. InvSc1 was grown as previously described in 1% Bacto yeast extract, 2% Bacto peptone, and 2% dextrose (61). Synthetic defined agar-uracil (4813-065; Qbiogene) was used for InvSc1 selections.
Species identification of streptococci.
Streptococcus spp. were isolated at the Dartmouth Hitchcock Medical Center in Lebanon, NH. The species of Streptococcus clinical isolates were determined using 16S rRNA gene sequencing. Genomic DNA (gDNA) was extracted from each strain from overnight cultures using the Gentra Puregene Yeast/Bact kit (Qiagen) followed by 16S-internal transcribed spacer (ITS) PCR as previously described (62) using the Strep16S-1471F and 6R-IGS primers (listed in Table S4 in the supplemental material). Streptococcus oralis, S. mitis, and S. pneumoniae were further differentiated by PCR of a region of the gdh gene and sequencing as previously described, using the Strep-gdhF and Strep-gdhR primers (listed in Table S4) (62). The Phusion polymerase PCR protocol (New England Biolabs) was followed for preparing 5-μl reaction mixtures, and the PCR conditions for the 16S-ITS region were as follows: 98°C for 30 s followed by 25 cycles of 98°C for 10 s, 61°C for 15 s, and 72°C for 30 s and a final extension at 72°C for 7 min. The PCR conditions for amplifying gdh were as follows: 98°C for 30 s, followed by 30 cycles of 98°C for 10 s, 57.9°C for 15 s, and 72°C for 30 s, followed by a final extension at 72°C for 7 min. The resulting PCR products were imaged on a 1% agarose gel with Sybr Safe (Thermo Fisher Scientific Inc.). The remaining PCR product was purified using the QIAquick PCR purification kit (Qiagen), and the purified DNA product was sequenced at the Dartmouth Molecular Biology Core Facility using the Applied Biosystems 3730 DNA Analyzer. Sequence results were analyzed using NCBI BLAST for species identification.
Mixed microbial coculture system.
Cocultures were conducted in the CFBE model system as previously described (8, 23, 65, 67) with some modifications. Overnight cultures of P. aeruginosa and Streptococcus spp. were individually centrifuged at 10,000 × g for 3 min, the cell pellet was washed with 1.5 ml minimal essential medium (MEM) supplemented with 2 mM l-glutamine (MEM+L-Gln) and centrifuged again, and the cell pellet was resuspended in 1.5 ml MEM+L-Gln. The optical density at 600 nm (OD600) of each culture was determined, and the P. aeruginosa cultures were adjusted in MEM+L-Gln to an OD600 of 0.05. The Streptococcus sp. cultures were adjusted to an OD600 of 0.1. S. sanguinis SK36 overnight cultures were adjusted to an OD600 of 0.1 and then further diluted 1:100 in MEM+L-Gln due to the robust growth that S. sanguinis SK36 exhibits in monoculture. A 1:1 mixture by volume of P. aeruginosa and Streptococcus spp. was prepared from the adjusted cultures. Three wells of a 96-well plate per monoculture and coculture condition were inoculated with 100 μl per well. The culture plates were then incubated statically for 1 h at 37°C with 5% CO2, at which point the unattached planktonic cells were aspirated with a multichannel pipette and replaced with 100 μl MEM supplemented with 2 mM l-glutamine and 0.4% l-arginine (MEM+L-Gln+L-Arg). The culture plates were incubated statically for an additional 5.5 h, at which point the supernatant was removed and replaced with 100 μl MEM+L-Gln+L-Arg. l-Arginine (0.4%) was added to the minimal medium at the 1- and 5.5-h medium changes to promote P. aeruginosa biofilm formation (63). At 21 h postinoculation, planktonic cells were removed to be plated, and biofilms were disrupted using a 96-pin replicator in 100 μl of MEM+L-Gln. Both planktonic and biofilm cells were 10-fold serially diluted and plated on selective media. PIA plates were grown overnight aerobically and blood agar plates were grown overnight in AnaeroPak-Anaero containers (Thermo Scientific) with GasPak sachets (BD) to selectively grow P. aeruginosa and Streptococcus spp., respectively. Following overnight incubation, colonies were counted and the CFU per milliliter of culture were determined.
Growth kinetics in mixed microbial coculture system.
