

Summary
Hepatic lipid metabolism is a series of complex processes that control influx and efflux of not only hepatic lipid pools, but also organismal pools. Lipid homeostasis is usually tightly controlled by expression, substrate supply, oxidation and secretion that keep hepatic lipid pools relatively constant. However, perturbations of any of these processes can lead to lipid accumulation in the liver. Although it is thought that these responses are hepatic arms of the ‘thrifty genome’, they are maladaptive in the context of chronic fatty liver diseases. Ethanol is likely unique among toxins, in that it perturbs almost all aspects of hepatic lipid metabolism. This complex response is due in part to the large metabolic demand placed on the organ by alcohol metabolism, but also appears to involve more nuanced changes in expression and substrate supply. The net effect is that steatosis is a rapid response to alcohol abuse. Although transient steatosis is largely an inert pathology, the chronicity of alcohol-related liver disease seems to require steatosis. Better and more specific understanding of the mechanisms by which alcohol causes steatosis may therefore translate into targeted therapies to treat alcohol-related liver disease and/or prevent its progression.
Keywords: Alcohol-related liver disease, Steatosis, Lipid homeostasis, Metabolism
Introduction
Alcohol is highly prevalent in most societies and more than 50% of Americans consume alcohol at least once a month.1 Heavy alcohol consumption associated with alcohol dependence and/or abuse (i.e., binge drinking) is well known to damage the liver. Alcohol-related liver disease (ALD) affects more than 10 million Americans each year, while treating the medical consequences of the disease costs more than $166 billion annually.2 Furthermore, alcohol consumption can enhance damage to the liver caused by other diseases (e.g., hepatitis virus infection) and drugs (e.g., acetaminophen).3,4 Although the progression of alcohol-induced liver injury is well characterised, there is no universally accepted therapy available to halt or reverse this process in humans. Therefore, there is an increasing focus on understanding the biochemical changes responsible for the development and progression of ALD. With better understanding of the mechanism(s) and risk factors that mediate the initiation and progression of this disease, rational targeted therapy can be developed to treat or prevent it in clinical practice.
The first and most common hepatic change caused by alcohol consumption is steatosis, or fatty liver (Fig. 1). The prevalence of steatosis is essentially 100% in those who consume alcohol at levels that increase their risk of liver disease.5 Fat accumulation can be both macrovesicular (having one large fat droplet per hepatocyte and lateral displacement of the nucleus) or microvesicular (many small fat droplets per hepatocyte) (Fig. 1).5 Alcohol-induced steatosis is rapidly and readily reversible upon cessation of alcohol consumption. Steatosis can also be clinically ‘silent,’ and can exist in the absence of increases in any other index of liver damage, such as plasma aminotransferases, for example. For these reasons, steatosis was originally viewed as an inert pathology in ALD (and in other fatty liver diseases). However, more recent studies have suggested that blunting or preventing steatosis could help attenuate the progression of ALD; in fact, the degree of steatosis is an early predictor of overall disease severity.6 These facts challenge the assumption that steatosis is an inert pathology. Hepatic fat accumulation can invoke metabolic changes that sensitise the liver to further injury (see below). Therefore, a full understanding of how alcohol induces steatosis could be key in preventing progression to later stages of ALD.
Fig. 1. Steatosis in alcohol-related liver disease.
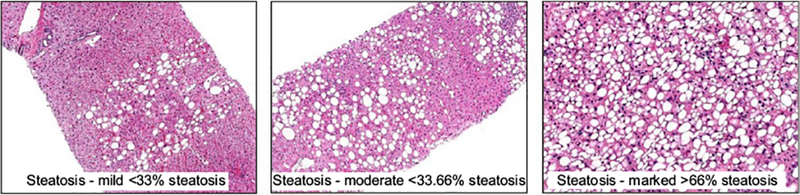
Representative pictures of liver biopsies from patients with ALD and different degrees of steatosis. In all cases macro- and microsteatosis are present. Photomicrographs courtesy of Dr. John Woosley, University of North Carolina at Chapel Hill.
The liver plays a central role in lipid metabolism for the entire organism. Hepatic free fatty acids (FAs) are not only directly synthesised from glycolytic end products and hepatic catabolism (e.g., autophagy), but are also actively taken up by the liver from dietary, and extrahepatic (e.g., adipose tissue lipolysis) sources. This pool of FAs can either be used for energy via β-oxidation, membrane synthesis or esterification into triglycerides by hepatocytes. Triglycerides are subsequently packaged as very low-density lipoproteins (VLDLs) that can be secreted into the bloodstream or serve as precursors for primary bile acids, which facilitate the emulsification of dietary lipids for delivery to the liver and extra-hepatic sites. There is intricate cross-talk between these systems. Hepatic lipid metabolism is controlled by a complex interplay of hormones, nuclear receptors, intracellular signalling pathways and transcription factors. Under homeostatic conditions, hepatic lipid flux maintains relatively low concentrations of hepatic lipid pools. However, dysregulation of this flux can cause lipids to accumulate in hepatocytes, leading to steatosis (Fig. 2).
Fig. 2. Intricate regulation of lipid metabolism, and the impact of ethanol exposure.
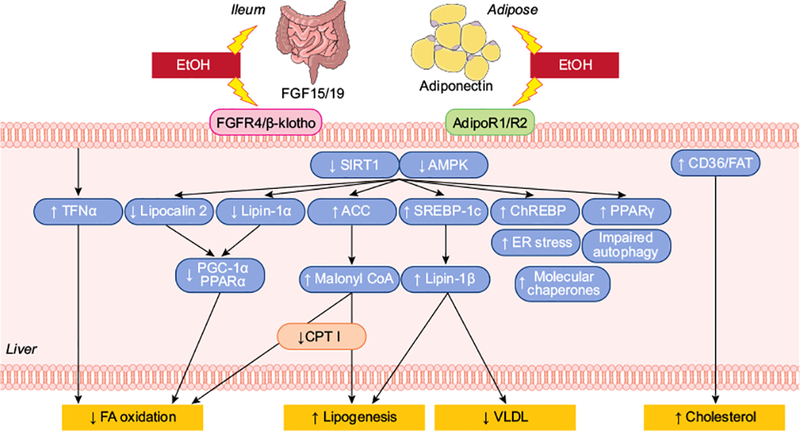
The liver plays a central role in lipid metabolism for the entire organism. Hepatic free FAs are not only directly synthesised (lipogenesis), but are also actively taken up by the liver. This pool of FAs can either be used for energy (FA oxidation), membrane synthesis or for esterification into triglycerides by hepatocytes. Triglycerides are subsequently packaged as VLDLs to be secreted. There is intricate cross-talk between these systems and hepatic lipid metabolism is controlled by a complex interplay of hormones, nuclear receptors, intracellular signalling pathways and transcription factors. Alcohol directly and indirectly impacts numerous aspects of hepatic lipid flux that ultimately leads to lipid accumulation. FA, fatty acid; VLDL, very low-density lipoprotein.
Alcohol directly and indirectly impacts numerous aspects of hepatic lipid flux that ultimately lead to lipid accumulation. The simplest example is that alcohol metabolism itself directly causes steatosis. Concentrations of alcohol can easily reach the mM range in the portal/hepatic circulation during alcohol consumption. In the process of metabolising ethanol to acetate, 2 equivalents of reduced NADH are generated per equivalent of ethanol oxidised. This metabolism robustly increases the ratio of NADH:NAD+ within the cell, which then favours inhibition of FA β-oxidation in the liver. Furthermore, ethanol metabolism also increases the rate of esterification of Fas.7 The net effect is to favour triglyceride accumulation in the hepatocytes. However, the impact of alcohol exposure on lipid metabolism is far more complex than simple redox inhibition of β-oxidation. The purpose of this review is to summarise the known impacts of ethanol on this process.
Effects of ethanol on fatty acid transporters
Circulating FAs are directly taken up by the liver, with a relatively high first pass extraction,8,9 and are the largest source of lipid for triglyceride synthesis.10 The liver also clears chylomicron-remnant triglyceride, which also contributes to the hepatic FA pool.8 FA transporters, including CD36/FA translocase (FAT) and FA transport protein (FATP encoded by SLC27A1) and FA binding proteins, play important roles in FA uptake.11,12 Although the liver is not the main site of CD36/FAT expression, stimulation of CD36/FAT promotes hepatic free FA uptake, which can lead to hepatic lipid accumulation and liver injury in rodents and humans.11–13 Ethanol exposure increases hepatic uptake of exogenous FAs and subsequent incorporation of FAs (e.g. palmitate) into triglycerides or total lipid in the liver.14–16 Ethanol-mediated upregulation of hepatic FA transporters, in particular, CD36/FAT, FATP1 and FATP5 promotes FA uptake, excessive fat accumulation, and development of steatosis in mice and rats.17–21 Co-administration of recombinant adiponectin to ethanol-fed mice markedly suppresses hepatic CD36/FAT expression and alleviates steatosis.22 Genetic ablation of mitoNEET (CISD1), a potential inducer of CD36/FAT, ameliorates experimental alcoholic steatohepatitis in mice, partially by downregulating CD36/FAT.23 These studies suggest the involvement of FA transporters, particularly CD36/FAT, in the pathogenesis of alcoholic fatty liver disease (AFLD).
