

Abstract
Purpose:
To estimate medical device utilization needed to detect safety differences among implantable cardioverter defibrillators (ICDs) generator models and compare these estimates to utilization in practice.
Methods:
We conducted repeated sample size estimates to calculate the medical device utilization needed, systematically varying device‐specific safety event rate ratios and significance levels while maintaining 80% power, testing 3 average adverse event rates (3.9, 6.1, and 12.6 events per 100 person‐years) estimated from the American College of Cardiology’s 2006 to 2010 National Cardiovascular Data Registry of ICDs. We then compared with actual medical device utilization.
Results:
At significance level 0.05 and 80% power, 34% or fewer ICD models accrued sufficient utilization in practice to detect safety differences for rate ratios <1.15 and an average event rate of 12.6 events per 100 person‐years. For average event rates of 3.9 and 12.6 events per 100 person‐years, 30% and 50% of ICD models, respectively, accrued sufficient utilization for a rate ratio of 1.25, whereas 52% and 67% for a rate ratio of 1.50. Because actual ICD utilization was not uniformly distributed across ICD models, the proportion of individuals receiving any ICD that accrued sufficient utilization in practice was 0% to 21%, 32% to 70%, and 67% to 84% for rate ratios of 1.05, 1.15, and 1.25, respectively, for the range of 3 average adverse event rates.
Conclusions:
Small safety differences among ICD generator models are unlikely to be detected through routine surveillance given current ICD utilization in practice, but large safety differences can be detected for most patients at anticipated average adverse event rates.
Keywords: implantable defibrillators, medical devices, pharmacoepidemiology, postmarketing product surveillance, sample size
1 |. INTRODUCTION
The Food and Drug Administration’s (FDA) developed an automated medical product surveillance system known as the Sentinel Initiative, consisting of a distributed database network of 18 health systems.1 While the Sentinel Initiative has been successful in performing analyses to investigate drug safety, a real‐time system that proactively monitors all approved drugs and devices has not been developed, but remains an aspiration.2,3
Surveillance methods that mirror automated systems have been developed for medical device surveillance that leverage medical product registry data,4–8 and FDA recently supported planning of the National Evaluation System for health Technology9 and proposed development of the National Medical Evidence Generation Collaborative.10 Key questions remain as to the potential effectiveness of a real‐time active surveillance system to detect safety differences among medical products. An effective system would provide timely access to data on medical product utilization and patient outcomes but would also need to capture sufficient medical product utilization to allow accurate comparisons between devices. Recently published analyses demonstrated that the Sentinel Initiative will have limited ability to detect large safety differences among drugs with background adverse event rates of 1/1000 person‐ years or lower, predominantly because of insufficient product utilization.11 The sample size estimates needed for postmarket surveillance initiatives to detect safety differences between medical devices, which are generally used less often than drugs, have not been characterized.
To better understand the expectations and limitations of a real‐time active surveillance system for postmarket medical device surveillance, we estimated the utilization needed to detect safety differences among implantable cardioverter defibrillators (ICDs) and compared these estimates to actual utilization in the United States from 2006 through 2010. Implantable cardioverter defibrillators offer a particularly relevant case study for this project. First, they represent a complex treatment requiring permanent implantation. Second, their use is relatively common12 in comparison to other implantable medical devices, as they are recommended for patients with heart failure and severe left ventricular systolic dysfunction, as well as for those who have sustained prior ventricular arrhythmias or cardiac arrest, to reduce mortality risk associated with these conditions.13 Finally, ICD failures are clinically important, as the risk of device failure includes death and device replacement involves significant risks and high costs. We conducted repeated sample size estimates to calculate the ICD utilization that would be required to detect various magnitudes of safety differences among device models. We then compared the sample size estimate to the actual medical device utilization from the American College of Cardiology’s National Cardiovascular Data Registry (NCDR) of ICDs, with the goal of informing expectations for ongoing and future medical device safety surveillance initiatives among regulators, manufacturers, and the public.
2 |. METHODS
2.1 |. Data source
To determine actual ICD utilization in practice, we used NCDR‐ICD data that include information on ICD implantations from January 1, 2006 through March 31, 2010 among all hospitals that perform the procedure in the United States, representing 100% of primary prevention implants reimbursed by Medicare, as well as approximately 80% of secondary prevention implants.14,15 These data include date for each ICD implant; ICD type implanted, including the ICD generator manufacturer and model, along with patient demographics and clinical comorbidities; the episode of care and procedure information; and postprocedure events and complications prior to discharge.14 NCDR‐ICD uses a multifaceted program to enhance data quality.15 The 2006 through 2010 years of data were used in part because they have been uniquely linked with Medicare fee‐for‐service claims data, offering longitudinal follow‐up that provides estimates of patient outcomes.
2.2 |. ICD generator models
To ensure the accuracy of assigned ICD type, manufacturer, and model, for each ICD generator implanted during the study period, the device type (single‐chamber, dual‐chamber, and cardiac resynchronization therapy‐defibrillator) and listed manufacturer and model name and number within the NCDR‐ICD registry were reviewed and verified, as described in prior work.16 Once accuracy of the ICD type, manufacturer, and model were confirmed, a study ID for the model was generated and used to ensure that the study team was blinded to the device manufacturer and model in accordance with our Data Use Agreement with the American College of Cardiology. Given our interest in medical device utilization needed to detect safety differences among device models, only models with at least 20 implantations were included in our study.