P. aeruginosa PA14 and PAO1 were grown in coculture with S. sanguinis SK36 as described above, with one 96-well plate per time point. Six time points were assessed: 0, 3, 5.5, 7.5, 10, and 24 h. The 0-h time point corresponds to the initial inoculum. At each time point, the planktonic and biofilm cells from the same wells were serially diluted and plated on PIA and blood agar. Cells were harvested at the 5.5-h time point plate prior to the 5.5-h medium exchange.
Coculture in rich media.
P. aeruginosa PA14 was grown in coculture with S. sanguinis SK36 as described above but with the following changes: at the 1-h medium change, the MEM+L-Gln was removed and replaced with 100 μl LB or THY. Fresh LB or THY (100 μl) was used at the 5.5-h medium change as well. This allows all of the culture conditions to originate from the same inocula.
Construction of P. aeruginosa PA14 deletion mutant strains.
The pMQ30 vector (Table S3) was used to generate the P. aeruginosa PA14 ΔkatA, ΔkatB, ΔkatA ΔkatB, and ΔdbpA mutant strains. The pMQ30-katA, pMQ30-katB, and pMQ30-dbpA deletion constructs were built using homologous recombination of the PCR products made with the respective “KO” primers (listed in Table S4) with the XbaI restriction enzyme-digested pMQ30 in yeast as previously reported (61). Plasmid integrants were isolated on LB agar supplemented with gentamicin and nalidixic acid, followed by counterselection on sucrose medium. Deletion mutants were confirmed by PCR with respective “conf.” primers (Table S4), followed by sequencing. Coculture was conducted as described above with the confirmed P. aeruginosa PA14 deletion mutant strains.
Genetic screen.
The Ausubel lab created a nonredundant P. aeruginosa PA14 transposon library (PA14NR set) in 96-well plate format (42). Initially, a 96-pin replicator was used to transfer inocula from the frozen library to a sterile 96-well plate containing 150 μl of LB per well. The plate was then incubated statically for 24 h at 37°C. S. constellatus 7155 frozen aliquots were made from 750 μl of overnight culture mixed with 750 μl of 40% glycerol. The day of the coculture experiment, frozen S. constellatus 7155 aliquots were thawed, 500 μl of aliquot was added to 4.5-ml THY cultures with 100 μl Oxyrase, and the cultures were grown for 6 to 8 h at 37°C with 5% CO2. The S. constellatus 7155 culture was then adjusted to an OD600 of 0.05 in MEM+L-Gln, and 100 μl of adjusted culture was added to each well of a sterile 96-well plate. The PA14NR set was grown in LB for 24 h in a 96-well plate format; each well contained a transposon mutant from the P. aeruginosa PA14NR set. A 96-pin replicator was then used to transfer 2 to 3 μl of culture from the transposon library plate into the plate containing S. constellatus 7155. The coculture plates were then incubated statically for 2 h at 37°C with 5% CO2. After 2 h, the supernatant and unattached bacteria were aspirated using a multichannel pipette, and 100 μl MEM+L-Gln with 5 μg/ml tobramycin to suppress P. aeruginosa PA14 rhamnolipid production was added to each well. The plates were then incubated statically for an additional 20 h at 37°C with 5% CO2. At 22 h postinoculation, the 96-pin replicator was used to disrupt the biofilms into the supernatant fraction. The 96-pin replicator was then used to spot culture onto large petri plates containing either PIA or Streptococcus selection agar. PIA plates were incubated overnight at 37°C, and Streptococcus selection agar plates were incubated for 24 h at 37°C with 5% CO2. In the initial screen, we identified P. aeruginosa mutants that showed low or undetectable S. constellatus 7155 growth. To confirm the phenotype, the candidate P. aeruginosa PA14 transposon mutant strain was picked from the PIA plate and grown statically overnight at 37°C in a sterile 96-well plate in 125 μl LB. The next morning, 125 μl of 40% glycerol was added to each well containing P. aeruginosa PA14 candidate mutants, and these “candidate mutant” plates were stored at −80°C for the next round of screening. For the second round of the screen, the coculture process described above was repeated with the plates containing candidate mutants.
If we had clean deletions of the candidate mutants, they were also tested in the assay described above. If the clean deletion did not recapitulate the original transposon mutant, that transposon mutant was eliminated from the list of candidate mutants. Table S2 shows the final list of P. aeruginosa PA14 transposon insertion mutant strains that yielded low or undetectable S. constellatus 7155 growth after rescreening.
