

This study examined the role of the two streptococcal histidine triad (Sht) proteins of Streptococcus agalactiae in zinc homeostasis and complement resistance. We showed that Sht and ShtII facilitate zinc homeostasis in conjunction with the metal-binding proteins Lmb and AdcAII. Here, we show that the Sht-family proteins are functionally redundant with overlapping roles in zinc uptake. Further, this work reveals that although the Sht-family proteins bind to factor H in vitro this did not influence survival in human blood.
KEYWORDS: Streptococcus agalactiae, metal homeostasis, zinc transporter
ABSTRACT
Streptococcus agalactiae is not only part of the human intestinal and urogenital microbiota but is also a leading cause of septicemia and meningitis in neonates. Its ability to cause disease depends upon the acquisition of nutrients from its environment, including the transition metal ion zinc. The primary zinc acquisition system of the pathogen is the Adc/Lmb ABC permease, which is essential for viability in zinc-restricted environments. Here, we show that in addition to the AdcCB transporter and the three zinc-binding proteins, Lmb, AdcA, and AdcAII, S. agalactiae zinc homeostasis also involves two streptococcal histidine triad (Sht) proteins. Sht and ShtII are required for zinc uptake via the Lmb and AdcAII proteins with apparent overlapping functionality and specificity. Both Sht-family proteins possess five-histidine triad motifs with similar hierarchies of importance for Zn homeostasis. Independent of its contribution to zinc homeostasis, Sht has previously been reported to bind factor H leading to predictions of a contribution to complement evasion. Here, we investigated ShtII to ascertain whether it had similar properties. Analysis of recombinant Sht and ShtII reveals that both proteins have similar affinities for factor H binding. However, neither protein aided in resistance to complement in human blood. These findings challenge prior inferences regarding the in vivo role of the Sht proteins in resisting complement‐mediated clearance.
IMPORTANCE This study examined the role of the two streptococcal histidine triad (Sht) proteins of Streptococcus agalactiae in zinc homeostasis and complement resistance. We showed that Sht and ShtII facilitate zinc homeostasis in conjunction with the metal-binding proteins Lmb and AdcAII. Here, we show that the Sht-family proteins are functionally redundant with overlapping roles in zinc uptake. Further, this work reveals that although the Sht-family proteins bind to factor H in vitro this did not influence survival in human blood.
INTRODUCTION
First-row transition metals, such as manganese, iron, and zinc, are essential for the viability of all organisms. Metal ions contribute to essential cellular processes, serving as structural and/or catalytic cofactors in many key proteins (1). During infection, pathogenic bacteria acquire essential metal ions from the host environment. The essentiality of metal ion acquisition by invading pathogens is exploited in the host via metal sequestration mechanisms. These antimicrobial mechanisms, collectively referred to as nutritional immunity, have been extensively studied in the context of iron (2). Nevertheless, recent studies have revealed that the host also alters the abundance of other essential metal ions, including manganese and zinc (3). To subvert host restriction of zinc bioavailability, pathogenic bacteria use highly efficient zinc uptake pathways to acquire this metal. The most prevalent zinc import pathways in prokaryotes are the ATP binding cassette (ABC) transporters (4). It therefore follows that these metal ion acquisition mechanisms are crucial virulence factors of pathogenic bacteria.
Streptococcus agalactiae (group B streptococcus [GBS]) is not only a human commensal of the digestive and genital tracts but also an opportunistic pathogen that causes invasive infections, including septicemia, pneumonia, and meningitis in human neonates (5, 6). Zinc acquisition is essential to these processes since our prior studies have established that zinc is required for fundamental processes, including S. agalactiae growth and morphology (7). In S. agalactiae, zinc acquisition involves the AdcCB transporter, which is comprised of the integral membrane protein, AdcB, and the nucleotide binding domain, AdcC. The transporter functions in concert with three membrane tethered solute-binding protein (SBPs) that have specificity for zinc (Fig. 1A). These SBPs are AdcA, which has an unusual two-domain structure (8), and the canonical cluster A-I proteins AdcAII and Lmb (7, 9). The presence of multiple zinc-specific SBPs is an unusual feature largely unique to the streptococci, since most prokaryotes employ only a single protein. This appears to have arisen due to the ancient acquisition of a genetic element containing Lmb (or AdcAII) and a histidine triad (HT) protein in the streptococcal genus (10). Subsequent gene duplication events have led to the emergence of multiple copies of the histidine triad proteins and Lmb/AdcAII-type SBPs. To date, 25 streptococcal species encode histidine triad proteins, with gene copy numbers between one and four, and at least one Lmb/AdcAII-type SBP. The most extensively characterized examples are the pneumococcal histidine triad (Pht) proteins from Streptococcus pneumoniae, which encodes four copies (PhtA, PhtB, PhtD, and PhtE) and one AdcAII protein. Here, the Pht proteins are shown to be cell wall-associated proteins that contribute to zinc acquisition via AdcAII, albeit with overlapping functionality (8, 11, 12). PhtD is the most highly conserved Pht family protein across pneumococcal strains and contains five HT motifs, with each motif predicted to bind zinc (13–15). Recent studies have shown that the HT1 motif, which is closest to the cell membrane, is the most important for the contribution of PhtD to zinc acquisition (15). The mechanistic basis for how the Pht proteins contribute to pneumococcal zinc acquisition remains contentious, but the current consensus model proposes that Pht proteins bind the extracellular zinc via their HT motifs, with the zinc then transferred to the AdcAII-type SBP for cytoplasmic import via the AdcCB translocon (16). Whether the SBP recruits the metal ion from the Pht proteins directly or indirectly remains to be established. Irrespective of how zinc is acquired, the role of the Pht proteins in supporting AdcAII zinc import during in vitro growth and pneumococcal infection has been demonstrated (8, 11, 15).
FIG 1.

S. agalactiae zinc acquisition. (A) Schematic of the Adc/Lmb system. The Adc permease comprises the ABC transporter AdcCB, three zinc-binding SBPs (Lmb, AdcA, and AdcAII), and the Sht and ShtII proteins. Cp, capsule; Cw, cell wall; Mb, membrane; Ct, cytoplasm. (B) Growth phenotypes of wild-type S. agalactiae A909 (WT) and ΔadcA, ΔadcA Δsht ΔshtII, Δlmb ΔadcAII Δsht ΔshtII, ΔadcA Δlmb ΔadcAII and ΔadcA Δlmb ΔadcAII Δsht ΔshtII mutant derivatives in zinc-restricted CDM. Growth was monitored by OD600 measurements every 60 min for 12 h. The data are presented as mean OD600 measurements ± the standard deviations from three independent experiments.
To date, no histidine triad proteins have been characterized in S. agalactiae, although two candidate genes were identified in our previous work (7). These streptococcal histidine triad proteins, Sht and ShtII, share 52 and 49% identity with PhtD of S. pneumoniae (in 375 and 387 overlapping residues, respectively). The sht gene is contained within an operon that contains the lmb gene. This operon is present on a mobile genetic element that is found in almost all human S. agalactiae isolates (96.8%) but in only 26.7% of the animal isolates (7, 17). In contrast, ShtII is encoded in an operon with adcAII, which is present in all sequenced S. agalactiae strains. Although their contribution, if any, to zinc homeostasis remains to be determined, Sht has previously been implicated in aiding resistance of S. agalactiae to the innate immune system by facilitating factor H deposition, a regulator promoting inactivation of C3b (18). Despite the similarity between Sht and ShtII (47% protein identity in 403 overlapping residues), the role of ShtII in factor H binding remains to be determined.
Here, we examined the role of Sht and ShtII in S. agalactiae zinc acquisition via the Adc/Lmb components of the Adc permease. Our results reveal that Sht and ShtII both contribute to zinc uptake with the two proteins showing overlapping functionality and specificity. We further show that Sht and ShtII are able to bind factor H in vitro, but this functionality is not protective for survival in human blood. Collectively, these findings provide further knowledge regarding the roles of streptococcal histidine triad proteins in zinc acquisition and highlights that the capacity to bind factor H may not provide physiologically significant protection from complement‐mediated clearance.
RESULTS
Sht and ShtII contribute to Lmb- and AdcAII-mediated zinc homeostasis.
