

Abstract
The termite gut microbiome is a model system to investigate microbial interactions and their associations with host. For decades, extensive research with molecular tools and conventional cultivation method has been carried out to define the microbial diversity in termite gut. Yet, many bacterial groups of the termite gut microbiome have not been successfully cultivated in laboratory. In this study, we adapted the recently developed microfluidic streak plate (MSP) technique for cultivation of termite gut microbial communities at both aerobic and anaerobic conditions. We found that 99 operational taxonomic units (OTUs) were cultivable by MSP approach and 18 OTUs were documented first time for termite gut microbiota. Further analysis of the bacterial diversities derived by culture‐dependent MSP approach and culture‐independent 16S rRNA gene typing revealed that both methods have bias in recovery of gut microbiota. In total 396 strains were isolated with MSP technique, and potential new taxa at species and/or genus levels were obtained that were phylogenetically related to Burkholderia, Micrococcus, and Dysgonomonas. Results from this study indicate that MSP technique is applicable for cultivating previously unknown and new microbial groups of termite gut microbiota.
Keywords: bacterial diversity, cultivation, gut microbiome, microfluidic streak plate (MSP), Reticulitermes chinensis
1. INTRODUCTION
The gut of termite harbors a dense and diverse microbiota of approximately 106–8 bacterial cells (Breznak, 1982, 2000). This microbiota and their symbiosis with host are essential for the efficient digestion of lignocellulose in termite gut (Brune & Dietrich, 2015; Ohkuma, 2003; Warnecke et al., 2007). For several decades, the gut microbiome of termites has been attracting interest from microbiologists and biotechnologists (Breznak, 1982; Brune & Friedrich, 2000; Ohkuma & Kudo, 1996), since termite gut microbiome not only plays important roles in carbon turnover in the environment but also is potential sources of biochemical catalysts converting wood into biofuels (Warnecke et al., 2007). Wood‐feeding termites can digest up to 83%–85% of glucosyl and xylosyl residues from lignocellulose (Bignell, 2011). Termite gut microbiomes have been exploited for production of carboxylates from low‐value biomass (Ali et al., 2017; Auer et al., 2017; Ni & Tokuda, 2013; Watanabe & Tokuda, 2010) as well as to discover commercially important enzymes (Cibichakravarthy, Abinaya, & Prabagaran, 2017; Liu et al., 2011; Martin & Martin, 1978; Matsuura, Yashiro, Shimizu, Tatsumi, & Tamura, 2009). Culture‐independent 16S rRNA gene typing and metagenomic tools have been extensively used for description of the termite gut microbial community (Huang, Bakker, Judd, Reardon, & Vivanco, 2013; Ohkuma & Brune, 2011; Tarayre et al., 2015).
Compared to culture‐independent methods, the culture‐dependent method would better serve the purpose to investigate host‐microbe interaction or to recover valuable microbial products (including commercial enzymes) (Keller & Zengler, 2004; Stewart, 2012). However, cultivation of microbes from various samples including termite gut is often hindered as many microbes in nature are resistant to be cultivated in laboratory conditions (Amann, Ludwig, & Schleifer, 1995; Hongoh, 2011; Ohkuma & Brune, 2011). To overcome this obstacle and to cultivate as yet not cultivated microorganisms in laboratory, techniques of high throughput and mimic natural conditions have been developed, such as the high‐throughput culturing procedures that utilize the concept of extinction culturing (Colin, Goñiurriza, Caumette, & Guyoneaud, 2013; Colin, Goñi‐Urriza, Caumette, & Guyoneaud, 2015; Connon & Giovannoni, 2002), the microencapsulation (Keller & Zengler, 2004; Zhou, Liu, Liu, Ma, & Su, 2008) and the isolation chip (Ichip) (Nichols et al., 2010). Microfluidic devices (Ma et al., 2014; Park, Kerner, Burns, & Lin, 2011; Tandogan, Abadian, Epstein, Aoi, & Goluch, 2014) were also developed for highly parallel cocultivation of symbiotic microbial communities and isolating pure bacterial cultures from samples containing multiple species. The microfluidic streak plate (MSP) technique (Jiang et al., 2016) exploits the advantages of microfluidics to manipulate tiny volume of liquid at several to hundred nanoliters and generate microdroplets for microbial single‐cell isolation and cultivation. Superior to the conventional agar plate cultivation, the MSP approach enabled higher throughput of bacterial isolation and better coverage of rare species in community (Jiang et al., 2016).
Reticulitermes chinensis (Snyder) (Isoptera: Rhinotermitidae) is wood‐feeding lower termite. In this study, we continued our efforts to cultivate microbes from the gut of from this termite (Chen, Wang, Hong, Yang, & Liu, 2012; Fang, Lv, Huang, Liu, & Yang, 2015; Fang et al., 2016), and adapted the MSP technique for cultivation of gut microbiome at both aerobic and anoxic conditions. With the MSP method, 99 OTUs representing Proteobacteria, Firmicutes, Actinobacteria, Bacteriodetes, Acidobacteria, and Verrucomicrobia were obtained, and 396 bacterial isolates were successfully cultivated in pure cultures. Our results demonstrated that MSP method significantly increased the recovery of various microbial groups and many of them were documented for the first time from termite gut.
2. MATERIALS AND METHODS
2.1. Termite cultivation and retrieving gut microbiota
The termite Reticulitermes chinensis colonies were collected and transferred to laboratory, and were maintained in glass containers on a diet of pinewood and water. Only worker termites were used in this study. The termite's surface was washed three times with 70% ethanol, rinsed with distilled water and blotted dry on sterilized filter papers. The guts from 40 termites were removed aseptically with fine‐tipped forceps onto a sterilized glass slide and the gut microbiota were squeezed out of the guts and were transferred into a tube with 1mL of PBS buffer (PBS buffer, g/L: NaCl, 8.00; KCl, 0.20; Na2HPO4.12H2O, 3.58; KH2PO4, 0.24; pH 7.2). The gut microbiota suspension in the PBS buffer was used subsequently for cell separation and cultivation.
2.2. Operation of microfluidic droplet arrays
Microfluidic streak plate (MSP) was operated according to previously described (Jiang et al., 2016), except that the automated dish driver and the microfluidic device were setup in an anaerobic chamber (ThermoScientific 1029). Droplets were arrayed onto surface‐modified Petri‐dish (Jiang et al., 2016), and about 3000 droplets were displayed onto the surface of 9‐cm Petri‐dish.
2.3. Dilution of gut microbiota samples and cultivation of microbes
Fivefold‐diluted (1/5) R2A medium (1/5 R2A, g/L: Yeast extract, 0.1; Peptone, 0.1; Casamino Acids, 0.1; Glucose, 0.1; Soluble starch, 0.1; Sodium pyruvate, 0.1; K2HPO4, 0.75; KH2PO4, 0.75; MgSO4·7H2O, 0.2; pH 7.2) was used as growth broth and for dilution of gut microbiota samples. In order to prepare samples for MSP, the gut microbiota suspension (see M&M section 1) was diluted with growth broth, either directly from the suspension or after three times washing with Cysteine‐reduced (1 g/L) PBS buffer (pH 7.2). The final concentration of diluted gut microbiota suspension was approximately 1 × 104–5 cells/ml. This diluted suspension was used for separation and cultivation of the gut microbiota with the MSP method. Petri dishes with droplet arrays were incubated at 30°C under both aerobic and anaerobic condition. After 72 hr incubation, the droplets were individually transferred into 96‐well cell‐culture plates, each well contained 80 μL of 1/5 R2A medium. After another 72 hr of cultivation at 30°C, the growth of bacterial cells was monitored with a Microplate reader (Biotek SynergyHT). The grown cells were streaked on R2A agar plates, and all bacterial strains obtained were stored at 10°C in cold room until further tests.