We then tested each individual P. aeruginosa PA14 transposon mutant listed in Table S2 in our standard 96-well coculture assay as described above with S. sanguinis SK36. The two mutants that yielded consistently low S. sanguinis SK36 growth in our standard coculture are in bold in Table S2.
P. aeruginosa ΔpqsL complementation.
The pMQ72 vector (Table S3) with an arabinose-inducible promoter was used to complement the P. aeruginosa PA14 ΔpqsL deletion mutant. The pMQ72-pqsL complementation plasmid was built using homologous recombination of the PCR product made with the pqsL comp 3′ and pqsL comp 5′ primers (listed in Table S4) with SacI restriction enzyme-digested pMQ72 in yeast as previously reported (61). P. aeruginosa PA14 ΔpqsL/pMQ72-pqsL and P. aeruginosa PA14 ΔpqsL/pMQ72-empty vector control strains were cocultured with S. sanguinis SK36 as described above with the following change: at 1 and 5.5 h postinoculation, MEM+L-Gln+L-Arg supplemented with l-arabinose at 0% and 0.2% final concentrations was added to the medium to induce pMQ72-pqsL gene expression.
P. aeruginosa ΔpchE complementation construct.
Due to the gene length (4.3 kb) and content of repetitive DNA, the pchE gene was amplified in two overlapping PCR fragments using Phusion polymerase (NEB). Fragment 1 was amplified using the primers pchE 5′0.2 and pchE int R, and fragment 2 was amplified with primers pchE int 1B F (see Table S4 for primer sequences). The resulting PCR fragments were cloned into pMQ72 by homologous recombination in yeast as described above.
Coculture with ferric chloride.
Coculture was conducted as described above but with the following change: at 1 and 5.5 h postinoculation, supernatants were aspirated with a multichannel pipette and replaced with MEM+L-Gln+L-Arg with or without freshly prepared, filter-sterilized ferric chloride hexahydrate (ranging from 5 to 50 μM).
Anaerobic coculture.
Coculture was conducted as described above but with the following alterations. Once the plates were inoculated, they were incubated in AnaeroPak-Anaero containers with a GasPak sachet. At each medium change (1 h and 5.5 h), a new sachet was added to the container to ensure anaerobic coculture conditions. The AnaeroPak container was incubated in the same incubator as the aerobic plate to control for any environmental effects.
Complementing mutations of S. sanguinis SK36.
The open reading frames (ORFs) of SSA_1742 and SSA_1744 were PCR amplified with PfuUltra II fusion HS DNA polymerase (Agilent Technologies) using primers F-1742-oe/R-1742-oe and F-1744-oe/R-1744-oe, respectively (Table S4). The PCR products and the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible plasmid pJFP126 were digested with SphI and/or HindIII, ligated, and electroporated into E. coli DH5α (64). Plasmid DNA was purified from DH5α cells using a Qiagen Miniprep kit (Qiagen). Transformation of S. sanguinis strains with each plasmid was carried out as previously described (28). The DNA fragment containing gene expression elements and the aad9 gene, encoding resistance to Spc, was transferred from the plasmid to the genomes of the resulting S. sanguinis strains.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ping Xu for providing his S. sanguinis SK36 mutant library and the pVA838 vector and Deborah Hogan, Nicholas Jacobs, and Dominique Limoli for providing bacterial strains. We thank Brian Jackson for quantifying the iron concentration in our media with ICP-MS.
This work was supported by the Cystic Fibrosis Foundation (OTOOLE16GO), a Molecular and Cellular Biology at Dartmouth training grant (T32GM008704), the Munck-Pfefferkorn Fund, an NIH grant (R37 AI83256-06) to G.A.O., and a China Scholarship Council (CSC) grant (201708330005) to K.L.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00014-19.