Effects of ethanol on FA and triglyceride synthesis: potential key players
As mentioned, the liver can generate FAs from non-lipid precursors via de novo lipogenesis. This process is predominantly regulated by insulin and glucose flux in the liver and serves to provide a storage source of energy during times of fasting. Pyruvate from glycolysis enters the citric acid cycle and is converted to citrate, which is subsequently converted to acetyl- and malonyl-CoA and used to synthesise FAs. Rate-limiting enzymes in this process include acetyl-CoA carboxylases 1 and 2 (ACC-1 and −2 which convert acetyl-CoA to malonyl-CoA), FA synthase (FASN which synthesise saturated FAs from malonyl-CoA), and steryl-CoA-desaturase-1 (SCD-1 which converts saturated FAs to monounsaturated FAs). The synthesis of glycerolipid (i.e., triglycerides) from FAs is mediated by key acetyltransferases (e.g., GPAT, AGPAT and DGAT) and phosphatidate phosphatases (e.g., lipin-1).
SREBP-1c and ChREBP and transcriptional control of lipogenesis
Although they are controlled at several levels, the dominant regulation of the lipogenic genes described above is transcriptional (Fig. 1). The most potent inducers of these genes are the transcription factors SREBP-1c and ChREBP. The canonical activator of SREBP-1c is insulin and its inhibitor is glucagon. In contrast, substrate supply (glucose and citrate) regulates the expression of ChREBP. Under normal conditions, lipogenesis is thus maximally induced after the intake of nutrients and is downregulated during fasting. Previous studies have indicated that both SREBP-1c and ChREBP are activated by alcohol exposure,24–27 which clearly explains the induction of lipogenic genes by alcohol. However, alcohol and/or its metabolites blunt glucose-induced insulin release from the pancreas and activate glucagon release.28 Furthermore, alcohol causes insulin resistance and inhibits gluconeogenesis, which should decrease intrahepatic glucose concentrations. These net effects should in principle disfavour activation of these transcription factors, suggesting that alternate activation pathways are in play, as discussed later.
Lipin-1
Lipin-1 protein plays a pivotal role in lipid synthesis as a mammalian Mg2+-dependent phosphatidic acid phosphohydrolase (PAP), which catalyses the penultimate step in triglyceride synthesis.29–34 In addition to PAP activity, lipin-1 contains a putative nuclear localisation signal, and acts as a transcriptional co-regulator of the expression of genes involved in lipid metabolism in the nucleus.29–34
Lipin-1 pre-mRNA alternative splicing generates 3 lipin-1 protein isoforms, lipin-1α, lipin-1β, and lipin-1γ.29,30,34 Lipin-1α and lipin-1β are expressed in various organs, such as the liver and adipose, while lipin-1γ is predominately expressed in the brain.29,30,34 The variant splicing of lipin-1α and lipin-1β is partially regulated by a splicing factor, arginine/serine-rich 10 (SFRS10 or TRA2B).35The consequent protein products exert different functions. Lipin-1β serves as a PAP enzyme, which catalyses phosphatidate to diacylglycerol, facilitating the synthesis of triglycerides and phospholipids at the endoplasmic reticulum.29,30,32 In contrast, lipin-1α is predominately localised to the nucleus, where it acts as a transcriptional co-regulator, activating PGC-1α, PPARα and inhibiting SREBP-1c.31–33 The overall effects of lipin-1α are to increase β-oxidation of free FAs and reduce lipid synthesis.
Aberrant lipin-1 contributes to the abnormalities in lipid metabolism associated with AFLD in rodents and in humans.29,33,36–45 Owing to its inhibition of AMPK activity and activation of SREBP-1c, ethanol upregulates lipin-1, induces accumulation of cytosolic lipin-1 protein, enhances PAP activity, and promotes triglyceride synthesis in the livers of rodents and human alcoholics.33,39–46 Ethanol also blocks lipin-1 nuclear entry, inhibits nuclear lipin-1-mediated FA oxidation and perturbs VLDL secretion in mouse liver.42 Furthermore, ethanol suppresses lipin-1 alternative pre-mRNA splicing and subsequently increases the ratio of lipin-1β/α by disrupting the SIRT1-SFRS10 axis.41,43 Abnormalities in lipin-1 are also involved in the ethanol-induced production of a panel of pro-inflammatory cytokines.45 These ethanol-mediated alterations in lipin-1 promote steatosis, exacerbate inflammation and cause liver injury.
ER stress and the UPR
The endoplasmic reticulum (ER) is critically involved in the proper folding and assembly of secreted and membrane proteins. Homeostasis between the protein load and the capacity of the ER to process this load must be maintained to ensure proper protein folding. Physiological and pathological stimuli can disrupt this homeostasis causing misfolded or unfolded proteins to accumulate, leading to ER stress. In attempts to reestablish homeostasis, the ER activates a signalling network known as the unfolded protein response (UPR). One downstream effect of activation of the UPR by ER stress is the insulin-independent proteolytic activation of SREBP-1c. This effect of the UPR makes teleological sense, in that increasing lipogenesis would increase lipid substrate supply to the ER for protein processing.47 It has been shown that alcohol induces ER stress in the liver, at least in part by causing hyperhomocysteinaemia.24,48
TNFα
It is well known that both the basal and lipopolysaccharide-stimulated production of TNFα (or TNF) are increased in humans consuming alcohol and in experimental ALD.49,50 The role of TNFα and other pro-inflammatory cytokines in hepatic inflammation is well known. However, studies in experimental ALD indicate that they may also contribute to lipogenesis. Specifically, genetic or pharmacologic inhibition of TNFα signalling blunted steatosis caused by alcohol.51–53 This effect of TNFα may be mediated at several levels of lipid metabolism. For example, TNFα increases free FA release from adipocytes in the periphery,54 increases lipogenesis in hepatocytes,55 and inhibits β-oxidation of Fas.56 Moreover, prooxidant production stimulated by TNFα in hepatocytes could impair mitochondrial electron flow and cause lipid peroxidation, processes that could also slow the metabolism of fat by mitochondria. Other studies demonstrated transcription and activation of SREBP-1c is enhanced by TNFα in hepatocytes,57,58 which yields another mechanistic link between TNFα and lipogenesis. Other cytokines induced by alcohol (e.g., IL-1 and IL-6) may also impair transport and secretion of triglycerides.59
PPARγ
Peroxisome proliferator-activated receptor gamma (PPARγ) is a nuclear hormone receptor that is known to impact on lipid metabolism and glucose homeostasis. The PPARG gene encodes 2 splice isoforms of the protein product, PPARγ1 and PPARγ2; the former is constitutively expressed at low levels in most tissues, whereas the latter is expressed predominantly in adipose tissue under basal conditions.60 Although the liver normally expresses low levels of PPARγ2, expression is elevated in steatotic livers, both alcoholic and non-alcoholic.60–62 The activation of PPARγ may be pleiotropic in fatty liver disease. Specifically, PPARγ agonists exert beneficial effects in both diet-induced and alcohol-induced fatty liver injury;63–65 these protective effects are largely attributed to increasing adiponectin production in adipocytes (66; see later). In contrast, studies in hepatocyte-specific knockout mice indicate that PPARγ2 activation is detrimental to the liver in experimental alcoholic and non-alcoholic liver disease.15 This hepatic effect of PPARγ appears to be mediated via induction of SREBP-1c and other genes key to lipogenesis.
AMPK and SIRT1
The protein kinase complex, AMPK, provides another level of control over lipid metabolism. AMPK acts as a “sensor” of cellular energy status and helps to maintain homeostasis.67 In general, the downstream effects of AMPK activation are considered catabolic and favour ATP generation during energy depletion. For example, glycolysis is enhanced by AMPK. Signalling downstream of AMPK also inhibits ATP-consuming processes, such as de novo lipogenesis.68 More specifically, AMPK phosphorylates a number of serine residues on both isoforms of ACC (ACC-1 and ACC-2), which inhibits their activity, even in the presence of citrate.69 In addition to blocking the activity of key lipogenic enzymes, AMPK indirectly decreases lipogenesis by phosphorylating ChREBP, thereby hindering its nuclear translocation and transcriptional activity.70 Likewise, AMPK directly phosphorylates SREBP-1c, which also causes an inhibition of this factor’s transcriptional activity.71 Ethanol has been demonstrated to inhibit AMPK phosphorylation, thereby inhibiting ACC, SREBP-1c and ChREBP.33,72,73,27 The mechanisms appear to involve activation of the dephosphorylase PP2A via aSMase-mediated ceramide signalling74,75 and and/or via inhibition of upstream activation pathways (e.g., LKB176).
