

Summary
Background
The purposes of this study were three‐fold: (i) to determine the contribution of known genes to the causation of a broad‐spectrum of pediatric drug‐resistant epilepsy (DRE), (ii) to compare the diagnostic yield and cost among different next‐generation sequencing (NGS) approaches, and especially (iii) to assess how NGS approaches can benefit patients by improving diagnosis and treatment efficiency.
Methods
This study enrolled 273 pediatric DRE patients with no obvious acquired etiology. Seventy‐four patients underwent whole‐exome sequencing (WES), 141 patients had epilepsy‐related gene panel testing, and another 58 patients had clinical WES gene panel testing. We obtained these patients’ seizure and hospitalization frequency by periodic follow‐up phone calls and outpatient visits.
Results
Genetic diagnosis was achieved in 86 patients (31.5%) and involved 93 likely disease‐causing mutations in 33 genes. In this study, the detection rates of the epilepsy‐related gene panel, the clinical WES gene panel, and WES were 32.6% (46/141), 44.8% (26/58), and 17.3% (13/74), respectively. Moreover, 34 patients accepted corrective therapy according to their mutant genes, after which 52.9% (18/34) became seizure‐free and 38.2% (13/34) achieved seizure reduction. In the end, patients with either positive or negative genetic results had significantly fewer hospitalization incidents (times/half year) than before (positive genetic results group 0.58 ± 1.14 vs 0.10 ± 0.26; negative genetic results group 0.72 ± 1.65 vs 0.12 ± 0.33).
Conclusions
These results offer further proof that NGS approaches represent powerful tools for establishing a definitive diagnosis. Moreover, this study indicated how NGS can improve treatment efficacy and reduce hospitalization in children with DRE.
Keywords: drug‐resistant epilepsy, hospitalization frequency, next‐generation sequencing, treatment efficacy
1. BACKGROUND
Approximately 40‐50 million people in the world have epilepsy,1 making it one of the most common neurological diseases globally and accounting for 1% of the global burden of disease.2 Although the number of available antiepileptic drugs (AEDs) has more than doubled in the past few decades, current medications fail to control seizures in 20%‐30% of patients 3—the same rate as in 1979 (30%).4 Drug‐resistant epilepsy (DRE) accounts for 80% of the cost of epilepsy treatment,5 and is therefore a major health concern not only for patients and their families, but also for society as a whole.
To a certain extent, antiepileptic drug treatment is still typically a matter of trial and error, and the guidelines for drugs of first and second choice have been largely based on current seizure‐type classifications and EEG results.6 However, it has been recognized that etiology is a major determinant of treatment, prognosis, and clinical course in epilepsy patients. The causes of epilepsy have been reclassified in the 2016 International League against Epilepsy (ILAE) classification of epilepsies. After carefully excluding secondary causes such as HIE, CNS infectious and immunity disease, and brain structure abnormality, evaluation of the genetic basis of DRE is one of the most frequent referral indications in a neurological clinic.
During the last few years, accumulated evidence supports the strong role of genetics in unexplained DRE patients.7, 8, 9, 10 Moreover, advances in clinical testing technology have increased the diagnostic yield from 10% a few years ago to 30%‐40%. This increase has primarily been technology‐driven, due to the development of next‐generation sequencing (NGS). There are several commercial NGS panels available, which vary in the number of genes included (from dozens to hundreds). The widespread use of NGS approaches has led to a paradigm change in clinical practice. Although most clinicians realize this technique is effective and could benefit epilepsy patients, there are little data quantifying how much NGS can improve treatment efficacy and disease burden in DRE patients by influencing treatment choices.
Thus, we screened a pilot cohort of 273 children with unexplained DRE and aimed to determine the contribution of known genes to the causation of a broad‐spectrum of this disease, comparing the diagnostic yield and cost among different NGS approaches. Moreover, we analyzed the diagnostic adjustment ratio and investigated the impact of NGS on improving treatment efficiency and reducing hospitalization in pediatric DRE patients.
2. METHODS
2.1. Patients
From 2012 to 2016, 273 pediatric DRE patients, with no obvious acquired brain abnormalities, infection, or autoimmune etiology, were recruited into our cohort. “Drug‐resistant epilepsy” was defined as Kwan et al previously reported.11 The study design was approved by the institutional review board of Xiangya Hospital of Central South University, China. Study procedures were carried out in accordance with human subject protection laws. After obtaining informed consent from the guardians of the participants, we recorded the clinical features of the patients and collected blood samples from the patients and their parents via venipuncture.