We hypothesized that the Sht and ShtII proteins contribute to S. agalactiae zinc homeostasis. To address this, we generated isogenic S. agalactiae deletion mutants of sht, shtII, or both sht and shtII, in isolation and in combination with the deletion of the zinc-recruiting SBPs adcA, lmb, and adcAII. The phenotypic impact of these deletions was assessed by growth assays in a zinc-restricted chemically defined medium (CDM) in which a functional Adc permease was shown to be essential for bacterial viability (7). Under these conditions, the three SBPs can be considered to be redundant suppliers of zinc, with the deletion of adcA, lmb, and adcAII recquired to abolish growth (7). We observed that, in zinc-restricted CDM, the loss of one or both of the sht and shtII genes had no phenotypic impact (see Fig. S1 in the supplemental material). Similarly, the AdcA-only strain (S. agalactiae Δlmb ΔadcAII Δsht ΔshtII), showed wild-type growth, indicating that the two-domain SBP was necessary and sufficient for survival in zinc restricted media (Fig. 1B). However, growth was strongly impaired in the ΔadcA Δsht ΔshtII strain while the ΔadcA strain grew normally. These results suggest that the Sht and ShtII proteins were necessary for zinc acquisition via the Lmb and/or AdcAII proteins in the absence of AdcA. This growth deficiency was similar to that observed for the ΔadcA Δlmb ΔadcAII mutant strain, which lacked all three SBPs or in the ΔadcA Δlmb ΔadcAII Δsht ΔshtII mutant strain, which lacked all the cell surface components of the zinc acquisition machinery (Fig. 1B).
These results are consistent with prior studies from S. pneumoniae, which revealed distinct zinc acquisition mechanisms for pneumococcal AdcA and AdcAII (8, 16): one wherein AdcA facilitates zinc acquisition independent of accessory protein partners and a second in which AdcAII/Lmb are dependent upon one or both of the Sht-family proteins to facilitate zinc recruitment.
To complement the growth analyses, whole-cell metal ion accumulation was analyzed for wild‐type S. agalactiae and the derived mutant strains. Here, we observed that zinc accumulation in the AdcA-only mutant strain (Δlmb ΔadcAII Δsht ΔshtII) was significantly reduced by comparison with the wild-type and the ΔadcA mutant strain (Fig. 2). This indicated that despite the lack of a growth phenotype, AdcA alone did not facilitate accumulation of the zinc to the same extent as wild-type bacteria or strains containing the Sht/ShtII and Lmb/AdcAII family of proteins. Further, as zinc accumulation was not significantly different between the AdcA-only mutant strain (Δlmb Δsht ΔadcAII ΔshtII) and the strain containing AdcA and both Sht proteins (Δlmb ΔadcAII), these data are consistent with the Sht proteins not contributing to AdcA-mediated zinc homeostasis (Fig. 2). The impacts of these mutations were restricted to zinc, since analysis of other first-row transition metal ions, i.e., manganese, cobalt, nickel and copper, did not show significant altered metal ions abundances (Table S1).
FIG 2.
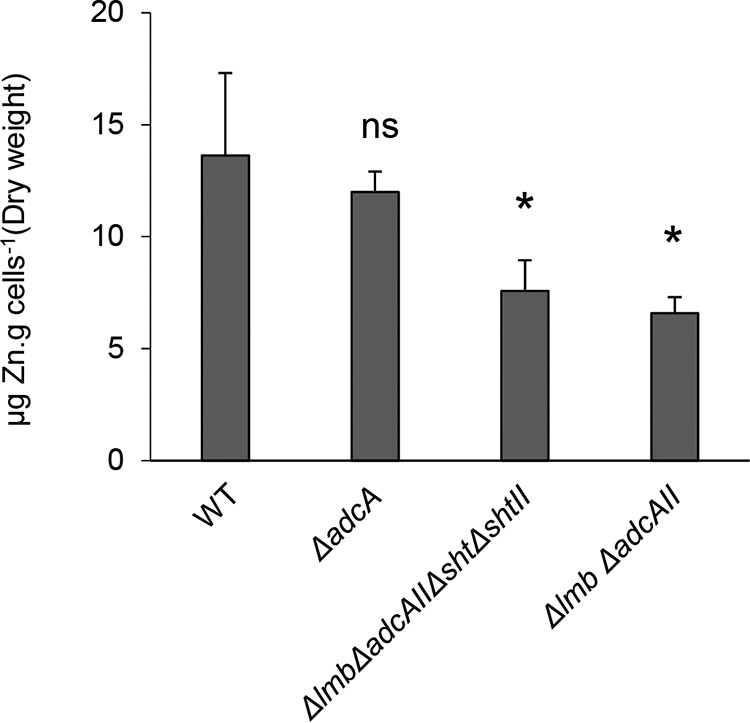
Zn2+ content of S. agalactiae grown in zinc-restricted conditions Whole-cell Zn2+ accumulation of WT and mutant strains determined by ICP-MS. The data represent the means ± the standard deviations from four biological replicates. The statistical significance of the differences in zinc concentrations compared to WT was determined by an unpaired Student t test (ns, not significant; *, P < 0.05).
Sht and ShtII both contribute to zinc homeostasis.
Building on the above findings, we sought to assess the relative contributions of the Sht and ShtII proteins to zinc acquisition. This was addressed in the ΔadcA genetic background to remove the contribution of AdcA, which does not interact with the Sht proteins. We first examined the impact of deleting a single Sht-family gene. Phenotypic growth analyses comparing S. agalactiae ΔadcA Δsht and ΔadcA ΔshtII mutant strains showed no significant difference in growth rates (Fig. 3A). This indicates that either sht gene is sufficient for growth with Lmb or AdcAII. Deletion of both sht genes abrogated bacterial growth, indicating that Lmb and AdcAII are unable to acquire sufficient zinc in the absence of these cell wall proteins (Fig. 3A). To complement this analysis, we examined the transcriptional responses in the S. agalactiae ΔadcA Δsht and ΔadcA ΔshtII mutant strains. Here, we observed that in the mutant strains, the remaining Sht-family gene was significantly upregulated by ∼3-fold (Fig. 3B). These findings indicate that a single Sht-family gene is sufficient for viability and growth. Nevertheless, the increased upregulation of the remaining Sht-family gene may show that an intracellular zinc deprivation is experienced by the mutant strains.
FIG 3.

Efficiency of the Sht-family proteins in zinc acquisition. (A) Growth of S. agalactiae A909 mutant strains in zinc-restricted CDM. The data are representative mean OD600 measurements ± the standard deviations from three independent experiments. (B) Transcriptional profiling of sht (black bars) and shtII (gray bars) in S. agalactiae A909 strains during growth in zinc-restricted CDM to mid-exponential phase (OD600 = 0.5 phase). Transcription of each gene was normalized to recA. The results are presented as means ± the standard deviations from three independent experiments. The asterisks indicate P values obtained using an unpaired Student t test to compare the level of expression of sht or shtII in the WT strain as a reference for each strain (nd, not detected; *, P < 0.05; **, P < 0.01).
To confirm that the abrogated growth phenotype of the S. agalactiae ΔadcA Δsht ΔshtII strain was due to zinc starvation, we supplemented the growth medium with increasing concentrations of metal ions. Here, we observed the recovery of growth with zinc but not with other metal ions (Fig. S2A and B). We then exploited the compromised zinc homeostasis of the ΔadcA Δsht ΔshtII strain to examine the relative efficiency of the Sht-family proteins to this process. This was addressed by ectopic expression of sht or shtII under the control of the constitutive promoter PTet (19). Transcriptional analyses of the Sht-family genes from pTCV-PTet::sht or pTCVPTet::shtII in S. agalactiae ΔadcA Δsht ΔshtII showed similar levels of expression (Fig. 3B). Phenotypic growth analyses of mutant strains containing the ectopic expression constructs in CDM showed growth comparable to the mutant ΔadcA strain (Fig. S3). This was in contrast to the parental strain, S. agalactiae ΔadcA Δsht ΔshtII, that was compromised for growth in this medium. Taken together, these findings indicate that a single Sht-family protein is necessary and sufficient to facilitate growth in zinc-limited media in the absence of AdcA. However, these findings also indicate that S. agalactiae experiences a greater level of intracellular zinc stress when only a single Sht-family protein is present, suggesting that two copies are optimally required.
Sht and ShtII have functionally redundant interactions with Lmb and AdcAII.