SIRT-1 is an NAD+-dependent protein deactylase. Targets of its deactylase activity include several key players in SREBP-1 and ChREBP-1 signalling.77–80 SIRT-1 also deacetylates histones, namely H3 and H4, which could epigenetically increase expression of lipogenic genes (e.g., SREBF178). Ethanol exposure downregulates expression of SIRT-1,78,81 likely at multiple levels of control.78 Additionally, the deactylase activity of SIRT-1 is sensitive to the NADH redox state of the cell.82 Thus, the increased ratio of NADH: NAD+ in the more reduced state caused by ethanol metabolism may not only blunt FA oxidation, but also directly contribute to increased de novo lipogenesis by blunting SIRT-1 activity. AMPK and SIRT-1 share many overlapping targets of regulation, the former via phosphorylation and the latter via deacetylation. Indeed, it is thought that these overlapping functions are at least permissive to each other and that maximal inhibition of lipogenesis is only affected when both AMPK and SIRT-1 are activated.83 Thus, the fact that both are inhibited by ethanol implies that lipogenesis will be effectively disinhibited.
Molecular chaperones
Stress induced heat shock proteins (Hsps) such as Hsp90, Hsp70, and Hsp72 are ubiquitous and highly conserved, and can be induced by a wide variety of physiological and environmental insults.84 Heat shock factors (HSFs) upregulate a family of Hsp genes by binding to the heat shock-binding element (HSE).85–87 Hsps serve as chaperones that maintain the function of signalling molecules in lipid metabolism. For instance, Hsp90 alters lipid homeostasis by regulating SREBP-1.86
Hsps play pivotal pathophysiological roles in AFLD.87 Like other stress signals, ethanol consumption results in accumulation of stress proteins such as hepatic Hsp70, Hsp72, Hsp90 and HSF-1 in human and experimental murine AFLD.87–97 For example, ethanol exposure induces hepatic Hsp90 in mice and contributes to the development of steatosis and liver injury via dysregulation of molecules important in lipid metabolism, including SREBP-1, SCD-1, FASN and ACC-1.97 Pharmacologic inhibition of Hsp90 ameliorates fatty liver injury during chronic or acute ethanol exposure in rodents. These studies have demonstrated the clear and direct regulation of hepatic lipid metabolism by Hsps in rodents in response to ethanol challenge. In addition to Hsps, sestrins are a family of stress-sensitive genes regulating lipid metabolism.97 The inhibitory effect of ethanol on sestrin 3 contributes to the development of steatosis by disrupting AMPK signalling, which leads to alterations in the genes involved in FA synthesis and oxidation.98 Future studies are needed to delineate the precise role of Hsps and sestrins in lipid metabolism and its contribution to alcoholic steatosis.
Adiponectin and FGF-15 axis
Adiponectin is an adipose-derived hormone that circulates in the plasma as low, middle, and high molecular weight multimers.99,100 Adiponectin is a pivotal player in the regulation of lipid metabolism (Fig. 1). After reaching the liver, adiponectin transduces signals via 2 major adiponectin receptors AdipoR1 and AdipoR2. Adiponectin inhibits lipid synthesis and stimulates FA oxidation, in part by activating SIRT1, AMPK, PGC-1α and PPARα, and suppressing SREBP-1.99,100 Fibroblast growth factor (FGF) 15 (human homolog FGF19), is a terminal small intestine (ileum)-derived hormone.101 Circulating FGF15/19 signalling regulates bile acid and lipid metabolism in the liver through activation of a receptor complex comprised of fibroblast growth factor receptor 4 (FGFR4)/β-Klotho.101
Ethanol impairs adiponectin synthesis and production in adipocytes and downregulates hepatic adiponectin receptors.39,44,97,100,102–106 Adiponectin elicits profound lipid lowing effects in rodents administered ethanol and in patients with AFLD.39,44,97,100,102–106 Aberrant hepatic adiponectin signalling is associated with lower activities of AMPK and SIRT1 and elevated levels of downstream molecules such as SREBP-1, ACC and lipin-1β in the livers of ethanol-fed rodents and patients with AFLD.39,44,97,100,102–106 These findings all point to a critical link between altered hepatic adiponectin signalling and AFLD.
Adipose-derived adiponectin and gut-derived FGF15/19 associate with each other, with the endocrine adiponectin-FGF15/19 axis a pivotal regulator of lipid metabolism.107,108 Chronic or chronic-binge ethanol feeding concomitantly reduces adiponectin and FGF15/19 levels in mice.23,109,110 Remarkably, the concurrent elevation of adiponectin and FGF15 is associated with inhibition of the genes involved in lipid uptake (e.g. CD36/FAT) and activation of the genes (e.g. PPARα and medium chain acyl-CoA dehydrogenase) implicated in lipid oxidation and the presence of ethanol-induced steatohepatitis in Cisd1 knockout mice.23 These findings suggest that endocrine adiponectin-FGF15/19 signalling protects against AFLD, at least in part by ameliorating the ethanol-induced abnormality in lipid metabolism.
Overall effect of ethanol exposure on lipogenesis
In summary, the net effect of ethanol is to activate (e.g., via ER stress, TNFα and/or hepatic PPARγ) de novo lipogenesis, while concomitantly inhibiting processes that block this response (e.g., AMPK and SIRT1). Although some of this net effect results from the direct action of ethanol on lipogenic enzymes (e.g., disinhibition of ACC by AMPK inhibition), it is primarily the result of ethanol activating the transcriptional activity of SREBP-1c and ChREBP. This explains why these transcription factors are activated even when ethanol decreases the canonical inducers of these pathways (see earlier). In NAFLD, a similar loss of negative regulation of SREBP-1c and ChREBP is hypothesised to contribute to de novo lipogenesis, even in the fasting state.111 Although the effect of ethanol on fasting de novo lipogenesis is less clear, a similar mechanism which contributes to the loss of diurnal regulation of lipid metabolism could be in play (see later).
Effects of ethanol on mitochondrial β-oxidation: potential key players
Mitochondrial β-oxidation shortens FAs into acetyl-CoA subunits, which can either enter the citric acid cycle, or be used to synthesise ketone bodies.112 Although short-chain FAs can readily cross the outer and inner mitochondrial membranes, medium- and long-chain FAs are actively transported into the inner mitochondrial space via the carnitine shuttle. The rate-limiting enzyme in this process is carnitine palmytoyl transferase I (CPTI), which is regulated both transcriptionally and post-transcriptionally. Ethanol causes several changes that can directly or indirectly impair β-oxidation.
Transcriptional inhibition of mitochondrial β-oxidation by ethanol
Despite a net increase in the supply of FAs for β-oxidation, there is no apparent induction of β-oxidation genes during alcohol exposure. The major mechanism of action underlying this effect is hypothesised to be the inhibition of peroxisome proliferator-activated receptor alpha (PPARα) signalling.113 PPARα is a nuclear hormone receptor that regulates expression of numerous genes involved in mitochondrial β-oxidation.114,115 Ethanol exposure decreases PPARα DNA binding activity, without decreasing PPARα expression;116 this effect is potentially mediated via decreasing protein levels of the retinoid X receptor (RXR), which heterodimerises with PPARα to bind to target DNA.116
Nutritional deficiencies
Alcoholics replace in excess of 50% of their total daily calories with ethanol.117 Furthermore, alcohol consumption often causes malabsorption,118 which may further exacerbate nutrient deficiencies. As the name implies, the carnitine shuttle requires carnitine as a cofactor. Roughly 25% of carnitine is synthesised endogenously from lysine and methionine, with the remainder derived from dietary sources.119 Several experimental lines of evidence support the hypothesis that nutritional deficiencies may lead to functional carnitine deficiency, via restricting precursor supply and/or carnitine proper.120,121 In contrast, the impact of alcohol on circulating levels of carnitine metabolites is equivocal at this time.122–124 Nevertheless, alcohol consumption may cause nutritional deficiencies that potentially impair mitochondrial β-oxidation.
Inhibition of β-oxidation activity
As mentioned, the increase in the NADH:NAD+-ratio caused by alcohol metabolism directly inhibits mitochondrial β-oxidation. This effect is thought to be predominantly mediated by the NAD+ reducing enzyme, 3-hydroxy-CoA dehydrogenase, the final step in generating acetyl-CoA during β-oxidation.125 Furthermore, the disinhibition of ACC caused by impairing AMPK activity (see earlier) increases the carboxylation of acetyl-CoA to malonyl-CoA, which inhibits CPTI activity.73,126 Coupled to the activity of CPTI, voltage-dependent anion channels (VDACs) are required to transport acyl-CoA esters through the outer membrane to the intermembrane space. Ethanol and acetaldehyde cause VDACs on hepatocyte mitochondria to close, which also impairs mitochondrial β-oxidation.127,128 Lastly, ethanol exposure damages the mitochondria and leads to mitochondria dysfunction;129 this impact on mitochondrial function can indirectly impair the ability of the organelles to oxidise free FAs. This latter point is likely exacerbated by the impaired autophagy of damaged mitochondria that is associated with alcohol exposure.130
Effect of ethanol exposure on mitochondrial β-oxidation
In summary, the net effect of ethanol is to inhibit mitochondrial β-oxidation by blunting the induction of β-oxidation genes, even in the context of increased FA supply (e.g., via inhibition of PPARα signalling), through potential functional deficiencies in critical cofactors for β-oxidation (e.g., carnitine), directly (e.g., increased NADH malonyl-CoA), and indirectly (via VDAC closure and mitochondrial dysfunction). Ethanol’s myriad of inhibitory effects on mitochondrial β-oxidation likely explain the continued inhibition of this process during chronic ethanol consumption, even after the ratio of NADH:NAD+ appears to normalise.131
Effects of ethanol on cholesterol synthesis and secretion
Another mechanism by which lipids can accumulate in the liver is via alterations in the packaging of triglycerides into lipoproteins to form cholesterol. Some studies have indicated that chronic experimental ethanol impairs hepatic cholesterol synthesis,20,132 whereas others have shown no effect.133,134 However, few studies suggest that hepatic cholesterol synthesis is increased by alcohol. In this context, the lack of response of this system to the increase in lipid flux through the hepatocyte may contribute indirectly to the steatosis caused by ethanol consumption. The rate of cholesterol synthesis and release is controlled predominantly by the supply of apoliprotein B and the activity of microsomal triglyceride transfer protein (MTTP). A key regulator of both processes is hepatocyte growth factor (HGF) signalling via its receptor c-Met.135 The activation of hepatic nuclear receptor 4α (HNF-4α) is also hypothesised to play a key role in this process.136,137
Activation of c-Met by HGF stimulates VLDL synthesis in hepatocytes through upregulation of apoliprotein B synthesis.138 HGF administration has also been shown to enhance the rate of recovery from experimental alcohol-induced fatty liver and is associated with increased synthesis and secretion of apolipoprotein B and subsequent formation of VLDL.139,140 The protective effect of medium chain triglycerides20 and the PPARγ agonist pioglitazone132 are hypothesised to be mediated, at least in part, by enhancing the capacity of hepatocytes to synthesise cholesterol. Enhancing the post-translational formation of HGF has also been shown to be protective against ethanol-induced steatosis. For example, although the canonical role of plasminogen activator inhibitor-1 (PAI-1) is to inhibit fibrinolysis by plasminogen activators, such as urokinase plasminogen activator (uPA),141 uPA also activates pro-HGF to mature HGF.142,143 Indeed, genetic or pharmacologic inhibition of PAI-1 prevents ethanol-induced steatosis, in part, by enhancing HGF-mediated VLDL synthesis.133
It is highly likely that other processes impacted by alcohol exposure (e.g., ER stress48) contribute to altered/impaired VLDL synthesis during ALD. This area of research has been somewhat underappreciated partly because of the difficultly in studying cholesterol metabolism in intact organisms. The advent of more advanced stable isotope labelling approaches and lipidomic analyses may now make this possible.144
Other mechanisms by which ethanol impacts lipid metabolism
Lipocalin-2
Lipocalin-2 is an important innate immune protein belonging to the lipocalin family.145 Emerging evidences demonstrate a pivotal and multifunctional role of lipocalin-2 in the early stages of ALD and in alcoholic steatosis.23,42–44,109,146,147 Ethanol administration in mice or rats markedly increases liver and adipose lipocalin-2 expression and elevates circulating lipocalin-2 levels.23,42–44,109,146,147 In a cellular model of alcoholic steatosis, recombinant lipocalin-2 or over-expression of lipocalin-2 exacerbates the ethanol-induced fat accumulation, whereas knocking down lipocalin-2 prevents steatosis in hepatocytes exposed to ethanol.147 Consistently, global ablation of lipocalin-2 partially but significantly prevents experimental alcoholic fatty liver injury in mice.147 Lipocalin-2 also promotes liver inflammation after alcohol intake by mediating neutrophil infiltration into liver and prolonging neutrophil lifespan in rodents and humans.148 Mechanistically, abnormally elevated lipocalin-2 plays a causative role in the experimental cellular and animal models of alcoholic steatosis by disrupting signalling cascades involved in lipid metabolism, including the phosphoribosyltransferase-SIRT1 axis, chaperone-mediated autophagy, FA oxidation and endocrine metabolic regulatory hepatic FGF15/19 signalling.147
Autophagy
Macroautophagy (herein, referred to as autophagy) is a genetically programmed and highly conserved intracellular lysosomal degradation mechanism.149,150 Autophagy maintains normal cellular functions and regulates lipid homeostasis, including lipid droplet turnover and formation. Aberrant autophagic machinery is associated with the development and progression of AFLD.147,149–161 However, because of the complexity of autophagic machinery and differences in animal AFLD models, experimental findings are controversial. The induction of autophagy by acute ethanol treatment eliminates hepatic intracellular lipid droplets and reduces lipid accumulation in rodents.151–153 However, chronic ethanol administration at higher dosages inhibits autophagy, coupled with accumulation of hepatic triglycerides in mice.147,154,155
In summary, regardless of acute or chronic ethanol exposure in animals, autophagy serves as a cellular adaptive mechanism and protects against ethanol-induced detrimental effects on lipid metabolism by removing lipid droplets and/or damaged mitochondria.147,151–161 Although the mechanisms by which ethanol regulates autophagic machinery are not fully understood, ethanol metabolism-induced oxidative stress is likely to participate in the activation of autophagy.149,150,158 In addition, regulation of autophagy by acute vs. chronic ethanol exposure may be determined by a gene transcription programme in liver.156,157
Circadian clock
The circadian clock regulates circadian rhythms and is maintained by a complex circuitry of transcriptional/translational regulatory loops at molecular levels.162,163 The circadian clock plays an essential role in orchestrating many physiological processes, including lipid metabolism. Derangements in the finely tuned circadian clock can contribute to dyslipidaemia and liver diseases.162,163
Circadian clock disruption is an important contributor to aberrant lipid metabolism and ethanol-induced steatosis.164 Chronic ethanol exposure results in the disturbance of the hepatic circadian clock and time-of-day specific regulation of lipid homeostasis in rodents.165–169 Large time-of day-dependent increases in triglyceride and cholesterol levels have been demonstrated in the livers of mice receiving chronic ethanol-administration.165–169 Changes in the diurnal oscillations of core clock genes (Arntl, Clock, Cry1, Cry2, Per1, Per2) and clock-controlled genes (e.g. Dbp, Hlf, Noct, Npas2, Nr1d1, Tef) were observed in the steatotic livers of ethanol-fed rodents.165 Per1 knockout mice have lower levels of triglyceride synthesis genes following acute alcohol administration.166 Chronic ethanol administration to mice disrupts diurnal rhythms in hepatic lipid metabolism at gene and protein levels.167,168,170 Ethanol-mediated alterations in the hepatic NAD+/NADH ratio are also under clock control.167
The exact underlying mechanisms through which ethanol negatively impacts circadian clock-mediated lipid metabolism and contributes to steatosis, remain to be elucidated. Ethanol-mediated alterations in 2 key energy sensing metabolites, NAD + and ATP, may disturb the liver circadian clock by disrupting post-transcriptional modification events (e.g. acetylation, and ADP-ribosylation, and phosphorylation) mediated by the molecules involved in lipid metabolism (e.g. SIRT1, AMPK and poly ADP-ribose polymerase 1).164,165,167 Further, deciphering the mechanisms that link ethanol, lipid metabolism and circadian responses will provide valuable insights for the development of innovative therapeutic strategies.
Emerging areas
There are several new areas, including long non-coding RNAs, pre-mRNA splicing, and gut micro-biota that deserve further investigation in the context of alcohol and steatosis.
Alternate mRNA processing
A regulatory role for microRNAs in AFLD has been suggested.171 For example, microRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by disrupting the SIRT1-lipin-1 axis.40 Whether and how ethanol-mediated alterations in specific microRNA expression are linked to dysregulated lipid metabolism in alcoholic steatosis will need further investigation. Long noncoding RNAs (lncRNAs) influence lipid homeostasis by controlling the lipid metabolism-related gene expression, either by base-pairing with RNA and DNA or by binding to proteins.172,173 Alterations in lncRNA expression have been linked to a number of liver diseases including ALD.173,174 It is worthwhile exploring whether, and how, ethanol disrupts hepatic IncRNAs and subsequently causes fatty liver injury. Alternative splicing of precursor messenger RNA (pre-mRNA) is a pivotal step in gene expression, eliminating the introns and ligating the exons to form mature mRNAs that can be translated into proteins.175,176 Defects in the pre-mRNA splicing machinery can impact on lipid homeostasis and contribute to steatosis.176–178 Ethanol exposure causes changes in pre-mRNA splicing.179 However, alternative pre-mRNA splicing is an underappreciated mechanism in the pathogenesis of AFLD.180 It will be of importance to investigate whether aberrant splicing machinery contributes to ethanol-mediated dysregulation of lipid metabolism and alcoholic steatosis.
Microbiome
Growing evidence demonstrates the involvement of gut microbiota in the development and progression of ALD.180 The influences of gut microbiota on ethanol-mediated dysregulation of lipid metabolism and the relationship between gut microbiota and AFLD warrant future investigation. Undoubtedly, illuminating the mechanistic connections between these newly understood machineries and ethanol will provide a more cohesive picture of how ethanol deranges hepatic lipid metabolism and results in steatosis and liver injury.
Concluding remarks
Hepatic lipid metabolism is a series of complex processes that control influx and efflux of not only hepatic lipid pools, but also organismal pools. As mentioned, lipid homeostasis is usually tightly controlled by expression, substrate supply, oxidation and secretion that keeps hepatic lipid poolsrelatively constant. However, perturbations of any of these processes can lead to lipid accumulation in the liver. Although it is thought that these responses are hepatic arms of the ‘thrifty genome’, they are maladaptive in the context of chronic fatty liver diseases.181,182 Ethanol is likely unique among toxins, in that it perturbs almost all aspects of hepatic lipid metabolism. This complex response is due in part to the large metabolic demand placed on the organ by alcohol metabolism, but also appears to involve more nuanced changes in expression and substrate supply. The net effect is that steatosis is a rapid response to alcohol abuse. Although transient steatosis is largely an inert pathology, the chronicity of ALD seems to require steatosis. Better and more specific understanding of the mechanisms by which alcohol causes steatosis may therefore translate into targeted therapies to treat ALD and/or prevent its progression.
Supplementary Material
Key point.
The initial hepatic change caused by excessive alcohol consumption is steatosis, which occurs in almost all patients who consume harmful levels of alcohol.
Key point.
There is evidence that stimulation of fatty acid transporters, particularly CD36/FAT, has an important role in alcoholic fatty liver disease.
Key point.
Ethanol activates de novo lipogenesis via a number of processes, leading to lipid accumulation in the liver.
Key point.
The net effect of ethanol is to inhibit mitochondrial β-oxidation, even in the context of increased fatty acid supply, reducing the utilisation of lipid.
Key point.
A number of emerging research areas deserve further investigation in the context of alcohol and steatosis, including long noncoding RNAs, pre-mRNA splicing and gut microbiota.
Financial support
Supported, in part, by grants (AA021978, AA013623 and AA015951) and centers (P50 AA024333 P50 AA024337) funded by National Institute of Alcohol Abuse and Alcoholism (NIAAA, USA).
Footnotes
Conflict of interest
Dr Arteel and Dr You report grants from National Institutes of Health, during the conduct of the study.
Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2018.10.037.
References
- [1].Services USDoHaH. Results from the 2010 National Survey on Drug Use and Health; 2010, 2010.
- [2].Nelson S, Kolls JK. Alcohol, host defence and society. Nat Rev Immunol 2002;2:205–209. [DOI] [PubMed] [Google Scholar]
- [3].Reuben A Alcohol and the liver. Curr Opin Gastroenterol 2007;23:283–291. [DOI] [PubMed] [Google Scholar]
- [4].Draganov P, Durrence H, Cox C, Reuben A. Alcohol-acetaminophen syndrome – even moderate social drinkers are at risk. Postgraduate Med 2000;107:189–195. [DOI] [PubMed] [Google Scholar]
- [5].Ishak KG, Zimmerman HJ, Ray MB. Alcoholic liver disease: pathologic, pathogenetic and clinical aspects. Alcohol Clin Exp Res 1991;15:45–66. [DOI] [PubMed] [Google Scholar]
- [6].Day CP, James OF. Hepatic steatosis: innocent bystander or guilty party? Hepatology 1998;27:1463–1466. [DOI] [PubMed] [Google Scholar]
- [7].Ontko JA. Effects of ethanol on the metabolism of free fatty acids in isolated liver cells. J Lipid Res 1973;14:78–86. [PubMed] [Google Scholar]
- [8].Hagenfeldt L, Wahren J, Pernow B, Raf L. Uptake of individual free fatty acids by skeletal muscle and liver in man. J Clin Invest 1972;51:2324–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aydin A, Sokal JE. Uptake of plasma free fatty acids by the isolated rat liver: effect of glucagon. Am J Physiol 1963;205:667–670. [DOI] [PubMed] [Google Scholar]
- [10].Barrows BR, Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab 2006;91:1446–1452. [DOI] [PubMed] [Google Scholar]
- [11].Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res 2014;53:124–144. [DOI] [PubMed] [Google Scholar]
- [12].He J, Lee JH, Febbraio M, Xie W. The emerging roles of fatty acid translocase/CD36 and the aryl hydrocarbon receptor in fatty liver disease. Exp Biol Med (Maywood) 2011;236:1116–1121. [DOI] [PubMed] [Google Scholar]
- [13].Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 2001;108:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhong W, Zhao Y, Tang Y, Wei X, Shi X, Sun W, et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol 2012;180:998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhou SL, Gordon RE, Bradbury M, Stump D, Kiang CL, Berk PD. Ethanol up-regulates fatty acid uptake and plasma membrane expression and export of mitochondrial aspartate aminotransferase in HepG2 cells. Hepatology 1998;27:1064–1074. [DOI] [PubMed] [Google Scholar]
- [16].Berk PD, Zhou S, Bradbury MW. Increased hepatocellular uptake of long chain fatty acids occurs by different mechanisms in fatty livers due to obesity or excess ethanol use, contributing to development of steatohepatitis in both settings. Trans Am Clin Climatol Assoc 2005;116:335–344. [PMC free article] [PubMed] [Google Scholar]
- [17].Ronis MJ, Hennings L, Stewart B, Basnakian AG, Apostolov EO, Albano E, et al. Effects of long-term ethanol administration in a rat total enteral nutrition model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 2011;300:G109–G119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Clugston RD, Huang LS, Blaner WS. Chronic alcohol consumption has a biphasic effect on hepatic retinoid loss. FASEB J 2015;29:3654–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Clugston RD, Yuen JJ, Hu Y, Abumrad NA, Berk PD, Goldberg IJ, et al. CD36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. J Lipid Res 2014;55:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li Q, Zhong W, Qiu Y, Kang X, Sun X, Tan X, et al. Preservation of hepatocyte nuclear factor-4alpha contributes to the beneficial effect of dietary medium chain triglyceride on alcohol-induced hepatic lipid dyshomeostasis in rats. Alcohol Clin Exp Res 2013;37: 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sun XH, Tang YN, Tan XB, Li Q, Zhong W, Sun XG, et al. Activation of peroxisome proliferator-activated receptor-gamma by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanolfed mice. Am J Physiol-Gastrointestinal Liver Physiol 2012;302: G548–G557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 2003;112:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hu X, Jogasuria A, Wang J, Kim C, Han Y, Shen H, et al. MitoNEET deficiency alleviates experimental alcoholic steatohepatitis in mice by stimulating endocrine adiponectin-Fgf15 axis. J Biol Chem 2016;291:22482–22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003;124:1488–1499. [DOI] [PubMed] [Google Scholar]
- [25].You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem 2002;277:29342–29347. [DOI] [PubMed] [Google Scholar]
- [26].Ji C, Shinohara M, Vance D, Than TA, Ookhtens M, Chan C, et al. Effect of transgenic extrahepatic expression of betaine-homocysteine methyltransferase on alcohol or homocysteine-induced fatty liver. Alcohol Clin Exp Res 2008;32:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liangpunsakul S, Ross RA, Crabb DW. Activation of carbohydrate response element-binding protein by ethanol. J Investig Med 2013;61:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].den Boer M, Voshol PJ, Kuipers F, Havekes LM, Romijn JA. Hepatic steatosis: a mediator of the metabolic syndrome. Lessons from animal models. Arteriosclerosis Thrombosis and Vascular Biology 2004;24:644–649. [DOI] [PubMed] [Google Scholar]
- [29].Bou KM, Blais A, Figeys D, Yao Z. Lipin – the bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochim Biophys Acta 2010;1801:1249–1259. [DOI] [PubMed] [Google Scholar]
- [30].You M, Jogasuria A, Lee K, Wu J, Zhang Y, Lee YK, et al. Signal transduction mechanisms of alcoholic fatty liver disease: emerging role of Lipin-1. Curr Mol Pharmacol 2017;10:226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 2010;40:310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ishimoto K, Nakamura H, Tachibana K, Yamasaki D, Ota A, Hirano K, et al. Sterol-mediated regulation of human lipin 1 gene expression in hepatoblastoma cells. J Biol Chem 2009;284:22195–22205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu M, Wang F, Li X, Rogers CQ, Liang X, Finck BN, et al. Regulation of hepatic lipin-1 by ethanol: role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology 2012;55:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang H, Zhang J, Qiu W, Han GS, Carman GM, Adeli K. Lipin-1gamma isoform is a novel lipid droplet-associated protein highly expressed in the brain. FEBS Lett 2011;585:1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pihlajamaki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab 2011;14:208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Savolainen MJ, Baraona E, Pikkarainen P, Lieber CS. Hepatic triacylglycerol synthesizing activity during progression of alcoholic liver injury in the baboon. J Lipid Res 1984;25:813–820. [PubMed] [Google Scholar]
- [37].Simpson KJ, Venkatesan S, Martin A, Brindley DN, Peters TJ. Activity and subcellular distribution of phosphatidate phosphohydrolase (EC 3.1.3.4) in alcoholic liver disease. Alcohol Alcohol 1995;30:31–36. [PubMed] [Google Scholar]
- [38].Brindley DN, Cooling J, Burditt SL, Pritchard PH, Pawson S, Sturton RG. The involvement of glucocorticoids in regulating the activity of phosphatidate phosphohydrolase and the synthesis of triacylglycerols in the liver. Effects of feeding rats with glucose, sorbitol, fructose, glycerol and ethanol. Biochem J 1979;180:195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shen Z, Liang X, Rogers CQ, Rideout D, You M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 2010;298:G364–G374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem 2012;287:9817–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Everitt H, Hu M, Ajmo JM, Rogers CQ, Liang X, Zhang R, et al. Ethanol administration exacerbates the abnormalities in hepatic lipid oxidation in genetically obese mice. Am J Physiol Gastrointest Liver Physiol 2013;304:G38–G47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hu M, Yin H, Mitra MS, Liang X, Ajmo JM, Nadra K, et al. Hepaticspecific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology 2013;58:1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014;146:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jiang Z, Zhou J, Zhou D, Zhu Z, Sun L, Nanji AA. The adiponectin-SIRT1-AMPK pathway in alcoholic fatty liver disease in the rat. Alcohol Clin Exp Res 2015;39:424–433. [DOI] [PubMed] [Google Scholar]
- [45].Yin H, Liang X, Jogasuria A, Davidson NO, You M. miR-217 regulates ethanol-induced hepatic inflammation by disrupting sirtuin 1-lipin-1 signaling. Am J Pathol 2015;185:1286–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen H, Shen F, Sherban A, Nocon A, Li Y, Wang H, et al. DEP domain-containing mTOR-interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology 2018;68:496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes Metab 2010;12(Suppl 2):83–92. [DOI] [PubMed] [Google Scholar]
- [48].Kaplowitz N, Ji C. Unfolding new mechanisms of alcoholic liver disease in the endoplasmic reticulum. J Gastroenterol Hepatol 2006;21(Suppl 3):S7–S9. [DOI] [PubMed] [Google Scholar]
- [49].McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 1989;9:349–351. [DOI] [PubMed] [Google Scholar]
- [50].Nanji A, Zhoa S, Sadrzadeh S, Waxman D. Use of reverse transcription-polymerase chain reaction to evaluate in vivo cytokin gene expression in rats fed ethanol for long periods. Hepatology 1994;21:1309. [PubMed] [Google Scholar]
- [51].Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG. Antibodies to tumor necrosis factor-a attenuate hepatic necrosis and inflammation due to chronic exposure to ethanol in the rat. Hepatology 1997;26:1530–1537. [DOI] [PubMed] [Google Scholar]
- [52].Ji C, Deng Q, Kaplowitz N. Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology 2004;40:442–451. [DOI] [PubMed] [Google Scholar]
- [53].Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999;117:942–952. [DOI] [PubMed] [Google Scholar]
- [54].Hardardottir I, Doerrler W, Feingold KR, Grunfeld C. Cytokines stimulate lipolysis and decrease lipoprotein lipase activity in cultured fat cells by a prostaglandin independent mechanism. Biochem Biophys Res Commun 1992;186:237–243. [DOI] [PubMed] [Google Scholar]
- [55].Feingold KR, Serio MK, Adi S, Moser AH, Grunfeld C. Tumor necrosis factor stimulates hepatic lipid synthesis and secretion. Endocrinology 1989;124:2336–2342. [DOI] [PubMed] [Google Scholar]
- [56].Nachiappan V, Curtiss D, Corkey BE, Kilpatrick L. Cytokines inhibit fatty acid oxidation in isolated rat hepatocytes: synergy among TNF, IL-6, and IL-1. Shock 1994;1:123–129. [DOI] [PubMed] [Google Scholar]
- [57].Lawler JF Jr, Yin M, Diehl AM, Roberts E, Chatterjee S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J Biol Chem 1998;273:5053–5059. [DOI] [PubMed] [Google Scholar]
- [58].Endo M, Masaki T, Seike M, Yoshimatsu H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c). Exp Biol Med (Maywood) 2007;232:614–621. [PubMed] [Google Scholar]
- [59].Navasa M, Gordon DA, Hariharan N, Jamil H, Shigenaga JK, Moser A, et al. Regulation of microsomal triglyceride transfer protein mRNA expression by endotoxin and cytokines. J Lipid Res 1998;39:1220–1230. [PubMed] [Google Scholar]
- [60].Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest 1997;99: 2416–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab 2011;96:1424–1430. [DOI] [PubMed] [Google Scholar]
- [62].Zhang W, Sun Q, Zhong W, Sun X, Zhou Z. Hepatic peroxisome proliferator-activated receptor gamma signaling contributes to alcohol-induced hepatic steatosis and inflammation in mice. Alcohol Clin Exp Res 2016;40:988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bouskila M, Pajvani UB, Scherer PE. Adiponectin: a relevant player in PPARgamma-agonist-mediated improvements in hepatic insulin sensitivity? Int J Obes (Lond) 2005;29(Suppl 1):S17–S23. [DOI] [PubMed] [Google Scholar]
- [64].Wang Y, Lam KS, Yau MH, Xu A. Post-translational modifications of adiponectin: mechanisms and functional implications. Biochem J 2008;409:623–633. [DOI] [PubMed] [Google Scholar]
- [65].Enomoto N, Takei Y, Hirose M, Konno A, Shibuya T, Matsuyama S, et al. Prevention of ethanol-induced liver injury in rats by an agonist of peroxisome proliferator-activated receptor-gamma, pioglitazone. J Pharmacol Exp Ther 2003;306:846–854. [DOI] [PubMed] [Google Scholar]
- [66].Rogers CQ, Ajmo JM, You M. Adiponectin and alcoholic fatty liver disease. IUBMB Life 2008;60:790–797. [DOI] [PubMed] [Google Scholar]
- [67].Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2008;8:774–785. [DOI] [PubMed] [Google Scholar]
- [68].Krause U, Bertrand L, Hue L. Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur J Biochem 2002;269:3751–3759. [DOI] [PubMed] [Google Scholar]
- [69].Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Phys (Bethesda, Md: 1985) 2002;92:2475–2482. [DOI] [PubMed] [Google Scholar]
- [70].Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem 2002;277:3829–3835. [DOI] [PubMed] [Google Scholar]
- [71].Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011;13:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol 2004;287:G1–6. [DOI] [PubMed] [Google Scholar]
- [73].You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004;127:1798–1808. [DOI] [PubMed] [Google Scholar]
- [74].Supakul R, Liangpunsakul S. Alcoholic-induced hepatic steatosis-role of ceramide and protein phosphatase 2A. Trans Res 2011;158:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yang L, Jin GH, Zhou JY. The role of ceramide in the pathogenesis of alcoholic liver disease. Alcohol Alcohol 2016;51:251–257. [DOI] [PubMed] [Google Scholar]
- [76].Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol 2008;295:G1173–G1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem 2010;285:33959–33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].You M, Jogasuria A, Taylor C, Wu J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr 2015;4:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bouras T, Fu MF, Sauve AA, Wang F, Quong AA, Perkins ND, et al. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem 2005;280:10264–10276. [DOI] [PubMed] [Google Scholar]
- [80].Wang RH, Li C, Deng CX. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int J Biol Sci 2010;6:682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lieber CS, Leo MA, Wang X, Decarli LM. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun 2008;370:44–48. [DOI] [PubMed] [Google Scholar]
- [82].Revollo JR, Li X. The ways and means that fine tune Sirt1 activity. Trends Biochem Sci 2013;38:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab 2010;298:E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Krijgsheld KR, Scholtens E, Mulder GJ. An evaluation of methods to decrease the availability of inorganic sulphate for sulphate conjugation in the rat in vivo. Biochem Pharmacol 1981;30:1973–1979. [DOI] [PubMed] [Google Scholar]
- [85].Henstridge DC, Whitham M, Febbraio MA. Chaperoning to the metabolic party: the emerging therapeutic role of heat-shock proteins in obesity and type 2 diabetes. Mol Metab 2014;3:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kuan YC, Hashidume T, Shibata T, Uchida K, Shimizu M, Inoue J, et al. Heat Shock protein 90 modulates lipid homeostasis by regulating the stability and function of sterol regulatory element-binding protein (SREBP) and SREBP cleavage-activating protein. J Biol Chem 2017;292:3016–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mandrekar P Signaling mechanisms in alcoholic liver injury: role of transcription factors, kinases and heat shock proteins. World J Gastroenterol 2007;13:4979–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Porras N, Strauss M, Rodriguez M, Anselmi G. Hsp70 accumulation and ultrastructural features of lung and liver induced by ethanol treatment with and without L-carnitine protection in rats. Exp Toxicol Pathol 2006;57:227–237. [DOI] [PubMed] [Google Scholar]
- [89].Mikami T, Sumida S, Ishibashi Y, Ohta S. Endurance exercise training inhibits activity of plasma GOT and liver caspase-3 of mice [correction of rats] exposed to stress by induction of heat shock protein 70. J Appl Physiol (Bethesda, Md: 1985) 2004;96:1776–1781. [DOI] [PubMed] [Google Scholar]
- [90].Ikeyama S, Kusumoto K, Miyake H, Rokutan K, Tashiro S. A non-toxic heat shock protein 70 inducer, geranylgeranylacetone, suppresses apoptosis of cultured rat hepatocytes caused by hydrogen peroxide and ethanol. J Hepatol 2001;35:53–61. [DOI] [PubMed] [Google Scholar]
- [91].Yao X, Bai Q, Yan D, Li G, Lu C, Xu H. Solanesol protects human hepatic L02 cells from ethanol-induced oxidative injury via upregulation of HO-1 and Hsp70. Toxicol In Vitro 2015;29:600–608. [DOI] [PubMed] [Google Scholar]
- [92].Kitam VO, Maksymchuk OV, Chashchyn MO. The possible mechanisms of CYP2E1 interactions with HSP90 and the influence of ethanol on them. BMC Struct Biol 2012;12:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Islam A, Abraham P, Hapner CD, Deuster PA, Chen Y. Tissue-specific upregulation of HSP72 in mice following short-term administration of alcohol. Cell Stress Chaperones 2013;18:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bukong TN, Hou W, Kodys K, Szabo G. Ethanol facilitates hepatitis C virus replication via up-regulation of GW182 and heat shock protein 90 in human hepatoma cells. Hepatology 2013;57:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther 2005;315:8–15. [DOI] [PubMed] [Google Scholar]
- [96].Mandrekar P, Catalano D, Jeliazkova V, Kodys K. Alcohol exposure regulates heat shock transcription factor binding and heat shock proteins 70 and 90 in monocytes and macrophages: implication for TNF-alpha regulation. J Leukoc Biol 2008;84:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ambade A, Catalano D, Lim A, Kopoyan A, Shaffer SA, Mandrekar P. Inhibition of heat shock protein 90 alleviates steatosis and macrophage activation in murine alcoholic liver injury. J Hepatol 2014;61:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kang X, Petyaykina K, Tao R, Xiong X, Dong XC, Liangpunsakul S. The inhibitory effect of ethanol on Sestrin3 in the pathogenesis of ethanol-induced liver injury. Am J Physiol Gastrointest Liver Physiol 2014;307: G58–G65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tao C, Sifuentes A, Holland WL. Regulation of glucose and lipid homeostasis by adiponectin: effects on hepatocytes, pancreatic beta cells and adipocytes. Best Pract Res Clin Endocrinol Metab 2014;28:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].You M, Rogers CQ. Adiponectin: a key adipokine in alcoholic fatty liver. Exp Biol Med (Maywood) 2009;234:850–859. [DOI] [PubMed] [Google Scholar]
- [101].Markan KR, Potthoff MJ. Metabolic fibroblast growth factors (FGFs): Mediators of energy homeostasis. Semin Cell Dev Biol 2016;53:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wang M, Zhang XJ, Feng K, He CW, Li P, Hu YJ, et al. Dietary alpha-linolenic acid-rich flaxseed oil prevents against alcoholic hepatic steatosis via ameliorating lipid homeostasis at adipose tissue-liver axis in mice. Sci Rep 2016;6:26826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Correnti JM, Juskeviciute E, Swarup A, Hoek JB. Pharmacological ceramide reduction alleviates alcohol-induced steatosis and hepatomegaly in adiponectin knockout mice. Am J Physiol Gastrointest Liver Physiol 2014;306:G959–G973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Shearn CT, Smathers RL, Jiang H, Orlicky DJ, Maclean KN, Petersen DR. Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J Nutr Biochem 2013;24:1436–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Esfandiari F, You M, Villanueva JA, Wong DH, French SW, Halsted CH. S-adenosylmethionine attenuates hepatic lipid synthesis in micropigs fed ethanol with a folate-deficient diet. Alcohol Clin Exp Res 2007;31:1231–1239. [DOI] [PubMed] [Google Scholar]
- [106].Xu J, Lai KKY, Verlinsky A, Lugea A, French SW, Cooper MP, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol 2011;55:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ge H, Zhang J, Gong Y, Gupte J, Ye J, Weiszmann J, et al. Fibroblast growth factor receptor 4 (FGFR4) deficiency improves insulin resistance and glucose metabolism under diet-induced obesity conditions. J Biol Chem 2014;289:30470–30480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Luo Y, Yang C, Ye M, Jin C, Abbruzzese JL, Lee MH, et al. Deficiency of metabolic regulator FGFR4 delays breast cancer progression through systemic and microenvironmental metabolic alterations. Cancer Metab 2013;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Wang J, Kim C, Jogasuria A, Han Y, Hu X, Wu J, et al. Myeloid cell-specific Lipin-1 deficiency stimulates endocrine adiponectin-FGF15 axis and ameliorates ethanol-induced liver injury in mice. Sci Rep 2016;6:34117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther 1995;67:101–154. [DOI] [PubMed] [Google Scholar]
- [113].Crabb DW, Galli A, Fischer M, You M. Molecular mechanisms of alcoholic fatty liver: role of peroxisome proliferator-activated receptor alpha. Alcohol 2004;34:35–38. [DOI] [PubMed] [Google Scholar]
- [114].Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med 2003;3:561–572. [DOI] [PubMed] [Google Scholar]
- [115].Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 1998;273:5678–5684. [DOI] [PubMed] [Google Scholar]
- [116].Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem 2003;278:27997–28004. [DOI] [PubMed] [Google Scholar]
- [117].Patek AJ Jr. Alcohol, malnutrition, and alcoholic cirrhosis. Am J Clin Nutr 1979;32:1304–1312. [DOI] [PubMed] [Google Scholar]
- [118].Bujanda L The effects of alcohol consumption upon the gastrointestinal tract. Am J Gastroenterol 2000;95:3374–3382. [DOI] [PubMed] [Google Scholar]
- [119].Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q. Role of carnitine in disease. Nutr Metab (Lond) 2010;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sachan DS, Rhew TH, Ruark RA. Ameliorating effects of carnitine and its precursors on alcohol-induced fatty liver. Am J Clin Nutr 1984;39:738–744. [DOI] [PubMed] [Google Scholar]
- [121].Bykov I, Jarvelainen H, Lindros K. L-carnitine alleviates alcohol-induced liver damage in rats: role of tumour necrosis factor-alpha. Alcohol Alcohol 2003;38:400–406. [DOI] [PubMed] [Google Scholar]
- [122].Alonso dlP, Rozas I, Alvarez-Prechous A, Pardinas MC, Paz JM, Rodriguez-Segade S. Free carnitine and acylcarnitine levels in sera of alcoholics. Biochem Med Metab Biol 1990;44:77–83. [DOI] [PubMed] [Google Scholar]
- [123].Fuller RK, Hoppel CL. Plasma carnitine in alcoholism. Alcohol Clin Exp Res 1988;12:639–642. [DOI] [PubMed] [Google Scholar]
- [124].Kepka A, Waszkiewicz N, Zalewska-Szajda B, Chojnowska S, Pludowski P, Konarzewska E, et al. Plasma carnitine concentrations after chronic alcohol intoxication. Postepy Hig Med Dosw (Online) 2013;67:548–552. [DOI] [PubMed] [Google Scholar]
- [125].Latipaa PM, Karki TT, Hiltunen JK, Hassinen IE. Regulation of palmitoylcarnitine oxidation in isolated rat liver mitochondria. Role of the redox state of NAD(H). Biochim Biophys Acta 1986;875:293–300. [DOI] [PubMed] [Google Scholar]
- [126].Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest 2012;122:1958–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Holmuhamedov EL, Czerny C, Beeson CC, Lemasters JJ. Ethanol suppresses ureagenesis in rat hepatocytes: role of acetaldehyde. J Biol Chem 2012;287:7692–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator–thinking outside the box. Biochim Biophys Acta 2006;1762:181–190. [DOI] [PubMed] [Google Scholar]
- [129].Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Role of mitochondria in alcoholic liver disease. Curr Pathobiol Rep 2013;1:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Williams JA, Ding WX. A mechanistic review of mitophagy and its role in protection against alcoholic liver disease. Biomolecules 2015;5:2619–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Salaspuro MP, Shaw S, Jayatilleke E, Ross WA, Lieber CS. Attenuation of the ethanol-induced hepatic redox change after chronic alcohol consumption in baboons: metabolic consequences in vivo and in vitro. Hepatology 1981;1:33–38. [DOI] [PubMed] [Google Scholar]
- [132].Tomita K, Azuma T, Kitamura N, Nishida J, Tamiya G, Oka A, et al. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology 2004;126:873–885. [DOI] [PubMed] [Google Scholar]
- [133].Bergheim I, Guo L, Davis MA, Lambert JC, Beier JI, Duveau I, et al. Metformin prevents alcohol-induced liver injury in the mouse: critical role of plasminogen activator inhibitor-1. Gastroenterology 2006;130:2099–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Lieber CS. Interference of ethanol in hepatic cellular metabolism. Ann N Y Acad Sci 1975;252:24–50. [DOI] [PubMed] [Google Scholar]
- [135].Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991;251:802–804. [DOI] [PubMed] [Google Scholar]
- [136].Sheena V, Hertz R, Nousbeck J, Berman I, Magenheim J, Bar-Tana J. Transcriptional regulation of human microsomal triglyceride transfer protein by hepatocyte nuclear factor-4alpha. J Lipid Res 2005;46:328–341. [DOI] [PubMed] [Google Scholar]
- [137].Yu D, Chen G, Pan M, Zhang J, He W, Liu Y, et al. High fat diet-induced oxidative stress blocks hepatocyte nuclear factor 4alpha and leads to hepatic steatosis in mice. J Cell Physiol 2017. [DOI] [PubMed] [Google Scholar]
- [138].Kaibori M, Kwon AH, Oda M, Kamiyama Y, Kitamura N, Okumura T. Hepatocyte growth factor stimulates synthesis of lipids and secretion of lipoproteins in rat hepatocytes. Hepatology 1998;27:1354–1361. [DOI] [PubMed] [Google Scholar]
- [139].Tahara M, Matsumoto K, Nukiwa T, Nakamura T. Hepatocyte growth factor leads to recovery from alcohol-induced fatty liver in rats. J Clin Invest 1999;103:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Sugimoto T, Yamashita S, Ishigami M, Sakai N, Hirano K, Tahara M, et al. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. J Hepatol 2002;36:157–162. [DOI] [PubMed] [Google Scholar]
- [141].Kruithof EK. Plasminogen activator inhibitors–a review. Enzyme 1988;40:113–121. [DOI] [PubMed] [Google Scholar]
- [142].Naldini L, Vigna E, Bardelli A, Follenzi A, Galimi F, Comoglio PM. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. J Biol Chem 1995;270:603–611. [DOI] [PubMed] [Google Scholar]
- [143].Taniyama Y, Morishita R, Nakagami H, Moriguchi A, Sakonjo H, Shokei K, et al. Potential contribution of a novel antifibrotic factor, hepatocyte growth factor, to prevention of myocardial fibrosis by angiotensin II blockade in cardiomyopathic hamsters. Circulation 2000;102:246–252. [DOI] [PubMed] [Google Scholar]
- [144].Wang HY, Quan C, Hu C, Xie B, Du Y, Chen L, et al. A lipidomics study reveals hepatic lipid signatures associating with deficiency of the LDL receptor in a rat model. Biology open 2016;5:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Asimakopoulou A, Weiskirchen S, Weiskirchen R. Lipocalin 2 (LCN2) expression in hepatic malfunction and therapy. Front Physiol 2016;7:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Glavind E, Vilstrup H, Gronbaek H, Hamilton-Dutoit S, Magnusson NE, Thomsen KL. Long-term ethanol exposure decreases the endotoxin-induced hepatic acute phase response in rats. Alcohol Clin Exp Res 2017;41:562–570. [DOI] [PubMed] [Google Scholar]
- [147].Cai Y, Jogasuria A, Yin H, Xu MJ, Hu X, Wang J, et al. The detrimental role played by lipocalin-2 in alcoholic fatty liver in mice. Am J Pathol 2016;186:2417–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wieser V, Tymoszuk P, Adolph TE, Grander C, Grabherr F, Enrich B, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol 2016;64:872–880. [DOI] [PubMed] [Google Scholar]
- [149].Wang L, Khambu B, Zhang H, Yin XM. Autophagy in alcoholic liver disease, self-eating triggered by drinking. Clin Res Hepatol Gastroenterol 2015;39(Suppl 1):S2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Manley S, Ding W. Role of farnesoid X receptor and bile acids in alcoholic liver disease. Acta Pharm Sin B 2015;5:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ding WX, Li M, Yin XM. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 2011;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010;139:1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol 2013;183:1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 2013;58:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Eid N, Ito Y, Maemura K, Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J Mol Histol 2013;44:311–326. [DOI] [PubMed] [Google Scholar]
- [156].Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr. Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB. Alcohol Clin Exp Res 2015;39:2354–2363. [DOI] [PubMed] [Google Scholar]
- [157].Li Y, Ding WX. A gene transcription program decides the differential regulation of autophagy by acute versus chronic ethanol? Alcohol Clin Exp Res 2016;40:47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lu Y, Cederbaum AI. Autophagy protects against CYP2E1/chronic ethanol-induced hepatotoxicity. Biomolecules 2015;5:2659–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Tang L, Yang F, Fang Z, Hu C. Resveratrol ameliorates alcoholic fatty liver by inducing autophagy. Am J Chin Med 2016;44:1207–1220. [DOI] [PubMed] [Google Scholar]
- [160].Kong X, Yang Y, Ren L, Shao T, Li F, Zhao C, et al. Activation of autophagy attenuates EtOH-LPS-induced hepatic steatosis and injury through MD2 associated TLR4 signaling. Sci Rep 2017;7:9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Cho HI, Seo MJ, Lee SM. 2-Methoxyestradiol protects against ischemia/reperfusion injury in alcoholic fatty liver by enhancing sirtuin 1-mediated autophagy. Biochem Pharmacol 2017;131:40–51. [DOI] [PubMed] [Google Scholar]
- [162].Mayeuf-Louchart A, Zecchin M, Staels B, Duez H. Circadian control of metabolism and pathological consequences of clock perturbations. Biochimie 2017;143:42–50. [DOI] [PubMed] [Google Scholar]
- [163].Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab 2011;13:125–137. [DOI] [PubMed] [Google Scholar]
- [164].Udoh US, Valcin JA, Gamble KL, Bailey SM. The molecular circadian clock and alcohol-induced liver injury. Biomolecules 2015;5:2504–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Filiano AN, Millender-Swain T, Johnson R Jr, Young ME, Gamble KL, Bailey SM. Chronic ethanol consumption disrupts the core molecular clock and diurnal rhythms of metabolic genes in the liver without affecting the suprachiasmatic nucleus. PLoS ONE 2013;8 e71684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Wang T, Yang P, Zhan Y, Xia L, Hua Z, Zhang J. Deletion of circadian gene Per1 alleviates acute ethanol-induced hepatotoxicity in mice. Toxicology 2013;314:193–201. [DOI] [PubMed] [Google Scholar]
- [167].Zhou P, Ross RA, Pywell CM, Liangpunsakul S, Duffield GE. Disturbances in the murine hepatic circadian clock in alcohol-induced hepatic steatosis. Sci Rep 2014;4:3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Udoh US, Swain TM, Filiano AN, Gamble KL, Young ME, Bailey SM. Chronic ethanol consumption disrupts diurnal rhythms of hepatic glycogen metabolism in mice. Am J Physiol Gastrointest Liver Physiol 2015;308, G964–G974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Tran M, Yang Z, Liangpunsakul S, Wang L. Metabolomics analysis revealed distinct cyclic changes of metabolites altered by chronic ethanol-plus-binge and Shp deficiency. Alcohol Clin Exp Res 2016;40:2548–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Summa KC, Voigt RM, Forsyth CB, Shaikh M, Cavanaugh K, Tang Y, et al. Disruption of the circadian clock in mice increases intestinal permeability and promotes alcohol-induced hepatic pathology and inflammation. PLoS ONE 2013;8 e67102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Natarajan SK, Pachunka JM, Mott JL. Role of microRNAs in alcohol-induced multi-organ injury. Biomolecules 2015;5:3309–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Progress Chen Z. and prospects of long noncoding RNAs in lipid homeostasis. Mol Metab 2016;5:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Smekalova EM, Kotelevtsev YV, Leboeuf D, Shcherbinina EY, Fefilova AS, Zatsepin TS, et al. lncRNA in the liver: prospects for fundamental research and therapy by RNA interference. Biochimie 2016;131:159–172. [DOI] [PubMed] [Google Scholar]
- [174].Mayfield RD. Emerging roles for ncRNAs in alcohol use disorders. Alcohol 2017;60:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Calandra S, Tarugi P, Bertolini S. Altered mRNA splicing in lipoprotein disorders. Curr Opin Lipidol 2011;22:93–99. [DOI] [PubMed] [Google Scholar]
- [176].Elizalde M, Urtasun R, Azkona M, Latasa MU, Goni S, Garcia-Irigoyen O, et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J Clin Invest 2014;124:2909–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Sen S, Jumaa H, Webster NJG. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun 2013;4:1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Sen S, Langiewicz M, Jumaa H, Webster NJ. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015;61:171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol Cell 2007;27:928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Starkel P, Schnabl B. Bidirectional communication between liver and gut during alcoholic liver disease. Semin Liver Dis 2016;36: 331–339. [DOI] [PubMed] [Google Scholar]
- [181].Chakravarthy MV, Booth FW. Eating exercise, and, “thrifty” genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J Appl Phys (Bethesda, Md: 1985) 2004;96:3–10. [DOI] [PubMed] [Google Scholar]
- [182].van Ginneken VJ. Liver fattening during feast and famine: an evolutionary paradox. Med Hypotheses 2008;70:924–928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.