We obtained information about hospitalization and seizure frequency in each case through periodic follow‐up, which has been taken by the way of follow‐up phone calls and outpatient visits.
2.2. Capture strategy and sequencing
Our study contains two types of NGS strategies: gene panel and whole‐exome sequencing (WES). For the first type, we developed two gene panels to test candidate variants in epilepsy‐related or Mendelian disease‐related patients, respectively. For the epilepsy‐related gene panel, we searched the OMIM and HGMD professional databases for epilepsy‐related known or candidate genes. The first version of the epilepsy‐related gene panel contained 308 genes, applied in the year 2012.12 We have continued updating the gene list of this panel, which now contains 540 genes with 2.0 Mb of sequence from the coding exons (GRCh37/hg19 human reference sequence, UCSC Genome Browser). One hundred and forty‐one patients were tested by this method. For 58 patients with atypical clinical manifestations, we developed a gene panel to test all the candidate or known Mendelian disease genes in humans according to OMIM and HGMD, which we called clinical WES. This target was 11.4 Mb of sequence from the coding exons (GRCh37/hg19 human reference sequence, UCSC Genome Browser) with 3994 Mendelian disease‐related genes. A custom‐based targeted Agilent SureSelect pull‐down array was designed with the SureDesign program (Agilent Technologies, Santa Clara, CA). Target enrichment and amplification were performed with the SureDesign target enrichment kit (Agilent, Santa Clara, CA) according to the manufacturer's instructions. The Illumina HiSeq 2500 platform (Illumina Inc., San Diego, CA) was used to sequence the exons from the targeted regions.
For the second type of NGS, we performed WES for another 74 patients. Exome capturing was performed to collect the target regions of human genome DNA using the Agilent SureSelect Human All Exon V5 enrichment platform (Agilent Technologies, Santa Clara, CA), according to the manufacturer's instructions.13 The DNA libraries were sequenced using the Illumina X Ten platform (Illumina, San Diego, CA), following the manufacturer's instructions.
2.3. Data quality control and filtering
For quality control, the Cutadapt and FastQC were used to remove 3′‐/5′‐ adapters and low‐quality reads, respectively. The clean reads were mapped to the reference human genome using the BWA (Burrows‐Wheeler Aligner) program with at most two mismatches. The alignment files (.bam) were generated with SAM tools and the reads of low mapping quality (<Q30) were filtered out. Clonal duplicated reads that may be derived from PCR artifacts were removed using Picard Tools by default parameters. Short read alignment and annotation visualization were performed using the IGV (Integrative Genomics Viewer). The percentage of alignment of the clean read to the exome regions was obtained using our custom Perl scripts on the base of alignment files. SNVs and indels were detected by GATK (Genome Analysis ToolKit). Comprehensive annotation of all of the detected SNVs and indels was annotated by ANNOVAR, including function implication (gene region, functional effect, mRNA GenBank accession number, amino acid change, cytoband, etc.) and allele frequency in 1000 Genomes, ExAc. Candidate variants were filtered out according to ACMG guidelines for interpretation of sequence variants.14 Candidate variants were confirmed by Sanger sequencing in patients and their parents.
2.4. Statistics
Comparisons of the number of hospitalization incidents between before and after taking NGS approaches were analyzed via the paired samples t test in patients with positive and negative genetic results, respectively.
3. RESULTS
3.1. Patient phenotyping
A total of 273 DRE patients were included in this cohort. The average age was 13.2 ± 20.8 months. The male‐to‐female ratio was 1:0.54. Spasms and focal seizures were the major seizure types in 51.6% (141/273) and 24.9% (68/273) of the patients, respectively. 17.2% (47/273) of the patients had more than one seizure type. Multifocal epileptiform discharges (107/273, 39.2%) and hypsarrhythmia (110/273, 40.3%) were the major abnormalities in EEG results.