A homology model of S. agalactiae AdcAII was generated based on the structure of S. pneumoniae AdcAII (Fig. 4). This allowed comparison of S. agalactiae Lmb (PDB 3HJT) and AdcAII, and the identification of any features that could suggest a preferential interaction with their respective Sht-family protein (i.e., Lmb-Sht versus AdcAII-ShtII). Structural comparisons of AdcAII with Lmb indicated that the two proteins had similar overall folds (Cα root mean square deviation, 0.6 Å) with metal-binding sites buried ca. 10 to 15 Å beneath the molecular surface of the protein and occluded from solvent in the metal-bound state. Surface charge calculations revealed a negatively charged region at the interdomain cleft proximal to the metal-binding site in both Lmb and the model of AdcAII (Fig. 4). Collectively, these analyses did not reveal any apparent structural distinctions between Lmb and AdcAII that would otherwise suggest a structural basis for a preferential interaction with either Sht-family protein. To probe the veracity of our insights, we constructed different combinations of S. agalactiae deletion strains that expressed either lmb (ΔadcA ΔadcAII Δsht ΔshtII) or adcAII (ΔadcA Δlmb Δsht ΔshtII) as the only cell surface component of the zinc homeostasis machinery. These mutant strains were incapable of growth in zinc-restricted CDM (Fig. 5). We then introduced the ectopic expression constructs for Sht (pTCV-PTet::sht) or ShtII (pTCVPTet::shtII) into the mutant strains. Irrespective of the combination of Sht-family protein with Lmb or AdcAII, growth was restored in zinc-restricted CDM (Fig. 5). Taken together, these data show that there is no significant preference of SBP for Sht-family proteins, indicating that, in vitro, there is functional redundancy in this aspect of S. agalactiae zinc homeostatic machinery.
FIG 4.

Structural modeling of Lmb and AdcAII. (A) Representation of the crystal structure of Lmb (3HJT; cyan) and the homology model of AdcAII (green). The bound Zn2+ is shown as a gray sphere. The surface electrostatic potentials of AdcAII (B) and Lmb (C) are shown in the same orientation as in panel A (right panel). Positive and negative potentials are shown in blue and red, respectively, colored continuously between −5 and 5 kT/e. The surface electrostatic potential was calculated using APBS (41).
FIG 5.

Sht-family proteins aid in S. agalactiae zinc acquisition via either Lmb and/or AdcAII. (A) Growth of the Lmb-only strain (S. agalactiae ΔadcA ΔadcAII Δsht ΔshtII) complemented with the pTCV-PTet vector containing either sht or shtII. The strain ΔadcA ΔadcAII Δsht ΔshtII containing the empty vector (ᴓ) was used as a negative control. (B) Growth of the AdcAII-only strain (S. agalactiae ΔadcA Δlmb Δsht ΔshtII) mutant (only AdcAII remains as the substrate binding protein) complemented with the pTCV-PTet vector containing either sht or shtII. The ΔadcA Δlmb Δsht ΔshtII strain containing the empty vector (ᴓ) was used as a negative control. Growth was monitored by OD600 measurements every 60 min for 12 h. The data are representative mean OD600 measurements from three independent experiments.
Role of the five-histidine triad motifs of Sht and ShtII proteins in zinc acquisition.
Primary sequence comparisons of Sht-family proteins and PhtD revealed that the S. agalactiae proteins also contain five HT motifs (Fig. 6). The N-terminal portion of the proteins (residues 1 to 380) containing the three first HT motifs is the most conserved. Here, we sought to examine the respective contributions of the HT motifs to zinc acquisition. To address this, the three His residues (HXXHXH) of each HT motif in Sht and ShtII were replaced by three Phe residues. These mutations were shown to abrogate the zinc binding capacity of the HT motifs in S. pneumoniae PhtD while preserving steric bulk to ensure conformational fidelity (15). The sht and shtII HT mutant alleles (designated of shtΔHT* or shtIIΔHT*, where “*” represents the HT motif designated in Fig. 6) were introduced into the ΔadcA Δsht ΔshtII strain via the pTCV-PTet vector. Phenotypic growth assays were then performed in zinc-limited CDM and compared to the ΔadcA Δsht ΔshtII strain complemented with the respective parental allele (Fig. 6). In zinc-limited CDM, growth was completely abolished for the ΔHT1 and ΔHT2 Sht and ShtII strains. In contrast, growth was compromised for the ΔHT3 and ΔHT5 Sht and Sht strains, albeit to a lesser extent (Fig. 7). The ΔHT4 Sht and ShtII strains were the least affected, with growth of the shtIIΔHT4 strain indistinguishable from that of the parental strain (Fig. 7). Interestingly, both Sht and ShtII had identical patterns of behavior, despite significant sequence divergence with respect to HT4 and HT5. Supplementation of the CDM with 1 μM zinc restored growth of all but the ΔHT1 mutant strains. Further supplementation (10 μM zinc) restored growth of all strains (Fig. 7). Collectively, these data suggest a hierarchy of the importance with respect to the Sht and ShtII HT motifs that follows the order of HT1 > HT2 > HT3 > HT5 > HT4.
FIG 6.

Comparison of amino acid sequences of the Sht and ShtII proteins of S. agalactiae and the PhtD protein of S. pneumoniae. The signal sequences are highlighted in gray, and the identified histidine triad motifs (HT) are indicated by black boxes. The protein sequences were aligned using BioEdit software.
FIG 7.

Contribution of the Sht and ShtII HT sites to zinc acquisition. Growth of the S. agalactiae ΔadcA Δsht ΔshtII mutant with the pTCV-PTet vector containing either wild-type sht or a mutant derivative, in which a single HT motif was mutated (A), or wild-type shtII or a mutant derivative, in which a single HT motif was mutated (B). The empty vector (ᴓ) was used as a negative control. Bacteria were grown in zinc-restricted CDM, 1 μM Zn2+, or 10 μM Zn2+, as shown. Growth was monitored based on OD600 measurements every 60 min for 18 h. The data are representative mean OD600 measurements from three independent experiments.
Sht and ShtII bind factor H in vitro.
Sht was first described as a factor H binding protein and a negative regulator of the alternative pathway of complement (18). Since Sht and ShtII share 43% of protein identity, we investigated whether the ShtII protein had the same ability to bind factor H. The factor H binding assay examined recombinant S. agalactiae A909 Sht, ShtII, and CcpA and S. pneumoniae PspC. CcpA is a cytoplasmic transcriptional regulator (i.e., negative control), whereas PspC is a pneumococcal surface protein that has been shown to interact strongly with factor H (i.e., positive control) (20).
Enzyme-linked immunosorbent assay (ELISA) analyses of inactivated serum containing factor H showed that both Sht and ShtII bound factor H and to a similar extent (Fig. 8). Notably, Sht-family protein binding of factor H was significantly more than that observed for CcpA but weaker than PspC (Fig. 8). These data suggest that the Sht-family proteins may also serve a protective role for evading complement-mediated killing during in vivo infection.
FIG 8.
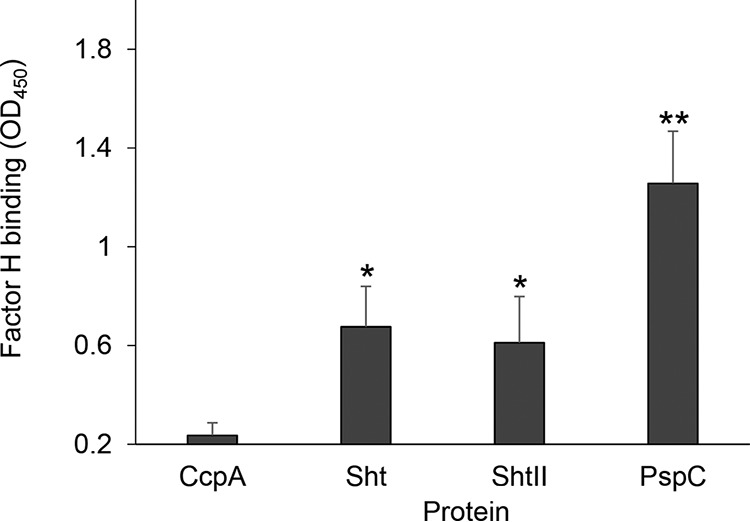
Factor H binding by the Sht-family proteins. Factor H binding to the streptococcal proteins CcpA, Sht, ShtII, and PspC was determined by ELISA. The data are representative mean OD450 measurements ± the standard deviations from three independent experiments. The asterisks indicate P values obtained using an unpaired Student t test to compare factor H binding by the Sht or ShtII proteins with the CcpA (negative control). *, P < 0.05; **, P < 0.01.
Sht and ShtII do not provide resistance against complement in human blood.