REFERENCES
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Elborn JS. 2016. Cystic fibrosis. Lancet 388:2519–2531. doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 3.Tavernier S, Crabbe A, Hacioglu M, Stuer L, Henry S, Rigole P, Dhondt I, Coenye T. 2017. Community composition determines activity of antibiotics against multispecies biofilms. Antimicrob Agents Chemother 61:e00302-17. doi: 10.1128/AAC.00302-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkins MD, Sibley CD, Surette MG, Rabin HR. 2008. The Streptococcus milleri group—an unrecognized cause of disease in cystic fibrosis: a case series and literature review. Pediatr Pulmonol 43:490–497. doi: 10.1002/ppul.20809. [DOI] [PubMed] [Google Scholar]
- 5.Cade A, Denton M, Brownlee KG, Todd N, Conway SP. 1999. Acute bronchopulmonary infection due to Streptococcus milleri in a child with cystic fibrosis. Arch Dis Child 80:278–279. doi: 10.1136/adc.80.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sibley CD, Duan K, Fischer C, Parkins MD, Storey DG, Rabin HR, Surette MG. 2008. Discerning the complexity of community interactions using a Drosophila model of polymicrobial infections. PLoS Pathog 4:e1000184. doi: 10.1371/journal.ppat.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sibley CD, Sibley KA, Leong TA, Grinwis ME, Parkins MD, Rabin HR, Surette MG. 2010. The Streptococcus milleri population of a cystic fibrosis clinic reveals patient specificity and intraspecies diversity. J Clin Microbiol 48:2592–2594. doi: 10.1128/JCM.00414-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filkins LM, Hampton TH, Gifford AH, Gross MJ, Hogan DA, Sogin ML, Morrison HG, Paster BJ, O'Toole GA. 2012. The prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J Bacteriol 194:4709–4717. doi: 10.1128/JB.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flight WG, Smith A, Paisey C, Marchesi JR, Bull MJ, Norville PJ, Mutton KJ, Webb AK, Bright-Thomas RJ, Jones AM, Mahenthiralingam E. 2015. Rapid detection of emerging pathogens and loss of microbial diversity associated with severe lung disease in cystic fibrosis. J Clin Microbiol 53:2022–2029. doi: 10.1128/JCM.00432-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acosta N, Heirali A, Somayaji R, Surette MG, Workentine ML, Sibley CD, Rabin HR, Parkins MD. 2018. Sputum microbiota is predictive of long-term clinical outcomes in young adults with cystic fibrosis. Thorax 73:1016–1025. doi: 10.1136/thoraxjnl-2018-211510. [DOI] [PubMed] [Google Scholar]
- 11.O'Toole GA. 2018. Cystic fibrosis airway microbiome: overturning the old, opening the way for the new. J Bacteriol 200:e00561-17. doi: 10.1128/JB.00561-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marshall B, Elbert A, Petren K, Rizvi S, Fink A, Ostrenga J, Sewall A, Loeffler D. 2016. Patient registry: annual data report 2015. Cyst Fibros Found Patient Regist 2015:1–94. [Google Scholar]
- 13.Rudkjobing VB, Thomsen TR, Alhede M, Kragh KN, Nielsen PH, Johansen UR, Givskov M, Hoiby N, Bjarnsholt T. 2012. The microorganisms in chronically infected end-stage and non-end-stage cystic fibrosis patients. FEMS Immunol Med Microbiol 65:236–244. doi: 10.1111/j.1574-695X.2011.00925.x. [DOI] [PubMed] [Google Scholar]
- 14.Sibley CD, Parkins MD, Rabin HR, Duan K, Norgaard JC, Surette MG. 2008. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci U S A 105:15070–15075. doi: 10.1073/pnas.0804326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan DA, Willger SD, Dolben EL, Hampton TH, Stanton BA, Morrison HG, Sogin ML, Czum J, Ashare A. 2016. Analysis of lung microbiota in bronchoalveolar lavage, protected brush and sputum samples from subjects with mild-to-moderate cystic fibrosis lung disease. PLoS One 11:e0149998. doi: 10.1371/journal.pone.0149998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whiley RA, Fleming EV, Makhija R, Waite RD. 2015. Environment and colonisation sequence are key parameters driving cooperation and competition between Pseudomonas aeruginosa cystic fibrosis strains and oral commensal streptococci. PLoS One 10:e0115513. doi: 10.1371/journal.pone.0115513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiley RA, Sheikh NP, Mushtaq N, Hagi-Pavli E, Personne Y, Javaid D, Waite RD. 2014. Differential potentiation of the virulence of the Pseudomonas aeruginosa cystic fibrosis liverpool epidemic strain by oral commensal Streptococci. J Infect Dis 209:769–780. doi: 10.1093/infdis/jit568. [DOI] [PubMed] [Google Scholar]
- 18.Duan K, Dammel C, Stein J, Rabin H, Surette MG. 2003. Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication. Mol Microbiol 50:1477–1491. doi: 10.1046/j.1365-2958.2003.03803.x. [DOI] [PubMed] [Google Scholar]
- 19.Waite RD, Qureshi MR, Whiley RA. 2017. Modulation of behaviour and virulence of a high alginate expressing Pseudomonas aeruginosa strain from cystic fibrosis by oral commensal bacterium Streptococcus anginosus. PLoS One 12:e0173741. doi: 10.1371/journal.pone.0173741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scoffield JA, Wu H. 2016. Nitrite reductase is critical for Pseudomonas aeruginosa survival during co-infection with the oral commensal Streptococcus parasanguinis. Microbiology 162:376–383. doi: 10.1099/mic.0.000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scoffield JA, Wu H. 2015. Oral streptococci and nitrite-mediated interference of Pseudomonas aeruginosa. Infect Immun 83:101–107. doi: 10.1128/IAI.02396-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waite RD, Qureshi MR, Whiley RA. 2017. Correction: modulation of behaviour and virulence of a high alginate expressing Pseudomonas aeruginosa strain from cystic fibrosis by oral commensal bacterium Streptococcus anginosus. PLoS One 12:e0176577. doi: 10.1371/journal.pone.0176577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price KE, Naimie AA, Griffin EF, Bay C, O'Toole GA. 2016. Tobramycin-treated Pseudomonas aeruginosa PA14 enhances Streptococcus constellatus 7155 biofilm formation in a cystic fibrosis model system. J Bacteriol 198:237–247. doi: 10.1128/JB.00705-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scoffield JA, Duan D, Zhu F, Wu H. 2017. A commensal streptococcus hijacks a Pseudomonas aeruginosa exopolysaccharide to promote biofilm formation. PLoS Pathog 13:e1006300. doi: 10.1371/journal.ppat.1006300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, Hendricks S, Wang Y, Chaplin MD, Akan D, Paik S, Peterson DL, Macrina FL, Buck GA. 2007. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol 189:3166–3175. doi: 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK-S, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 27.Winsor GL, Van Rossum T, Lo R, Khaira B, Whiteside MD, Hancock RE, Brinkman FS. 2009. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res 37:D483–D488. doi: 10.1093/nar/gkn861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L, Ge X, Xu P. 2015. Identifying essential Streptococcus sanguinis genes using genome-wide deletion mutation. Methods Mol Biol 1279:15–23. doi: 10.1007/978-1-4939-2398-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu P, Ge X, Chen L, Wang X, Dou Y, Xu JZ, Patel JR, Stone V, Trinh M, Evans K, Kitten T, Bonchev D, Buck GA. 2011. Genome-wide essential gene identification in Streptococcus sanguinis. Sci Rep 1:125. doi: 10.1038/srep00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maeda Y, Elborn JS, Parkins MD, Reihill J, Goldsmith CE, Coulter WA, Mason C, Millar BC, Dooley JS, Lowery CJ, Ennis M, Rendall JC, Moore JE. 2011. Population structure and characterization of viridans group streptococci (VGS) including Streptococcus pneumoniae isolated from adult patients with cystic fibrosis (CF). J Cyst Fibros 10:133–139. doi: 10.1016/j.jcf.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Toyofuku M, Nakajima-Kambe T, Uchiyama H, Nomura N. 2010. The effect of a cell-to-cell communication molecule, Pseudomonas quinolone signal (PQS), produced by P. aeruginosa on other bacterial species. Microbes Environ 25:1–7. doi: 10.1264/jsme2.ME09156. [DOI] [PubMed] [Google Scholar]
- 33.Morales DK, Hogan DA. 2010. Candida albicans interactions with bacteria in the context of human health and disease. PLoS Pathog 6:e1000886. doi: 10.1371/journal.ppat.1000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen AI, Dolben EF, Okegbe C, Harty CE, Golub Y, Thao S, Ha DG, Willger SD, O'Toole GA, Harwood CS, Dietrich LE, Hogan DA. 2014. Candida albicans ethanol stimulates Pseudomonas aeruginosa WspR-controlled biofilm formation as part of a cyclic relationship involving phenazines. PLoS Pathog 10:e1004480. doi: 10.1371/journal.ppat.1004480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filkins LM, Graber JA, Olson DG, Dolben EL, Lynd LR, Bhuju S, O'Toole GA. 2015. Coculture of Staphylococcus aureus with Pseudomonas aeruginosa drives S. aureus towards fermentative metabolism and reduced viability in a cystic fibrosis model. J Bacteriol 197:2252–2264. doi: 10.1128/JB.00059-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown SM, Howell ML, Vasil ML, Anderson AJ, Hassett DJ. 1995. Cloning and characterization of the katB gene of Pseudomonas aeruginosa encoding a hydrogen peroxide-inducible catalase: purification of KatB, cellular localization, and demonstration that it is essential for optimal resistance to hydrogen peroxide. J Bacteriol 177:6536–6544. doi: 10.1128/jb.177.22.6536-6544.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malhotra S, Limoli DH, English AE, Parsek MR, Wozniak DJ. 2018. Mixed communities of mucoid and nonmucoid Pseudomonas aeruginosa exhibit enhanced resistance to host antimicrobials. mBio 9:e00275-18. doi: 10.1128/mBio.00275-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassett DJ, Alsabbagh E, Parvatiyar K, Howell ML, Wilmott RW, Ochsner UA. 2000. A protease-resistant catalase, KatA, released upon cell lysis during stationary phase is essential for aerobic survival of a Pseudomonas aeruginosa oxyR mutant at low cell densities. J Bacteriol 182:4557–4563. doi: 10.1128/JB.182.16.4557-4563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jakubovics NS, Yassin SA, Rickard AH. 2014. Community interactions of oral streptococci. Adv Appl Microbiol 87:43–110. doi: 10.1016/B978-0-12-800261-2.00002-5. [DOI] [PubMed] [Google Scholar]
- 40.Zhu L, Kreth J. 2012. The role of hydrogen peroxide in environmental adaptation of oral microbial communities. Oxid Med Cell Longev 2012:717843. doi: 10.1155/2012/717843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JS, Heo YJ, Lee JK, Cho YH. 2005. KatA, the major catalase, is critical for osmoprotection and virulence in Pseudomonas aeruginosa PA14. Infect Immun 73:4399–4403. doi: 10.1128/IAI.73.7.4399-4403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shajani Z, Sykes MT, Williamson JR. 2011. Assembly of bacterial ribosomes. Annu Rev Biochem 80:501–526. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- 44.Gentry RC, Childs JJ, Gevorkyan J, Gerasimova YV, Koculi E. 2016. Time course of large ribosomal subunit assembly in E. coli cells overexpressing a helicase inactive DbpA protein. RNA 22:1055–1064. doi: 10.1261/rna.055137.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fergie N, Bayston R, Pearson JP, Birchall JP. 2004. Is otitis media with effusion a biofilm infection? Clin Otolaryngol Allied Sci 29:38–46. doi: 10.1111/j.1365-2273.2004.00767.x. [DOI] [PubMed] [Google Scholar]
- 46.Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J Bacteriol 184:6472–6480. doi: 10.1128/JB.184.23.6472-6480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D'Argenio DA, Calfee MW, Rainey PB, Pesci EC. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol 184:6481–6489. doi: 10.1128/JB.184.23.6481-6489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pesci EC, Milbank JBJ, Pearson JP, McKnight S, Kende AS, Greenberg EP, Iglewski BH. 1999. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96:11229–11234. doi: 10.1073/pnas.96.20.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maura D, Hazan R, Kitao T, Ballok AE, Rahme LG. 2016. Evidence for direct control of virulence and defense gene circuits by the Pseudomonas aeruginosa quorum sensing regulator, MvfR. Sci Rep 6:34083. doi: 10.1038/srep34083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wade DS, Calfee MW, Rocha ER, Ling EA, Engstrom E, Coleman JP, Pesci EC. 2005. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J Bacteriol 187:4372–4380. doi: 10.1128/JB.187.13.4372-4380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rampioni G, Falcone M, Heeb S, Frangipani E, Fletcher MP, Dubern JF, Visca P, Leoni L, Camara M, Williams P. 2016. Unravelling the genome-wide contributions of specific 2-alkyl-4-quinolones and PqsE to quorum sensing in Pseudomonas aeruginosa. PLoS Pathog 12:e1006029. doi: 10.1371/journal.ppat.1006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diggle SP, Matthijs S, Wright VJ, Fletcher MP, Chhabra SR, Lamont IL, Kong X, Hider RC, Cornelis P, Camara M, Williams P. 2007. The Pseudomonas aeruginosa 4-quinolone signal molecules HHQ and PQS play multifunctional roles in quorum sensing and iron entrapment. Chem Biol 14:87–96. doi: 10.1016/j.chembiol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 53.Bredenbruch F, Geffers R, Nimtz M, Buer J, Haussler S. 2006. The Pseudomonas aeruginosa quinolone signal (PQS) has an iron-chelating activity. Environ Microbiol 8:1318–1329. doi: 10.1111/j.1462-2920.2006.01025.x. [DOI] [PubMed] [Google Scholar]
- 54.Sonderholm M, Kragh KN, Koren K, Jakobsen TH, Darch SE, Alhede M, Jensen PO, Whiteley M, Kuhl M, Bjarnsholt T. 2017. Pseudomonas aeruginosa aggregate formation in an alginate bead model system exhibits in vivo-like characteristics. Appl Environ Microbiol 83:e00113-17. doi: 10.1128/AEM.00113-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haussler S, Becker T. 2008. The Pseudomonas quinolone signal (PQS) balances life and death in Pseudomonas aeruginosa populations. PLoS Pathog 4:e1000166. doi: 10.1371/journal.ppat.1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Filkins LM, O’Toole GA. 2015. Cystic fibrosis lung infections: polymicrobial, complex, and hard to treat. PLoS Pathog 11:e1005258. doi: 10.1371/journal.ppat.1005258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stites SW, Plautz MW, Bailey K, O'Brien-Ladner AR, Wesselius LJ. 1999. Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. Am J Respir Crit Care Med 160:796–801. doi: 10.1164/ajrccm.160.3.9811018. [DOI] [PubMed] [Google Scholar]
- 58.Stites SW, Walters B, O'Brien-Ladner AR, Bailey K, Wesselius LJ. 1998. Increased iron and ferritin content of sputum from patients with cystic fibrosis or chronic bronchitis. Chest 114:814–819. doi: 10.1378/chest.114.3.814. [DOI] [PubMed] [Google Scholar]
- 59.Moreau-Marquis S, Bomberger JM, Anderson GG, Swiatecka-Urban A, Ye S, O'Toole GA, Stanton BA. 2008. The DeltaF508-CFTR mutation results in increased biofilm formation by Pseudomonas aeruginosa by increasing iron availability. Am J Physiol Lung Cell Mol Physiol 295:L25–L37. doi: 10.1152/ajplung.00391.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coss-Bu JA, Sachdeva RC, Bricker JT, Harrison GM, Jefferson LS. 1997. Hemoptysis: a 10-year retrospective study. Pediatrics 100:E7. doi: 10.1542/peds.100.3.e7. [DOI] [PubMed] [Google Scholar]
- 61.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen XC, Justesen US, Dargis R, Kemp M, Christensen JJ. 2009. Identification of clinically relevant nonhemolytic streptococci on the basis of sequence analysis of 16S-23S intergenic spacer region and partial gdh gene. J Clin Microbiol 47:932–939. doi: 10.1128/JCM.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson GG, Moreau-Marquis S, Stanton BA, O'Toole GA. 2008. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect Immun 76:1423–1433. doi: 10.1128/IAI.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rhodes DV, Crump KE, Makhlynets O, Snyder M, Ge X, Xu P, Stubbe J, Kitten T. 2014. Genetic characterization and role in virulence of the ribonucleotide reductases of Streptococcus sanguinis. J Biol Chem 289:6273–6287. doi: 10.1074/jbc.M113.533620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orazi G, O'Toole G. 2017. Pseudomonas aeruginosa alters Staphylococcus aureus sensitivity to vancomycin in a biofilm model of cystic fibrosis infection. mBio 8:e00873-17. doi: 10.1128/mBio.00873-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sibley CD, Grinwis ME, Field TR, Parkins MD, Norgaard JC, Gregson DB, Rabin HR, Surette MG. 2010. McKay agar enables routine quantification of the “Streptococcus milleri” group in cystic fibrosis patients. J Med Microbiol 59:534–540. doi: 10.1099/jmm.0.016592-0. [DOI] [PubMed] [Google Scholar]
- 67.Limoli DH, Whitfield GB, Kitao T, Ivey ML, Davis MR Jr, Grahl N, Hogan DA, Rahme LG, Howell PL, O'Toole GA, Goldberg JB. 2017. Pseudomonas aeruginosa alginate overproduction promotes coexistence with Staphylococcus aureus in a model of cystic fibrosis respiratory infection. mBio 8:e00186-17. doi: 10.1128/mBio.00186-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.