Eighty‐six DRE patients (31.5%) had likely disease‐causing mutations with a broad range of phenotypes. Their clinical diagnoses before NGS included Dravet syndrome (31/86, 36%), West syndrome (19/86, 22.1%), epilepsy combined with global developmental delay (GDD) (14/86, 16.3%), epilepsy with focal seizures (10/86, 11.6%), MMSPI (3/86, 3.5%), PME(3/86, 3.5%), EOEE((2/86, 2.3%), OS(2/86, 2.3%), EIEE19(1/86,1.2%), and epilepsy with GTCS(1/86,1.2%) (Table S1).
3.2. Patient genotyping
In this study, genetic diagnosis was achieved in 86 patients, corresponding to the diagnostic yield of 31.5%. Ninety‐three disease‐related mutations were detected in 33 different genes in this study. Sixty‐two mutations were de novo. (Table S1). The most frequently mutated gene was SCN1A (24.4%,21/86), followed by TSC2 (8.1%, 7/86), SCN8A (5.8%, 5/86), CDKL5 (5.8%, 5/86), KCNMA1 (4.6%, 4/86), TSC1 (4.6%, 4/86), KCNQ2 (3.5%, 3/86), MECP2 (3.5%, 3/86), PCDH19 (3.5%, 3/86), STXBP1(3.5%, 3/86), and et al (Table S2). The gene function classification and the number of genes in each group are shown in Figure 1.
Figure 1.
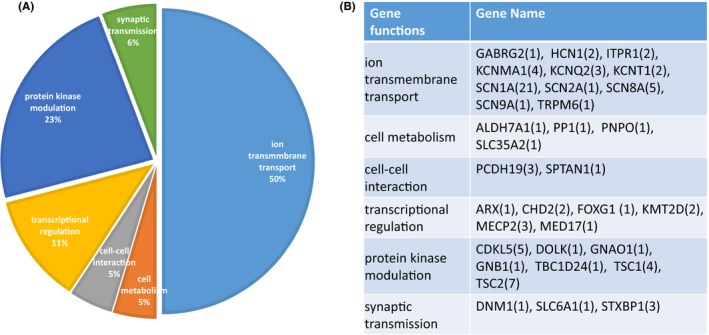
The gene function classification of genetic drug‐resistant epilepsy patients. A, The percentage of each group relevant different gene function classification. B, The number of genes in each groups
3.3. The effects of NGS approaches in improving diagnosis accuracy and treatment efficacy in DRE patients
Among the NGS approaches applied in our cohort, panels containing all of the previously reported genes for monogenic diseases (clinical WES) showed the highest detection rate (44.8%, 26/58), followed by panels containing the most relevant epilepsy genes (epilepsy‐related gene panel) (32.6%, 46/141) and WES (17.3%, 13/74). Moreover, by applying NGS approaches, 67.4% (58/86) of patients attained a more precise final diagnosis, as listed in Table S1.
In this study, we found 20 “actionable” genes—those that, if found, can direct therapy (Table S2). Sixty‐two patients had “actionable” genes, and more than half of them (34/62) accepted the immediate treatment implications. After 6 months of corrective therapy, 18 patients became seizure‐free and 13 patients achieved seizure reduction (Figure 2).
Figure 2.
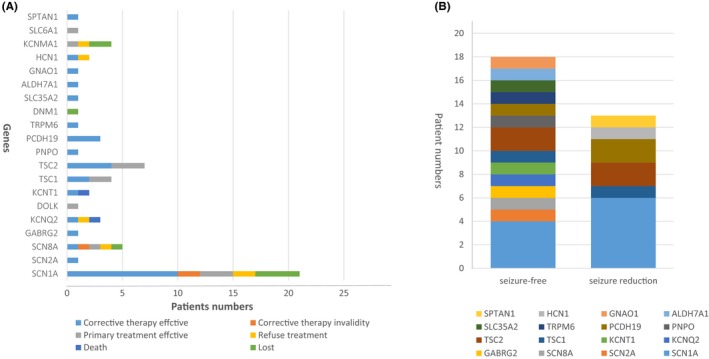
The efficacy of adjustment treatment in genetic drug‐resistant epilepsy (DRE) patients with “actionable” genes . A, The follow‐up of 62 DRE patients with 23 “actionable” genes. B, After receiving corrective therapy, 18 DRE patients became seizure‐free and 13 DRE patients achieved seizure reduction
3.4. The effects of NGS approaches in reducing hospitalization in DRE patients
Patients with positive genetic results presented with 0.58 ± 1.14 hospitalization incidents per half year previously, which was reduced to 0.10 ± 0.26 hospitalization incidents per half year after taking NGS approaches (P < 0.05). At the same time, NGS approaches also helped to reduce hospitalization in patients with negative genetic results, in which the average number of hospitalization incidents per half year before and after NGS were 0.72 ± 1.65 and 0.12 ± 0.33, respectively (P < 0.05).