It has been proposed that the capacity of Sht to recruit factor H decreases C3b deposition on the surface of S. agalactiae, thereby contributing to evasion of bacterial lysis mediated by the membrane attack complex (18). However, direct experimental evidence remains lacking, and thus the relevance of the in vivo binding capacities of the Sht-family proteins for factor H is unclear. We examined the survival of S. agalactiae A909 and the Δsht ΔshtII mutant strain in whole human blood and heat-inactivated blood. In whole blood, wild-type bacteria were nearly completely eliminated (∼20% survival) after 3 h. In contrast, survival of the wild-type strain was unimpeded in heat-inactivated blood, highlighting the contribution of complement-mediated killing (Fig. 9A). Further, loss of the Sht-family proteins did not affect the relative survival of the bacteria in whole blood (Fig. 9A). The same experiment was performed in human serum and provided the same results (Fig. S4). Taken together, these data indicate that the Sht proteins provide no apparent protection against complement-mediated killing under these conditions. Given the discrepancy in our observations by comparison to studies of Sht-family orthologs (18), we analyzed transcription of sht and shtII in human serum (Fig. 9B and C). Indeed, we have previously shown that the lmb-sht, adcAII-shtII, and adcA encoding genes were fully repressed as soon as bacteria are cultured in medium containing more than 10 μM zinc, for example, in TH medium containing 20 μM zinc (7). In serum, we observed that the Sht-family genes were also significantly repressed (Fig. 9B and C), most likely attributable to bioavailable zinc in the human serum (14 μM in average [21]). Consequently, Sht and ShtII are unlikely to have been expressed at significant levels under the experimental conditions investigated.
FIG 9.

Expression and contribution of the Sht-family proteins in complement resistance. (A) Bacterial survival, determined as the percent CFU per milliliter relative to input of WT (white) and Δsht ΔshtII (black) mutant strains after 3 h of incubation in whole blood or heat-inactivated blood. The results represent means ± the standard deviations of three independent experiments. (B and C) Transcription of sht (B) and shtII (C) after 90 min of incubation in zinc-restricted CDM (black), CDM supplemented with 10 μM Zn2+ (white), or heat-inactivated serum (gray). Transcripts of each gene were normalized to recA with a reference value of 100 for sht and shtII transcripts after bacterial growth in zinc-restricted CDM. The results are means ± the standard deviations from three independent experiments. The statistical significance of the differences was determined by a two-tailed unpaired Student t test comparing gene expression levels to the zinc-restricted CDM control (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
DISCUSSION
In this study, we examined the role of the Sht-family proteins in zinc acquisition and complement resistance. We showed that Sht and ShtII facilitate zinc uptake via the Lmb and AdcAII, with no discernible preference or dependency on either SBP. This highlights two distinct routes of zinc acquisition in S. agalactiae mediated by either AdcA alone, or via Lmb/AdcAII in concert with the Sht-family proteins. These modes of zinc acquisition have previously been reported in S. pneumoniae and attributed to the distinct structural features of AdcA and AdcAII (8, 16). In S. agalactiae the zinc-specific SBPs all contain a common high-affinity metal-binding site, which is comprised of three histidine residues and a glutamic acid in Lmb and predicted by sequence alignments to be the same in AdcA and AdcAII (7, 22). However, AdcA also contains a short region enriched for histidine residues (residues 129 to 139) and a C-terminal extension (residues 319 to 502) that has homology to the periplasmic metal-binding protein ZinT. These accessory regions potentially aid in zinc acquisition for S. agalactiae AdcA, as has been proposed for orthologs in S. pneumoniae or S. pyogenes (7, 8, 16, 23). Further, these accessory domains provide a plausible explanation for the apparent lack of dependency of AdcA on Sht-family proteins to capture environmental zinc. The overlapping roles of these zinc acquisition systems in S. agalactiae raise the question of the reason for the basis for the conservation of multiple zinc import pathways. Recently, it has been proposed that the Sht-family genes, which are frequently acquired as operons that also contain lmb and adcAII genes, were acquired prior to the emergence of the Streptococcus genus (10). As a consequence, maintenance or loss of Sht-family genes are presumed to arise from the competing selective pressures associated with their physiological function, such as zinc acquisition and evasion of host immune responses. The absence of such determinants provides a plausible explanation for the loss of the Sht-family proteins in nonpathogenic species, such as Streptococcus thermophilus, a nonpathogenic bacterium widely used in starter cultures for cheese and yogurt production and in oral streptococci. Notably, these bacteria encode only a single SBP, AdcA, and no Sht-family proteins. In contrast, pathogenic streptococci, such as S. pneumoniae, S. pyogenes, S. suis, and S. agalactiae, frequently encode one or more Sht-family proteins (7, 14, 24, 25). Consistent with these inferences, it has been shown in S. pneumoniae that, despite the overlapping functionality of AdcA and AdcAII in vitro, both proteins were required for optimal in vivo infection (26).
Gene duplication events have led to emergence of streptococcal strains with multiple copies of the Sht-family genes. In S. agalactiae, this has also led to the presence of the two additional SBPs, Lmb and AdcAII, whose genes are in operons with sht and shtII, respectively. Of the streptococci, S. pneumoniae contains the greatest number of Sht-family genes with four orthologs. Examination of their role in pneumococcal zinc homeostasis revealed that their roles were not completely redundant since strains containing only a single pht gene did not grow as effectively as strains containing two or more copies (12). Here, we have shown that in contrast to S. pneumoniae, a single sht gene was sufficient for growth under zinc-limiting conditions. Sht and ShtII also share a strong sequence similarity (43% identity) in contrast to the divergent sequences of the S. pneumoniae Pht proteins. Consistent with this observation, the highly similar SBPs, Lmb and AdcAII (58% sequence identity), can interact with either Sht-family protein to facilitate zinc homeostasis with equivalent levels of efficiency.
The Sht-family proteins contain five HT motifs, similar to the pneumococcal ortholog PhtD. Although the Pht proteins have been refractory to high-resolution structure determination of the full-length protein, truncated domains containing individual HT motifs (HT1, HT2, and HT3) have been revealed (13, 16, 27). Structural analyses have suggested that each of the HT motifs of PhtD adopt a similar fold, with an equivalent capacity for Zn2+ binding (13). Nonetheless, functional studies have shown that HT1 has the most critical role in zinc acquisition (15). Here, we examined the contribution of the HT motifs to Zn2+ acquisition by the Sht-family proteins. The HT1 motif was essential during growth in Zn2+-limited conditions, while the other HT mutants showed intermediate phenotypes. The HT4 motif was the only exception, with little to no apparent phenotypic impact associated with its loss. Sequence alignments show that motifs HT1 to -3 of Sht and ShtII align closely with the equivalent motifs in PhtD (Fig. 6). These data suggest that the amino-terminal portions of the proteins are structurally and functionally conserved between S. agalactiae and S. pneumoniae. However, further mutagenesis and structural analyses are required to assess the strength of this inference.
Despite the original description of Sht as a complement resistance protein (18), the role of ShtII in this process has remained unaddressed. Here, we showed that both of the Sht-family proteins contribute to bind factor H in vitro, albeit to a lesser extent than PspC. However, the Sht-family proteins were not protective in human blood, most likely due to their lack of expression. Confounding results have also been reported for the S. pneumoniae Pht proteins and their role in factor H recruitment (28, 29). However, the bioavailability of zinc can alter significantly over the course of infection, with serum levels decreasing as a component of the hypozincemic effect. Tissue abscesses caused by Staphylococcus aureus infection have also been shown to be devoid of detectable Zn2+ (30). As such, the in vivo role of the Sht proteins and their contribution to both zinc recruitment and resisting complement-mediated killing cannot be discounted and warrant further investigation.