2.4. Total DNA extraction, amplification of 16S rRNA genes, and DNA sequencing
Cells of termite gut samples and from MSP droplet arrays were collected by centrifugation. Metagenomic DNA was extracted with E.Z.N.A Meg‐Bind Soil DNA Kit (Omega Bio‐tek, GA, USA) using a KingFisher Flex Magnetic Particle Processor (Thermo Scientific, MA, USA). Extractions were performed according to Kit and instrument protocols. Purified DNA were used for 16S rRNA gene amplification with the PCR primers (targeted the V4 region) U515F (5′‐GTGCCAGCMGCCGCGGTAA‐3′) and 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′) containing barcodes at the 5′ end of the front primer (Werner, Zhou, Caporaso, Knight, & Angenent, 2012). PCR reactions were proceeded in 50 μL volumes, each containing 1.5 μL of 10 μM forward and reverse primers, respectively, 25 μL of 2× KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Inc., MA, USA), and up to 22 μL of purified DNA as template. The thermocycling was performed as follows: 30 cycles (98°C, 20 s; 54°C, 15 s; 72°C, 15 s) after an initial denaturation at 95°C for three min, following a final extension at 72°C for 60 s. Triplicate PCR products for each sample were purified using E.Z.N.A Gel Extraction Kit (Omega Bio‐Tek, Inc.) and then quantified using Qubit dsDNA HS Assay Kit (Invitrogen, CA, USA). Equal amounts of PCR products were mixed to produce equivalent sequencing depth from all samples. After purification using Agencount AMPure XP KIT, the pooled‐PCR products were used to construct a DNA library using NEB E7370L DNA Library Preparation Kit. The libraries were sequenced on an Illumina MiSeq 2500 platform at BGI GENE (Wuhan, China). Complete data with 250 bp reads had been submitted to the NCBI Short Read Archive database under accession No. SRP133587
The full length of 16S rRNA gene from each bacterial strain obtained in this study was amplified with the 27F and 1492R primers (Edwards, Rogall, Blöcker, Emde, & Böttger, 1989; Weisburg, Bars, Pelletier, & Lane, 1991). The 16S rRNA gene sequences of the isolates in this study have been deposited in GenBank databases under the accession numbers MG984070‐MG984092.
2.5. 16S rRNA gene‐based metagenomic analysis and phylogenetic tree construction
The raw sequences were assigned to individual samples by their unique barcodes. The 16S rDNA primers and barcodes were then removed to generate pair‐end (PE) reads. Raw tags were then generated by merging PE reads with FLASH (Magoč & Salzberg, 2011), the raw tags were then filtered and analyzed using QIIME software package (Quantitative Insights Into Microbial Ecology) (Bokulich et al., 2013). Reads from all samples were quality filtered using an average quality value of 20 (Q20) during demultiplexing, sequences with a mean quality score 20 were excluded from analysis, and chimeras were also excluded. For species analysis, 16S rRNA sequences with ≥97% similarity were assigned to the same OTUs using Uparse v7.0.1001 (Edgar, 2013), and similarity hits below 97% were not considered for classification purpose. A representative sequence of each OTU was picked out and the taxonomic information was annotated using RDP classifier (version 2.2) (Wang, Garrity, Tiedje, & Cole, 2007) and GreenGene database (Desantis et al., 2006). Sequences obtained were compared with the published sequences in GenBank using Blast from NCBI (http://www.ncbi.nlm.nih.gov/BLAST).
The 16S rRNA sequences of all the published termite‐gut‐derived bacteria were mined from NCBI. The OTU sequences of MSP pool sample were blasted with the GenBank of NCBI and the 16S rRNA sequences of type species with the highest similarity to our OTUs were selected. Those sequences together with the extracted termite‐gut‐derived bacterial 16S rRNA gene sequences were used for the construction of phylogenetic tree. The OTUs from MSP pool samples (accession numbers MH152413‐MH152511), the 16S rRNA gene sequences of isolated strains (accession numbers MG984070‐MG984092) and the reference sequences (the accession number was available in phylogenetic tree) were aligned using ClustalW (Thompson, Gibson, & Higgins, 2002). Phylogenetic trees were constructed with MEGA6 package based on the alignments of sequences using Neighbor‐joining method with p‐distance. Bootstrap analysis with 1000 replicates was performed to determine the statistical significance of the branching order.
3. RESULTS
3.1. Termite gut microbial community revealed with MSP technique and comparison to metagenomic method
We sequenced both the partial 16S RNA gene of the original microbiota from gut sample (hereafter called OMG sample) and DNA extracted from the pooled droplets from cultured MSP plates (hereafter called MSP pool). A total of 38,056 and 37,137 Pair‐end reads were retrieved, and after filtering and removing potential erroneous sequences, a total of 28,422 and 29,778 effective tags were obtained from OMG sample and MSP pool, respectively. These sequences represented 58,200 taxon tags that covered 141 genera, 102 families, 57 orders, or 33 classes of 15 phyla. As shown in Figure 1a, the rarefaction curves of OMG and MSP pool reached plateau after 10,000 and 5000 sequences per sample, respectively, indicating that the sequencing depth was adequate to reflect the bacterial diversity in both samples. Data analysis showed that OMG sample had much higher OTU richness than the MSP samples, At the phylum level, the relative abundances of five phyla in OMG samples and two phyla in MSP pool sample were higher than 1% (Figure 1b, for details please see Tables S1, S2 and S3). To be specific, Spirochaetes (44.3%), Proteobacteria (14.7%), Firmicutes (13.9%), Elusimicrobia (13.8%), and Bacteroidetes (10.0%) were the top five phyla in the OMG sample, whereas Proteobacteria (69.9%) and Firmicutes (29.2%), were the top two phyla in the MSP pool sample. We found that six phyla (Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, Planctomycetes, and Verrucomicrobia) presented in both OMG sample and MSP pool, suggesting members of those phyla were culturable with the MSP technique when the 1/5 R2A medium was used. Furthermore, Proteobacteria and Firmicutes were among the dominant phyla in both OMG sample and MSP pool, indicating they were well represented in the MSP pool. Significant differences were also observed: the phyla of Acidobacteria, Fusobacteria, Nitrospirae, and Thermi were only observed with MSP pool, whereas the phyla of Spirochaetes, Elusimicrobia, Synergistetes, Tenericutes, and ZB3 were only observed with OMG sample. When analyzed at Family level (Figure 1b), 19 of the total 102 families were found in both OMG sample and MSP pool and they accounted for 31.1% of the total taxon tags. The Spirochaetaceae (44.3%), Endomicrobiacea (13.8%), Porphyromonadaceae (7.5%), Rhodocyclaceae (4.5%), and Lachnospiraceae (3.8%) were the dominant families of OMG sample, whereas the Enterobacteriaceae (33.6%), Staphylococcaceae (27.4%), and Sphingomonadaceae (22.8%), Alcaligenaceae (9.7%)were the dominant families in MSP pool (Figure 1c).
Figure 1.