2.3 |. ICD adverse event and complication rates
To establish average rates of adverse events and complications for ICDs, we used event rates published by Ranasinghe et al,17 who studied initial ICD implantations from the NCDR‐ICD registry linked with Medicare fee‐for‐service claims data. Over a median follow‐up of 2.7 years, Ranasinghe et al observed 12.6 deaths per 100 person‐years of follow‐up. Furthermore, after accounting for risk for death, Ranasinghe et al observed 6.1 ICD‐related complications per 100 person‐years that required reoperation or hospitalization and 3.9 reoperations per 100 person‐years for reasons other than complications. While Medicare beneficiaries may have higher rates of safety events than younger patients receiving ICDs, they represent nearly two‐thirds of patients undergoing ICD implantation whose data are included in the NCDR‐ICD registry.18 Furthermore, long‐term data on younger patients are not available and we tested a range of rates, examining 3 reasonably estimated, referent adverse event and complication rates to provide insights into the impact of the anticipated safety event rate on estimates of the ICD utilization needed to detect differences. For our analyses, these rates are less important than the use of reasonable referent rates.
2.4 |. Statistical analysis
For our repeated sample size estimates, we calculated estimates of the sample number n of implanted individuals who would need to be followed for a 2‐year period to detect risk associated with receiving one specific ICD generator model relative to a contemporaneous control group of individuals receiving any of the other ICD models, censoring at the time of event. The 2‐year time period was used to conform with previously published estimates of ICD adverse event and complication rates.17 To estimate the medical device utilization needed to detect safety differences among ICD models, we set power to 80% (1 − β=0.80) and used a 2‐sided 5% significance level (α=0.05). We examined 3 rates of adverse events and complications for ICDs: 3.9, 6.1, and 12.6 events per 100 person‐years, as described above. We examined a range of safety difference magnitudes among device models, using rate ratios ranging from 0.50 (50% lower rates) to 2.0 (2‐fold higher rates).
Surveillance scenario parameters that we considered included the adverse event and complication rate and rate ratio, as described above (Table 1). Additionally, to better understand the impact of accepting higher chances of detecting false differences when the ICD model safety is identical on safety surveillance, we repeated all analyses using 2‐sided significance thresholds of 10% and 20% (α=0.10, 0.20).
TABLE 1.
Sample size parameters used
| Parameter | Values Considered |
|---|---|
| Adverse event and complication rate γ0 | 3.9, 6.1, 12.6 events per 100 person‐years |
| Significance α | 0.05, 0.10, 0.20 |
| Rate ratio ρ = γ1/γ0 | 1.05, 1.15, 1.25, 1.5, 2 |
| Power 1 - β | 0.80 |
| Per-person exposure time t0 in years | 2 |
Details of the estimated utilization needed to detect safety differences for a single ICD model are as follows. First, we assumed that all patients who receive the ICD model are prospectively matched 1:1 to a control group from all other individuals who underwent implantation of the same ICD type (single‐chamber, dual‐chamber, or cardiac resynchronization therapy‐defibrillator). Each individual is followed for a minimum of 2 years after receiving the implant, approximating the adverse event and complication rates characterized by Ranasinghe et al. We assumed that the number of adverse events in those 2 years followed a Poisson distribution with rate γ1 for the treatment of interest and γ0 for the control. Moreover, we assumed that the Poisson distributions were independent. The total exposure times for treatment (the ICD model of interest) and control (all other ICDs implanted) groups are denoted t1 and t0, respectively. We assumed t1 = t0 = 2n years, where n is the sample size. The criterion corresponding to a test of the hypothesis H0 : γ0 = γ1 against HA : γ0 ≠ γ1 based on Huffman’s asymptotic test is given by
for event rate ratio ρ = γ1/γ0 and d = t1/t0.19 In our scenario d= 1. We chose to use Huffman’s asymptotic test, since in a Monte Carlo simulation study, it was found to achieve closest to nominal significance and power relative to other asymptotic tests.20
After estimating the number of ICD implantations needed, we visualized the relationship between the estimated and actual utilization in practice of each ICD model across different combinations of surveillance scenario parameters. First, we graphed a timeline of time to accrual of actual utilization needed to detect safety differences for all ICDs, marking both the date of the first implant and the date that the utilization threshold is reached. Second, we plotted a histogram of the time to accrual of actual utilization needed (in years) for all ICDs. Third, we plotted over time the number of individuals who were implanted with an ICD model that accrued sufficient actual utilization as of 2 years’ postdata availability.
A copy of our original study proposal is available in the appendix. Analyses were conducted in Python 3.5 (Python Software Foundation), and the code for this work is available at https://github.com/JonathanRBates/PostmarketSurveillance/tree/master/SampleSizeProject.
3 |. RESULTS
From January 1, 2006 through March 31, 2010, 141 ICD models were implanted at least 20 times, accounting for 523,529 total procedures (Figures 1 and 2). Among the 141 ICD models, the median number of implants was 1053 (IQR: 176–4132). We focus the following results on analyses for the traditional significance level of 0.05; those based on significance levels of 0.10 and 0.20 are provided in the Appendix Table 1.
FIGURE 1.
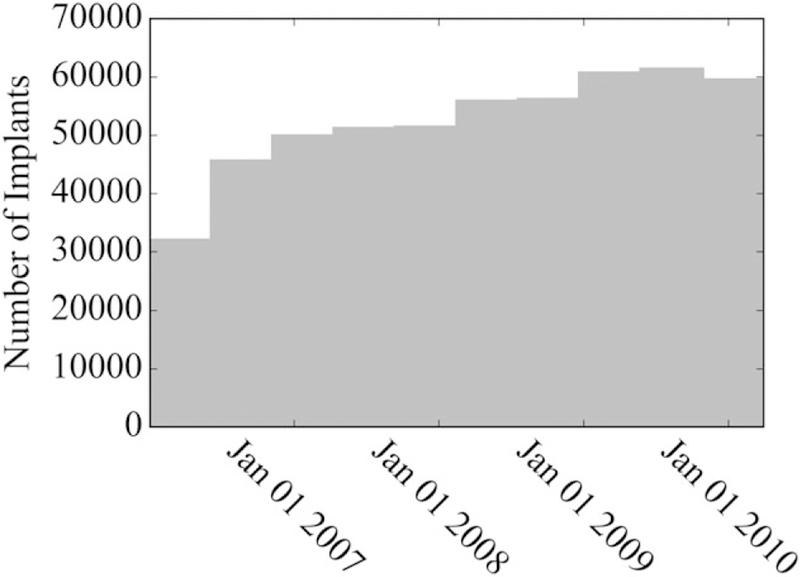
Overall utilization of implantable cardioverter defibrillators observed within the National Cardiovascular Data Registry, January 1, 2006 through March 31, 2010
FIGURE 2.
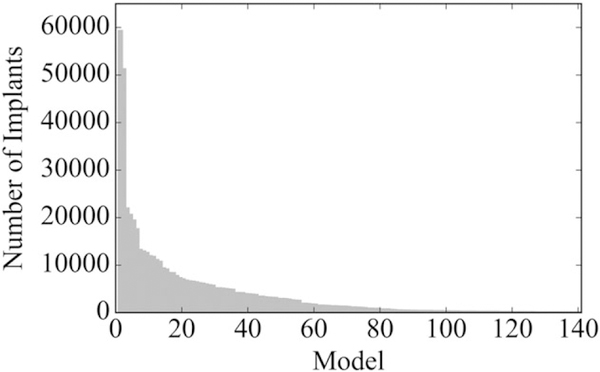
Utilization of each implantable cardioverter defibrillator model observed within the National Cardiovascular Data Registry, January 1, 2006 through March 31, 2010 (n = 141 unique models)
Figure 3 illustrates the estimated utilization needed to detect safety differences among ICD models with respect to the rate ratio scenarios considered for each of the 3 adverse event and complication rates. Qualitatively, as expected, there is a vertical asymptote where the rate ratio is 1.0; between rate ratios of 1.0 and 1.25, approximately, the estimated utilization needed drops sharply; and for larger rate ratios, drops more moderately thereafter. Thus, to detect rate ratios near 1.0, the adverse event and complication rates strongly influence the estimated utilization needed. For larger rate ratios, the difference in estimated utilization needed is less pronounced across the adverse event and complication rates under consideration. Exact estimates of the utilization needed to detect safety differences among ICD models are provided in Table 2.
FIGURE 3.
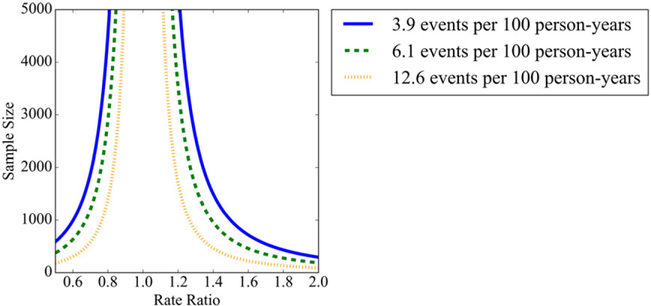
Sample size requirements needed to detect safety performance differences among implantable cardioverter defibrillators at varying rate ratios at 80% power for the following adverse event and complication rates: 3.9 (solid blue), 6.1 (dashed green), and 12.6 (dotted orange) events per 100 person‐years [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 2.
The percentage of ICD models that exceeded, and the percentage of patients who received ICD models that exceeded, and the estimated utilization required to detect safety difference for multiple safety rate ratios and adverse event (AE) and complication rates
| Safety Rate Ratio ρ | AE and Complication Rate, Reference γ0 | AE and Complication Rate, ICD Under Surveillance γ10 | Estimated Sample Size Required n (α = 0.05) | Percentage of ICD models With Utilization Exceeding Estimate | Percentage of Patients Who Received ICD Models With Utilization Exceeding Estimate |
|---|---|---|---|---|---|
| 1.05 | 3.9 | 4.10 | 82 502 | 0% | 0% |
| 1.05 | 6.1 | 6.41 | 52 747 | 1% | 0% |
| 1.05 | 12.6 | 13.23 | 25 537 | 1% | 21% |
| 1.15 | 3.9 | 4.49 | 9 604 | 9% | 32% |
| 1.15 | 6.1 | 7.02 | 6 141 | 18% | 51% |
| 1.15 | 12.6 | 14.49 | 2 973 | 34% | 70% |
| 1.25 | 3.9 | 4.88 | 3 612 | 30% | 67% |
| 1.25 | 6.1 | 7.63 | 2 309 | 39% | 74% |
| 1.25 | 12.6 | 15.75 | 1 118 | 50% | 84% |
| 1.5 | 3.9 | 5.85 | 997 | 52% | 84% |
| 1.5 | 6.1 | 9.15 | 637 | 57% | 89% |
| 1.5 | 12.6 | 18.90 | 309 | 67% | 91% |
| 2 | 3.9 | 7.80 | 294 | 67% | 91% |
| 2 | 6.1 | 12.20 | 188 | 74% | 92% |
| 2 | 12.6 | 25.20 | 91 | 83% | 93% |
Notes: All estimates assume a significance level of 0.05 and a power of 1 − β=0.80. Adverse event and complication rates are reported as events per 100 person‐years. “Utilization” refers to actual ICD model utilization in practice.
Abbreviations: AE, adverse event; ICD, implantable cardioverter defibrillator.
We compared the estimated utilization required to detect safety differences to the actual ICD utilization observed for each ICD model within NCDR‐ICD data. Appendix Figure 1 represents a timeline illustrating the date of the first observed model implant to the date when actual utilization surpassed the estimated utilization required to detect a safety difference, using a rate ratio of 1.25 for the magnitude of the safety difference among device models. This figure shows that the adverse event and complication rates significantly influence both the proportions of models for which actual utilization surpasses the estimated utilization required to detect a safety difference, while also demonstrating that many ICD models, even when assuming adverse event and complication rates of 12.6 events per 100 person‐years, never reach sufficient utilization to detect safety differences. Furthermore, independent of the scenario parameters, the time between the first implant and accrual of sufficient utilization to discern safety differences, which we henceforth refer to as the time to sufficient utilization, varies greatly across models.
Figure 4 plots a histogram of the time to sufficient utilization for all ICDs, illustrating what number of ICD models reaches the estimated utilization required to detect safety differences for a range of adverse event and complication rates and safety rate ratios, in addition to the distribution of the time to accrue sufficient utilization. For rate ratios of 1.05, no more than 1% of ICD models accrued sufficient utilization to detect safety differences, whereas for a rate ratio of 1.15 at the highest safety event rate of 12.6 per 100 person‐years, only 34% accrued sufficient utilization (Table 2). Even for a rate ratio of 1.25, the proportion of ICD models accruing sufficient utilization ranged between 30% and 50%, depending on the safety event rate. However, at a rate ratio of 1.5, between 52% and 67%, of ICD models accrued sufficient utilization, many of these within 2 years after the first implant. This is similarly true for rate ratios larger than 1.5.
FIGURE 4.
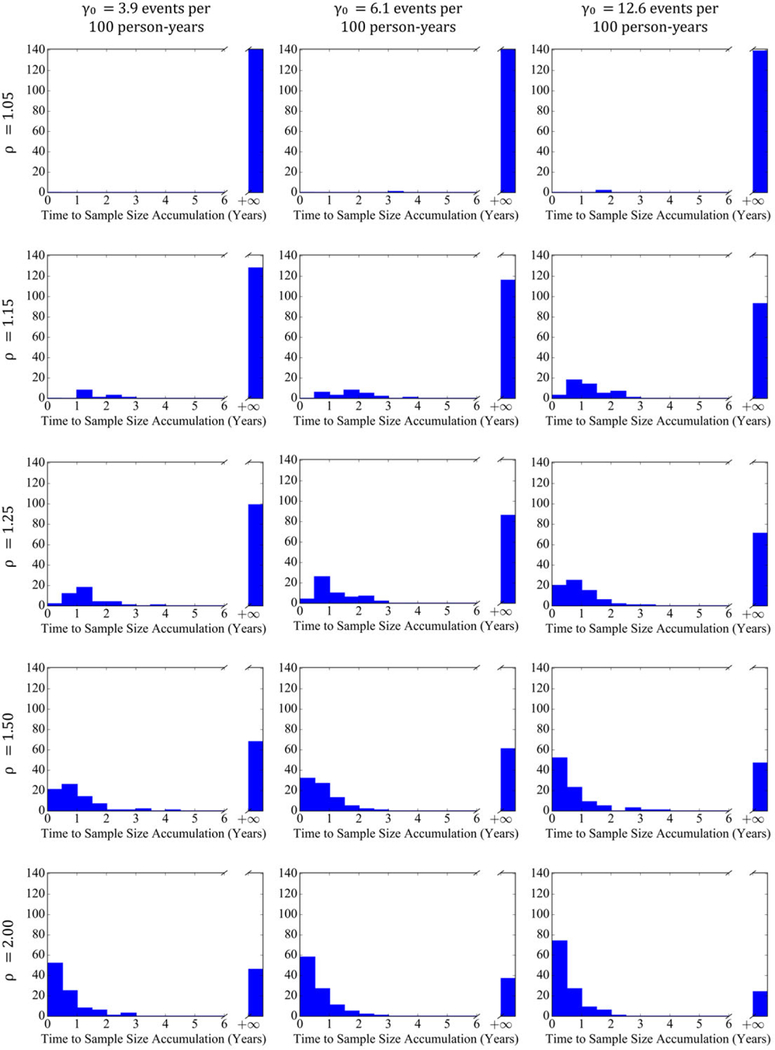
Histograms of time to sufficient accrued utilization in years to detect safety differences for implantable cardioverter defibrillators across adverse event and complication rates of 3.9, 6.1, and 12.6 events per 100 person‐years and rate ratios of 1.05, 1.15, 1.25, 1.50, and 2.00 for significance level α=0.05 [Colour figure can be viewed at wileyonlinelibrary.com]
Because actual ICD utilization is not equally distributed, of all individuals implanted with an ICD, the proportion who received an ICD model that reached the estimated utilization required to detect safety differences was consistently greater than our estimates. Figure 5 plots the number of individuals who received ICD models that reached the estimated utilization required to detect safety differences, for a range of adverse event and complication rates and safety rate ratios, after 2 years. For a rate ratio of 1.05, only a small fraction of patients, at most 21% at the highest safety event rate, received an ICD that accrued sufficient utilization to detect safety differences (Table 2). However, for a rate ratio of 1.15, between 32% and 70% of patients received ICD models that accrued sufficient utilization, depending on the safety event rate, and for rate ratios of 1.25 and higher, the percentage of patients exceeds 67% and approaches 93%.
FIGURE 5.
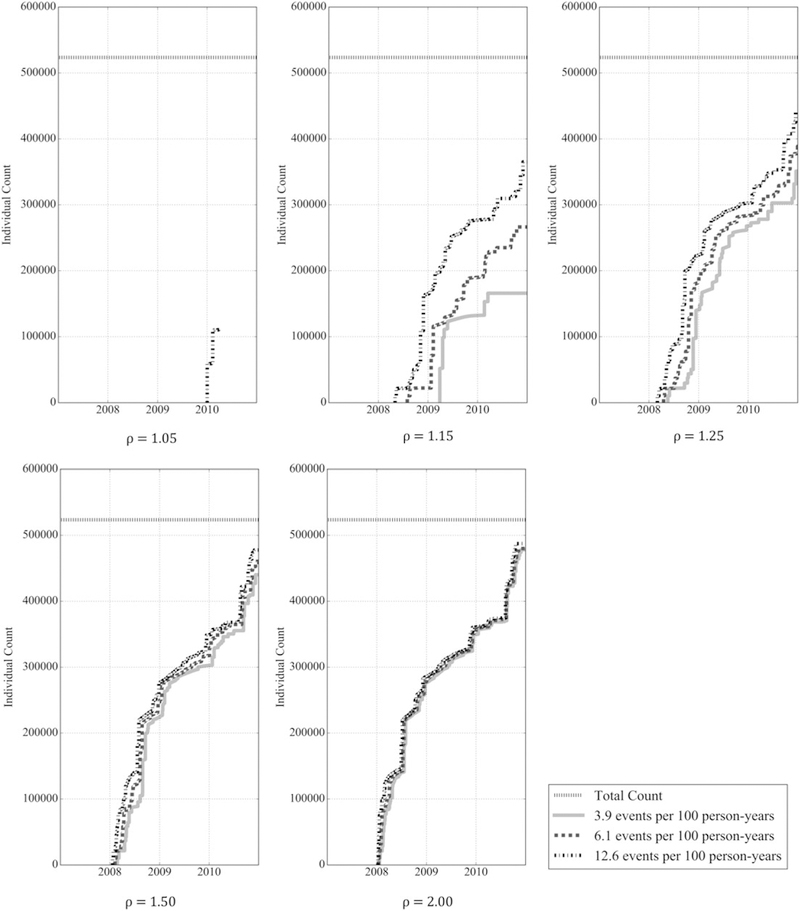
Number of patients receiving any implantable cardioverter defibrillator who received a model that accrued sufficient utilization 2 years post‐first implantation to detect safety differences for adverse event and complication rates of 3.9, 6.1, and 12.6 events per 100 person‐years and rate ratios of 1.05, 1.15, 1.25, 1.50, and 2.00 at significance level α=0.05
4 |. DISCUSSION
Using ICDs as a case study to better understand the expectations and limitations of a real‐time active postmarket safety surveillance system, we demonstrate that smaller safety differences between ICD generator models, including differences of 15% or less, would likely remain undetected for most ICDs because of insufficient medical device utilization to support reliable comparisons, even among otherwise optimistic assumptions with respect to patient follow‐up. However, we also demonstrate that because some devices are used far more frequently than others, medical device utilization is sufficient to detect larger safety differences, including differences of 25% or greater, for approximately half or more of patients receiving ICDs at what are likely to be reasonably anticipated safety event rates. Thus, even for a high‐risk device used as routinely as an ICD, our estimates demonstrate that only the most commonly used medical device models are likely to accrue sufficient utilization to detect real‐world safety difference should a postmarket surveillance system be established.
An automated and real‐time active adverse event surveillance system that relies on statistical tests alone is substantially influenced by population utilization rates and the magnitude of the safety performance difference. While our estimates evaluated several parameters, including adverse event and complication rate, significance level, and the rate ratio, we found that, qualitatively, the most influential parameter was the rate ratio for the range of safety event rates considered. For instance, no matter whether the safety event rate was 3.9 or 12.6 events per 100 person‐years, performance differences among most models would not be expected to be detected if the safety difference among models was less than 25%. Even then, for a rate ratio of 1.25, we would expect that at least 3 years of consistent use would need to occur before a performance difference could be detected for most models. Detecting differences of 15% increased risk or less among most ICD models with a power of 80% is practically infeasible.
Our study suggests that given actual rates of ICD utilization observed in practice within the NCDR‐ICD, a real‐time active postmarket safety surveillance system would be able to detect safety differences among ICD models for approximately 90% or more patients receiving ICDs if the safety difference among models was 50% or greater or if the safety event rate exceeded 6 per 100 person‐years. However, while detecting modest safety differences for any medical device may be difficult, if not impossible, such differences in safety may be acceptable to patients, the clinical community, regulators, and manufacturers. These determinations may depend upon the relative benefit of the device; the severity of the medical device safety risk; disease prevalence and severity for which the device is used; its anticipated length of use, including whether it is implanted for life‐time use; and the availability of other therapeutic options. If small differences in risk are considered acceptable, medical device model selection can be made based on patient and physician preference, known benefits, costs, or other criteria.
The more challenging issue to consider is how to design ongoing and future medical device safety surveillance initiatives to ensure that routinely collected data can be used to reliably identify medical device utilization and monitor safety over time while collecting feedback to promote future device iterations and innovation. At the time of approval of high‐risk medical devices, approximately 300 patients on average are exposed to the device during clinical trial evaluation,21 fewer for moderate‐risk devices. Thus, to ensure that a sufficient number of patients can be observed to detect safety differences, manufacturers and regulators will need to take advantage of larger, real‐world data sources, such as those being proposed for the National Evaluation System for health Technology.9 Similarly, the FDA recently supported the launch of the National Medical Evidence Generation (EvGen) Collaborative,10 bringing together efforts such as National Evaluation System for health Technology, the Sentinel Initiative, and the National Patient‐Centered Clinical Research Network (PCORnet, created by the Patient‐Centered Outcomes Research Institute).22 However, the utility of data from many of these efforts for medical device safety surveillance will be dependent upon the incorporation of the Unique Device Identifier into electronic medical record and administrative claims data.23,24 The EvGen Collaborative has great potential for promoting the generation of high‐quality scientific evidence to support the best choices for individual patients and populations, including gathering reliably collected data from real‐world settings for rigorous analysis to monitor medical device safety after approval.25 An infrastructure that aggregates data from the United States and international health systems may also be needed.26
There are several limitations to our study. First, while this case study is intended to offer insights into the expectations and limitations of real‐time active postmarket medical device surveillance, our focus on ICDs may not be generalizable to other medical products, including pharmaceuticals that are used much more commonly or to many other high‐risk medical devices that are used far less commonly and for which safety‐related risk predominantly occurs within the first 24 to 48 hours after use, as opposed to gradually over time as with ICDs. Second, for our analyses, we considered 2 safety event rates that accounted for risk of death, but otherwise assumed 100% patient follow‐up. Thus, our estimates may over‐count the number of individuals who received an ICD model for which sufficient utilization accrued, as we expect real‐world follow‐up may be less than 2 years for many patients, as they change insurance plans or move, lessening observed exposures. Related to this point, our estimates do not take unequal loss to follow‐up among compared devices into account; if we assume more loss to follow‐up among patients who received the device of interest, then the sample size estimate would increase; conversely, if we assume less loss to follow‐up, then the sample size estimate would decrease. Third, the adverse event and complication rates used were derived from a population of Medicare beneficiaries, who are 65 years and older and may be more likely to experience deaths than a younger population of patients receiving ICDs. However, other adverse event and complication rates may not be substantially different. Nevertheless, as noted earlier, for our analyses, these rates are less important than the use of reasonable referent rates to provide insights into the impact of the anticipated safety event rate on estimates of the ICD utilization needed to detect differences.
Fourth, for our analyses, we assumed that there was always a sufficient number of control patients receiving other ICD models to match. For ICDs, this is a reasonable assumption, though still likely optimistic, given the large number of available models. However, for other medical devices, this assumption may be less reasonable, leading to an insufficient pool of controls and greater difficulty in conducting medical device surveillance using existing data sources. We also assumed that all ICD models were independent in their characteristics, safety, and efficacy, although it might be possible that certain models would have similar risk profiles. For instance, manufacturers may use similar parts or manufacturing processes for multiple marketed models, as was the case involving a Class I recall of St. Jude manufactured ICDs.27 In such a case, grouping similar models will lead to greater sample sizes and sufficient levels of evidence. Fifth, assessments of ICD safety were always a comparison of one generator model to the average of all other models, as we assumed that it would not be known a priori which ICD was more likely to be associated with greater risk. However, this approach, while realistic, biases the average toward the null, as the average may represent many higher‐risk medical devices. Sixth, we used 2‐sided statistical tests, a conservative approach for safety surveillance, when 1‐sided tests might be used. However, since it is unlikely to know a priori which devices might be identified as either safer or riskier, a 2‐sided test was thought to be more appropriate to assess differences in either direction. Finally, using cross‐sectional observational data poses challenges, both with respect to establishing the “beginning” and “end” of the study period, since a real‐time active postmarket safety surveillance system would have neither a beginning nor an end.
In conclusion, our case study of ICD postmarket safety surveillance suggests that smaller safety differences of 15% or less in the first 2 years after implantation will not be detected for most ICDs given current rates of utilization, but that safety differences of 25% or more could be detected for the majority of patients receiving ICDs at anticipated safety event rates. While postmarket surveillance is unlikely to be perfect in its detection of safety risks, large differences in medical devices can reasonably be detected, at least for the most commonly used medical device models, provided adequate data are available that includes information on medical device models and relevant short‐term and long‐term information on adverse events and complications. Our findings should inform the expectations and limitations of real‐time active postmarket safety surveillance systems.
Supplementary Material
KEY POINTS.
Questions remain as to the potential effectiveness of a real‐time active surveillance system to detect safety differences among medical devices.
Any system is substantially influenced by population utilization rates and the magnitude of the safety performance difference.
Using implantable cardioverter defibrillators (ICDs) as an illustrative example, we characterized the sample size estimates needed to detect safety differences for postmarket surveillance initiatives.
Smaller safety differences among ICDs are unlikely to be detected through routine postmarket surveillance, whereas larger safety differences would be detected for most patients receiving ICDs at anticipated adverse event rates.
Only the most commonly used ICDs are likely to accrue sufficient utilization to detect real‐world safety differences.
ACKNOWLEDGEMENTS
The authors acknowledge Dr. Isuru Ranasinghe for the helpful discussion of ICD complications and event rates and Dr. Jerome Kassirer for helpful comments on the manuscript. Neither Drs. Ranasinghe nor Kassirer received compensation for their efforts nor have any potential competing interests to disclose.
NCDR ICD Registry is an initiative of the American College of Cardiology Foundation with partnering support from the Heart Rhythm Society. The views expressed in this manuscript represent those of the authors and do not necessarily represent the official views of the NCDR or its associated professional societies identified at CVQuality.ACC. org/NCDR.
Funding information
Medtronic, Inc.; U.S. Food and Drug Administration (FDA), Grant/Award Number: U01FD004585
FUNDING/SUPPORT AND ROLE OF THE SPONSOR
This project was jointly funded by the U.S. Food and Drug Administration (FDA) and Medtronic, Inc. to develop methods for postmarket surveillance of medical devices (U01FD004585). Members of the sponsoring organizations contributed directly to the project, participating in study conception and design, analysis and interpretation of data, and critical revision of the manuscript; the authors made the final decision to submit the manuscript for publication. In addition, the project was approved by but did not receive financial support from the American College of Cardiology’s National Cardiovascular Data Registry (NCDR). The NCDR research committee reviewed the final manuscript prior to submission but otherwise had no role in the design, conduct, or reporting of the study. Drs. Masoudi and Shaw receive support from the American College of Cardiology for roles within the National Cardiovascular Data Registry. Dr. Dhruva is supported by the National Clinician Scholars Program and the Department of Veterans Affairs. The authors assume full responsibility for the accuracy and completeness of the ideas presented, which do not represent the views of the Department of Veterans Affairs or any other supporting intuitions.
CONFLICT OF INTEREST
In the past 36 months, Dr. Ross has received research support through Yale University from the U.S. Food and Drug Administration to establish Yale‐Mayo Clinic Center for Excellence in Regulatory Science and Innovation (CERSI) program (U01FD005938), from the Laura and John Arnold Foundation to support the Collaboration for Research Integrity and Transparency (CRIT) at Yale, from the Agency for Healthcare Research and Quality (R01HS022882), and from the Blue Cross Blue Shield Association to better understand medical technology evidence generation; Drs. Krumholz and Ross have received research support from Johnson and Johnson to develop methods of clinical trial data sharing; and Drs. Li, Coppi, Warner, Krumholz, and Ross and Mr. Parzynski worked under contract to the Centers for Medicare and Medicaid Services (CMS) to develop and maintain performance measures that are used for public reporting. Dr. Kuntz is an employee of Medtronic, Inc. Dr. Marinac‐Dabic is an employee of the U.S. Food and Drug Administration. Dr. Krumholz chairs a cardiac scientific advisory board for UnitedHealth, is a participant/participant representative of the IBM Watson Health Life Sciences Board, is a member of the Advisory Board for Element Science and the Physician Advisory Board for Aetna, and is the founder of Hugo, a personal health information platform.
Footnotes
Prior presentation: Preliminary methods were presented at the November 2016 Medical Device Epidemiology Network (MDEpiNet) methodology portfolio meeting, coordinated by the Division of Epidemiology within the Center for Devices and Radiological Health of the U.S. Food and Drug Administration, and a summary of this work was presented for a May 2017 MDEpiNet midyear webcast and a July 2017 MDEpiNet annual meeting. This manuscript has not been published elsewhere and is not under consideration by another journal.
ETHICS STATEMENT
The Yale University Human Investigation Committee approved the study.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.U.S. Food and Drug Administration. FDA’s Sentinel Initiative 2016; https://www.fda.gov/Safety/FDAsSentinelInitiative/ucm2007250.htm (accessed 13 March 2017).
- 2.Harpaz R, DuMochel W, Shah NH. Big data and adverse drug reaction detection. Clin Pharmacol Ther 2016;99(3):268–270. 10.1002/cpt.302 [DOI] [PubMed] [Google Scholar]
- 3.Sentinel Coordinating Center. Sentinel 2017; https://www.sentinelinitiative.org/ (accessed 13 March 2017).
- 4.Resnic FS, Gross TP, Marinac‐Dabic D, et al. Automated surveillance to detect postprocedure safety signals of approved cardiovascular devices. JAMA 2010;304(18):2019–2027. 10.1001/jama.2010.1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Resnic FS, Majithia A, Marinac‐Dabic D, et al. Registry‐based prospective, active surveillance of medical‐device safety. N Engl J Med 2017;376(6):526–535. 10.1056/NEJMoa1516333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A, Matheny ME, Ho KK, et al. The data extraction and longitudinal trend analysis network study of distributed automated postmarket cardiovascular device safety surveillance. Circ Cardiovasc Qual Outcomes 2015;8(1):38–46. 10.1161/CIRCOUTCOMES.114.001123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matheny ME, Normand SL, Gross TP, et al. Evaluation of an automated safety surveillance system using risk adjusted sequential probability ratio testing. BMC Med Inform Decis Mak 2011;11(1):75 10.1186/1472‐6947‐11‐75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vidi VD, Matheny ME, Donnelly S, Resnic FS. An evaluation of a distributed medical device safety surveillance system: the DELTA network study. Contemp Clin Trials 2011;32(3):309–317. 10.1016/j.cct.2011.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shuren J, Califf RM. Need for a national evaluation system for health technology. JAMA 2016;316(11):1153–1154. 10.1001/jama.2016.8708 [DOI] [PubMed] [Google Scholar]
- 10.Califf RM, Robb MA, Bindman AB, et al. Transforming evidence generation to support health and health care decisions. N Engl J Med 2016;375(24):2395–2400. 10.1056/NEJMsb1610128 [DOI] [PubMed] [Google Scholar]
- 11.Mott K, Graham DJ, Toh S, et al. Uptake of new drugs in the early post‐approval period in the Mini‐Sentinel distributed database. Pharmacoepidemiol Drug Saf 2016;25(9):1023–1032. 10.1002/pds.4013 [DOI] [PubMed] [Google Scholar]
- 12.Masoudi FA, Ponirakis A, de Lemos JA, et al. Executive summary: trends in U.S. cardiovascular care: 2016 report from 4 ACC National Cardiovascular Data Registries. J Am Coll Cardiol 2016;69(11): 1424–1426. 10.1016/j.jacc.2016.12.004 [DOI] [PubMed] [Google Scholar]
- 13.Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device‐based therapy of cardiac rhythm abnormalities: executive summary. J Am Coll Cardiol 2008;51(21):2085–2105. 10.1016/j.jacc.2008.02.033 [DOI] [Google Scholar]
- 14.Masoudi FA, Ponirakis A, Yeh RW, et al. Cardiovascular care facts: a report from the national cardiovascular data registry: 2011. J Am Coll Cardiol 2013;62(21):1931–1947. 10.1016/j.jacc.2013.05.099 [DOI] [PubMed] [Google Scholar]
- 15.Messenger JC, Ho KK, Young CH, et al. The National Cardiovascular Data Registry (NCDR) data quality brief: the NCDR data quality program in 2012. J Am Coll Cardiol 2012;60(16):1484–1488. 10.1016/j.jacc.2012.07.020 [DOI] [PubMed] [Google Scholar]
- 16.Ross JS, Bates J, Parzynski CS, et al. Can machine learning complement traditional medical device surveillance? A case study of dual‐chamber implantable cardioverter–defibrillators. Med Devices 2017;10: 165–188. 10.2147/MDER.S138158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranasinghe I, Parzynski CS, Freeman JV, et al. Long‐term risk for device‐related complications and reoperations after implantable cardioverter‐defibrillator implantation: an observational cohort study. Ann Intern Med 2016;165(1):20–29. 10.7326/M15‐2732 [DOI] [PubMed] [Google Scholar]
- 18.Masoudi FA, Ponirakis A, de Lemos JA, et al. Trends in U.S. cardiovascular care: 2016 report from 4 ACC National Cardiovascular Data Registries. J Am Coll Cardiol 2017;69(11):1427–1450. 10.1016/j.jacc.2016.12.005 [DOI] [PubMed] [Google Scholar]
- 19.Huffman MD. An improved approximate two‐sample poisson test. J R Stat Soc Ser C Appl Stat 1984;33(2):224–226. 10.2307/2347448 [DOI] [Google Scholar]
- 20.Gu K, Ng HKT, Tang ML, Schucany WR. Testing the ratio of two poisson rates. Biom J 2008;50(2):283–298. 10.1002/bimj.200710403 [DOI] [PubMed] [Google Scholar]
- 21.Rathi VK, Krumholz HM, Masoudi FA, Ross JS. Characteristics of clinical studies conducted over the total product life cycle of high‐risk therapeutic medical devices receiving FDA premarket approval in 2010 and 2011. JAMA 2015;314(6):604–612. 10.1001/jama.2015.8761 [DOI] [PubMed] [Google Scholar]
- 22.The National Patient‐Centered Clinical Research Network (PCORnet). About PCORnet 2017; http://www.pcornet.org/about‐pcornet/. (accessed 13 March 2017).
- 23.Rising J, Moscovitch B. The Food and Drug Administration’s unique device identification system: better postmarket data on the safety and effectiveness of medical devices. JAMA Intern Med 2014;174(11):1719–1720. 10.1001/jamainternmed.2014.4195 [DOI] [PubMed] [Google Scholar]
- 24.Wilson NA, Drozda J. Value of unique device identification in the digital health infrastructure. JAMA 2013;309(20):2107–2108. 10.1001/jama.2013.5514 [DOI] [PubMed] [Google Scholar]
- 25.Sherman RE, Anderson SA, Dal Pan GJ, et al. Real‐world evidence: what is it and what can it tell us? N Engl J Med 2016;375(23): 2293–2297. 10.1056/NEJMsb1609216 [DOI] [PubMed] [Google Scholar]
- 26.Sedrakyan A, Marinac‐Dabic D, Holmes DR, et al. The international registry infrastructure for cardiovascular device evaluation and surveillance. JAMA 2013;310(3):257–259. 10.1001/jama.2013.7133 [DOI] [PubMed] [Google Scholar]
- 27.U.S. Food and Drug Administration. St. Jude Medical recalls implantable cardioverter defibrillators (ICD) and cardiac resynchronization therapy defibrillators (CRT‐D) due to premature battery depletion 2016; https://www.fda.gov/MedicalDevices/Safety/ListofRecalls/ucm526317.htm (accessed 13 March 2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.