The contribution of the Sht-family proteins to the virulence of S. agalactiae can be linked to the necessity for efficacious Zn2+ acquisition mechanisms during growth in human amniotic and cerebrospinal fluids, which are tightly restricted for Zn2+ abundance (zinc concentrations of about 1.5 and 2.3 μM, respectively) (31–33). These two latter fluids are highly relevant for S. agalactiae, since it remains the main causative agent of neonatal meningitidis arising from the vertical transmission through contaminated amniotic fluid (5). Intriguingly, human-pathogenic S. agalactiae contains an additional lmb-sht operon, suggesting that the additional Zn2+-recruiting machinery plays a role in the context of human colonization and/or infection. Further analysis of the zinc acquisition pathways of S. agalactiae during in vivo infection will elucidate the roles of the various components and their relative contribution to human disease.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
S. agalactiae (GBS) and E. coli strains used in this study are listed in Table 1. E. coli XL1-Blue served as the host for recombinant plasmid pG+host1TS, and E. coli BL21(DE3) was used for the expression of recombinant protein from plasmid pET28a (Table 1). S. agalactiae strains were cultured on 5% horse blood Trypticase soy agar plates (1.5% agar; bioMérieux) on Todd-Hewitt broth (TH) agar (Sigma-Aldrich) or in liquid TH medium at 37°C without agitation. Complemented mutant strains harboring pTCV-PTet plasmid were maintained in medium containing 10 μg ml−1 of erythromycin (Ery). E. coli strains were cultured on Luria-Bertani (LB) agar plates or in liquid LB medium at 37°C with agitation (200 rpm). E. coli strains harboring pG+host1TS or pTCV-PTet plasmids were maintained in medium containing 150 μg ml−1 of Ery. E. coli strains harboring pET28a plasmid were maintained in medium containing 25 μg ml−1 of kanamycin and 25 μg ml−1 of chloramphenicol.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Source or reference |
|---|---|---|
| Strains | Strains | |
| Escherichia coli | ||
| XL1-Blue | endA1 gyrA96 (Nalr) thi-1 recA1 relA1 lac glnV44 hsdR17(rK– mK+) F′ [::Tn10 (Tetr) proAB+ lacIqZΔM15] | Stratagene |
| BL21 codon + (DE3)-RIL | F– dcm ompT hsdS(rB– mB–) gal [malB+] K-12 (λS) | Novagen |
| Streptococcus agalactiae | ||
| A909 (WT) | Isolated from a septic human neonate in 1934 | 42 |
| Δlmb ΔadcAII strain | Isogenic lmb (sak_1319) and adcAII (sak_1898) deletion double mutant of A909 | 7 |
| ΔadcA strain | Isogenic adcA (sak_0685) deletion mutant of A909 | 7 |
| ΔadcA Δlmb ΔadcAII strain | Isogenic adcA, lmb, and adcAII deletion triple mutant of A909 | 7 |
| Δsht strain | Isogenic sht (sak_1318) deletion mutant of A909 | This study |
| ΔshtII strain | Isogenic shtII (sak_1897) deletion mutant of A909 | This study |
| Δsht ΔshtII strain | Isogenic sht and shtII deletion double mutant of A909 | This study |
| ΔadcA Δsht strain | Isogenic adcA and sht deletion double mutant of A909 | This study |
| ΔadcA ΔshtII strain | Isogenic adcA and shtII deletion double mutant of A909 | This study |
| ΔadcA Δsht ΔshtII strain | Isogenic adcA, sht, and shtII deletion triple mutant of A909 | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::sht strain | sht (sak_1318) plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::sht ΔHT1 strain | sht containing a point mutation in the HT1 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::sht ΔHT2 strain | sht containing a point mutation in the HT2 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::sht ΔHT3 strain | sht containing a point mutation in the HT3 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::sht ΔHT4 strain | sht containing a point mutation in the HT4 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::sht ΔHT5 strain | sht containing a point mutation in the HT5 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::shtII strain | shtII (sak_1897) plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::shtII ΔHT1 strain | shtII containing a point mutation in the HT1 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::shtII ΔHT2 strain | shtII containing a point mutation in the HT2 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::shtII ΔHT3 strain | shtII containing a point mutation in the HT3 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::shtII ΔHT4 strain | shtII containing a point mutation in the HT4 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| ΔadcA Δsht ΔshtII/pTCV-PTet::shtII ΔHT5 strain | shtII containing a point mutation in the HT5 motif plasmid complementation of A909 ΔadcA Δsht ΔshtII | This study |
| Δlmb Δsht ΔadcAII ΔshtII strain | Isogenic lmb, sht, adcAII, and shtII deletion quadruple mutant of A909 | This study |
| ΔadcA Δlmb Δsht ΔadcAII ΔshtII strain | Isogenic adcA, lmb, sht, adcAII shtII deletion quintuple mutant of A909 | This study |
| ΔadcA ΔadcAII ΔshtII Δsht strain | Isogenic adcA, adcAII, shtII, and sht deletion quadruple mutant of A909 | This study |
| ΔadcA ΔadcAII ΔshtII Δsht/pTCV-PTet::sht strain | sht plasmid complementation of A909 ΔadcA ΔadcAII ΔshtII Δsht | This study |
| ΔadcA ΔadcAII ΔshtII Δsht/pTCV-PTet::shtII strain | shtII plasmid complementation of A909 ΔadcA ΔadcAII ΔshtII Δsht | This study |
| ΔadcA Δlmb Δsht ΔshtII strain | Isogenic adcA, lmb, sht, and shtII deletion quadruple mutant of A909 | This study |
| ΔadcA Δlmb Δsht ΔshtII/pTCV-PTet::sht strain | sht plasmid complementation of A909 ΔadcA Δlmb Δsht ΔshtII | This study |
| ΔadcA Δlmb Δsht ΔshtII/pTCV-PTet::shtII strain | shtII plasmid complementation of A909 ΔadcA Δlmb Δsht ΔshtII | This study |
| Plasmids | ||
| pG+host1TS | Replication-thermosensitive shuttle (TS) plasmid; Eryr | 43 |
| pTCV-PTet | Mob+ (IncP); oriR pACYC184; oriR pAM_1; complementation vector, promoter PTet | 19 |
| pTCV-PTet::sht | sht complementation vector, promoter PTet | This study |
| pTCV-PTet::shtΔHT1 | sht containing a point mutation in the HT1 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtΔHT2 | sht containing a point mutation in the HT2 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtΔHT3 | sht containing a point mutation in the HT3 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtΔHT4 | sht containing a point mutation in the HT4 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtΔHT5 | sht containing a point mutation in the HT5 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtII | shtII complementation vector, promoter PTet | This study |
| pTCV-PTet::shtIIΔHT1 | shtII containing a point mutation in the HT1 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtIIΔHT2 | shtII containing a point mutation in the HT2 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtIIΔHT3 | shtII containing a point mutation in the HT3 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtIIΔHT4 | shtII containing a point mutation in the HT4 motif complementation vector, promoter PTet | This study |
| pTCV-PTet::shtIIΔHT5 | shtII containing a point mutation in the HT5 motif complementation vector, promoter PTet | This study |
| pET28a | Vector for expression of His-tagged proteins; Kanr | EMD Biosciences |
| pET28a::sht | pET28a containing the sht gene of S. agalactiae A909 in the EcoRI/SalI sites | This study |
| pET28a::shtII | pET28a containing the shtII gene of S. agalactiae A909 in the EcoRI/SalI sites | This study |
| pET28a::pspC | pET28a containing a part of the pspC gene of S. pneumoniae D39 in the EcoRI/SalI sites | This study |
| pET28a::ccpA | pET28a containing the ccpA gene of S. agalactiae A909 in the EcoRI/SalI sites | This study |
Construction of deletion mutants and complementation strains.
Nonpolar deletion mutants strains of A909 S. agalactiae with a single or combined deletions of the entire coding sequences of the lmb, adcA, adcAII, sht, or shtII genes had been generated as previously described (7). PCR and DNA sequencing were used to confirm that the desired mutations had been introduced. The primers used to generate mutant strains are listed in Table 2. To complement the Δsht ΔshtII ΔadcA mutant strain, the entire coding sequence of sht or shtII gene was amplified using Q5 high-fidelity DNA polymerase (New England BioLabs) and cloned into the pTCV-PTet vector using appropriate restriction sites as previously described (7). The cloned sequences into the pTCV-PTet vectors were confirmed by PCR and DNA sequencing. The oligonucleotides (Sigma-Aldrich) used for the complementation constructs are listed in Table 2.
TABLE 2.
Oligonucleotides used in this study
| Function and primer | Sequence (5′–3′)a |
|---|---|
| Primers used for deletion of sht | |
| OAH 107 | TTTGTTGGTACCCCATTCACATACCTTAGAAGC |
| OAH 108 | ATATGTCCATGGCACTAATAATCTCCTTTACTTC |
| OAH 109 | CAACAACCATGGCCTTAACCAAAAGAAGATCTC |
| OAH 110 | ATGTCTCCCGGGGGAACACCTGCAGATAATGCCTG |
| Primers used for deletion of shtII | |
| OAH 123 | ACTCTAGAATTCCTGTAATTGAAGCTTCAAAAG |
| OAH 124 | TTTACGGGTACCCCTCGCTCCTTTCTTTATATA |
| OAH 125 | CTAGCAGGTACCAAATAATAAAAGAGTTGAGTA |
| OAH 126 | TCAACACCCGGGCTTATCCTTGTAGGAGGAGAA |
| Primers used for deletion of lmb | |
| OAH 24 | AGGCTGGAATTCTGGAAGGCGCTACTGTTCC |
| OAH 25 | CTCCTTTACTTCAACCCTTTTTTCATAGTACCTCCTCAATT |
| OAH 26 | TACTATGAAAAAAGGGTTGAAGTAAAGGAGATTATTAGTGAAG |
| OAH 27 | GGCTTGGGATCCAGCTAGCTCACTTGGAGAC |
| Primers used for deletion of adcAII | |
| OAH 40 | AAAATCGAATTCCAACGTGTTAATCAAGCAAGTG |
| OAH 62 | TAAGGTACCTCCGTATCCTTTTCATTAAACCTCC |
| OAH 63 | TTAGGTACCTAGGTAGTTATATAAAGAAAGGACG |
| OAH 43 | TACTAAGGATCCATCTCCCATGTCATCAATGAC |
| Primers used for deletion of adcA | |
| OAH 86 | AATCGTGGTACCGCAGCTCTAGCAGATCCACAC |
| OAH 76 | ATAAAGCTTGAAATTTCTTTCTCATTTTTTCTCC |
| OAH 77 | ATGAAGCTTCATTAATATTTAAAAGATGATATCGG |
| OAH 87 | CATCCAGGATCCCGTCCAGTTGTTTTCTTAGATAC |
| Primers used for complementation of sht | |
| OAH 248 | TTTTTTAGATCTGGAGGTACTGTGAAGAAAACATATGGT |
| OAH 284 | TTTCTGCAGAAAAACTTTGAGCAATTTGCTCAAAGTTTTTTTAAGGGTTTATTTGTTGAAGTGTCTTGAT |
| Primers used for HT site-directed mutagenesis of sht | |
| OAH235 HT1 | TGGTCTCGAAAATAAAAGTCACCGAATGAGGTCACATAGCC |
| OAH236 HT1 | TGGTCTCGTTTTTTTTACAATGGGAAAGTTCCTTATGATGCG |
| OAH250 HT2 | TGGTCTCGGGAATATAAAAATAGAAATTACCAAAAGG |
| OAH251 HT2 | TGGTCTCGTTCCTAAAAAGGATTTGTCTCCAAGTG |
| OAH254 HT3 | TGGTCTCGGGGATAATAAAATAAAAATCTCCAAAAGGC |
| OAH255 HT3 | TGGTCTCGTCCCAAGAAGTCAGTTATCACCTC |
| OAH258 HT4 | TGGTCTCGTAGAAGAAAAAATCTCCGAATTTAGCTGTA |
| OAH259 HT4 | TGGTCTCGTCTATATAGGATTTGGAGAACTTGAAC |
| OAH262 HT5 | TGGTCTCGCGGAACGACAAAGATAAAATCAATAAATGGGATAAC |
| OAH263 HT5 | TGGTCTCGTCCGTATTCATGGTTGACGCGCGATCAG |
| Primers used for complementation of shtII | |
| OAH 249 | TTTTTTGGATCCGGAGGTACTATGAATCGTAAAAAAACAGTT |
| OAH 241 | TTTGCATGCAAAAACTTTGAGCAATTTGCTCAAAGTTTTTTTATTTCACTTCTGCTAGTGTTTTAATATC |
| Primers used for HT site-directed mutagenesis of shtII | |
| OAH237 HT1 | TGGTCTCGAAAGTAAAAGTCTCCAAAAGAAGTTACATAACC |
| OAH238 HT1 | TGGTCTCGCTTTTATTACAATGGGAAAGTGCCATATGATGCC |
| OAH252 HT2 | TGGTCTCGGGATATAAAAGAAAAAATCGCCAAATGG |
| OAH253 HT2 | TGGTCTCGATCCCGAAAGCTGATTTATCTCCATCA |
| OAH256 HT3 | TGGTCTCGAGAAGAAAAAATTACCGAACGGAATAGAAAC |
| OAH257 HT3 | TGGTCTCGTTCTTTATTCACTATAAGGATATGTCTCC |
| OAH260 HT4 | TGGTCTCGGGTACCCAAAACTCAAATGTCATAAATGGAATG |
| OAH261 HT4 | TGGTCTCGTACCAAAGAAAGATTTATCAGAGTCGG |
| OAH264 HT5 | TGGTCTCGCCACATAAAAGTAAAAATCTTTAAAAGGAATTAC |
| OAH265 HT5 | TGGTCTCGTCCGTATTCATGGTTGACGCGCGATCAGAT |
| Primers used for recombinant protein expression | |
| OAH 173 Sht | CATATTGAATTCTACCAACTTGGTAAGCATCATATGGGT |
| OAH 174 Sht | ATCTTCGTCGACTTA AGGGTTTATTTGTTGAAGTGT |
| OAH 175 ShtII | TACCAAGAATTCAGCTATAATGCCCAAAAATCAGAC |
| OAH 176 ShtII | TCA ACTGTCGACTTATTTCACTTCTGCTAG |
| OLM 186 CcpA | AAAATCGGATCCATACAGATGATACGATTACGATTTA |
| OLM 181 CcpA | ACTCTAGAATTCATTATTTGTTGTGCACGTTTAAC |
| OAH 287 PspC | CTTGTTGGATCCAGTGTGCTTCATGCGACA |
| OAH 288 PspC | AGCTTGGTCGACATCTGTTTTTTCTGCTTTTGGTTG |
| Primers used for qRT-PCR | |
| OAH223 sht | TATCCATGTCGTTCCGTATTCATGGTT |
| OAH192 sht | CTTAGACCATACATCCGGACG |
| OAH224 shtII | TGCACCCAGAAAAACGTCCTAAAGTTG |
| OAH194 shtII | GGTGCAGGACTTGGTTTATCT |
| OLM321 recA | CTGGTGGTCGTGCTTTGAAA |
| OLM322 recA | TATGCTCACCAGTCCCCTTG |
Added restriction site sequences are indicated in boldface. Nucleotides targeting histidine residues for mutation to phenylalanine to generate HT variants are underlined.
Construction of HT motifs variants.
Site-tagged mutagenesis was performed to obtain the five HT* motif variants of Sht and ShtII. The primers were designed with mutations to substitute phenylalanine residues in place of each histidine residue of the relevant triad (Table 2). For each construction, two DNA fragments were generated by PCR. The point mutations were obtained using oligonucleotides containing the designed mismatches and also carrying BsaI restriction sites, a type IIS restriction endonuclease, which cleaves after its restriction site, generating DNA fragments with tetranucleotide cohesive ends (Table 2). After digestion with BsaI (1 h, 37°C; New England BioLabs), the two fragments were purified with a NucleoSpin PCR cleanup kit (Macherey-Nagel) and ligated using the sticky-end instant ligase (New England BioLabs), seamlessly fusing the fragments together without adding any additional nucleotides. The resulting fragments were reamplified by PCR using the external oligonucleotides OAH248 and OAH284 for sht and OAH249 and OAH241 for shtII and cloned in the pTCV-PTet vector (Tables 1 and 2).
GBS culture in chemically defined medium.
S. agalactiae strains (Table 1) were cultured in zinc-restricted chemically defined medium (CDM) as previously described (7). Briefly, S. agalactiae was grown in TH until it reached stationary phase and was then inoculated, at an optical density at 600 nm (OD600) of 0.005, into zinc-restricted CDM supplemented with 0.2% (w/vol) glucose [d-(+)-glucose; Sigma-Aldrich] and 500 μM EDTA and grown overnight. S. agalactiae was then inoculated at an OD600 of 0.005 into zinc-restricted CDM supplemented with 1% glucose (w/vol), 500 μM EDTA, and ZnSO4 concentrations as specified. S. agalactiae strains were then grown at 37°C for 12 h in microtiter plates (Greiner Bio-One; Cellstar) with OD600 measurements recorded using an Eon spectrophotometer (BioTek).
Whole-cell metal ion accumulation analyses.
Whole-cell metal ion accumulation was determined by inductively coupled plasma-mass spectrometry (ICP-MS) essentially as previously described (34, 35). Briefly, S. agalactiae strains were grown to mid-log phase (OD600 of 0.4 to 0.5) into CDM plus 1% glucose plus 500 μM EDTA. Cells were harvested by centrifugation, washed twice with PBS plus 5 mM EDTA, and then washed twice with PBS prior to desiccation at 95°C for 18 h. Metal ions were released by treatment with 0.5 ml of 35% HNO3 at 95°C for 60 min. Samples were then diluted to a final concentration of 3.5% HNO3 and metal content determined on an Agilent 8900 QQQ ICP-MS. The data represent four biological replicates, and the statistical difference was assessed using an unpaired Student t test (GraphPad Prism 7.0c).
RNA extraction.
For analysis of the sht and shtII expression in CDM and in serum, cells were grown in zinc-restricted CDM until reaching an OD600 of 0.5 and resuspended either in zinc-restricted CDM, in CDM supplemented with 100 μM zinc, or in inactivated serum (90 min). Cells were harvested (10 ml), and the pellets were frozen and stored at –80°C. For all other analyses, CDM-grown cells were harvested to mid-exponential phase (OD600 = 0.5). Bacteria were mechanically lysed by glass beads in a FastPrep-24 instrument, and total RNA was extracted using a phenol/TRIzol-based purification method as previously described (36). The concentration and purity of RNA were assessed with a NanoDrop Lite spectrophotomer (Thermo Fisher Scientific) with subsequent treatment using DNase (Turbo DNA-free DNase; Ambion). The absence of DNA contamination was confirmed by PCR using 50 ng of the purified RNA.
Reverse transcription and qRT-PCR.
RNA was reverse transcribed by using an iScript cDNA synthesis kit (Bio-Rad), according to the manufacturer’s instructions. Primers (Table 2) were selected with Primer3web software (http://bioinfo.ut.ee/primer3/) in order to design 100- to 200-bp amplicons. Quantitative reverse-transcriptase PCRs (qRT-PCRs) were performed in a 20-μl reaction volume containing 40 ng of cDNA, 1 μl of gene-specific primers (10 μM), and 10 μl of LightCycler 480 SYBR Green I mix (Roche). PCR amplification, detection, and analysis were performed with a LightCycler 480 PCR detection system and LightCycler 480 software (Roche). PCR conditions included an initial denaturation step at 95°C for 2 min, followed by a 45-cycle amplification (95°C for 5 s and 60°C for 20 s). The specificity of the amplified product and the absence of primer dimer formation were verified by generating a melting curve (65 to 95°C). The crossing point (CP) was defined for each sample. The expression levels of the tested genes were normalized using the recA (primers OLM321 and OLM322) gene of S. agalactiae as an internal standard whose transcript level did not vary under our experimental conditions. Each assay was performed in duplicate and repeated with at least three independent RNA samples.
Purification of recombinant Sht, ShtII, CcpA, and PspC proteins.
S. agalactiae A909 DNA was used to amplify the sht, shtII, and ccpA genes by PCR, and the S. pneumoniae D39 DNA was used to amplify the pspC genes with the Q5 high-fidelity DNA polymerase (New England BioLabs) using the OAH173-174, OAH175-176, OLM186-181, and OAH287-288 primers, respectively (Table 2). For Sht and ShtII, the primers were designed to remove the predicted sequence signal of each proteins (residues 1 to 23 for Sht and residues 1 to 27 for ShtII). For PspC, only the adhesive fraction of the protein was expressed (residues 33 to 445) (20). PCR amplifications were cloned into pET28a(+) vector (EMD Biosciences) in E. coli BL21 codon + (DE3)-RIL (Novagen) for high-level expression and addition of an amino-terminal 6×His tag. The derived expression constructs are listed in Table 1. Protein expression was conducted in E. coli BL21(DE3). Here, the expression construct-containing strains were grown in LB broth to an OD600 of 0.5 at 37°C, with shaking at 200 rpm in an orbital shaking incubator. Protein expression was induced by addition of IPTG (isopropyl-β-d-thiogalactopyranoside), and the cells were then grown for a further 5 h at 37°C at 200 rpm. Postinduction, the cells were pelleted at 10,000 × g, and the supernatant was decanted. The cells were then resuspended in 1 ml of lysis buffer (20 mM Tris [pH 8.0], 300 mM NaCl, 10% [wt/vol] glycerol) and lysed mechanically with glass beads in a FastPrep-24 instrument. Recombinant proteins were then purified by immobilized metal affinity chromatography (His-Select nickel affinity gel; Sigma-Aldrich) and eluted under native conditions according to the manufacturer’s instructions. Eluates were analyzed by SDS-PAGE and Coomassie blue staining to assess protein purity, as described by Laemmli (37).
Homology modeling.
The homology model of AdcAII was constructed using the SwissModel webserver (38), using S. pneumoniae AdcAII (PDB 3CX3) as a template. The resulting model of AdcAII was energy minimized in SwissPDBViewer (39) using the inbuilt 43B1 vacuum force field (40). Surface electrostatic potentials for the Lmb and AdcAII structures were calculated using APBS (41).
Factor H binding assays.
A 96-well microtiter plate (Sarstedt) was coated with 1 μg of the purified recombinant proteins (PspC, Sht, ShtII, or CcpA) overnight at 4°C. The wells were washed with 200 μl of PBS and then blocked with 200 μl of PBS containing 5% of skim milk (w/vol) for 1 h at room temperature. Human serum, provided by healthy donors of the Etablissement Français du Sang (EFS Centre Atlantique, France), was inactivated by treatment at 56°C for 30 min and used as a source of factor H. Then, 200 μl of inactivated serum was added to the wells for 2 h, followed by incubation at 37°C. The wells were then washed with PBS and incubated with 200 μl of a mouse antibody against factor H (1:2,000 [vol/vol]; Sigma-Aldrich) in PBS for 1 h at 37°C. After the wells were washed, 100 μl of peroxidase-conjugated anti-mouse IgG (1:2,000 [vol/vol]; Sigma-Aldrich) in 0.05 M carbonate-bicarbonate buffer (pH 9.6) was added, and the plate was incubated for 1 h at room temperature. After a final wash, 200 μl of Sigma Fast OPD substrate (Sigma-Aldrich) was added, and the absorbance at 450 nm was read using an Eon spectrophotometer (BioTek). The readings were background corrected by subtracting the absorbance of wells coated with bovine serum albumin. All assays were repeated three times in duplicate.
Complement resistance assays.
Human whole blood or serum was provided by healthy donors of the Etablissement Français du Sang who were not taking medication. Blood and serum were then used directly or heat inactivated by treatment at 56°C for 30 min. Bacterial strains, both wild-type (WT) and mutant strains, were grown for 8 h in TH broth and then inoculated at an OD600 of 0.005 into zinc-restricted CDM and grown for a further 12 h. The cells were harvested by centrifugation at 12,000 × g, washed, and resuspended in PBS. Bacteria were then inoculated into 1 ml of blood or heat-inactivated blood at a concentration of 106 CFU ml−1. The samples were gently mixed by rotation at 37°C for 3 h. Bacterial counts were performed after resuspension in blood (T0) or after incubation (T3) by dilution plating on TH agar. The survival rate represents the ratio between the number of cells at T3 and T0. The data represent the means of three independent experiments.
Statistical analyses.
The data represent means ± the standard errors of the mean. Statistical analyses were performed using a two-tailed unpaired Student t test. A probability value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elise Borezée-Durant, Franck Biet, Arnaud Firon, and Philippe Gilot for helpful discussions. We also thank Kevin Patron and Claudia Schneider for the purified CcpA and Daniel Niquet for technical assistance.
P.M. was supported by a doctoral fellowship of the Région Centre (France). C.A.M. is an Australian Research Council Future Fellow (FT170100006).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00757-18.
REFERENCES
- 1.Andreini C, Bertini I, Cavallaro G, Holliday GL, Thornton JM. 2008. Metal ions in biological catalysis: from enzyme databases to general principles. J Biol Inorg Chem 13:1205–1218. doi: 10.1007/s00775-008-0404-5. [DOI] [PubMed] [Google Scholar]
- 2.Schaible UE, Kaufmann SHE. 2004. Iron and microbial infection. Nat Rev Microbiol 2:946–953. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- 3.Kehl-Fie TE, Skaar EP. 2010. Nutritional immunity beyond iron: a role for manganese and zinc. Curr Opin Chem Biol 14:218–224. doi: 10.1016/j.cbpa.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hantke K. 2005. Bacterial zinc uptake and regulators. Curr Opin Microbiol 8:196–202. doi: 10.1016/j.mib.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Joubrel C, Tazi A, Six A, Dmytruk N, Touak G, Bidet P, Raymond J, Trieu Cuot P, Fouet A, Kernéis S, Poyart C. 2015. Group B streptococcus neonatal invasive infections, France 2007–2012. Clin Microbiol Infect 21:910–916. doi: 10.1016/j.cmi.2015.05.039. [DOI] [PubMed] [Google Scholar]
- 6.Schrag SJ, Verani JR. 2013. Intrapartum antibiotic prophylaxis for the prevention of perinatal group B streptococcal disease: experience in the United States and implications for a potential group B streptococcal vaccine. Vaccine 31:D20–D26. doi: 10.1016/j.vaccine.2012.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moulin P, Patron K, Cano C, Zorgani MA, Camiade E, Borezée-Durant E, Rosenau A, Mereghetti L, Hiron A. 2016. The Adc/Lmb system mediates zinc acquisition in Streptococcus agalactiae and contributes to bacterial growth and survival. J Bacteriol 198:3265–3277. doi: 10.1128/JB.00614-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plumptre CD, Eijkelkamp BA, Morey JR, Behr F, Couñago RM, Ogunniyi AD, Kobe B, O’Mara ML, Paton JC, McDevitt CA. 2014. AdcA and AdcAII employ distinct zinc acquisition mechanisms and contribute additively to zinc homeostasis in Streptococcus pneumoniae. Mol Microbiol 91:834–851. doi: 10.1111/mmi.12504. [DOI] [PubMed] [Google Scholar]
- 9.Berntsson RP-A, Smits SHJ, Schmitt L, Slotboom D-J, Poolman B. 2010. A structural classification of substrate-binding proteins. FEBS Lett 584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 10.Shao Z-Q, Zhang Y-M, Pan X-Z, Wang B, Chen J-Q. 2013. Insight into the evolution of the histidine triad protein (HTP) family in Streptococcus. PLoS One 8:e60116. doi: 10.1371/journal.pone.0060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayle L, Chimalapati S, Schoehn G, Brown J, Vernet T, Durmort C. 2011. Zinc uptake by Streptococcus pneumoniae depends on both AdcA and AdcAII and is essential for normal bacterial morphology and virulence. Mol Microbiol 82:904–916. doi: 10.1111/j.1365-2958.2011.07862.x. [DOI] [PubMed] [Google Scholar]
- 12.Plumptre CD, Hughes CE, Harvey RM, Eijkelkamp BA, McDevitt CA, Paton JC. 2014. Overlapping functionality of the Pht proteins in zinc homeostasis of Streptococcus pneumoniae. Infect Immun 82:4315–4324. doi: 10.1128/IAI.02155-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo Z, Pederick VG, Paton JC, McDevitt CA, Kobe B. 2018. Structural characterization of the HT3 motif of the polyhistidine triad protein D from Streptococcus pneumoniae. FEBS Lett 592:2341–2350. doi: 10.1002/1873-3468.13122. [DOI] [PubMed] [Google Scholar]
- 14.Loisel E, Chimalapati S, Bougault C, Imberty A, Gallet B, Di Guilmi AM, Brown J, Vernet T, Durmort C. 2011. Biochemical characterization of the histidine triad protein PhtD as a cell surface zinc-binding protein of pneumococcus. Biochemistry 50:3551–3558. doi: 10.1021/bi200012f. [DOI] [PubMed] [Google Scholar]
- 15.Eijkelkamp BA, Pederick VG, Plumptre CD, Harvey RM, Hughes CE, Paton JC, McDevitt CA. 2016. The first histidine triad motif of PhtD is critical for zinc homeostasis in Streptococcus pneumoniae. Infect Immun 84:407–415. doi: 10.1128/IAI.01082-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bersch B, Bougault C, Roux L, Favier A, Vernet T, Durmort C. 2013. New insights into histidine triad proteins: solution structure of a Streptococcus pneumoniae PhtD domain and zinc transfer to AdcAII. PLoS One 8:e81168. doi: 10.1371/journal.pone.0081168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franken C, Haase G, Brandt C, Weber-Heynemann J, Martin S, Lämmler C, Podbielski A, Lütticken R, Spellerberg B. 2001. Horizontal gene transfer and host specificity of beta-haemolytic streptococci: the role of a putative composite transposon containing scpB and lmb. Mol Microbiol 41:925–935. [DOI] [PubMed] [Google Scholar]
- 18.Maruvada R, Prasadarao NV, Rubens CE. 2009. Acquisition of factor H by a novel surface protein on group B streptococcus promotes complement degradation. FASEB J 23:3967–3977. doi: 10.1096/fj.09-138149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Firon A, Tazi A, Da Cunha V, Brinster S, Sauvage E, Dramsi S, Golenbock DT, Glaser P, Poyart C, Trieu-Cuot P. 2013. The Abi-domain protein Abx1 interacts with the CovS histidine kinase to control virulence gene expression in group B streptococcus. PLoS Pathog 9:e1003179. doi: 10.1371/journal.ppat.1003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duthy TG, Ormsby RJ, Giannakis E, Ogunniyi AD, Stroeher UH, Paton JC, Gordon DL. 2002. The human complement regulator factor H binds pneumococcal surface protein PspC via short consensus repeats 13 to 15. Infect Immun 70:5604–5611. doi: 10.1128/IAI.70.10.5604-5611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rink L, Gabriel P. 2000. Zinc and the immune system. Proc Nutr Soc 59:541–552. doi: 10.1017/S0029665100000781. [DOI] [PubMed] [Google Scholar]
- 22.Ragunathan P, Spellerberg B, Ponnuraj K. 2009. Structure of laminin-binding adhesin (Lmb) from Streptococcus agalactiae. Acta Crystallogr D Biol Crystallogr 65:1262–1269. doi: 10.1107/S0907444909038359. [DOI] [PubMed] [Google Scholar]
- 23.Cao K, Li N, Wang H, Cao X, He J, Zhang B, He Q-Y, Zhang G, Sun X. 2018. Two zinc-binding domains in the transporter AdcA from Streptococcus pyogenes facilitate high-affinity binding and fast transport of zinc. J Biol Chem 293:6075–6089. doi: 10.1074/jbc.M117.818997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunitomo E, Terao Y, Okamoto S, Rikimaru T, Hamada S, Kawabata S. 2008. Molecular and biological characterization of histidine triad protein in group A streptococci. Microbes Infect 10:414–423. doi: 10.1016/j.micinf.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Shao Z, Pan X, Li X, Liu W, Han M, Wang C, Wang J, Zheng F, Cao M, Tang J. 2011. HtpS, a novel immunogenic cell surface-exposed protein of Streptococcus suis, confers protection in mice. FEMS Microbiol Lett 314:174–182. doi: 10.1111/j.1574-6968.2010.02162.x. [DOI] [PubMed] [Google Scholar]
- 26.Brown LR, Gunnell SM, Cassella AN, Keller LE, Scherkenbach LA, Mann B, Brown MW, Hill R, Fitzkee NC, Rosch JW, Tuomanen EI, Thornton JA. 2016. AdcAII of Streptococcus pneumoniae affects pneumococcal invasiveness. PLoS One 11:e0146785. doi: 10.1371/journal.pone.0146785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riboldi-Tunnicliffe A, Isaacs NW, Mitchell TJ. 2005. 1.2 Angstrom crystal structure of the S. pneumoniae PhtA histidine triad domain a novel zinc binding fold. FEBS Lett 579:5353–5360. doi: 10.1016/j.febslet.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 28.Ogunniyi AD, Grabowicz M, Mahdi LK, Cook J, Gordon DL, Sadlon TA, Paton JC. 2009. Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor H. FASEB J 23:731–738. doi: 10.1096/fj.08-119537. [DOI] [PubMed] [Google Scholar]
- 29.Melin M, Di Paolo E, Tikkanen L, Jarva H, Neyt C, Käyhty H, Meri S, Poolman J, Väkeväinen M. 2010. Interaction of pneumococcal histidine triad proteins with human complement. Infect Immun 78:2089–2098. doi: 10.1128/IAI.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP. 2008. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 31.Meret S, Henkin RI. 1971. Simultaneous direct estimation by atomic absorption spectrophotometry of copper and zinc in serum, urine, and cerebrospinal fluid. Clin Chem 17:369–373. [PubMed] [Google Scholar]
- 32.Tamura GS, Kuypers JM, Smith S, Raff H, Rubens CE. 1994. Adherence of group B streptococci to cultured epithelial cells: roles of environmental factors and bacterial surface components. Infect Immun 62:2450–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shafeeq S, Kuipers OP, Kloosterman TG. 2013. The role of zinc in the interplay between pathogenic streptococci and their hosts. Mol Microbiol 88:1047–1057. doi: 10.1111/mmi.12256. [DOI] [PubMed] [Google Scholar]
- 34.Eijkelkamp BA, Morey JR, Ween MP, Ong CY, McEwan AG, Paton JC, McDevitt CA. 2014. Extracellular zinc competitively inhibits manganese uptake and compromises oxidative stress management in Streptococcus pneumoniae. PLoS One 9:e89427. doi: 10.1371/journal.pone.0089427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Begg SL, Eijkelkamp BA, Luo Z, Couñago RM, Morey JR, Maher MJ, Ong C-LY, McEwan AG, Kobe B, O’Mara ML, Paton JC, McDevitt CA. 2015. Dysregulation of transition metal ion homeostasis is the molecular basis for cadmium toxicity in Streptococcus pneumoniae. Nat Commun 6:6418. doi: 10.1038/ncomms7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamy M-C, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, Glaser P, Kunst F, Msadek T, Trieu-Cuot P, Poyart C. 2004. CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol 54:1250–1268. doi: 10.1111/j.1365-2958.2004.04365.x. [DOI] [PubMed] [Google Scholar]
- 37.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 38.Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 40.van Gunsteren WF. 1996. Biomolecular simulation: the GROMOS96 manual and user guide. Verlag der Fachvereine Hochschulverlag AG an der ETH Zurich, Groningen, Germany. [Google Scholar]
- 41.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O’Connor KJB, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc Natl Acad Sci U S A 102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biswas I, Gruss A, Ehrlich SD, Maguin E. 1993. High-efficiency gene inactivation and replacement system for gram-positive bacteria. J Bacteriol 175:3628–3635. doi: 10.1128/jb.175.11.3628-3635.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.