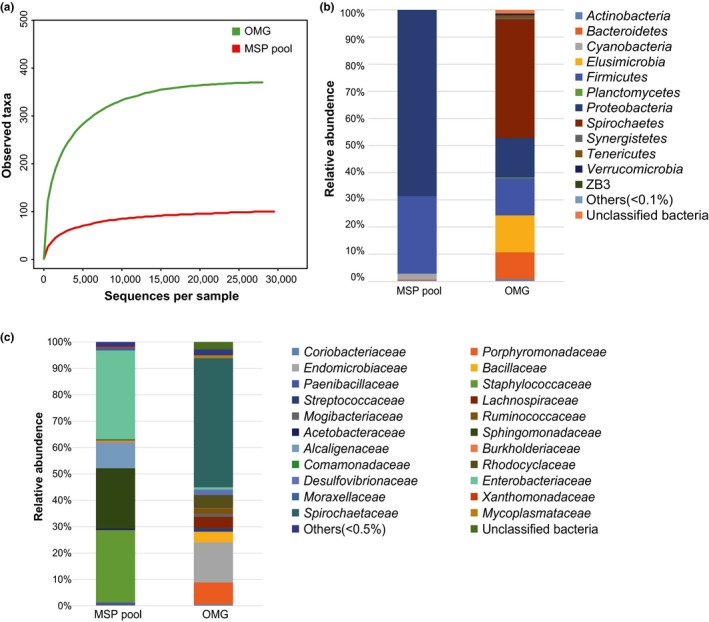
Rarefaction curves of 16S rDNA sequences of the samples (a), the relative abundances of the dominant Phylum in all samples indicated and the rest being labeled as “Others” (b) and the relative abundances of the dominant Families in all samples (c). Curves were calculated based on OTUs at 97% similarity
3.2. Identification of yet‐to‐be cultured microbial OTUs/taxa from MSP pool
With a cutting edge of 97% sequence similarity, 99 and 353 OTUs from MSP pool and OMG sample, respectively, were recognized. Venn diagram showed that OMG and MSP shared 24 OTUs, but more OTUs were uniquely in either MSP pool or OMG sample (Figure 2). This is one more example representing that the microbial diversities was differentially reflected with culture‐dependent and ‐independent methods, which is generally acknowledged for that none of the current tools is able to disclose the whole picture of microbial diversity in environments (Lagier et al., 2012; Rettedal, Gumpert, & Sommer, 2014; Sommer, 2015).
Figure 2.
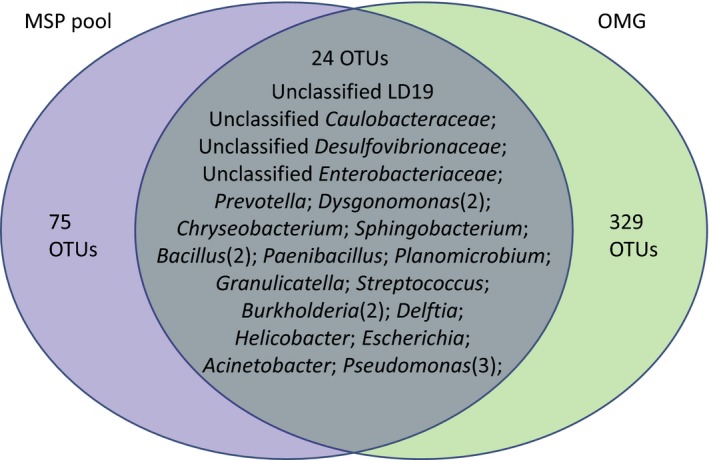
Venn diagram of OTUs in the two samples. Unique and shared OTUs in the two samples are based on 97% similarity. The numbers inside the diagram indicate the numbers of OTUs
Phylogenetic trees were constructed based on the 99 OTUs from MSP pool (Figure 3a–e). Meanwhile, our data mining of public databases (Ribosomal Database Project, GreenGenes database, GenBank) revealed that 81 of the 99 OTUs (Figure 3, asterisk), representing 55 bacterial genera, had been well cultivated. But there were still 18 of the 99 OTUs, which had been previously not detected and not cultured (Figure 3, solid circle). The detection of these 18 OTUs in MSP pool indicated that they could grow in 1/5 R2A broth with MSP method. Indeed, we isolated and cultivated 396 bacterial strains with MSP method, and these strains covered 9.1% of the OTUs from MSP pool (Figure 3a–e). The identification of these bacterial strains is discussed in the following sections.
Figure 3.
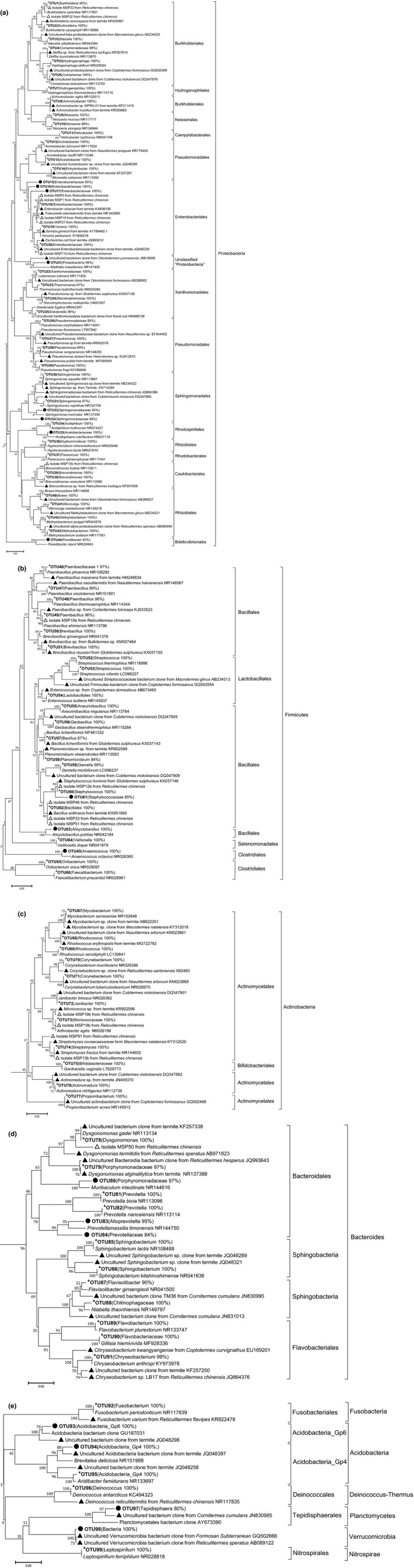
Phylogenetic trees of 99 OTUs from MSP pool. (a) Proteobacteria; (b) Firmicutes; (c) Actinobacteria; (d) Bacteroides; (e) OTUs belonging to other Phyla as indicated. The 16S rRNA sequences of all the published termite‐gut‐derived bacteria were mined from NCBI. The OTU sequences of MSP pool sample were blasted with the GenBank of NCBI and the 16S rRNA sequences of type species with the highest similarity to our OTUs were selected, together with previous mined termite gut derived bacterial 16s rRNA sequences, as reference sequences in phylogenetic tree. Tree viewing was inferred by the Neighbor‐joining method of Mega 6 based on the 16S rRNA gene sequences. OTUs obtained from this MSP pool were showed in bold. The most likely taxon category and its confidence value was listed in the bracket behind each OTU, the confidence threshold was set to be ≥80%. Symbols: ▲: Terminate sequence obtained from GenBank, accession numbers are shown at the end; ●: Sequence with 16S rRNA gene similarity lower than 97% compared with the other isolates in the environments; △: Strains isolated in this study, *: Cultivable taxa.
3.3. Isolation and cultivation of members from Proteobacteria
Analysis of the MSP pool data with RDP databank showed that there were 44 OTUs from Proteobacteria (Figure 3a). These 44 OTUs were assigned to 24 genera. We found that 12 of the 24 genera had been previously observed (Butera, Ferraro, Alonzo, Colazza, & Quatrini, 2016; Chou, Chen, Arun, & Young, 2007; Fall et al., 2007; Hongoh et al., 2006; Liu et al., 2013; Shinzato, Muramatsu, Matsui, & Watanabe, 2007; Thong‐On et al., 2012) but other 12 genera (Massilia, Hydrogenophaga, Hydrogenophilus, Neisseria, Helicobacter, Thermomonas, Dokdonella, Acidiphilium, Hyphomicrobium, Paracoccus, Microvirga, and Peredibacter) had not been reported previously for termite gut microbiota. We also found that there were eight of the 44 OTUs represented possible new taxa, as their 16S rRNA similarities to that of the currently known species were less than 97%. We obtained 351 isolates of Proteobacteria, from which four isolates belong to Alphaproteobacteria, 37 isolates belong to Betaproteobacteria, and 310 isolates belongs to Gammaproteobacteria. The members of family Enterobacteriaceae (307 isolates) and Burkholderiaceae (37 isolates) accounted for 98% (344/351) and they were the most abundant cultivable Proteobacteria in termite gut. As shown in Table 1, representative strains of the total isolates were good reflection of the MSP pool OTUs (See also Figure 3a), and two strains (MSP23, MSP32) that represented possible new species and/or genus were obtained.
Table 1.
Bacterial strains isolated from the gut of Reticulitermes chinensis with MSP method
| Phylogenetic affiliation | Strains | GenBank acc. No | Total isolates | Relatedness to known species |
|---|---|---|---|---|
| Proteobacteria | ||||
| Brevundimonas | MSP15 | MG984086 | 4 | Brevundimonas terrae, NR043726, 99% |
| Burkholderia | MSP23a | MG984075 | 25 | Burkholderia sacchari, NR025097, 97% |
| MSP32a | MG984074 | 12 | Burkholderia acidipaludis, NR113024, 98% | |
| Luteibacter | MSP17b | MG984089 | 2 | Luteibacter anthropi NR116911, 99% |
| Frateuria | MSP1b | MG984088 | 1 | Frateuria aurantia NR040947.1, 99% |
| Escherichia | MSP11b | MG984083 | 2 | Escherichia fergusonii MF678858, 100% |
| Citrobacter | MSP27 | MG984072 | 7 | Citrobacter farmeri KT313001, 99% |
| Trabulsiella | MSP19 | MG984073 | 16 | Trabulsiella odontotermitis NR043860, 99% |
| Enterobacter | MSP1 | MG984091 | 183 | Enterobacter amnigenus DQ481471, 99% |
| MSP6 | MG984071 | 99 | Enterobacter amnigenus DQ481471, 99% | |
| Subtotal | 10 | 351 | ||
| Firmicutes | ||||
| Bacillus | MSP33 | MG984080 | 10 | Bacillus cereus, KP694231, 100% |
| MSP51 | MG984081 | 10 | Bacillus thuringiensis, EU647704, 99% | |
| MSP46 | MG984082 | 6 | Bacillus wiedmannii, MG780249, 100% | |
| Staphylococcus | MSP12b | MG984084 | 2 | Staphylococcus epidermidis, MG725753, 99% |
| Lysinibacillus | MSP8 | MG984078 | 1 | Lysinibacillus xylanilyticus, KY038731, 99% |
| MSP58 | MG984079 | 1 | Lysinibacillus macroides, KF053268, 100% | |
| MSP14 | MG984077 | 1 | Lysinibacillus macrolides, KF053268, 99% | |
| Paenibacillus | MSP10b | MG984070 | 1 | Paenibacillus ginsengagri, AB245383, 99% |
| Subtotal | 8 | 32 | ||
| Actinobacteria | ||||
| Streptomyces | MSP91 | MG984076 | 4 | Streptomyces nigrogriseolus, KF782837, 99% |
| MSP13b | MG984085 | 3 | Streptomyces aureus, NR025663, 99% | |
| Arthrobacter | MSP18b | MG984087 | 1 | Arthrobacter cumminsii, EU086827, 99% |
| Micrococcus | MSP19ba | MG984090 | 2 | Micrococcus luteus, NR075062, 98% |
| Subtotal | 4 | 10 | ||
| Bacteroidetes | ||||
| Dysgonomonas | MSP50a | MG984092 | 3 | Dysgonomonas gadei, NR113134, 95% |
| Subtotal | 1 | 3 | ||
| Total | 23 | 396 | ||
Possible new species or genus.
Phylogenetic analysis also showed that most bacterial isolates were affiliated with several subgroups of Proteobacteria especially Gammaproteobacteria. Enterobacter strain MSP6 and MSP1 shared sequence similarity higher than 99% in 16S rRNA genes with those of the previously reported bacteria TSB7 and TSB75 (Fang et al., 2016) from R. chinensis. The Citrobacter strain MSP27 was closely related to the Citrobacter strains isolated from R. chinensis (Fang et al., 2016) and R. speratus (Cho, Kim, Kim, Kim, & Kim, 2010), whereas the Trabulsiela strain MSP19 was closely related to Trabulsiella strain LB10 (Fang et al., 2016) isolated from R. chinensis and O. formosanus (Chou et al., 2007). In addition, the 16S rRNA genes of strain MSP1b and MSP17b were similar to those of Dyella strains isolated from R. chinensis with sequence divergence less than 2%, and Burkholderia strains MSP23 and MSP32 were similar to strains TM6 and TSB14 from R. chinensis (Fang et al., 2016).
3.4. Isolation and cultivation of members from Firmicutes
Firmicutes was the secondly dominant phylum in the MSP pool of termite gut microbiota. Analysis of the MSP pool data with RDP databank showed that 21 OTUs were classified into Firmicutes (Figure 3b). These 21 OTUs were assigned to 14 genera. Closely related members (16S RNA similarity >97%) of the genera Anaerococcus, Aneurinibacillus, Geobacillus, Gemella, Alicyclobacillus, Veillonella, Oribacterium, and Faecalibacterium were the first time observed in termite gut microbiota. Totally 32 isolates were obtained with MSP method, and phylogenetic analysis showed that all the strains in the phylum Firmicutes were affiliated with four genera (Bacillus, Lysinibacillus, Paenibacillus, and Staphylococcus) (Table 1 and Figure 3b). Further 16S rRNA gene analysis showed that the majority of our strains shared high similarities to bacterial strains previously isolated from R. chinensis or other wood‐feeding termites. For example, strains MSP33, MSP46, MSP12b, and MSP10b shared 99% sequence similarities to members of Bacillus, Staphaylococcus, and Paenibacillus isolated from R. chinensis (Cibichakravarthy et al., 2017; Fang et al., 2016; Tarayre et al., 2013), whereas strains MSP14, MSP58, and MSP8 shared higher than 99% in 16S rRNA genes to members of Lysinibacillus isolated from G. sulphureus (Hussin & Majid, 2017).
3.5. Isolation and cultivation of members from Actinobacteria, Bacteroidetes, and other phyla
As showed in Table 1, there were 11 OTUs that corresponding to seven genera of Actinobacteria (Figure 3c), and 14 OTUs that corresponding to seven genera of Bacteroidetes (Figure 3d). Four strains were obtained (Table 1) and representing members of Micrococcus (MSP19b), Arthrobacter (MSP18b), and Streptomyces (MSP91, MSP13b). The 16S rRNA genes of strains MSP19b of Actinobacteria had similarities less than 98% to the previously reported microbial species, suggesting that it was possibly new species of Micrococcus. Compared to Actinobacteria, only three isolates belonging to Bacteriodetes (represented by strain MSP50) were obtained (Table 1), although more OTUs were recognized from the MSP pool (Figure 3d). Members of the genera Prevotella, Alloprevotella, Flavisolibacter, and Flavobacterium (Figure 3d) were the first time to be documented for termite gut microbiota. Figure 3d also shows that OTUs (such as OTU80, OTU83, OTU84, Similarity <97%) represented unclassified and possibly new taxa occurred in termite gut. For example, strain MSP50 shared only 95% of 16S rRNA gene sequence similarity to Dysgonomonas gadi, and represented a new member of Bacteroidetes and was closely associated with genus Dysgonomonas.
4. DISCUSSION
In this study, we continued our previous efforts to cultivate microbes from the termite gut (Chen et al., 2012; Fang et al., 2015, 2016) by application of the newly developed MSP method (Jiang et al., 2016). As a microfluidic technology (Ma et al., 2014; Tandogan et al., 2014), MSP method enables high‐throughput single‐cell cultivation of diverse bacterial groups and even rare species from environmental samples (Jiang et al., 2016). Comparing with other cultivation tools such as extinction‐culturing‐based method (Colin et al., 2013, 2015; Connon & Giovannoni, 2002), MSP technology has higher throughput as one culture‐plate can harbor thousands of droplets, whereas the extinction cultivation method carried only a few hundreds of wells. Other methods such as the microcapsulation and the Ichip (Keller & Zengler, 2004; Nichols et al., 2010) was reported to be applicable under exclusively aerobic condition, whereas the MSP approach can be used both aerobically and anaerobically. The MSP technique can be further exploited for extended applicability as (a) cocultivation of different microorganisms for the study of symbiotic interaction (Park et al., 2011), (b) recovering functional and rare biosphere members and (c) single‐cell sequencing.
We successfully cultivated a range of bacterial strains belonging to the Delftia, Comamonas, Acinetobacter, Moraxella, Luteimona, Sphingomonas, Bosea, Methylobacterium, Corynebacterium, Janibacter, Propionibacterium, and Sphingobacterium with MSP method in this study. Of note, the occurrence of these bacterial taxa in termite gut had been previously detected with molecular tools but they had not been cultivated (Butera et al., 2016; Diouf et al., 2015; Fall et al., 2007; Hongoh et al., 2005; Husseneder, Berestecky, & Grace, 2009; Matsui, Tanaka, Namihira, & Shinzato, 2012; Nakajima, Hongoh, Usami, Kudo, & Ohkuma, 2005; Thong‐On et al., 2012; Visser, Nobre, Currie, Aanen, & Poulsen, 2012; Zhu et al., 2012). The members of Massilia, Hydrogenophaga, Hydrogenophilus, Neisseria, Helicobacter, Thermomonas, Dokdonella, Acidiphilium, Hyphomicrobium, Paracoccus, Microvirga, Peredibacter, Anaerococcus, Aneurinibacillus, Geobacillus, Gemella, Alicyclobacillus, Veillonella, Oribacterium, Faecalibacterium, Prevotella, Alloprevotella, Flavisolibacter, and Flavobacterium were the first time to be documented for association with termite gut. Several possible new taxa were obtained with MSP method. The OTU21 represented an unclassified member of Proteobacteria, and its 16S RNA gene showed 90% similarity to Mailhella massiliensis (Ndongo et al., 2017). The OTU93 recovered in MSP pool represented a member of Acidobacteria, and its 16S RNA gene showed 94% similarity to Vicinamibacter silvestris (Huber et al., 2016); the OTU97 represented an unclassified member of Phycisphaerae and its closest relative is Tepidisphaera mucosa (Kovaleva et al., 2015) (Their 16S rRNA gene similarity is 91%); More interestingly, the OTU98 recovered from MSP pool in this study together with 16S rRNA gene sequences detected in Formosan subterranean termite (Husseneder et al., 2009) and Reticulitermes speratus (Hongoh, Ohkuma, & Kudo, 2003) clustered to a unique lineage of Verrucomicrobia (Figure 3e). So far, there is not any bacterial culture showing 16S rRNA gene sequence similarity higher than 80% to this unique lineage. Although we had not obtained pure cultures of those above OTUs, their occurrence in MSP pool indicated they did grow in MSP droplets. Further efforts to optimize their growth in droplets would result in finally obtaining their pure cultures.
Several strains that represent potential novel taxa were isolated with MSP method. Dysgonomonas (Hofstad et al., 2000) belongs to Bacteroidetes, and four species of Dysgonomonas were isolated from clinical specimen. Recently, two new species were isolated and characterized from termite guts, Dysgonomonas macrotermitis (Yang et al., 2014) and Dysgonomonas termitidis (Pramono, Sakamoto, Iino, Hongoh, & Ohkuma, 2015). In this study, we obtained three isolates, as represented by strain MSP50, and they are phylogenetically close to Dysgonomonas. The 16S rRNA gene of MSP50 showed 95% similarity to Dysgonomonas gadei. Whether MSP50 represents a novel species within Dysgonomonas or a novel genus within Bacteroidetes needs additional taxonomic studies.
There are many other bacterial taxa that existed in termite gut but have not been successfully cultivated in this study, such as the members of Spirochaetes and Elusimicrobia (see Supplementary material Table S1). Spirochaetes widely occur in wood‐feeding termites and are the most abundant bacterial symbionts in Reticulitermes (Brune & Dietrich, 2015; Graber & Breznak, 2005; Noda, Ohkuma, Yamada, Hongoh, & Kudo, 2003). Elusimicrobia are found almost exclusively in the intestinal tract of animals and are particularly abundant in lower termites, where they reside as intracellular symbionts in the cellulolytic gut flagellates (Geissinger, Herlemann, Mörschel, Maier, & Brune, 2009; Ikeda‐Ohtsubo, Faivre, & Brune, 2010; Zheng, Dietrich, Radek, & Brune, 2016). The rarefaction curves of observed taxa in Figure 1a indicated that the achieved isolates can only recover a fraction of total species in termite gut. However, the observed recovery rate might be further improved by utilization of various cultural media and different culture conditions. Specifically, further efforts should be made to apply low but diverse nutrients in culture broth, besides 1/5 R2A, at various aerobic and anoxic levels for harvest of additional bacterial groups that are fastidious to nutrients and sensitive to oxidoreductive states.
In this study, 99 OTUs were identified to be cultivable with culture‐dependent MSP method, whereas 353 OTUs were detected from the termite gut microbiota sample (OMG) using the culture‐independent metagenomic method. This result showed that, only a fraction of the cultivable taxa (24 of 99 OTUs) was detectable with metagenomic method. Similar observation was reported by Lagier et al. (2012). In their study, 340 bacterial species were cultured from human gut using the MALDI‐TOF‐based culturomic strategy, and only 15% (51 species) of these cultivable isolates were detected by metagenomic pyrosequencing. The results manifested that culturomics complemented metagenomic by overcoming the depth bias inherent in metagenomic approaches (Lagier et al., 2012). Later on, Rettedal et al. (2014) demonstrated that the recovery rate and representativeness of culture‐dependent approaches in gut microbiota could be further improved by careful design of culture conditions. It is believed that, by optimization of the culture conditions, MSP method would have better performance in microbiome recovery.
Supporting information
ACKNOWLEDGMENT
This research is supported by grant from National Nature Science Foundation of China (Grant No. 31670102).
CONFLICT OF INTEREST
None declared.
Zhou N, Sun Y‐T, Chen D‐W, Du W, Yang H, Liu S‐J. Harnessing microfluidic streak plate technique to investigate the gut microbiome of Reticulitermes chinensis . MicrobiologyOpen. 2019;8:e654 10.1002/mbo3.654
Contributor Information
Hong Yang, Email: hyang@mail.ccnu.edu.cn.
Shuang‐Jiang Liu, Email: liusj@im.ac.cn.
REFERENCES
- Ali, S. S. , Wu, J. , Xie, R. , Zhou, F. , Sun, J. , & Huang, M. (2017). Screening and characterizing of xylanolytic and xylose‐fermenting yeasts isolated from the wood‐feeding termite, Reticulitermes chinensis . PLoS ONE, 12(7), e0181141 10.1371/journal.pone.0181141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann, R. I. , Ludwig, W. , & Schleifer, K. H. (1995). Phylogenetic identification of individual microbial cells without cultivation. Microbiological Reviews, 59(1), 143–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer, L. , Lazuka, A. , Sillamdussès, D. , Miambi, E. , O'Donohue, M. , & Hernandezraquet, G. (2017). Uncovering the potential of termite gut microbiome for lignocellulose bioconversion in anaerobic batch bioreactors. Frontiers in Microbiology, 8, 2623 10.3389/fmicb.2017.02623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell, D. E. (2011). Morphology, physiology, biochemistry and functional design of the termite gut: An evolutionary wonderland In Bignell D. E., Roisin Y., & Lo N. (Eds.), Biology of termites: A modern synthesis (pp. 375–412). Netherland: Springer; 10.1007/978-90-481-3977-4 [DOI] [Google Scholar]
- Bokulich, N. A. , Subramanian, S. , Faith, J. J. , Gevers, D. , Gordon, J. I. , Knight, R. , … Caporaso, J. G. (2013). Quality‐filtering vastly improves diversity estimates from illumina amplicon sequencing. Nature Methods, 10(1), 57 10.1038/nmeth.2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breznak, J. A. (1982). Intestinal microbiota of termites and other xylophagous insects. Annual Review of Microbiology, 36(1), 323–343. 10.1146/annurev.mi.36.100182.001543 [DOI] [PubMed] [Google Scholar]
- Breznak, J. A. (2000). Ecology of prokaryotic microbes in the guts of wood‐ and litter‐feeding termites In Abe Y., Bignell D. E., & Higashi T. (Eds.), Termites: Evolution, Sociality, Symbioses, Ecology (pp. 209–231). Netherlands: Kluwer Academic Publishers; 10.1007/978-94-017-3223-9 [DOI] [Google Scholar]
- Brune, A. , & Dietrich, C. (2015). The gut microbiota of termites: Digesting the diversity in the light of ecology and evolution. Annual Review of Microbiology, 69(1), 145 10.1146/annurev-micro-092412-155715 [DOI] [PubMed] [Google Scholar]
- Brune, A. , & Friedrich, M. (2000). Microecology of the termite gut: Structure and function on a microscale. Current Opinion in Microbiology, 3(3), 263–269. 10.1016/S1369-5274(00)00087-4 [DOI] [PubMed] [Google Scholar]
- Butera, G. , Ferraro, C. , Alonzo, G. , Colazza, S. , & Quatrini, P. (2016). The gut microbiota of the wood‐feeding termite Reticulitermes lucifugus (isoptera; Rhinotermitidae). Annals of Microbiology, 66(1), 253–260. 10.1007/s13213-015-1101-6 [DOI] [Google Scholar]
- Chen, W. , Wang, B. , Hong, H. , Yang, H. , & Liu, S. J. (2012). Deinococcus reticulitermitis sp. nov., isolated from a termite gut. International Journal of Systematic & Evolutionary Microbiology, 62(1), 78–83. 10.1099/ijs.0.026567-0 [DOI] [PubMed] [Google Scholar]
- Cho, M. J. , Kim, Y. K. , Kim, Y. K. , Kim, Y. S. , & Kim, T. J. (2010). Symbiotic adaptation of bacteria in the gut of Reticulitermes speratus: Low endo‐beta‐1,4‐glucanase activity. Biochemical & Biophysical Research Communications, 395(3), 432–435. 10.1016/j.bbrc.2010.04.048 [DOI] [PubMed] [Google Scholar]
- Chou, J. H. , Chen, W. M. , Arun, A. B. , & Young, C. C. (2007). Trabulsiella odontotermitis sp. nov., isolated from the gut of the termite Odontotermes formosanus shiraki. International Journal of Systematic & Evolutionary Microbiology, 57(4), 696–700. 10.1099/ijs.0.64632-0 [DOI] [PubMed] [Google Scholar]
- Cibichakravarthy, B. , Abinaya, S. , & Prabagaran, S. R. (2017). Syntrophic association of termite gut bacterial symbionts with bifunctional characteristics of cellulose degrading and polyhydroxyalkanoate producing bacteria. International Journal of Biological Macromolecules, 103, 613–620. 10.1016/j.ijbiomac.2017.05.100 [DOI] [PubMed] [Google Scholar]
- Colin, Y. , Goñiurriza, M. , Caumette, P. , & Guyoneaud, R. (2013). Combination of high throughput cultivation and dsrA sequencing for assessment of sulfate‐reducing bacteria diversity in sediments. FEMS Microbiology Ecology, 83(1), 26–37. 10.1111/j.1574-6941.2012.01452.x [DOI] [PubMed] [Google Scholar]
- Colin, Y. , Goñi‐Urriza, M. , Caumette, P. , & Guyoneaud, R. (2015). Contribution of enrichments and resampling for sulfate reducing bacteria diversity assessment by high‐throughput cultivation. Journal of Microbiol Methods, 110, 92–97. 10.1016/j.mimet.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Connon, S. A. , & Giovannoni, S. J. (2002). High‐throughput methods for culturing microorganisms in very‐low‐nutrient media yield diverse new marine isolates. Applied & Environmental Microbiology, 68(8), 3878–3885. 10.3410/f.1008856.113558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis, T. Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E. L. , Keller, K. , … Andersen, G. L. (2006). Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Applied & Environmental Microbiology, 72(7), 5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diouf, M. , Roy, V. , Mora, P. , Frechault, S. , Lefebvre, T. , Hervé, V. , … Miambi, E. (2015). Profiling the succession of bacterial communities throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) using 16S rRNA gene pyrosequencing. PLoS ONE, 10(10), e0140014 10.1371/journal.pone.0140014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. 10.1038/NMETH.2604 [DOI] [PubMed] [Google Scholar]
- Edwards, U. , Rogall, T. , Blöcker, H. , Emde, M. , & Böttger, A. E. C. (1989). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16s ribosomal RNA. Nucleic Acids Research, 17(19), 7843–7853. 10.1093/nar/17.19.7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fall, S. , Hamelin, J. , Ndiaye, F. , Assigbetse, K. , Aragno, M. , Chotte, J. L. , & Brauman, A. (2007). Differences between bacterial communities in the gut of a soil‐feeding termite (Cubitermes niokoloensis) and its mounds. Applied & Environmental Microbiology, 73(16), 5199–5208. 10.1128/AEM.02616-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, H. , Chen, W. , Wang, B. J. , Li, X. J. , Liu, S. J. , & Yang, H. (2016). Cultivation and characterization of symbiotic bacteria from the gut of Reticulitermes chinensis . Applied Environmental Biotechnology, 1(1), 3–12. 10.18063/AEB.2016.01.004 [DOI] [Google Scholar]
- Fang, H. , Lv, W. , Huang, Z. , Liu, S. J. , & Yang, H. (2015). Gryllotalpicola reticulitermitis sp. nov., isolated from a termite gut. International Journal of Systematic & Evolutionary Microbiology, 65(1), 85–89. 10.1099/ijs.0.062984-0 [DOI] [PubMed] [Google Scholar]
- Geissinger, O. , Herlemann, D. P. , Mörschel, E. , Maier, U. G. , & Brune, A. (2009). The ultramicrobacterium “Elusimicrobium minutum” gen. nov., sp. nov., the first cultivated representative of the termite group 1 phylum. Applied & Environmental Microbiology, 75(9), 2831–2840. 10.1128/AEM.02697-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber, J. R. , & Breznak, J. A. (2005). Folate cross‐feeding supports symbiotic homoacetogenic spirochetes. Applied & Environmental Microbiology, 71(4), 1883–1889. 10.1128/AEM.71.4.1883-1889.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstad, T. , Olsen, I. , Eribe, E. R. , Falsen, E. , Collins, M. D. , & Lawson, P. A. (2000). Dysgonomonas gen. nov. to accommodate Dysgonomonas gadei sp. nov., an organism isolated from a human gall bladder, and Dysgonomonas capnocytophagoides (formerly CDC group DF‐3). International Journal of Systematic & Evolutionary Microbiology, 50(6), 2189–2195. 10.1099/00207713-50-6-2189 [DOI] [PubMed] [Google Scholar]