4. DISCUSSION
In our cohort, we achieved genetic diagnosis in 31.5% of all cases by applying NGS approaches, and noted the most common phenotypes in these patients. Moreover, we compared both the diagnostic yield and the cost among different NGS approaches to make it easier for clinicians to balance efficiency and cost‐effectiveness. Last, and also important, this study reports specific data quantifying how NGS can improve treatment efficiency and reduce hospitalization in pediatric DRE patients.
4.1. Next‐generation sequencing has significant diagnostic utility in pediatric DRE patients
The total diagnostic yield was 31.5% in this pediatric DRE patient cohort, demonstrating that NGS has significant diagnostic utility in these patients. In our study, more than 60% of mutations were found in cases where the phenotype was either not easily distinguishable from that caused by a number of other genes or was atypical for the previously reported phenotypes. For example, among the 86 patients with a positive genetic cause, 19 patients had a primary diagnosis of West syndrome, according to the clinical features and EEG. However, after applying NGS, these patients had a different final molecular diagnosis and prognosis, including EIEE1, 2, 4, 7, 13, 31, CDG2M, GEPD, MRD42, and so on. This illustrates the challenges in recognizing the causative gene based on phenotype, and emphasizes the benefit of an NGS approach over targeted single‐gene testing.
However, the diagnostic rate in our study was relatively high compared with similar studies,7 which might be attributed to several reasons. First, according to previous electroclinical investigations, some cases had sufficient clinical certainty before genetic testing, such as Dravet syndrome. Moreover, a large number of previous investigations, including metabolic screening and cranial MRI, were applied to exclude secondary causes. Finally, NGS approaches applied in our cohort covered 308‐20 000 genes, representing dramatically expanded gene coverage compared with previously reported epilepsy panels.
4.2. What kinds of genes contribute to a possible common monogenic mechanism in pediatric DRE patients?
To understand a possible common genetic mechanism of DRE, 33 genes were classified into groups according to gene function. We found that ion channel genes had the largest percentage of occurrence at 50% (43/86), followed by 20 cases with genes related to protein kinase modulation at 23% (20/86). Other identified genes were classified as having functions in transcriptional regulation, synaptic transmission, cell metabolism, and cell‐cell interaction.
It has been shown that in a heterogeneous cohort of pediatric drug‐resistant epilepsies, the possibility of harboring mutations reaches 7.4% for SCN1A, 2.5% for TSC2, 1.8% for both SCN8A and CDKL5, 1.4% for both KCNMA1 and TSC1, and 1.1% for either KCNQ2, MECP2, PCDH19, or STXBP1. However, the SCN2A gene was detected in only 1 case (1/86, 1.2%) in our cohort, far below a previous report (9/73,12.9%).7 This is likely due to many contributing factors: (i) the race difference among different cohorts, (ii) in Parrini's study, the proportion of SCN2A might have been underestimated as they have used Sanger sequencing instead of NGS approaches to detect it.
4.3. What is the best NGS approach in a clinical setting?
Since 2012, NGS has become a widespread diagnostic tool in neurology, and there are more than 30 diagnostic laboratories in China offering gene panels for patients with epilepsy. Therefore, we compared the cost and diagnostic yield of three kinds of NGS techniques (epilepsy‐related gene panel, clinical WES, and WES), to make it easier for busy clinicians to determine the best NGS approach.
In our cohort, the epilepsy‐related gene panel, clinical WES, and WES cost $570, $720, and $1450, respectively. None of these tests would be covered by common insurances in our country. The diagnostic yield of each NGS technique was 32.6% (46/141), 44.8% (26/58), and 17.6% (13/74). Compared with the epilepsy‐related gene panel, clinical WES showed a higher diagnostic rate and detected more kinds of mutations, especially in the genes involved in protein kinase modulation (such as DOLK, MECP2, and so on) and transcriptional regulation (such as FOXG1 and MED17), indicating that larger panels may be more effective in diagnosing refractory epilepsy patients with no suggestive phenotype. However, as previously reported, having more genes on the panel is not always ideal, especially when the phenotype of the patient is well defined, as in the case of tuberous sclerosis. In addition, at first sight, the detection rate of targeted NGS panels (72/199, 36.1%) was higher than WES. That could be due to the fact that some DRE patients with positive panel results did not undergo WES, thereby reducing the WES detection rate. Moreover, the subsequent work of identifying novel epilepsy candidate gene mutations has been in progress (data not shown), so indicating the exact detection rate of WES should be higher now.
Considering both cost and detection rate, we believe that a larger gene panel approach, such as clinical WES, can be a comparable alternative to WES analysis in most cases. However, reflex testing to WES is an option when a gene panel is negative. It is also important to realize that there is no single best NGS approach. Instead, the NGS approach should be selected based on each patient's presentation, while respecting the wishes of patients and/or parents.
4.4. Can applying NGS in a clinical setting actually benefit DRE patients?
Once an epilepsy gene is discovered, the most exciting aspect is the possibility for the patient to have personalized medicine, or at least optimized drug therapy. However, the idea of personalized medicine has been met with some skepticism recently. A few months ago, articles in the New England Journal of Medicine15 and Nature16 indicted “applying precision medicine,” or in other words, using DNA sequence information from individual patients to tailor what therapy they should get. These articles noted that such therapy had not been shown to work in cancer patients, and suggested that the clinical benefit of personalized medicine will be limited. Therefore, we aimed to gather and analyze data to confirm whether applying NGS in a clinical setting could actually benefit DRE patients. We decided to approach the issue in two ways: on the one hand, how applying NGS could enable us to improve total treatment efficacy and on the other hand, whether it could reduce the economic burden of treatment.
In our cohort, more than 50% of cases had “actionable” genes (listing in Table S2) with immediate treatment implications. Eighteen of these patients underwent targeted therapies, such as vitamin B6 for patients with PNPO (Case 23) and ALDH7A1 (Case 68) mutations, rapamune for patients with TSC1 (Case 49) and TSC2 (Case 53 and 56) mutations, magnesium sulfate for patients with TRPM6 mutations (Case 47), and quinidine for patients with KCNT1 mutations (Case16). All of them achieved complete seizure freedom. Other patients with “actionable” genes, such as SCN1A (Case 29, 32, 35 and 38) and PCDH19 (Case 22), had already received corrective therapy and also presented with no convulsions for at least 6 months. Overall, applying NGS in this cohort guided 12% (34/283) of patients to the correct therapy, and the number of patients who attained seizure freedom for at least 6 months increased 6.4% in the whole group, indicating that applying NGS can in fact improve treatment efficacy in DRE patients.
To answer the second question of cost, the patients who were followed up with for more than 6 months were divided into 2 groups: cases with positive genetic diagnosis and cases with negative genetic diagnosis. We found that patients with either negative or positive genetic results were hospitalized less frequently after undergoing NGS. Some patients with particular mutations did not end up changing their epilepsy management, but NGS did help with anticipatory guidance for the doctor and the family, helping to avoid unnecessary duplication of examinations. For some epilepsy patients with metabolic disorders, such as vitamin B6 dependency, timely personalized treatment would help them not only attain seizure freedom, but also avoid disability. Moreover, more specific genetic diagnosis leads to less but more effective therapy. For example, such as case 85, who was previously diagnosed as West syndrome and have tired 4 kinds of AEDs including ACTH, VPA, TPM and VGB; however, all of these therapies were failed. After the genetic results came out (SCN2A mutation), this patient was given OXC and received excellent effect, for now, he has taken OXC alone at the dose of 60 mg/kg/d and kept seizure‐free for more than 6 months. In these cases, the social and economic benefits for the patients and their families cannot be measured simply by P value.
5. CONCLUSIONS
Overall, NGS approaches represent a powerful tool for establishing a definitive diagnosis in DRE patients. Although a large sample of multicenter studies is needed, this study gives data confirming that NGS can benefit patients by improving diagnosis accuracy and treatment efficiency, and reducing hospitalization.
DISCLOSURES
None of the authors has any conflict of interest to disclose.
Supporting information
ACKNOWLEDGMENT
This work was supported by grants from the National Natural Science Foundation of China (NO. 81771409, NO. 81771408, NO. 81701541, NO. 81370771, NO. 81671297, NO. 81301031, NO. 81371434) as well as The National Key Research and Development Program of China (NO. 2016YFC1306202; NO. 2016YFC0904400).
Peng J, Pang N, Wang Y, et al. Next‐generation sequencing improves treatment efficacy and reduces hospitalization in children with drug‐resistant epilepsy. CNS Neurosci Ther. 2019;25:14–20. 10.1111/cns.12869
Search Terms: Clinical neurology examination; All Epilepsy/Seizures; All Genetics; All Pediatric
Contributor Information
Fang He, Email: bubbly_ho@163.com.
Fei Yin, Email: yf2323@hotmail.com.
REFERENCES
- 1. Birbeck GL. Epilepsy care in developing countries: part I of II. Epilepsy Curr. 2010;10:75‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reynolds EH. The ILAE/IBE/WHO global campaign against epilepsy: bringing epilepsy “out of the shadows”. Epilepsy Behav. 2000;1:S3‐S8. [DOI] [PubMed] [Google Scholar]
- 3. Löscher W, Klitgaard H, Twyman RE, Schmidt D. New avenues for anti‐epileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757‐776. [DOI] [PubMed] [Google Scholar]
- 4. Annegers JF, Hauser WA, Elveback LR. Remission of seizures and relapse in patients with epilepsy. Epilepsia. 1979;20:729‐737. [DOI] [PubMed] [Google Scholar]
- 5. Moshe SL, Perucca E, Ryvlin P, et al. Epilepsy: new advances. Lancet. 2015;385:884‐898. [DOI] [PubMed] [Google Scholar]
- 6. Lindhout D. Epilepsy treatment: precision medicine at a crossroads. Lancet Neurol. 2015;14:1148‐1149. [DOI] [PubMed] [Google Scholar]
- 7. Parrini E, Marini C, Mei D, et al. Diagnostic targeted resequencing in 349 patients with drug‐resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. 2017;38:216‐225. [DOI] [PubMed] [Google Scholar]
- 8. Lv N, Qu J, Long H, et al. Association study between polymorphisms in the CACNA1A, CACNA1C, and CACNA1H genes and drug‐resistant epilepsy in the Chinese Han population. Seizure. 2015;30:64‐69. [DOI] [PubMed] [Google Scholar]
- 9. Qu J, Zhang Y, Yang ZQ, et al. Gene‐wide tagging study of the association between KCNT1 polymorphisms and the susceptibility and efficacy of genetic generalized epilepsy in Chinese population. CNS Neurosci Ther. 2014;20:140‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu SZ, Ye H, Yang XG, Lu ZL, Qu Q, Qu J. Case‐control pharmacogenetic study of HCN1/HCN2 variants and genetic generalized epilepsies. Clin Exp Pharmacol Physiol. 2018;45:226‐233. [DOI] [PubMed] [Google Scholar]
- 11. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 2010;51:1069‐1077. [DOI] [PubMed] [Google Scholar]
- 12. Arafat A, Jing P, Ma Y, et al. Unexplained early infantile epileptic encephalopathy in Han Chinese children: next‐generation sequencing and phenotype enriching. Sci Rep. 2017;7:46227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gai N, Jiang C, Zou YY, Zheng Y, Liang DS, Wu LQ. Novel SIL1 nonstop mutation in a Chinese consanguineous family with Marinesco‐Sjögren syndrome and Dandy‐Walker syndrome. Clin Chim Acta. 2016;458:1‐4. [DOI] [PubMed] [Google Scholar]
- 14. Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294‐300. [DOI] [PubMed] [Google Scholar]
- 15. Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med. 2016;375:1289‐1294. [DOI] [PubMed] [Google Scholar]
- 16. Prasad V. Perspective: The precision‐oncology illusion. Nature. 2016;537:S63. [DOI] [PubMed] [Google Scholar]
- 17. Hayashi K, Ueshima S, Ouchida M, et al. Therapy for hyperthermia‐induced seizures in Scn1a mutant rats. Epilepsia. 2011;52:1010‐1017. [DOI] [PubMed] [Google Scholar]
- 18. Lin GW, Lu P, Zeng T, et al. GAPDH‐mediated posttranscriptional regulations of sodium channel Scn1a and Scn3a genes under seizure and ketogenic diet conditions. Neuropharmacology. 2017;113:480‐489. [DOI] [PubMed] [Google Scholar]
- 19. Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun. 2013;4:2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dilena R, Striano P, Gennaro E, et al. Efficacy of sodium channel blockers in SCN2A early infantile epileptic encephalopathy. Brain Dev. 2017;39:345‐348. [DOI] [PubMed] [Google Scholar]
- 21. Wagnon JL, Barker BS, Hounshell JA, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 2016;3:114‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Larsen J, Carvill GL, Gardella E, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84:480‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen D, Hernandez CC, Shen W, et al. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain. 2017;140:49‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pisano T, Numis AL, Heavin SB, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. 2015;56:685‐691. [DOI] [PubMed] [Google Scholar]
- 25. Ihara Y, Tomonoh Y, Deshimaru M, et al. Retigabine, a kv7.2/kv7.3‐channel opener, attenuates drug‐induced seizures in knock‐in mice harboring kcnq2 mutations. PLoS ONE. 2016;11:e150095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Helander A, Stödberg T, Jaeken J, Matthijs G, Eriksson M, Eggertsen G. Dolichol kinase deficiency (DOLK‐CDG) with a purely neurological presentation caused by a novel mutation. Mol Genet Metab. 2013;110:342‐344. [DOI] [PubMed] [Google Scholar]
- 27. Mikati MA, Jiang YH, Carboni M, et al. Quinidine in the treatment of KCNT1‐positive epilepsies. Ann Neurol. 2015;78:995‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Camposano SE, Major P, Halpern E, Thiele EA. Vigabatrin in the treatment of childhood epilepsy: a retrospective chart review of efficacy and safety profile. Epilepsia. 2008;49:1186‐1191. [DOI] [PubMed] [Google Scholar]
- 29. Krueger DA, Wilfong AA, Holland‐Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679‐687. [DOI] [PubMed] [Google Scholar]
- 30. Mills PB, Camuzeaux SS, Footitt EJ, et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain. 2014;137:1350‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lotte J, Bast T, Borusiak P, et al. Effectiveness of antiepileptic therapy in patients with PCDH19 mutations. Seizure. 2016;35:106‐110. [DOI] [PubMed] [Google Scholar]
- 32. Chen BB, Prasad C, Kobrzynski M, Campbell C, Filler G. Seizures related to hypomagnesemia: a case series and review of the literature. Child Neurol Open. 2016;3 10.1177/2329048X16674834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. EuroEPINOMICS‐RES Consortium . Epilepsy Phenome/Genome Project, and Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2017;100:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dorre K, Olczak M, Wada Y, et al. A new case of UDP‐galactose transporter deficiency (SLC35A2‐CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. 2015;38:931‐940. [DOI] [PubMed] [Google Scholar]
- 35. Marguet F, Barakizou H, Tebani A, et al. Pyridoxine‐dependent epilepsy: report on three families with neuropathology. Metab Brain Dis. 2016;31:1435‐1443. [DOI] [PubMed] [Google Scholar]
- 36. Ananth AL, Robichaux‐Viehoever A, Kim YM, et al. Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr Neurol. 2016;59:81‐84. [DOI] [PubMed] [Google Scholar]
- 37. Reid CA, Phillips AM, Petrou S. HCN channelopathies: pathophysiology in genetic epilepsy and therapeutic implications. Brit J Pharmacol. 2012;165:49‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sheehan JJ, Benedetti BL, Barth AL. Anticonvulsant effects of the BK‐channel antagonist paxilline. Epilepsia. 2009;50:711‐720. [DOI] [PubMed] [Google Scholar]
- 39. Palmer S, Towne MC, Pearl PL, et al. SLC6A1 mutation and ketogenic diet in epilepsy with myoclonic‐atonic seizures. Pediatr Neurol. 2016;64:77‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tohyama J, Nakashima M, Nabatame S, et al. SPTAN1 encephalopathy: distinct phenotypes and genotypes. J Hum Genet. 2015;60:167‐173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials