

Abstract
The implementation of surveillance biopsies in pediatric kidney transplantation remains controversial. Surveillance biopsies detect subclinical injury prior to clinical dysfunction, which could allow for early interventions that prolong allograft survival. We conducted a single-center retrospective cohort study of 120 consecutive pediatric kidney recipients, of whom 103 had surveillance biopsies ≤6 months posttransplant. We tested the hypothesis that subclinical inflammation (borderline or T cell–mediated rejection without clinical dysfunction) is associated with a 5-year composite endpoint of acute rejection and allograft failure. Overall, 36% of subjects had subclinical inflammation, which was associated with increased hazard for the composite endpoint (adjusted hazard ratio 2.89 [1.27, 6.57]; P < .01). Subjects with treated vs untreated subclinical borderline rejection had a lower incidence of the composite endpoint (41% vs 67%; P < .001). Subclinical vascular injury (subclinical inflammation with Banff arteritis score > 0) had a 78% incidence of the composite endpoint vs 11% in subjects with no major surveillance abnormalities (P < .001). In summary, we showed that subclinical inflammation phenotypes were prevalent in pediatric kidney recipients without clinical dysfunction and were associated with increased acute rejection and allograft failure. Once prospectively validated, our data would support implementation of surveillance biopsies as standard of care in pediatric kidney transplantation.
Keywords: biopsy, clinical research/practice, graft survival, kidney transplantation/nephrology, pathology/histopathology, pediatrics, protocol biopsy, rejection: subclinical, rejection: vascular
1 |. INTRODUCTION
Kidney transplantation remains the optimal treatment for end-stage renal disease (ESRD). However, improvements in short-term acute rejection rates and 1-year allograft survival have not led to improved long-term allograft survival.1,2 Specific causes of late allograft failure include antibody-mediated injury, glomerular diseases, and late acute rejection.3 These are diagnosed by kidney biopsy, usually after substantial allograft damage produces a decline in estimated glomerular filtration rate (eGFR). Unfortunately, by the time of apparent clinical dysfunction, inflammation and fibrosis are present, which are often resistant to treatment and associated with allograft failure.3–5 Detection of subclinical allograft injury prior to clinical dysfunction could allow for early interventions that prevent or interrupt processes leading to allograft failure, thereby improving life expectancy in people with ESRD. This is particularly relevant for pediatric kidney recipients, in whom creatinine-based eGFR is more imprecise than in adult recipients because of a greater mismatch between patient size and nephron mass.6 Therefore, pediatric recipients can incur substantially more acute and chronic allograft injury before overt clinical dysfunction occurs.
Surveillance biopsies can detect subclinical allograft injury at prespecified time points when allograft function is stable and histology findings are potentially treatable.7,8 These findings include subclinical borderline T cell–mediated rejection (B-TCMR), subclinical acute T cell–mediated rejection (SC-TCMR), or subclinical antibody-mediated rejection (SC-ABMR). However, despite their utility in detecting early subclinical injury, surveillance biopsies are not part of routine care in most transplant centers. Mehta et al recently illustrated that 17%−21% of US transplant centers performed surveillance biopsies in the first year posttransplant, and that many did not practice universal surveillance of all recipients.9 Previous reports have shown that surveillance biopsies detect SC-TCMR in 2.6%−61% and B-TCMR in 7%−50% of pediatric and adult kidney recipients.8,10–13 In recent surveillance studies in which more potent maintenance immunosuppression was used (eg, tacrolimus and mycophenolate mofetil with depletional induction), the incidence of SC-TCMR declined to 2.6%−25% in the first posttransplant year.14–17 Similar declines in SC-TCMR rates have not been shown in pediatric kidney transplantation,7,11 which may be attributed to more robust alloimmune responses, medication nonadherence, and lower sensitivity of creatinine-based eGFR to detect clinical dysfunction.6,18 The impact of detecting and treating subclinical allograft injury is still under debate, with some studies showing improved long-term outcomes and others showed no therapeutic benefit in children or adults.11,13,15,17,19
For the past decade our center has performed universal surveillance biopsies at 3 and/or 6 months posttransplant in order to detect subclinical allograft injury or other relevant pathology. We have gathered a racially diverse retrospective pediatric kidney transplant cohort treated with the same modern immunosuppression protocol. The purpose of this study was to test the hypothesis that early subclinical inflammation, defined as B-TCMR or SC-TCMR without clinical dysfunction, is associated with increased late acute rejection and allograft loss after pediatric kidney transplantation.
2 |. MATERIALS AND METHODS
2.1 |. Study design
We performed a retrospective cohort study of 120 consecutive pediatric kidney transplant recipients at our center from July 1, 2008 to December 31, 2014. Subjects were included if they were 1–21 years at the time of transplant and had a surveillance biopsy performed at 3 and/or 6 months posttransplant. Subjects were excluded if we found no record of a surveillance biopsy within 6 months posttransplant, or if records showed that only indication biopsies were performed to investigate allograft dysfunction within the first 6 months. Subjects were also excluded if they had clinical acute rejection prior to the surveillance period. The institutional review board (IRB) at the University of Alabama approved this study (IRB-150825006), and all study procedures adhered to the guidelines set forth in the Declaration of Helsinki.
2.2 |. Immunosuppression and surveillance
All subjects were treated with a similar triple immunosuppressive protocol and received nondepletional induction with basiliximab on day 0 and day 4 posttransplant, with maintenance immunosuppression consisting of tacrolimus, mycophenolate mofetil (MMF), and corticosteroids. All subjects were confirmed to have a negative virtual and flow crossmatch against a potential donor before proceeding with the transplant. We adjusted tacrolimus dosing to achieve the following target trough levels: 10–12 ng/mL through week 4, followed by 8–10 ng/mL through week 12, and then 5–8 ng/ mL thereafter; all subjects remained on calcineurin inhibitor–based immunosuppression. The dose of MMF was 600 mg/m2 every 12 hours for the first 48 hours and then adjusted to 450 mg/m2 every 12 hours thereafter. Corticosteroids were initially given as intravenous pulse methylprednisolone followed by an oral taper to achieve a maintenance dose of 0.1 mg/kg/day by 12 weeks posttransplant. Maintenance immunosuppression dosing was adjusted for leukopenia, gastroenteritis, or viral reactivation. Allograft function was assessed serially using the bedside Chronic kidney disease (CKD)-Schwartz equation (subjects aged <18 years) or the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) calculation (subjects aged ≥18 years), choosing the appropriate equation for age at the time of each assessment.6,20,21 Of note, we used the CKD-Schwartz calculation for 1-year eGFR and the CKD-EPI calculation for 5-year (or last follow-up) eGFR in 6/103 (6%) subjects.
Surveillance biopsies were planned at 3 and 6 months posttransplant in each subject, with procedural sedation and ultrasound guidance of a disposable 16-gauge biopsy device. In some subjects either the 3-or 6-month surveillance biopsy was not done because an indication biopsy for allograft dysfunction was performed instead, or because of transportation or other logistical issues. Biopsies were identified as surveillance if they were explicitly recorded as such in the medical record, or occurred within 4 weeks of the intended surveillance time points and were performed when eGFR was within 25% of recent baseline values. Surveillance biopsies were scored by 3 renal pathologists (D.R.K., E.C.M., and F.R.) according to Banff 2007 and 2013 criteria, as relevant to the timing of each biopsy.22,23 D.R.K. and E.C.M. had access to clinical information about each biopsy, whereas F.R. was blinded to clinical data. C4d staining of peritubular capillaries was assessed using both immunohistochemistry and immunofluorescence techniques as part of routine clinical care.
HLA laboratory monitoring was performed at the discretion of the treating physician, as there was no established protocol for donor-specific antibody (DSA) surveillance during the study period. DSA testing was routinely performed at the time of indication biopsies for allograft dysfunction but rarely at the time of surveillance biopsies. DSA were assessed using Luminex single-antigen bead assays for class I (A, B, and C loci) and II (DR and DQ loci) HLA antigens (One Lambda, Canoga Park, CA). DSAs against class II DP loci were not routinely performed during the study period. Our HLA laboratory uses a mean fluorescence intensity (MFI) cutoff of >1500 MFI for a positive DSA, but reports “weak” or “probable” DSA between 500 and 1500 MFI. In this study, we considered a positive DSA as >1500 MFI. These cutoffs were developed internally by our HLA laboratory director (V.H.D.) and validated longitudinally against flow-cytometry crossmatch testing, consistent with recent consensus guidelines.24,25
2.3 |. Exposures and outcomes
The primary exposure was subclinical inflammation on either a 3-or 6-month surveillance biopsy, defined as B-TCMR or SC-TCMR using Banff criteria and modeled as a categorical variable. We did not include SC-ABMR, since DSAs were not routinely assessed at the time of surveillance biopsies. Biopsies without subclinical inflammation and no other major surveillance abnormalities were classified as no major surveillance abnormalities (NOMOA).4 Secondary exposures modeled as continuous variables included age at transplant, cold ischemia time, transplant vintage, tacrolimus trough levels, and the number of antigen mismatches at HLA-A, -B, -C, -DR, and -DQ loci. Secondary exposures modeled as categorical variables included sex, donor type (deceased or living donor), race (black or non-black), repeat transplantation, de novo class I and II DSA during year-1 posttransplant (present or absent), C4d staining in peritubular capillaries (present or absent), medication nonadherence in the first 6 months posttransplant (defined as nonadherence listed in the medical record or 2 undetectable tacrolimus trough levels), and treated (increased immunosuppression) vs untreated (observed without change in immunosuppression) B-TCMR and SC-TCMR. Specifically, immunosuppression was augmented at the discretion of the treating physician, including increased maintenance immunosuppression, pulse intravenous corticosteroids, or occasionally antithymocyte globulin. In addition to the overall Banff classification we included individual Banff severity scores (t, i, ti, v, g, ptc, ct, ci, cg, cv; each score ranging 0–3) as secondary exposures modeled as continuous variables, using the highest score from any surveillance biopsy in each subject for analysis.
The primary endpoint was prespecified a priori as a composite outcome of clinical acute rejection (TCMR, ABMR, or mixed rejection) and death-censored allograft loss within 5 years posttransplant. Clinical acute rejection was defined using Banff consensus criteria in indication biopsies performed for allograft dysfunction, and was considered as an endpoint after the latest surveillance biopsy in each subject, so that a temporal relationship between surveillance findings and subsequent rejection could be ascertained. Early acute rejection was defined as occurring after the surveillance period but less than 1 year posttransplant, whereas late acute rejection was defined as occurring between 1 and 5 years posttransplant. Secondary endpoints were prespecified a priori as each component of the primary composite endpoint (presented without correction for multiplicity testing), death with a functioning allograft, eGFR at 1 year posttransplant, eGFR at last follow-up, and annualized change in eGFR. For subjects with death-censored allograft loss, the last follow-up eGFR was defined as 10 mL/min/1.73 m2 for statistical analysis. We also examined subclinical inflammation as a secondary endpoint in order to study demographic and clinical determinants of B-TCMR and SC-TCMR.
2.4 |. Statistical analysis
Continuous variables were assessed for normality using the Kolmogorov-Smirnov test, and comparisons between groups were made using Student’s t-test, Mann-Whitney U-test, or one-way analysis of variance (ANOVA) as appropriate for the normality of the data distribution and the number of comparator groups. Categorical variables were compared using a chi-square test or Fisher’s exact test as appropriate for the number of subjects. Survival distributions for the primary composite endpoint and each component were compared between groups using Kaplan-Meier methods and the log-rank test. The primary composite endpoint was also modeled using Cox proportional hazards regression. Any covariates that were associated with the outcome of interest by univariable analysis at P < .10 were entered into a multivariable Cox model. Visual inspection of log-log plots was used to confirm the proportionality of hazards assumption. All statistical tests were two-tailed with statistical significance defined as P < .05. All analyses were performed using SPSS Statistics version 23 (IBM, Armonk, NY, USA).
3 |. RESULTS
3.1 |. Characterization of the cohort
Of the 120 consecutive transplants performed during the study period, 103 had at least one surveillance biopsy at 3 and/or 6 months posttransplant. Of these, 49 (48%) had biopsies at both 3 and 6 months posttransplant. Surveillance biopsies had an excellent safety profile with no reports of significant bleeding, acute kidney injury, or allograft failure attributable to the procedure. One-year patient and allograft survival were 99% and 100%, respectively. No deaths occurred between 1 and 5 years posttransplant, but there were 8 instances of allograft loss. In all, 94 subjects (91%) survived with functioning allografts until 5 years posttransplant or until administrative censoring at the end of the study (Figure 1).
FIGURE 1.
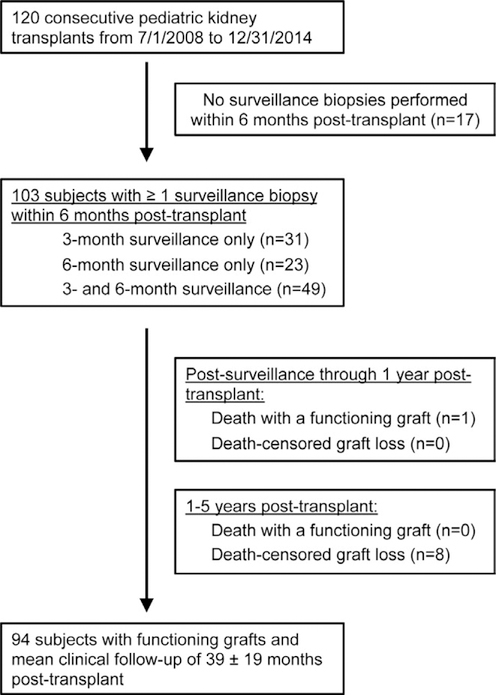
CONSORT diagram
3.2 |. Determinants of subclinical inflammation
Of 103 subjects with surveillance biopsies at 3 and/or 6 months posttransplant, 37 (36%) had at least one biopsy with subclinical inflammation including 24 subjects with B-TCMR and 13 subjects with SC-TCMR. Subclinical inflammation was detected in 19% and 31% of surveillance biopsies at 3 and 6 months posttransplant, respectively. Our cohort was racially diverse and enriched for immunologic risk, as 45% were of black race and 69% had deceased donor transplants. DSAs were assessed at least once during year-1 posttransplant in 79/103 (77%), with 9/79 (11%) testing positive for class I and/or II DSA. We did not detect differences in early de novo DSA rates between those with subclinical inflammation and those with NOMOA. Nonadherence was present in 16% of subjects, with a trend toward more nonadherence in the subclinical inflammation group that was not statistically significant. We also observed trends for higher proportions of deceased donor transplants and more HLA-DQ mismatches in those with subclinical inflammation vs NOMOA, but other baseline demographic and clinical characteristics were similar between surveillance groups (Table 1). As expected, acute Banff severity scores (t, i, v, and ti) and the chronic tubular atrophy (ct) score were significantly higher in those with subclinical inflammation compared to NOMOA. Of interest, chronic Banff severity scores (ct, ci, and cv) were similar between B-TCMR and SC-TCMR at 6 months, indicating that early chronic injury and fibrosis were present regardless of the severity of subclinical inflammation and despite the absence of clinical dysfunction (Table S1).
TABLE 1.
Demographic data
| Subclinical Inflammation (n = 37) |
|||||
|---|---|---|---|---|---|
| B-TCMR (n = 24) | SC-TCMR (n = 13) | Total (n = 37) | NOMOA (n = 66) | P-value | |
| Age at transplant (y) | 12.6 ± 5.2 | 6.9 ± 6.3 | 10.6 ± 6.2 | 12.1 ± 5.2 | .17a |
| Sex (male/female) | 12/12 | 9/4 | 21/16 | 49/17 | .07 |
| Donor type (living [related or unrelated]/deceased) | 4/20 | 3/10 | 7/30 | 25/41 | .05 |
| Race (black/nonblack) | 14/10 | 4/9 | 18/19 | 28/38 | .54 |
| Cold ischemia time (h) | 17.1 ± 10.1 | 12.1 ± 9.8 | 15.3 ± 10.1 | 14.8 ± 9.2 | .85 |
| Transplant vintage (year) | 2.9 ± 1.7 | 2.8 ± 1.8 | 2.9 ± 1.7 | 3.4 ± 1.5 | .14 |
| Repeat kidney transplant (no., %) | 1 (4%) | 0 (0%) | 1 (3%) | 2 (3%) | 1.00 |
| Tacrolimus trough at 3 mo posttransplant (ng/mL) | 8.6 ± 2.3 | 8.1 ± 2.6 | 8.4 ± 2.4 | 8.8 ± 3.1 | .53 |
| Tacrolimus trough at 6 mo posttransplant (ng/mL) | 7.7 ± 3.0 | 7.0 ± 2.4 | 7.4 ± 2.8 | 7.3 ± 2.3 | .92 |
| HLA mismatches (no./10 total) | 7.4 ± 1.4 | 7.2 ± 2.1 | 7.3 ± 1.6 | 6.9 ± 2.3 | .30b |
| HLA-A (no.) | 1.4 ± 0.7 | 1.4 ± 0.7 | 1.4 ± 0.7 | 1.4 ± 0.7 | .74 |
| HLA-B (no.) | 1.7 ± 0.5 | 1.6 ± 0.5 | 1.7 ± 0.5 | 1.6 ± 0.6 | .52 |
| HLA-C (no.) | 1.4 ± 0.6 | 1.3 ± 0.8 | 1.4 ± 0.7 | 1.4 ± 0.6 | .87 |
| HLA-DR (no.) | 1.5 ± 0.5 | 1.6 ± 0.5 | 1.5 ± 0.5 | 1.4 ± 0.6 | .52 |
| HLA-DQ (no.) | 1.5 ± 0.5 | 1.4 ± 0.5 | 1.4 ± 0.5 | 1.2 ± 0.7 | .07 |
| DSA during year-1 posttransplant (no./assessed, %) | 3/18 (17%) | 1/10 (10%) | 4/28 (14%) | 5/51 (10%) | .55 |
| Class I only (no., %) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1.00 |
| Class II only (no., %) | 1 (6%) | 0 (0%) | 1 (4%) | 3 (6%) | .79 |
| Class I and II (no., %) | 2 (11%) | 1 (10%) | 3 (11%) | 2 (4%) | .35 |
| Nonadherence ≤6 mo posttransplant (no, %) | 4 (17%) | 4 (31%) | 8 (22%) | 8 (12%) | .20 |
| Subclinical inflammation at 3 mo (no., %) | 10 (42%) | 5 (38%) | 15 (41%) | N/A | — |
| Subclinical inflammation at 6 mo (no., %) | 14 (58%) | 8 (62%) | 22 (59%) | N/A | — |
| Treated surveillance findings (no., %) | 8 (33%) | 13 (100%) | 21 (57%) | N/A | — |
Comparisons were made between the combined subclinical inflammation (n = 37) and no major abnormalities (n = 66) groups using Student’s t-test, chi-square test, or Fisher’s exact test, as appropriate.
B-TCMR, borderline T cell–mediated rejection; DSA, donor-specific antibody; NOMOA, no major abnormalities; SC-TCMR, subclinical T cell–mediated rejection.
P-value represents comparison of mean age at transplant between subclinical inflammation and normal surveillance groups. The mean age at transplant was significantly different between B-TCMR and SC-TCMR subgroups at P < .01.
HLA mismatch data were available in 96 subjects.
3.3 |. Outcomes after subclinical inflammation
The incidence of the primary composite endpoint was significantly higher in subjects with subclinical inflammation in both chi-square and Kaplan-Meier analyses, with the greatest effect derived from 3-fold higher rates of the acute rejection component of the endpoint (Table 2 and Figure 2). Outcomes were similar between B-TCMR and SC-TCMR subgroups, except trends for higher rates of late acute rejection and lower eGFR in the B-TCMR subgroup that did not reach statistical significance. Notably, all 7 cases of late acute rejection in the subclinical inflammation group occurred in those with B-TCMR (Table 2). Kaplan-Meier plots confirmed a similar incidence of the 5-year composite endpoint in those with B-TCMR and SC-TCMR (Figure 3A). Cox regression modeling showed that subclinical inflammation was independently associated with a 3.4-fold greater hazard for the composite endpoint after adjusting for DSA during year-1 posttransplant, nonadherence, race, and donor type in multivariable analysis (Table 3). In separate yet similarly constructed multivariable Cox models, we found that both B-TCMR (adjusted hazards ratio [HR] 260, 95% confidence interval [CI] 1.03–6.54) and SC-TCMR (adjusted HR 3.54, 95% CI 1.18–10.62) were independently associated with increased hazard for the primary endpoint as well.
TABLE 2.
Primary and secondary outcomes
| Subclinical inflammation (n = 37) |
|||||
|---|---|---|---|---|---|
| B-TCMR (n = 24) | SC-TCMR (n = 13) | Total (n = 37) | NOMOA (n = 66) | P-value | |
| Composite endpoint (no., %) | 13 (54%) | 6 (46%) | 19 (51%) | 11 (17%) | <.001 |
| Acute rejection, postsurveil- lance (no., %) | 13 (54%) | 5 (39%) | 18 (49%) | 11 (17%) | .001 |
| Death-censored graft loss (no., %) | 3 (13%) | 2 (15%) | 5 (14%) | 4 (6%) | .20 |
| Early rejection (<1 y)/late rejection (1–5 y) | 6/7 | 5/0 | 11/7 | 5/6 | .47 |
| Estimated GFR at 1 y (mL/min/1.73 m2) | 61 ± 25 | 79 ± 37 | 67 ± 30 | 74 ± 34 | .35 |
| eGFR at last follow-up (mL/min/1.73 m2) | 48 ± 25 | 63 ± 36 | 53 ± 29 | 58 ± 22 | .36 |
| Annualized change in eGFR (mL/min/1.73 m2/y) | −10.04 | −8.31 | −9.46 | −8.57 | .85 |
The primary outcome is a 5-year composite endpoint of acute rejection after surveillance and death-censored graft failure. The P-value represents the comparison between the subclinical inflammation and no major abnormalities groups, with significant values noted in bold type.
B-TCMR, borderline T cell–mediated rejection; NOMOA, no major abnormalities; SC-TCMR, subclinical T cell–mediated rejection.
FIGURE 2.
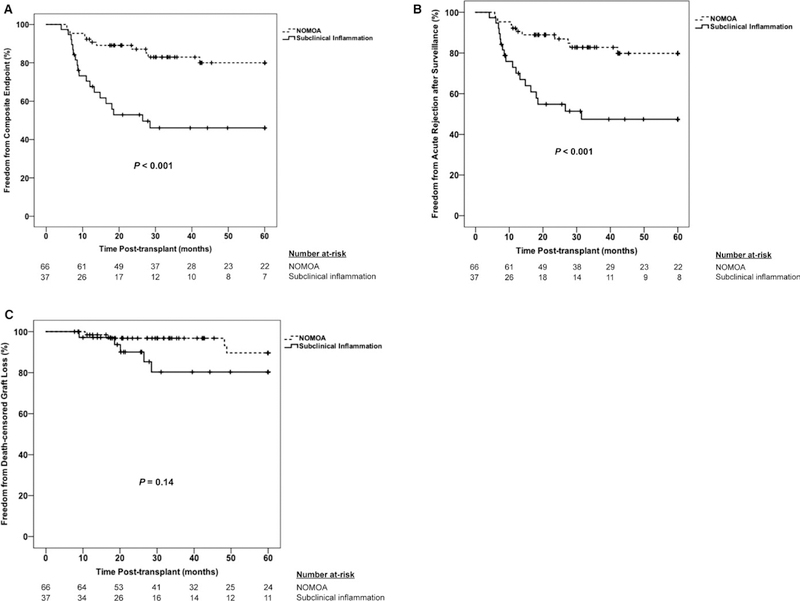
Kaplan-Meier plot of 5-year incidence of primary (A) and secondary (B, C) outcomes between the subclinical inflammation and no major abnormalities groups. (A) Composite endpoint of acute rejection after surveillance and death-censored graft loss. (B) Acute rejection after surveillance component of the composite endpoint. (C) Death-censored graft loss component of the composite endpoint. Comparisons of time-to-event data between groups were made using the log-rank test. NOMOA, no major abnormalities
FIGURE 3.
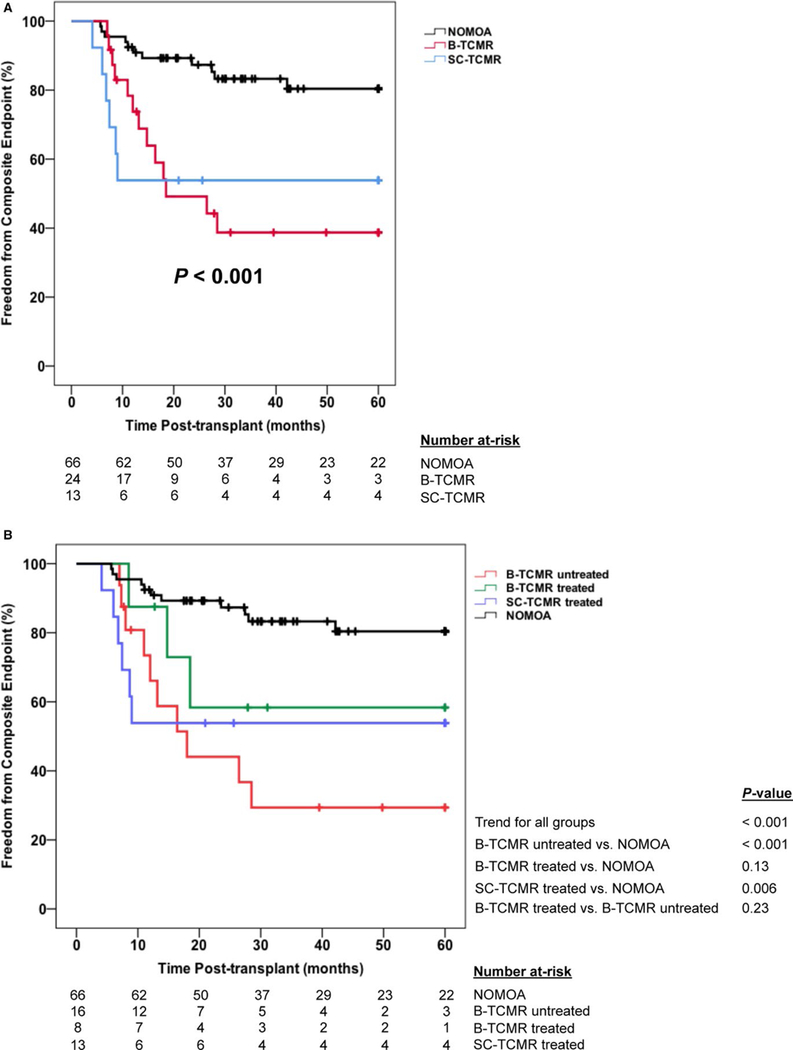
Kaplan-Meier plots of the 5-year incidence of the primary composite endpoint between the borderline and subclinical TCMR subgroups and the no major abnormalities group. (A) Five-year incidence of acute rejection after surveillance and graft loss between groups. (B) Five-year incidence of acute rejection after surveillance and graft loss between treated B-TCMR (n = 8), untreated B-TCMR (n = 16), SC-TCMR (all were treated, n = 13), and NOMOA (n = 66) groups. Comparisons of time-to-event data between groups were made using the log-rank test. In (B), the P-values for the overall trend plus key between-group comparisons are presented. B-TCMR, borderline T cell–mediated rejection; NOMOA, no major abnormalities; SC-TCMR, subclinical T cell–mediated rejection
TABLE 3.
Cox proportional hazards model of the primary composite endpoint
| Parameter | Univariable HR (95% CI) | P-value | Multivariable HR (95% CI) | P-value |
|---|---|---|---|---|
| Subclinical Inflammation (vs NOMOA) | 4.13 (1.96, 8.72) | <.001 | 2.89 (1.27, 6.57) | .01 |
| DSA during year-1 posttransplant (vs no DSA) | 3.87 (1.60, 9.35) | .003 | 3.14 (1.17, 8.39) | .02 |
| Total HLA mismatch (per no. out of 10 possible) | 1.18 (0.83, 1.68) | .37 | ||
| Nonadherence <6 mo posttransplant (yes vs no) | 2.04 (0.87, 4.77) | .10 | 2.34 (0.93, 5.89) | .07 |
| Age at transplant (per year) | 1.01 (0.94, 1.08) | .85 | ||
| Cold ischemia time (per hour) | 1.00 (0.99, 1.00) | .66 | ||
| Black race (vs nonblack race) | 2.43 (1.16, 5.10) | .02 | 1.88 (0.72, 4.90) | .20 |
| Deceased donor (vs living donor) | 2.56 (0.98, 6.70) | .06 | 0.75 (0.18, 3.11) | .69 |
| Male sex (vs female sex) | 0.78 (0.37, 1.64) | .52 |
The primary exposure was subclinical inflammation, defined as either borderline or subclinical T cell–mediated rejection. Covariates that were significantly associated with the primary endpoint at P < .10 by univariable analysis were entered into a multivariable model using forced entry. The final model was significant at P < .001, df = 6, χ2 = 29.59.
CI, confidence interval; DSA, donor-specific antibodies; HR, hazard ratio; NOMOA, no major abnormalities.
3.4 |. Outcomes by treatment of subclinical inflammation
We further explored outcomes by treatment of subclinical inflammation phenotypes. All subjects with SC-TCMR were treated with increased immunosuppression, typically intravenous pulse corticosteroids. However, one-third of those with B-TCMR were similarly treated with increased immunosuppression and two-thirds were observed without changes to immunosuppression (untreated B-TCMR). In Kaplan-Meier analyses, untreated B-TCMR and treated SC-TCMR had a significantly higher incidence of the composite endpoint compared to NOMOA, whereas treated B-TCMR did not (Figure 3B).
3.5 |. Outcomes by subclinical vascular injury
We built a second Cox model to test for associations between Banff injury scores in surveillance biopsies and the 5-year composite endpoint, using the highest Banff score for each lesion in those who had biopsies at 3 and 6 months. Only peritubular capillaritis (ptc) and arteritis (v) scores were associated with increased hazard for the composite endpoint, indicating that subclinical vascular injury had a significant impact on the composite endpoint in our cohort (Table S2).
Based on these results, we then reclassified each subject based on early subclinical vascular injury (v score > 0 or = 0). We focused on the Banff v score since the ptc score had a much less precise point estimate in our multivariable Cox models (Table S2). Specifically, each subject was categorized as having subclinical inflammation with v > 0, subclinical inflammation with v = 0, or NOMOA. Subjects with subclinical vascular injury were younger and more likely to have C4d+ staining of peritubular capillaries in surveillance biopsies (Table S3), but none met Banff diagnostic criteria for SC-ABMR. Those with subclinical vascular injury had the worst outcomes in our cohort, as nearly 80% reached the 5-year composite endpoint. Time-to-event analyses confirmed that the incidence of acute rejection and allograft failure after surveillance were significantly greater in subclinical vascular injury compared to subclinical inflammation (v = 0) and NOMOA (Figure 4). Furthermore, all episodes of subsequent clinical acute rejection in the subclinical vascular injury group occurred less than 1 year posttransplant, and all episodes of allograft failure occurred <2 years posttransplant. Taken together, these data show that subclinical vascular injury is an important, albeit relatively infrequent, surveillance phenotype in pediatric kidney recipients that is resistant to treatment.
FIGURE 4.
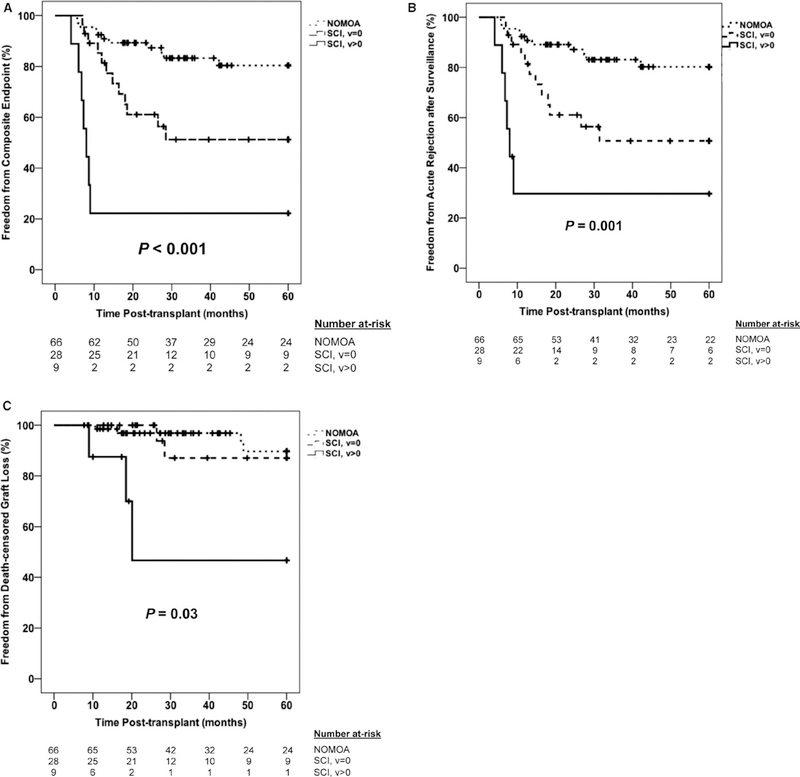
Kaplan-Meier plots of 5-year incidence of the primary composite endpoint (A), the acute rejection after surveillance component (B), and the death-censored graft failure component (C) between subjects with subclinical inflammation (SCI) and v > 0, SCI and v = 0, and no major abnormalities (NOMOA). Comparisons of time-to-event data between groups were made using the log-rank test
4 |. DISCUSSION
This represents one of the largest pediatric studies to investigate 5-year outcomes across a variety of subclinical inflammation phenotypes. Our major findings were the following: (1) subclinical inflammation, either B-TCMR or SC-TCMR, was detected in 36% of pediatric kidney transplant recipients by 6 months; (2) subclinical inflammation was associated with increased risk for acute rejection and graft failure by 5 years; and (3) among patients with subclinical inflammation, those with untreated B-TCMR and vascular injury had the worst outcomes.
Our rate of subclinical inflammation was higher than in recent pediatric surveillance studies, which found B-TCMR and SC-TCMR in 19%−29% of subjects.11,13,26 These differences may reflect important population differences in demographics and immunologic risk. Birk et al studied 21 children with serial surveillance biopsies over 3 years, with fewer African-American and deceased donor recipients than our study. They did not increase immunosuppression for B-TCMR.13 Hymes et al evaluated 89 children with 3-month surveillance biopsies. They had similar proportions of subclinical vascular injury compared to our cohort, but did not report differential outcomes by subclinical phenotypes.11,27 They detected more subsequent acute rejection overall, despite treating all B-TCMR cases with pulse intravenous methylprednisolone. This difference from our study may be attributable to their conversion from tacrolimus to sirolimus and a lower maintenance MMF dose in patients with NOMOA.28–30 Finally, the steroid-based arm (n = 70) of the multicenter SNS01-NIH-CCTPT study found a 35% lower rate of subclinical inflammation in 6-month surveillance biopsies, but was also less racially diverse than our study.26 Our data from a diverse pediatric cohort provide a significant addition to the existing literature on outcomes after surveillance biopsies.
Our improved outcomes with treatment of B-TCMR are interesting when placed in context with recent adult surveillance studies. Mehta et al showed that B-TCMR at 3 months was associated with reduced eGFR and increased incidence of de novo DSA by 1 year, as well as increased clinical acute rejection after surveillance.15 However, they used no maintenance prednisone, set a lower MFI cutoff for identifying DSA, and did not increase immunosuppression for B-TCMR. Our B-TCMR cases treated with increased immunosuppression had outcomes similar to NOMOA by Kaplan-Meier analysis, although this was an exploratory analysis with small sample sizes in these subgroups. These findings should be validated in a comparative effectiveness study of treatment strategies for B-TCMR. Collectively, our studies identify B-TCMR as a significant surveillance finding that is perhaps equally hazardous as SC-TCMR, with a negative impact on kidney transplant outcomes that may benefit from treatment.
We found that subclinical vascular injury was associated with the highest rates of subsequent clinical acute rejection and allograft failure by 5 years, despite the fact that all cases were treated with increased immunosuppression. Although more subjects with subclinical vascular injury had C4d+ peritubular capillaries and subsequent DSA during year-1 posttransplant, they did not have significant glomerulitis or peritubular capillaritis, and their DSA status was unknown at the time of biopsy (see Materials and Methods). It is unknown whether additional therapies targeting humoral immunity would have improved outcomes in this high-risk subgroup. In addition, many of these subclinical vascular injury cases were diagnosed prior to the Banff 2013 update, which first recognized C4d-negative ABMR as a pathological entity and included all grades of arteritis (v > 0) as diagnostic for ABMR.22 Therefore, it is possible that all our subclinical vascular injury cases actually represent existing high-risk phenotypes such as SC-ABMR or subclinical mixed rejection, which have been associated with an over 4-fold risk of allograft failure in recent studies.14 Collectively, our data indicate that subclinical vascular injury is prevalent in pediatric surveillance biopsies despite the absence of clinical dysfunction, is resistant to treatment, and is associated with increased acute rejection and graft failure after surveillance. Prospective surveillance studies are needed with universal assessment of C4d staining, thoroughly validated endothelial injury transcripts, and concurrent de novo DSA testing (or non-HLA antibody testing)31, 32 in the context of updated Banff criteria in order to properly characterize subclinical vascular injury and determine the optimal management of this important surveillance phenotype.
Our study had several strengths, which include the use of a relatively large single-center pediatric cohort of consecutive kidney recipients treated with the same modern immunosuppression regimen. Our cohort was racially diverse compared to previous studies, which not only enriched our cohort for immunologic risk but also allowed multivariable adjustment of our results for race. Unlike previous surveillance studies, we were able to demonstrate the benefit of treatment vs nontreatment of B-TCMR on long-term outcomes within a single study.
Despite these strengths, our study had limitations common to retrospective cohort studies, including a lack of randomization of treatment of B-TCMR, limited assessment of DSA at the time of surveillance, and dated Banff diagnostic criteria. A similar retrospective analysis of treatment at the clinician’s discretion has been used successfully in a recent subclinical pathology study.19 We added analysis of pathology by a third individual blinded to the clinical status of each case and had remarkable concurrence with clinical reads from the other 2 pathologists without substantial revisions to any original clinical diagnoses (data not shown). We also recognize that our prespecified composite endpoint was unbalanced, in that acute rejection after surveillance and allograft failure rates for the cohort were 49% and 14%, respectively. We presented Kaplan-Meier plots of each component to illustrate their relative contributions to the composite endpoint, and to align with recent US Food and Drug Administration (FDA) guidance on multiple endpoints.33 However, we concede that conclusions regarding the composite endpoint might be influenced by the relatively high rate of acute rejection after surveillance compared to allograft failure. There may have been selection bias in how clinicians chose to treat or not treat B-TCMR. Our multivariable Cox models included prespecified exposures but likely excluded some potential confounders. We also recognize that although our study cohort was relatively large compared to published pediatric studies, it was still small compared to larger adult studies on similar topics.14 Our small sample size may limit the interpretation of exploratory analyses involving treatment of subclinical inflammation and outcomes after subclinical vascular injury.
In summary, we demonstrated that B-TCMR, SC-TCMR, and subclinical vascular injury were prevalent in pediatric kidney transplant recipients without clinical dysfunction, and that these phenotypes were associated with increased acute rejection and allograft failure after surveillance. Moreover, our data suggest that treating children with subclinical B-TCMR may improve their risk for subsequent acute rejection and allograft failure. Paired with a low rate of adverse events, our data indicate that surveillance biopsies can safely and effectively diagnose subclinical inflammation phenotypes in children and are a potentially valuable tool for monitoring early allograft injury. Given the current low rate of surveillance biopsy performance nationally, once our data are prospectively validated they would support a paradigm shift to perform surveillance biopsies as standard of care in pediatric kidney transplantation.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank Dr. Miguel Melendez-Ferro for critical review of the manuscript and Dr. Gary Cutter for biostatistical guidance. This study was supported by K23 DK101690 and the Angus Cooper Award in Transplant Investigation from the UAB Comprehensive Transplant Institute (both to M.E.S.). These data were originally presented in abstract form at the 2016 American Transplant Congress in Boston, MA.
Funding information
National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: K23DK101690 and L40DK099748
Abbreviations:
- B-TCMR
borderline T cell–mediated rejection
- CADI
chronic allograft disease index
- DSA
donor-specific antibody
- eGFR
estimated glomerular filtration rate
- ESRD
end-stage renal disease
- IRB
institutional review board
- MFI
mean fluorescence intensity
- MMF
mycophenolate mofetil
- NOMOA
no major surveillance abnormalities
- SC-ABMR
subclinical antibody-mediated rejection
- SC-TCMR
subclinical T cell–mediated rejection
Footnotes
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Lodhi SA, Lamb KE, Meier-Kriesche HU. Solid organ allograft survival improvement in the United States: the long-term does not mirror the dramatic short-term success. Am J Transplant 2011;11(6):1226–1235. [DOI] [PubMed] [Google Scholar]
- 2.Meier-Kriesche HU, Schold JD, Srinivas TR, Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant 2004;4(3):378–383. [DOI] [PubMed] [Google Scholar]
- 3.El-Zoghby ZM, Stegall MD, Lager DJ, et al. Identifying specific causes of kidney allograft loss. Am J Transplant 2009;9(3):527–535. [DOI] [PubMed] [Google Scholar]
- 4.Famulski KS, Reeve J, de Freitas DG, Kreepala C, Chang J, Halloran PF. Kidney transplants with progressing chronic diseases express high levels of acute kidney injury transcripts. Am J Transplant 2013;13(3):634–644. [DOI] [PubMed] [Google Scholar]
- 5.Walsh RC, Brailey P, Girnita A, et al. Early and late acute antibody-mediated rejection differ immunologically and in response to proteasome inhibition. Transplantation 2011;91(11):1218–1226. [DOI] [PubMed] [Google Scholar]
- 6.de Souza V, Cochat P, Rabilloud M, et al. Accuracy of different equations in estimating GFR in pediatric kidney transplant recipients. Clin J Am Soc Nephrol 2015;10(3):463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birk PE. Surveillance biopsies in children post-kidney transplant. Pediatr Nephrol 2012;27(5):753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosio FG, Grande JP, Wadei H, Larson TS, Griffin MD, Stegall MD. Predicting subsequent decline in kidney allograft function from early surveillance biopsies. Am J Transplant 2005;5(10):2464–2472. [DOI] [PubMed] [Google Scholar]
- 9.Mehta R, Cherikh W, Sood P, Hariharan S. Kidney allograft surveillance biopsy practices across US transplant centers: a UNOS survey. Clin Transplant 2017;31:e12945 10.1111/ctr.12945 [DOI] [PubMed] [Google Scholar]
- 10.Gonzales MM, Bentall A, Kremers WK, Stegall MD, Borrows R. Predicting individual renal allograft outcomes using risk models with 1-year surveillance biopsy and alloantibody data. J Am Soc Nephrol 2016;27(10):3165–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hymes LC, Warshaw BL, Hennigar RA, Amaral SG, Greenbaum LA. Prevalence of clinical rejection after surveillance biopsies in pediatric renal transplants: does early subclinical rejection predispose to subsequent rejection episodes? Pediatr Transplant 2009;13(7):823–826. [DOI] [PubMed] [Google Scholar]
- 12.Harmon W, Meyers K, Ingelfinger J, et al. Safety and efficacy of a calcineurin inhibitor avoidance regimen in pediatric renal transplantation. J Am Soc Nephrol 2006;17(6):1735–1745. [DOI] [PubMed] [Google Scholar]
- 13.Birk PE, Stannard KM, Konrad HB, et al. Surveillance biopsies are superior to functional studies for the diagnosis of acute and chronic renal allograft pathology in children. Pediatr Transplant 2004;8(1):29–38. [DOI] [PubMed] [Google Scholar]
- 14.Loupy A, Vernerey D, Tinel C, et al. Subclinical rejection phenotypes at 1 year post-transplant and outcome of kidney allografts. J Am Soc Nephrol 2015;26(7):1721–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehta R, Bhusal S, Randhawa P, et al. Short-term adverse effects of early subclinical allograft inflammation in kidney transplant recipients with a rapid steroid withdrawal protocol [published online ahead of print 2018]. Am J Transplant 2018;00:1–8. 10.1111/ajt.14627 [DOI] [PubMed] [Google Scholar]
- 16.Gloor JM, Cohen AJ, Lager DJ, et al. Subclinical rejection in tacrolimus-treated renal transplant recipients. Transplantation 2002;73(12):1965–1968. [DOI] [PubMed] [Google Scholar]
- 17.Mehta R, Sood P, Hariharan S. Subclinical rejection in renal transplantation: reappraised. Transplantation 2016;100(8):1610–1618. [DOI] [PubMed] [Google Scholar]
- 18.Dharnidharka VR, Fiorina P, Harmon WE. Kidney transplantation in children. N Engl J Med 2014;371(6):549–558. [DOI] [PubMed] [Google Scholar]
- 19.Orandi BJ, Chow EH, Hsu A, et al. Quantifying renal allograft loss following early antibody-mediated rejection. Am J Transplant 2015;15(2):489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz GJ, Schneider MF, Maier PS, et al. Improved equations estimating GFR in children with chronic kidney disease using an immunonephelometric determination of cystatin C. Kidney Int 2012;82(4):445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150(9):604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas M, Sis B, Racusen LC, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant 2014;14(2):272–283. [DOI] [PubMed] [Google Scholar]
- 23.Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant 2008;8(4):753–760. [DOI] [PubMed] [Google Scholar]
- 24.Reed EF, Rao P, Zhang Z, et al. Comprehensive assessment and standardization of solid phase multiplex-bead arrays for the detection of antibodies to HLA. Am J Transplant 2013;13(7):1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tait BD, Susal C, Gebel HM, et al. Consensus guidelines on the testing and clinical management issues associated with HLA and non-HLA antibodies in transplantation. Transplantation 2013;95(1):19–47. [DOI] [PubMed] [Google Scholar]
- 26.Naesens M, Salvatierra O, Benfield M, Ettenger RB, Dharnidharka V, Harmon W, et al. Subclinical inflammation and chronic renal allograft injury in a randomized trial on steroid avoidance in pediatric kidney transplantation. Am J Transplant 2012;12(10):2730–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hymes LC, Greenbaum L, Amaral SG, Warshaw BL. Surveillance renal transplant biopsies and subclinical rejection at three months post-transplant in pediatric recipients. Pediatr Transplant 2007;11(5):536–539. [DOI] [PubMed] [Google Scholar]
- 28.Knechtle SJ, Pascual J, Bloom DD, et al. Early and limited use of tacrolimus to avoid rejection in an alemtuzumab and sirolimus regimen for kidney transplantation: clinical results and immune monitoring. Am J Transplant 2009;9(5):1087–1098. [DOI] [PubMed] [Google Scholar]
- 29.Filler G, Todorova EK, Bax K, Alvarez-Elias AC, Huang SH, Kobrzynski MC. Minimum mycophenolic acid levels are associated with donor-specific antibody formation. Pediatr Transplant 2016;20(1):34–38. [DOI] [PubMed] [Google Scholar]
- 30.Torres IB, Reisaeter AV, Moreso F, et al. Tacrolimus and mycophenolate regimen and subclinical tubulo-interstitial inflammation in low immunological risk renal transplants. Transpl Int 2017;30(11):1119–1131. [DOI] [PubMed] [Google Scholar]
- 31.Pearl MH, Zhang Q, Palma Diaz MF, et al. Angiotensin II Type 1 receptor antibodies are associated with inflammatory cytokines and poor clinical outcomes in pediatric kidney transplantation. Kidney Int 2018;93(1):260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seifert ME, Gunasekaran M, Horwedel TA, et al. Polyomavirus reactivation and immune responses to kidney-specific self-antigens in transplantation. J Am Soc Nephrol 2017;28(4):1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.U.S. Department of Health and Human Services Food and Drug Administration. Multiple Endpoints in Clinical Trials: Guidance for Industry https://www.fda.gov/downloads/drugs/guidance-complianceregulatoryinformation/guidances/ucm536750.pdf. Accessed on April 1, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.