- Hongoh, Y. (2011). Toward the functional analysis of uncultivable, symbiotic microorganisms in the termite gut. Cellular and Molecular Life Sciences: CMLS, 68(8), 1311–1325. 10.1007/s00018-011-0648-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongoh, Y. , Deevong, P. , Inoue, T. , Moriya, S. , Trakulnaleamsai, S. , Ohkuma, M. , … Kudo, T. (2005). Intra‐ and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Applied & Environmental Microbiology, 71(11), 6590–6599. 10.1128/AEM.71.11.6590-6599.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongoh, Y. , Ekpornprasit, L. , Inoue, T. , Moriya, S. , Trakulnaleamsai, S. , Ohkuma, M. , … Kudo, T. (2006). Intracolony variation of bacterial gut microbiota among castes and ages in the fungus‐growing termite Macrotermes gilvus . Molecular Ecology, 15(2), 505–516. 10.1111/j.1365-294X.2005.02795.x [DOI] [PubMed] [Google Scholar]
- Hongoh, Y. , Ohkuma, M. , & Kudo, T. (2003). Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus (Isoptera; Rhinotermitidae). FEMS Microbiology Ecology, 44(2), 231–242. 10.1016/S0168-6496(03)00026-6 [DOI] [PubMed] [Google Scholar]
- Huang, X. F. , Bakker, M. G. , Judd, T. M. , Reardon, K. F. , & Vivanco, J. M. (2013). Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microbial Ecology, 65(3), 531–536. 10.1007/s00248-013-0219-y [DOI] [PubMed] [Google Scholar]
- Huber, K. J. , Geppert, A. M. , Wanner, G. , Fösel, B. U. , Wüst, P. K. , & Overmann, J. (2016). The first representative of the globally widespread subdivision 6 Acidobacteria, Vicinamibacter silvestris gen. nov., sp. nov., isolated from subtropical savannah soil. International Journal of Systematic & Evolutionary Microbiology, 66(8), 2971–2979. 10.1099/ijsem.0.001131 [DOI] [PubMed] [Google Scholar]
- Husseneder, C. , Berestecky, J. M. , & Grace, J. K. (2009). Changes in composition of culturable bacteria community in the gut of the Formosan subterranean termite depending on rearing conditions of the host. Annals of the Entomological Society of America, 102(3), 498–507. 10.1603/008.102.0321 [DOI] [Google Scholar]
- Hussin, N. A. , & Majid, A. H. A. (2017). Inter and intra termites colonies comparisons of gut microbial diversity from worker and soldier caste of Globitermes sulphureus (Blattodea: Termitidae) using 16S rRNA gene. Malaysian Journal of Microbiology, 13(3), 228–234. [Google Scholar]
- Ikeda‐Ohtsubo, W. , Faivre, N. , & Brune, A. (2010). Putatively free‐living ‘Endomicrobia’‐ ancestors of the intracellular symbionts of termite gut flagellates? Environmental Microbiology Reports, 2(4), 554–559. 10.1111/j.1758-2229.2009.00124.x [DOI] [PubMed] [Google Scholar]
- Jiang, C. Y. , Dong, L. , Zhao, J. K. , Hu, X. , Shen, C. , Qiao, Y. , … Du, W. (2016). High‐throughput single‐cell cultivation on microfluidic streak plates. Applied & Environmental Microbiology, 82(7), 2210–2218. 10.1128/AEM.03588-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, M. , & Zengler, K. (2004). Tapping into microbial diversity. Nature Reviews Microbiology, 2(2), 141–150. 10.1038/nrmicro819 [DOI] [PubMed] [Google Scholar]
- Kovaleva, O. L. , Merkel, A. Y. , Novikov, A. A. , Baslerov, R. V. , Toshchakov, S. V. , & Bonch‐Osmolovskaya, E. A. (2015). Tepidisphaera mucosa gen. nov., sp. nov., a moderately thermophilic member of the class Phycisphaerae in the phylum Planctomycetes, and proposal of a new family, Tepidisphaeraceae fam. nov., and a new order, Tepidisphaerales ord. nov. International Journal of Systematic & Evolutionary Microbiology, 65(2), 549–555. 10.1099/ijs.0.070151-0 [DOI] [PubMed] [Google Scholar]
- Lagier, J. C. , Armougom, F. , Million, M. , Hugon, P. , Pagnier, I. , Robert, C. , … Raoult, D. (2012). Microbial culturomics: Paradigm shift in the human gut microbiome study. Clinical Microbiology & Infection, 18(12), 1185–1193. 10.1111/1469-0691.12023 [DOI] [PubMed] [Google Scholar]
- Liu, N. , Yan, X. , Zhang, M. , Xie, L. , Wang, Q. , Huang, Y. , … Zhou, Z. (2011). Microbiome of fungus‐growing termites: A new reservoir for lignocellulase genes. Applied & Environmental Microbiology, 77(1), 48–56. 10.1128/AEM.01521-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N. , Zhang, L. , Zhou, H. , Zhang, M. , Yan, X. , Wang, Q. , … Zhou, Z. (2013). Metagenomic insights into metabolic capacities of the gut microbiota in a fungus‐cultivating termite (Odontotermes yunnanensis). PLoS ONE, 8(7), e69184 10.1371/journal.pone.0069184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. , Kim, J. , Hatzenpichler, R. , Karymov, M. A. , Hubert, N. , Hanan, I. M. , … Ismagilov, R. F. (2014). Gene‐targeted microfluidic cultivation validated by isolation of a gut bacterium listed in Human Microbiome Project's Most Wanted taxa. Proceedings of the National Academy of Sciences of the United States of America, 111(27), 9768–9773. 10.1073/pnas.1404753111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč, T. , & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21), 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. M. , & Martin, J. S. (1978). Cellulose digestion in the midgut of the fungus‐growing termite Macrotermes natalensis: The role of acquired digestive enzymes. Science, 199(4336), 1453–1455. 10.1126/science.199.4336.1453 [DOI] [PubMed] [Google Scholar]
- Matsui, T. , Tanaka, J. , Namihira, T. , & Shinzato, N. (2012). Antibiotics production by an Actinomycete isolated from the termite gut. Journal of Basic Microbiology, 52(6), 731–735. 10.1002/jobm.201100500 [DOI] [PubMed] [Google Scholar]
- Matsuura, K. , Yashiro, T. , Shimizu, K. , Tatsumi, S. , & Tamura, T. (2009). Cuckoo fungus mimics termite eggs by producing the cellulose‐digesting enzyme β‐glucosidase. Current Biology, 19(1), 30–36. 10.1016/j.cub.2008.11.030 [DOI] [PubMed] [Google Scholar]
- Nakajima, H. , Hongoh, Y. , Usami, R. , Kudo, T. , & Ohkuma, M. (2005). Spatial distribution of bacterial phylotypes in the gut of the termite Reticulitermes speratus and the bacterial community colonizing the gut epithelium. FEMS Microbiology Ecology, 54(2), 247–255. 10.1016/j.femsec.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Ndongo, S. , Cadoret, F. , Dubourg, G. , Delerce, J. , Fournier, P. E. , Raoult, D. , & Lagier, J. C. (2017). ‘Collinsella phocaeensis’ sp. nov., ‘Clostridium merdae’ sp. nov., ‘Sutterella massiliensis’ sp. nov., ‘Sutturella timonensis’ sp. nov., ‘Enorma phocaeensis’ sp. nov., ‘Mailhella massiliensis’ gen. nov., sp. nov., ‘Mordavella massiliensis’ gen. nov., sp. nov. and ‘Massiliprevotella massiliensis’ gen. nov., sp. nov., 9 new species isolated from fresh stool samples of healthy French patients. New Microbes and New Infections, 17, 89–95. 10.1016/j.nmni.2017.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, J. , & Tokuda, G. (2013). Lignocellulose‐degrading enzymes from termites and their symbiotic microbiota. Biotechnology Advances, 31(6), 838–850. 10.1016/j.biotechadv.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Nichols, D. , Cahoon, N. , Trakhtenberg, E. M. , Pham, L. , Mehta, A. , Belanger, A. , … Epstein, S. S. (2010). Use of ichip for high‐throughput in situ cultivation of “uncultivable” microbial species. Applied & Environmental Microbiology, 76(8), 2445–2450. 10.1128/aem.01754-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda, S. , Ohkuma, M. , Yamada, A. , Hongoh, Y. , & Kudo, T. (2003). Phylogenetic position and in situ identification of ectosymbiotic spirochetes on protists in the termite gut. Applied & Environmental Microbiology, 69(1), 625–633. 10.1128/AEM.69.1.625-633.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma, M. (2003). Termite symbiotic systems: Efficient bio‐recycling of lignocellulose. Applied Microbiology & Biotechnology, 61(1), 1–9. 10.1371/10.1007/s00253-002-1189-z [DOI] [PubMed] [Google Scholar]
- Ohkuma, M. , & Brune, A. (2011). Diversity, structure, and evolution of the termite gut microbial community In Bignell D. E., Roisin Y., & Lo N. (Eds.), Biology of termites: A modern synthesis (pp. 413–438). Netherlands: Springer. [Google Scholar]
- Ohkuma, M. , & Kudo, T. (1996). Phylogenetic diversity of the intestinal bacterial community in the termite Reticulitermes speratus . Applied & Environmental Microbiology, 62(2), 461–468. 10.1128/JB.00345-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. , Kerner, A. , Burns, M. A. , & Lin, X. N. (2011). Microdroplet‐enabled highly parallel co‐cultivation of microbial communities. PLoS ONE, 6(2), e17019 10.1371/journal.pone.0017019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramono, A. K. , Sakamoto, M. , Iino, T. , Hongoh, Y. , & Ohkuma, M. (2015). Dysgonomonas termitidis sp. nov., isolated from the gut of the subterranean termite Reticulitermes speratus . International Journal of Systematic & Evolutionary Microbiology, 65(2), 681–685. 10.1099/ijs.0.070391-0 [DOI] [PubMed] [Google Scholar]
- Rettedal, E. A. , Gumpert, H. , & Sommer, M. O. (2014). Cultivation‐based multiplex phenotyping of human gut microbiota allows targeted recovery of previously uncultured bacteria. Nature Communications, 5, 4714 10.1038/ncomms5714 [DOI] [PubMed] [Google Scholar]
- Shinzato, N. , Muramatsu, M. , Matsui, T. , & Watanabe, Y. (2007). Phylogenetic analysis of the gut bacterial microflora of the fungus‐growing termite Odontotermes formosanus . Bioscience, Biotechnology, and Biochemistry, 71(4), 906–915. 10.1271/bbb.60540 [DOI] [PubMed] [Google Scholar]
- Sommer, M. O. (2015). Advancing gut microbiome research using cultivation. Current Opinion in Microbiology, 27, 127–132. 10.1016/j.mib.2015.08.004 [DOI] [PubMed] [Google Scholar]
- Stewart, E. J. (2012). Growing unculturable bacteria. Journal of Bacteriology, 194(16), 4151–4160. 10.1128/JB.00345-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandogan, N. , Abadian, P. N. , Epstein, S. , Aoi, Y. , & Goluch, E. D. (2014). Isolation of microorganisms using sub‐micrometer constrictions. PLoS ONE, 9(6), e101429 10.1371/journal.pone.0101429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarayre, C. , Bauwens, J. , Mattéotti, C. , Brasseur, C. , Millet, C. , Massart, S. , … Delvigne, F. (2015). Multiple analyses of microbial communities applied to the gut of the wood‐feeding termite Reticulitermes flavipes, fed on artificial diets. Symbiosis, 65(3), 143–155. 10.1007/s13199-015-0328-0 [DOI] [Google Scholar]
- Tarayre, C. , Brognaux, A. , Brasseur, C. , Bauwens, J. , Millet, C. , Mattéotti, C. , … Thonart, P. (2013). Isolation and cultivation of a xylanolytic Bacillus subtilis extracted from the gut of the termite Reticulitermes santonensis . Applied Biochemistry & Biotechnology, 171(1), 225–245. 10.1007/s12010-013-0337-5 [DOI] [PubMed] [Google Scholar]
- Thompson, J. D. , Gibson, T. J. , & Higgins, D. G. (2002). Multiple sequence alignment using ClustalW and ClustalX. Current Protocols in Bioinformatics, 2, Unit 2.3 10.1002/0471250953.bi0203s00 [DOI] [PubMed] [Google Scholar]
- Thong‐On, A. , Suzuki, K. , Noda, S. , Inoue, J. I. , Kajiwara, S. , & Ohkuma, M. (2012). Isolation and characterization of anaerobic bacteria for symbiotic recycling of uric acid nitrogen in the gut of various termites. Microbes and Environments, 27(2), 186–192. 10.1264/jsme2.ME11325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser, A. A. , Nobre, T. , Currie, C. R. , Aanen, D. K. , & Poulsen, M. (2012). Exploring the potential for actinobacteria as defensive symbionts in fungus‐growing termites. Microbial Ecology, 63(4), 975–985. 10.1007/s00248-011-9987-4 [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied & Environmental Microbiology, 73(16), 5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke, F. , Luginbühl, P. , Ivanova, N. , Ghassemian, M. , Richardson, T. H. , Stege, J. T. , … Leadbetter, J. R. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood‐feeding higher termite. Nature, 450(7169), 560–565. 10.1038/nature06269 [DOI] [PubMed] [Google Scholar]
- Watanabe, H. , & Tokuda, G. (2010). Cellulolytic systems in insects. Annual Reviews of Entomology, 55, 609–632. 10.1146/annurev-ento-112408-085319 [DOI] [PubMed] [Google Scholar]
- Weisburg, W. G. , Bars, S. M. , Pelletier, D. A. , & Lane, D. J. (1991). 16S Ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology, 173(2), 697–703. 10.1128/jb.173.2.697-703.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, J. J. , Zhou, D. , Caporaso, J. G. , Knight, R. , & Angenent, L. T. (2012). Comparison of Illumina paired‐end and single‐direction sequencing for microbial 16S rRNA gene amplicon surveys. The ISME Journal, 6(7), 1273–1276. 10.1038/ismej.2011.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. J. , Zhang, N. , Ji, S. Q. , Lan, X. , Zhang, K. D. , Shen, Y. L. , … Ni, J. F. (2014). Dysgonomonas macrotermitis sp. nov., isolated from the hindgut of a fungus‐growing termite. International Journal of Systematic & Evolutionary Microbiology, 64(9), 2956–2961. 10.1099/ijs.0.061739-0 [DOI] [PubMed] [Google Scholar]
- Zheng, H. , Dietrich, C. , Radek, R. , & Brune, A. (2016). Endomicrobium proavitum, the first isolate of Endomicrobia class. nov. (phylum Elusimicrobia)–an ultramicrobacterium with an unusual cell cycle that fixes nitrogen with a Group IV nitrogenase. Environmental Microbiology, 18(1), 191–204. 10.1111/1462-2920.12960 [DOI] [PubMed] [Google Scholar]
- Zhou, Q. Z. , Liu, X. Y. , Liu, S. J. , Ma, G. H. , & Su, Z. G. (2008). Preparation of uniformly sized agarose microcapsules by membrane emulsification for application in sorting bacteria. Industrial & Engineering Chemistry Research, 47(17), 6386–6390. 10.1021/ie800011r [DOI] [Google Scholar]
- Zhu, Y. H. , Li, J. , Liu, H. H. , Yang, H. , Xin, S. , Zhao, F. , … Lu, X. Y. (2012). Phylogenetic analysis of the gut bacterial microflora of the fungus‐growing termite macrotermes barneyi. African Journal of Microbiology Research, 6(9), 2071–2078. 10.5897/AJMR11.1345 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials