

Abstract
Protein–ligand interaction plays a critical role in regulating the biochemical functions of proteins. Discovering protein targets for ligands is vital to new drug development. Here, we present a strategy that combines experimental and computational approaches to identify ligand-binding proteins in a proteomic scale. For the experimental part, we coupled pulse proteolysis with filter-assisted sample preparation (FASP) and quantitative mass spectrometry. Under denaturing conditions, ligand binding affected protein stability, which resulted in altered protein abundance after pulse proteolysis. For the computational part, we used the software Patch-Surfer2.0. We applied the integrated approach to identify nicotinamide adenine dinucleotide (NAD)-binding proteins in the Escherichia coli proteome, which has over 4200 proteins. Pulse proteolysis and Patch-Surfer2.0 identified 78 and 36 potential NAD-binding proteins, respectively, including 12 proteins that were consistently detected by the two approaches. Interestingly, the 12 proteins included 8 that are not previously known as NAD binders. Further validation of these eight proteins showed that their binding affinities to NAD computed by AutoDock Vina are higher than their cognate ligands and also that their protein ratios in the pulse proteolysis are consistent with known NAD-binding proteins. These results strongly suggest that these eight proteins are indeed newly identified NAD binders.
Keywords: pulse proteolysis, protein–ligand interaction, quantitative mass spectrometry, tandem mass tags labeling, Patch-Surfer2.0, binding pocket screening, binding pocket comparison, structural bioinformatics
Graphical Abstract
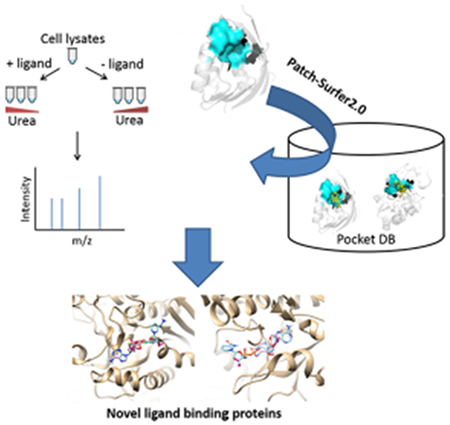
INTRODUCTION
Protein–ligand interactions play critical roles in regulating biochemical functions of proteins. The study of protein–ligand interactions is important for understanding various vital processes in a cell, which include transport pathways, metabolic processes, regulation of biological processes, and cell signaling. The discovery of a ligand that binds a targeted protein is also a major focus of early stage drug discovery.1
Recently, high-throughput discovery methods to study ligand-binding proteins, such as affinity chromatography2–4 and genetic array,5,6 have gained popularity. However, they both have inherent problems; these methods permit large amounts of nonspecific binding and can be time-consuming and costly.7 As protein–ligand interaction is governed by general thermodynamics, proteins are stabilized by ligands that bind preferentially to their native conformations.8,9 Profiling proteins based on the change in thermodynamic stability upon ligand binding have become popular as well, such as pulse proteolysis,10,11 stability of proteins from rates of oxidation (SPROX),12 Domain Architecture Retrieval Tools (DARTS),13 and stable isotope labeling with amino acids in cell culture (SILAC) coupled with pulse proteolysis.14 In two recent large-scale quantitative mass spectrometry based proteomic studies,15,16 researchers profiled the changes in resistance of proteins to aggregation due to ligand binding in elevated temperatures.
Pulse proteolysis is a method with which to monitor protein unfolding by exploiting the change in proteolytic susceptibility upon unfolding.10,11 The method employs a brief incubation (pulse) with a nonspecific protease, which digests denatured proteins but not the native ones in a complex biological sample. Because ligand binding to the native form stabilizes a protein, pulse proteolysis is able to monitor ligand binding to a protein by comparing proteolysis results with and without the target ligand. Compared to the method based on thermal denaturation, pulse proteolysis is indeed based on the equilibrium property of the target proteins. So far, this method has been coupled with gel electrophoresis and matrix-assisted laser desorption ionization (MALDI) mass spectrometry17 or with the SILAC technique.14 Although coupling SILAC to advanced mass-spectrometry-enabled high-throughput explorations demonstrates that, in principle, unknown ligand-binding proteins could be identified, the study only carried out targeted analyses. Thus, large-scale methods remain to be developed.
Besides experimental methods, computational approaches have also been used in protein–ligand interaction studies.18–21 Computational methods can be roughly classified into two categories. The first category predicts the binding ligand of a target protein by inferring from similar proteins in a database. The similarity between a target and proteins in a database can be calculated by comparing their structures and sequences.22–27 The second category directly docks a ligand molecule to a binding site of a target protein.28–30 These two categories have different advantages. The former category would be appropriate for a proteomic-scale screening because they are computationally less expensive, particularly for searching novel proteins that bind to known ligands that are abundant in a database. However, the advantages of the latter approach include that it usually provides ligand-binding poses, and thus, the binding affinity can be estimated.
Here, we developed a strategy that combines experimental and computational methods to investigate protein–ligand interactions and discover novel ligand-binding proteins (Figure 1). We used nicotinamide adenine dinucleotide (NAD) as the ligand to test the strategy. For the experimental part, we coupled pulse proteolysis with filter-assisted sample preparation (FASP) and quantitative mass spectrometry. In contrast to the previous applications of pulse proteolysis for target identification, which employed gel electrophoresis to separate pulse-digested peptides and intact proteins,14 we utilized FASP31 to filter out the digested peptides. Stabilization from ligand-binding leads to a change in protein abundance after pulse proteolysis. The change in abundance was measured by tandem mass tags (TMT)-labeling coupled quantitative mass spectrometry. Unlike the previous work,14 this approach allows us to monitor overall protein abundance change in a proteomics scale.
Figure 1.
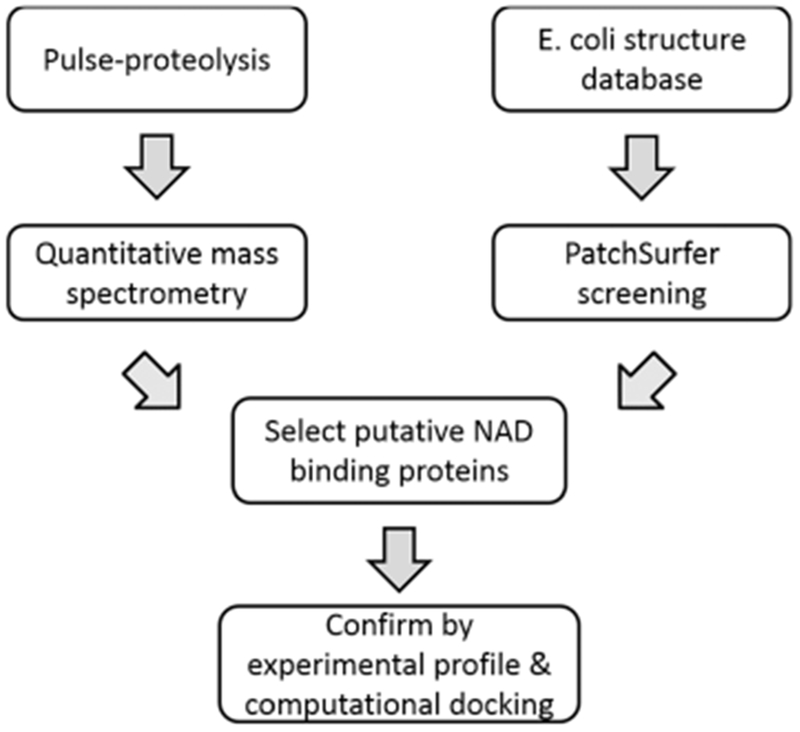
Overview of the combined approach with pulse proteolysis–quantitative mass spectrometry and computational screening with Patch-Surfer2.0.
We generated a list of proteins that showed a change in stability in the presence of NAD and identified known NAD-binding proteins in the list. Putative NAD-binding proteins were also examined using Patch-Surfer2.0,25,26 a computational program that predicts protein–ligand binding. Patch-Surfer2.0 predicts ligand binding of a query protein by comparing its pocket to known ligand-binding pockets in a database. Pockets are compared on a local surface patch basis so that the similarities in local pocket regions that are critical in binding can be identified. A surface patch is characterized by four physicochemical properties: shape, electrostatic potential, hydrophobicity, and visibility (i.e., concavity). The properties are described in a compact, rotation-invariant fashion using three-dimensional Zernike descriptors (3DZD), a mathematical series expansion of 3D functions.32
By combining the experimental results and prediction by Patch-Surfer2.0, we identified known NAD-binding proteins, and moreover, we also predicted novel NAD-binding proteins. The potential novel NAD-binding proteins that were commonly identified by pulse proteolysis and Patch-Surfer2.0 were further examined for their binding pose and affinity by a protein–ligand docking method to conclude that the NAD-binding is highly likely.
METHODS
Escherichia coli Cell Lysate Preparation
E. coli K12 cell lysates were grown in Luria–Bertani (LB) medium (10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl in 1 L of H2O) at 37 °C until OD 600 reaches 0.6. Cells were collected and lysed in lysis buffer (20 mM Tris pH 8.0, 1 mM EDTA, and 1 mM TCEP) with sonication. Whole-cell lysates were centrifuged at 16100g for 30 min, and supernatant was taken and centrifuged at 16100g for 15 min. Protein concentration was measured by Bradford assay (Bio-Rad). Before pulse proteolysis, cell lysates were denatured in 50 mM Tris HCl (pH 8.0) with 8 M urea at 37 °C for 30 min. The samples were then buffer-exchanged and renatured in 10 000 molecular weight cutoff filters with 50 mM Tris HCl (pH 8.0).
Ligand-Binding Equilibrium and Pulse Proteolysis
Denatured and renatured E. coli cell lysates were incubated with or without 1.0 mM NAD in the reaction buffer (20 mM Tris HCl (pH 8.0), 100 mM NaCl, and 1 mM DTT) with a certain concentration of urea for 2 h at room temperature. The process of denaturing and renaturing was performed to discard the endogenous small molecules that had already bound to the proteins. We denatured the cell lysate in 8 M urea first and then centrifuged the samples in 10K filters to filter out the small molecules and urea. For pulse proteolysis, thermolysin (from Bacillus thermoproteolyticus rokko, Sigma Chemical) stock solution in 2.5 M NaCl containing 10 mM CaCl2 was added to the sample and incubated at room temperature. The reaction was quenched by 600 μM phosphoramidon and 500 mM EDTA.
Peptide Sample Preparation and TMT Labeling
After pulse proteolysis, samples were transferred to a 30 000 or 100 000 molecular weight cutoff filter. Samples were buffer exchanged with 200 mM HEPES (pH 8.0). Protein samples in the filter were reduced in 8 M urea in 50 mM Tris HCl and 10 mM DTT at 37 °C for 30 min. Then samples were alkylated in 30 mM IAA at room temperature, protected from light, for 30 min. Next, samples were buffer exchanged with 200 mM HEPES (pH 8.0) and digested with trypsin at 37 °C overnight. On the following day, the digested peptides were collected by centrifugation and labeled with TMT reagents (dissolved in acetonitrile, ACN) at room temperature for 1 h. Reactions were quenched by 5% hydroxylamine for 15 min. Labeled peptides were acidified with 10 μL of 10% trifluoroacetic acid (TFA) and enough 0.1% TFA to make sure the final ACN concentration was less than 5%. Finally, samples were combined according to the experiment design, desalted using NuTips (Glygen) and lyophilized in a centrifugal vacuum concentrator (SpeedVac).
Liquid Chromatography–Mass Spectrometry Analysis
Peptide samples were redissolved in 10 μL of 0.1% formic acid and injected into an EASY-nLC 1000 liquid chromatograph. Reverse-phase separation was performed using an in-house C18 capillary column (75 μm i.d. and 15 cm bed length) packed with 3 μm C18 ProntoSIL AQ bead resin (Bischoff Chromatography, Germany). The mobile-phase buffer consisted of 0.1% formic acid in ultrapure water with the elution buffer of 0.1% formic acid in 80% ACN run over a shallow linear gradient (from 2% to 40% ACN) over 90 or 120 min with a flow rate of 300 nL/min. The electrospray ionization emitter tip was generated on the packed column with a laser puller (Model P-2000, Sutter Instrument Co.). The EASY-nLC 1000 liquid chromatograph was coupled with a hybrid linear ion trap orbitrap mass spectrometer (LTQ-Orbitrap Velos; Thermo Fisher). The mass spectrometer was operated in the data-dependent mode in which a full MS scan was done from m/z 300–1700 (resolution of 30 000). The top 12 most abundant ions from the full scan were selected for higher-energy collision dissociation (HCD; 40% energy; activation time, 30 ms; max injection time, 500 ms; AGC target, 50 000) with tandem mass spectrometry (MS/MS) detection in the Orbitrap (from m/z 100–2000; resolution, 7500). Ions with a charge state of +1 or with undetermined charge states were excluded. The mass exclusion time was 90 s.
Mass Spectrometry Data Analysis
The LTQ-Orbitrap raw files were searched directly against an E. coli K12 database using SEQUEST on Proteome Discoverer (Version 1.4, Thermo Fisher). Proteome Discoverer created DTA files from raw data files with a minimum ion threshold of 15 and an absolute intensity threshold of 50. Peptide precursor mass tolerance was set to 10 ppm, and MS/MS tolerance was set to 0.6 Da. Cysteine carbamidomethylation (+57.0214 Da), and TMT modifications (N-terminus and lysine residues, +229.2634 Da) were defined as fixed modifications and methionine oxidation (+15.9949Da) was variable modifications. Searches were performed with full tryptic digestion and allowed a maximum of two missed cleavages on the peptides analyzed from the sequence database. At least one peptide with high confidence was used for identification and quantification. Only quantifiable proteins were used for the data analysis. False discovery rates (FDR) were set to 1% for each analysis. For TMT quantitation, the ratios of TMT reporter ion abundances in MS/MS spectra generated by HCD (reporter ions of m/z 126.2193 or 127.2127 for duplex TMT) from raw data sets were used to calculate fold changes in protein abundances between samples with NAD and samples without NAD. After we obtained the data of protein identification and corresponding ratio of NAD-treated sample and control, we chose the protein ratio larger than 1.25 for at least two out of three replications.
Binding Ligand Prediction Using Patch-Surfer2.0
NAD-binding proteins in E. coli were computationally predicted using a ligand-binding prediction program, Patch-Surfer2.0.25,26 Here we briefly explain the Patch-Surfer2.0 algorithm. For more details and its performance, refer to the original papers.25,26
Patch-Surfer2.0 predicts the binding ligand of a query pocket by comparing the query with a precompiled database of known ligand-binding pockets in proteins. A pocket region for a known ligand binding protein is defined as a set of atoms that are closer than 4.5 Å to the co-crystallized ligand. The pocket surface is characterized by four properties: surface shape, electrostatic potential, hydrophobicity, and visibility. The molecular surface and electrostatic potentials of a query protein are generated by the Adaptive Poisson–Boltzmann Solver (APBS) program,33 and the hydrophobicity is assigned according to the Kyte–Doolittle scale.34 Visibility quantifies the concavity of a surface region by counting the number of visible directions from each point on the surface.35 The surface of a pocket is divided into a number of overlapping patches, which are characterized by the four physicochemical properties. A comparison between pockets is performed by identifying similar patches between them. The patch-based comparison enables the identification of corresponding critical binding regions in two pockets even in cases in which the pocket shapes are globally different from each other. The physicochemical properties of a patch are converted into 3DZD, which is a compact, rotation-invariant descriptor of the properties.
A query pocket is compared with each of the known pockets in a preconstructed pocket database, and the known pockets are ranked by a similarity score to the query. The score is computed using the modified auction algorithm,36 which optimizes matching surface patches between two pockets so that the score, which considers the similarity of individual matching patch pairs, relative locations of corresponding patches, and pocket size, is minimized (smaller score is more similar). Then, the known pockets in the database are sorted by the score to the query pocket, and finally, the ligands that bind the query pocket are predicted according to the pocket score:
| (1) |
where n is the number of pockets in the database, k is the number of the top-scoring ligands to use for computing the pocket score, and ω1(i),F is a ligand similarity between the ligand F and the ligand of the kth pocket calculated by SIMCOMP.37 Intuitively, the pocket score for a ligand F for a query pocket becomes larger if many ligands of top-hit pockets are similar to F (the sum of the ligand similarity between F and the binding ligand of top hits; the left part of the equation), and the score is normalized by the abundance of similar ligands to F in the database (fraction of the sum of the ligand similarity of up to the kth most similar pockets relative to the ligand similarity to the entire database; the right part of the equation).
Figure S1 illustrates how Patch-Surfer2.0 was applied to predict NAD-binding proteins in E. coli. Crystal structures of E. coli proteins were collected from the PDB database (December of 2014).38 Structures were selected from PDB using the following two conditions: (1) the protein is from E. coli, and (2) to reduce the query structures to scan, homologous proteins were filtered out with a sequence identity cutoff of 90% to each other. Once a query protein is predicted to bind NAD, its homologous proteins are also examined to confirm the possibility of NAD binding. These two conditions identified 1918 PDB entries. From these entries, individual protein chains were extracted.
If a protein structure was co-crystallized with a ligand, the pocket was defined as the set of atoms that are, at most, 4.5 Å from any atom of the ligand. If there was no ligand in the crystal structure, a pocket was predicted by LigSiteCSC39 and VisGrid.35 After pocket information was obtained from either method, binding ligand prediction was performed by Patch-Surfer2.0. If NAD or NADP was ranked within the top ten scoring ligand types, the protein was denoted as a putative NAD binder.
Reverse Screening by AutoDock Vina
For predicted NAD-binding proteins that are not known, AutoDock Vina40 was employed to further examine the binding affinity and the binding pose of NAD in the proteins’ binding pockets of their co-crystallized ligands. For each protein, NAD and the cognate ligand from the crystal structure were redocked to the binding pocket, and their affinity values were compared.
The docking box of the protein was defined as a cube with 25 Å length on each side at the binding pocket. The protonation state and the Gasteiger charge were assigned on the receptor structure by using the DockPrep module of UCSF Chimera.41 The flexible ligand mode was used for docking.
RESULTS
Pulse-Proteolysis Condition Optimization
We used E. coli K12 cell lysates as the protein pool for finding NAD-binding proteins. In the pulse proteolysis, we used 0.20 mg/mL thermolysin for 1 min, selected on the basis of the test performed with different concentrations of thermolysin and urea (Figure S2). This condition is consistent with our previous study.11 To filter out digested peptides, 30 000 and 100 000 molecular weight cutoff filters were examined (Figure S3). After pulse proteolysis and filtering, digested peptides were detected in the collection tubes of the 100 000 group but not the 30 000 group. This result indicated that the 100 000 filter was more efficient than the 30 000 filter in filtering out the digested peptides.
Quantitative Mass Spectrometry analysis
For quantitative MS, we used TMT-duplex labeling. The workflow that incorporates the TMT-duplex strategy is shown in Figure S4. A total of three biological replicates were conducted. Histograms of protein counts for each experiment across log2 protein ratio (+NAD/−NAD) are shown in Figure S5. The numbers of all the proteins detected in the three replicates are provided in Figure S6. Among the proteins detected, we further selected proteins with a large stability change upon NAD binding (namely, those that had a protein ratio, +NAD/−NAD, of 1.25 or larger; see Table 1). This ratio is chosen on the basis of a previous study,42 which showed that in a mixture sample where co-isolation interference occurs,43 a reporter ion ratio of 1.25 or higher is likely to be biologically meaningful. Approximately 4% to 8.5% among the detected proteins had a protein ratio of 1.25 or higher. The average fraction of the proteins with a ratio ≥1.25 at a urea concentration of 3.5, 4.0, and 4.5 M was 6.77%, 4.31%, and 6.81%, respectively. Figure 2 illustrates the numbers of proteins above this threshold across the triplicate experiments using three urea concentrations: 3.5, 4.0, and 4.5 M. If a protein showed a protein ratio of ≥1.25 for at least two replicates for a urea concentration, it is considered significant.
Table 1.
Number of Identified Proteins by Quantitative Mass Spectrometry Analysis
| proteins with protein ratios of
≥1.25 (with NAD/without NAD) when divided by total number of
detected proteins |
|||
|---|---|---|---|
| urea concentration (M) | replicate 1 | replicate 2 | replicate 3 |
| 3.5 | 50/800 | 64/880 | 58/856 |
| 4.0 | 38/882 | 40/985 | 40/875 |
| 4.5 | 48/818 | 76/910 | 49/788 |
Figure 2.
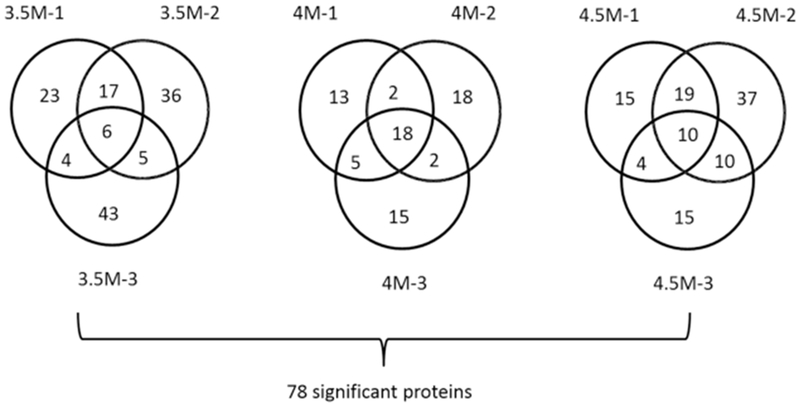
Number of proteins detected as NAD binding by the experiments. A total of three urea concentrations (3.5, 4.0, and 4.5 M) were used, and for each, three biological replicates were produced. We considered that NAD significantly changed the stability of a protein if the ratio of the protein measured by quantitative mass spectrometry with and without NAD changed 1.25-fold or more. Then, a protein is as NAD binding for a certain urea concentration if the protein ratio was 1.25 or higher in at least two out of the three replicates. The number of proteins with a significant protein ratio detected by each replicate is shown in the Venn diagram. For example, 3.5M-1 indicates the first replicate of a 3.5 M urea concentration. In total, 32, 27, and 43 proteins were detected as NAD binding at a 3.5, 4.0, and 4.5 M urea concentration, respectively, with 78 unique proteins in total.
There were 32, 27, and 43 proteins, respectively, for 3.5, 4.0, and 4.5 M urea concentrations, which include 78 unique proteins. These proteins include 4 (out of 32), 5 (out of 27), and 4 (out of 43) known NAD-binding proteins that are annotated with a Gene Ontology (GO) term of “NAD binding” (GO: 0051288), respectively. When we compare this ratio (13/78 = 16.7%) to the ratio of known NAD binders to total E. coli proteome (about 1%) as well as the ratio of known NAD binders among 1229 proteins detected by MS (28/1229 = 2.3%), these are high enrichments of known NAD binders. DeArmond et al. used SPROX experiments to identify NAD-binding proteins in the yeast proteome and identified 11 NAD-binding proteins.12 Among the 11 proteins they identified, 2 do not have obvious homologs in E. coli. Compared with the remaining 9 proteins, the 28 proteins we identified in E. coli include 2 proteins in common glyceraldehyde 3-phosphate dehydrogenase and isocitrate dehydrogenase. We have observed some apparent destabilizations, which may result from NAD binding to non-native forms or NAD-induced dissociation of some proteins.17,44 Applying the criterion of a decreased protein ratio ≥1.25 with NAD in 2 replicates, 53 proteins were identified. Although these observations are worth further investigation, we focused on stabilized proteins in this study because functional binding of NAD should occur to native proteins, and binding to native proteins must stabilize the target proteins.
Figure 3 shows a breakdown of the GO terms of the 78 proteins, which are significant proteins from the three urea concentrations. For the Molecular Function category (Figure 3A), the majority of the proteins have catalytic activity. For the Biological Process category (Figure 3B), the majority of the proteins are involved in metabolic processes. We have also examined the protein classes of the significant proteins using the Panther database45 (Figure 3C). A total of 14 out of the 78 proteins (29.2%) were oxidoreductases, which use NAD as the coenzyme in redox reactions in metabolism.
Figure 3.
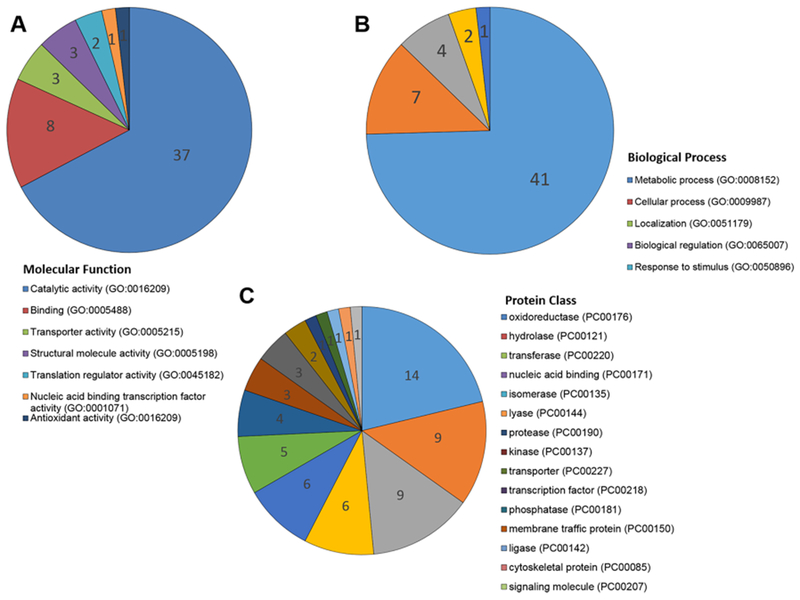
Functional classification of proteins detected as NAD-binding by pulse proteolysis. (A) The 55 proteins that have GO terms in the Molecular Function category; (B) classification of the 55 proteins in the Biological Process GO; (C) the 66 proteins that have Panther Protein Class assignment.
NAD-Binding Protein Prediction by Patch-Surfer2.0
Now we turn our attention to computational prediction of NAD-binding proteins by Patch-Surfer2.0 (Supplemental Figure S-1). As explained in the Methods section, a protein is predicted as an NAD-binding protein if either NAD or NADP is predicted within the top 10 scoring binding ligands for the protein. We included predicted NADP-binding proteins in the list because PatchSurfer2.0 often had difficulty in distinguishing NAD-binding and NADP-binding proteins in the previous benchmark study.26
As listed in Table 2, 36 proteins were predicted as NAD binders. The 36 proteins include 20 that have a high similarity score to a known NAD-binding protein (the first half in the table) and 22 that have high similarity to NADP-binding pockets (the bottom half in the table). There were six proteins (indicated with asterisks in the table), which were predicted as both NAD- and NADP-binding proteins. The prediction results by Patch-Surfer2.0 are reasonably specific because the many of the top hits are proteins that were co-crystallized with NAD or NADP. For example, four out of the top five predicted NAD binders and NADP binders are co-crystallized with NAD or NADP. Also, when the similarity of the co-crystallized ligands of the predicted proteins to NAD was considered, it was found that 19 out of 36 them have a SIMCOMP similarity score of 0.4 or higher, which indicates that the ligands share common chemical groups with NAD.26
Table 2.
Predicted NAD-Binding Proteins by PatchSurfer2.0a
| proteinb | gene ID | PDB ID | co-crystallized ligand | ligand similarity to NAD (SIMCOMP score)c | GO term annotatedd | overlap with experimental resultsee |
|---|---|---|---|---|---|---|
| predicted NAD-binding proteins | ||||||
| glyceraldehyde 3-phosphate dehydrogenase | gapA | 1GAD | NAD | 1.000 | × | × |
| dTDB–glucose 4,6-dehydrogenase | rffG | 1BXK | NAD | 1.000 | ||
| formate dehydrogenase H | fdhF | 2IV2 | molybdopterin guanine dinucleotide | 0.517 | ||
| β-ketoacyl–ACP reductase* | fabG | 1Q7B | NADP | 0.878 | × | |
| 2,4-dienoyl–CoA reductase* | fadH | 1PS9 | NADP | 0.878 | ||
| quinone oxidoreductase* | qorA | 1QOR | NADP | 0.878 | ||
| glutathione transferase | yghU | 3C8E | glutathione | 0.085 | × | |
| glycerol–3-phosphate dehydrogenase* | glpD | 2QCU | FAD | 0.470 | ||
| methionine synthase | metH | 1BMT | co-methylcobalamin | 0.193 | ||
| 7-α-hydroxysteroid dehydrogenase* | hdhA | 1FMC | NAD | 1.000 | ||
| nucleotidyltrasferase | galT | 1GUQ | uridine-5′-diphosphate-glucose | 0.483 | × | |
| purine nucleoside phosphorylase | deoD | 1K9S | N7-methyl-formycin A | 0.306 | × | |
| 3-isopropylmalate dehydrogenase | leuB | 1CM7 | × | × | ||
| GTP cyclohydrolase | folE | 1A8R | GTP | 0.462 | ||
| malonyl–CoA/ACP transcyclase | fabD | 1MLA | × | |||
| 2-methylcitrate dehydratase | prpD | 1SZQ | ||||
| glutathione reductase | gor | 1GES | NAD | 1.000 | ||
| alkyl hydroperoxide reductase | ahpF | 4O5Q | FAD | 0.470 | × | |
| UDP–N-acetylmyramyl tripeptide synthase* | murE | 1E8C | uridine-5′-diphosphate-N-acetylmuramoyl-l-alanine-d-glutamate | 0.384 | ||
| dihydrolipoamide hydrogenase | lpd | 4JDR | FAD | 0.470 | × | × |
| predicted NADP-binding proteins | ||||||
| GDP 4-keto-6-de-oxy-d-mannose epimerase reductase | fcl | 1E6U | NADP | 0.878 | ||
| dihydrofolate reductase | folA | 1RA9 | NADP | 0.878 | ||
| 2,4-dienoyl–CoA reductase* | fadH | 1PS9 | NADP | 0.878 | ||
| β-ketoacyl–ACP reductase* | fabG | 1Q7B | NADP | 0.878 | ||
| GAR transformylase | purN | 1JKX | N-[5′-O-phosphono-ribofuranosyl]-2-[2-hydroxy-2-[4-[glutamic acid]-N-carbonylphenyl]-3-[2-amino-4-hydroxy-quinazolin-6-Yl]-propanylamino]-acetamide | 0.256 | ||
| microcin C7 self-immunity protein | mccF | 4IIY | 5′-O-(α-glutamylsulfamoyl) inosine | 0.333 | ||
| UDP–N-acetylmyramyl tripeptide synthase* | murE | 1E8C | uridine-5′-diphosphate-N-acetylmuramoyl-l-alanine-d-glutamate | 0.384 | ||
| glutamate racemase | murI | 2JFN | uridine-5′-diphosphate-N-acetylmuramoyl-l-alanine | 0.409 | ||
| UDP–N-acetylmuramoyl l-alanine ligase | murC | 2F00 | ||||
| glycerol-3-phosphate dehydrogenase* | glpD | 2QCU | FAD | 0.470 | ||
| 7-α-hydroxysteroid dehydrogenase* | hdhA | 1FMC | NAD | 1.000 | ||
| UDP–l-Ara4N formyltransferase | arnA | 1Z75 | × | |||
| auccinyl–ALA–PRO–ALA–P-nitroanilide | ppiA | 1V9T | succinyl–ALA–PRO–ALA–P-nitroanilide | 0.098 | × | |
| threonyl–tRNA synthetase | thrS | 1EVL | 5′-O-(N-(l-threonyl)-sulfamoyl)adenosine | 0.396 | ||
| enoyl–ACP reductase | fabI | 4JQC | NAD | 1.000 | × | |
| MTA/SAH nucleosidase | mtnN | 3O4V | (3R,4S)-4-(4-chlorophenylthiomethyl)-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine | 0.320 | × | |
| predicted NADP-binding proteins | ||||||
| 2,3-diketo-l-gulonate reductase | slr | 1S2O | × | |||
| quinone oxidoreductase* | qorA | 1QOR | NADP | 0.878 | ||
| ketopantoate hydroxymethyltransferase | panB | 1M3U | ketopantoate | 0.058 | × | |
| putative N-acetylmannosamin kinase | nanK | 2AA4 | ||||
| pyridine nucleotide transhydrogenase | pntA | 1X15 | NAD | 1.000 | × | |
| UDP–sugar hydrolase | ushA | 1HP1 | ATP | 0.531 | × |
A protein was predicted as NAD-binding if either NAD or NADP was predicted within the top 10 scoring ligands for the protein.
The proteins marked with asterisks are predicted both as NAD-binding (top half of the table) and NADP-binding (bottom half). There are six such proteins
SIMCOMP is software that compares and quantifies similarity between two chemical compounds by considering two-dimensional chemical structures of the compounds. The score ranges from 0.0 to 1.0, with 1.0 indicating the identical compounds.
The GO term for NAD binding (GO: 0051288) in annotation in the UniProt database was examined.
A protein was checked if it was included in the list of the 78 detected NAD-binding proteins by pulse proteolysis.
We also compared the Patch-Surfer2.0s prediction with E. coli proteins that are annotated with the GO term for NAD binding (GO: 0051288). It turned out that the information on co-crystallized ligands and the GO annotations do not always agree, i.e., there are proteins that are co-crystallized with NAD but are not annotated with GO: 00051288 and vice versa (Table 2). Out of the 36 proteins in Table 2, 8 are annotated as NAD-binding in GO. Considering all factors together, 16 out of the 36 proteins are either co-crystallized with NAD or NADP or annotated as NAD binding in GO annotation, and many other bound ligands in the predicted proteins have sufficient similarity to NAD.
When further comparisons were made among the GO annotations of all of the E. coli proteins, there were 46 proteins in the E. coli proteome that are annotated as NAD-binding, of which 8 overlapped with the computational prediction. Thus, the remaining 38 GO-annotated NAD-binding proteins were not predicted by Patch-Surfer2.0. Among the 38 proteins, 27 proteins have not been crystallized yet and thus could not be handled by Patch-Surfer2.0 because Patch-Surfer2.0 needs the 3D structure of a protein. In the remaining 11,7 proteins were crystallized in an apo form, where the binding pockets needed to be predicted by LigSiteCSC and VisGrid, which apparently did not work well. The predicted binding pockets were smaller in size, about 29.3% of the average size of known NAD-binding pockets. The remaining four proteins were predicted as flavin adenine dinucleotide (FAD) binding proteins but not as NAD-binding. FAD and NAD are similar (0.470 SIMCOMP score), sharing an adenine dinucleotide moiety.
Predicted Novel NAD-Binding Proteins by Both Methods
Figure 4 summarizes the overlap between NAD-binding proteins identified by pulse proteolysis, those that were predicted by Patch-Surfer2.0 and those that are annotated with the GO term of NAD-binding. A total of three proteins were common in the three groups. They are 3-isopropylmaltate dehydrogenase (leuB), glyceraldehyde-3-phosphate dehydrogenase A (gapA), and dihydrolipoamide hydrogenase (lpd).
Figure 4.
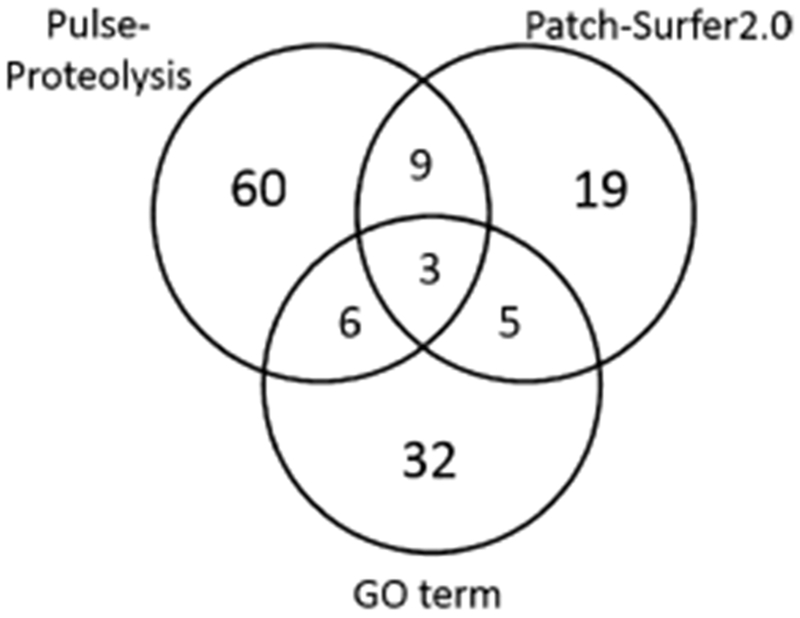
Commonly detected NAD-binding proteins by pulse proteolysis, Patch-Surfer2.0, and GO term annotation shown in a Venn diagram.
There were nine proteins that were shared between the pulse-proteolysis results and the computational prediction but not included in the GO-annotated protein list. We are interested in these proteins because they are potentially novel NAD-binding proteins. These proteins are Enoyl–ACP reductase (fabI), 3-methyl-2-oxobutanoate hydroxymethyltransferase (panB), 5′-methylthioadenosine–S-adenosylhomocysteine nucleosidase (mtn). disulfide reductase–organic hydroperoxide reductase (yghU), galactose-1-phosphate uridylyltransferase (galT), malonyl–CoA–ACP transacylase (fabD), peptidyl–prolyl cis–trans isomerase A (ppiA), UDP–sugar hydrolase (ushA), and purine nucleoside phosphorylase (deoD). To validate further, we have predicted binding affinity and binding pose of NAD to the proteins using AutoDock Vina, a commonly used ligand-protein docking method, except for fabI, which is crystallized with NAD.
The predicted NAD-binding affinities of the eight proteins are listed in Table 3. For comparison, the cognate ligands of the crystal structures of the proteins were redocked, and their binding affinities are compared (the last column of Table 3). Remarkably, for all but one case (galT), NAD had a stronger binding affinity than the cognate ligands, suggesting that these proteins could bind NAD. Computed NAD-binding poses of the eight proteins are provided in Figure 5. The binding poses show that NAD fits inside the binding pockets in a physically feasible fashion, suggesting that the molecule could bind to the pocket. The eight identified proteins do not share high sequence identity between each other, indicating that they are not homologous (Table S1). The highest sequence identity was 24.39%, which was obtained between panB and ushA.
Table 3.
Predicted NAD-Binding Affinity of the Eight Putative NAD Binders
| protein name | gene ID | PDB ID | NAD-binding affinity (kcal/mol) | binding affinity of the cognate ligand (kcal/mol) |
|---|---|---|---|---|
| galactose-1-phosphate uridylyltransferase | galT | 1GUQ | −11.0 | −12.5 |
| 5′-methylthioadenosine and S-adenosylhomocysteine nucleosidase | mtn | 3O4V | −10.6 | −10.4 |
| purine nucleoside phosphorylase | deoD | 1K9S | −10.5 | −9.4 |
| disulfide reductase and organic hydroperoxide reductase | yghU | 3C8E | −10.2 | −6.2 |
| peptidyl–prolyl cis–trans isomerase A | ppiA | 1V9T | −8.2 | −7.7 |
| 3-methyl-2-oxobutanoate hydroxymethyltransferase | panB | 1M3U | −8.2 | −6.5 |
| malonyl–coA–ACP transacylase | fabD | 1MLA | −7.3 | −6.0 |
| UDP–sugar hydrolase | ushA | 1HP1 | −7.2 | −6.6 |
The affinity was computed with Autodock Vina.
Figure 5.
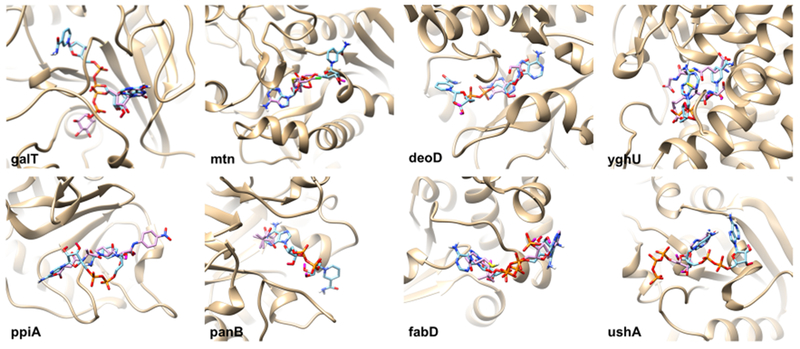
Predicted NAD-binding pose of the eight predicted novel NAD-binding proteins. NAD is colored in cyan, and the crystal structure of the cognate ligand is shown in magenta.
We also evaluated the predicted NAD binders from the experimental side by inspecting the profile of the protein ratio with and without NAD. Because NAD binding will increase protein stability in denaturing conditions, we expected to see that NAD binders will have higher abundance after pulse proteolysis with NAD than without NAD at a certain urea concentration. In Figure 6, we show the protein ratio (+NAD/−NAD) profile as a function of the three urea concentrations for proteins classified into four categories, predicted novel binders (Figure 6A), known NAD binders that are detected as included in the list of significant proteins (experimentally identified as NAD binders) (Figure 6B), known NAD binders not included in the list of significant proteins (Figure 6C), and those are neither among known NAD binders nor listed as significant proteins (Figure 6D).
Figure 6.
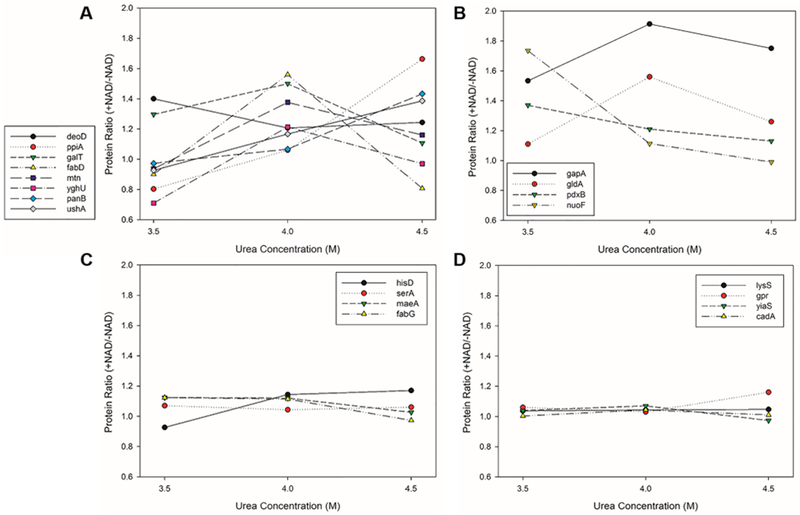
Protein ratio profile under three urea concentrations for the predicted novel NAD-binding proteins and other representative proteins. (A) The eight predicted novel NAD-binding proteins. (B) Examples of known NAD-binding proteins that are detected as significant proteins by the pulse-proteolysis experiments. (C) Examples of known NAD-binding proteins that are not detected as significant proteins by the experiments. (D) Examples of proteins that are neither known as NAD-binding proteins nor detected as significant proteins by the experiments. Via the quantitative mass spectrometry analysis following pulse proteolysis, a protein is considered NAD-binding if its protein ratio with and without NAD is 1.25 or more for at least two out of three biological replicates. The protein ratio values plotted are the average of three biological replicates.
A clear difference was observed between known NAD binders detected by pulse proteolysis (Figure 6B) and those that were not (Figure 6C,D). The former proteins showed a pattern of increased ratios at certain urea concentrations. In contrast, the latter proteins showed an almost 1:1 ratio for all the three urea concentrations, indicating that the protein stability did not change by adding NAD for the three urea concentrations tested. Notably, the predicted novel NAD binders (Figure 6A) follow the same trend as the detected, known NAD binders (Figure 6B), indicating that NAD does increase stability of the proteins.
DISCUSSION
We have presented the first study that combines experimental and computational methods to identify NAD-binding proteins in a proteomic scale. To experimentally identify NAD-binding proteins in a high-throughput fashion, we coupled pulse proteolysis with quantitative mass spectrometry. Combining with Patch-Surfer2.0, we identified not only known NAD-binding proteins but also eight potential novel ones.
Target identification by pulse proteolysis was previously coupled with gel electrophoresis for the detection of the change in protein stability. Due to the limited coverage of gel electrophoresis, the previous approaches had low sensitivity and throughput.14,17 In this work, we used the FASP method to filter digested peptides and, subsequently, trypsin-digested the remaining proteins. Quantitative mass spectrometry was then applied to observe the protein stability changes due to NAD binding. FASP is more efficient and convenient than in-gel digestion,46 leading to higher identification numbers with shorter analysis time. In addition, TMT labeling-based quantitative mass spectrometry allowed us to accurately detect protein abundance changes that are not visible in the gel.
In our experiment, we tested three urea concentrations ranging from 3.5 to 4.5 M. These are the urea concentrations at which the majority of proteins would be denatured, while some known NAD binders remain intact upon NAD binding. Because we did not cover a wider range of urea concentrations, some known NAD-binding proteins may not show a change in abundance in our experiment. Pulse proteolysis is not able to identify about 20% of E. coli soluble proteins because some proteins are susceptible to pulse proteolysis even under native conditions, and also quite a few proteins still remain folded, even in 8 M urea.17 Our method does not allow us to detect those proteins due to the inherent limitations of pulse proteolysis. As a future work, it would be interesting to use a wider range of urea concentrations to detect a larger number of proteins that bind NAD or other ligand molecules. Our lysis condition is relatively mild; thus, the extracted proteins are probably mainly cytoplasm proteins. Because this work is intended to be a nonbiased discovery method, the whole-cell lysates were used. However, the method can be also applied to specific cellular organelles by using lysates after cellular fractionation to identify organelle-specific ligand–protein interactions.
Computational methods are, in general, very suitable for a large-scale analysis of proteins. Patch-Surfer2.0 is one of the most recent binding ligand prediction methods and was shown to perform better than other existing methods.25,26,36 However, it showed two weaknesses in this study. First, as the method requires the 3D structures of ligand-binding sites, it clearly could not make predictions for proteins whose structure has not yet been solved. Second, the binding of NAD was not correctly predicted for some known NAD-binding proteins because their structures are of an apo form. Binding site predictions for these proteins were not successful. The first problem can be accommodated by using protein structure prediction methods47–49 to build structure models of proteins in case they do not have solved structures. To deal with the second problem, binding-site prediction needs to be improved, which may be addressed by explicitly considering conformational change of proteins between the holo and apo forms.50 These two weaknesses actually stem from the same issue, which is how to effectively use predicted structures that potentially have some errors.
Despite the limitations of the methods, we were able to predict eight novel NAD-binding proteins. Those proteins were further examined both computationally using AutoDock Vina and experimentally by inspecting the protein ratio profile with and without NAD. The computational docking showed that the binding affinity computed for NAD was higher than that for the co-crystallized ligand for all eight proteins except for one, suggesting that the remaining seven proteins actually bind NAD.
Methods for systematic detection of ligand-protein binding are not well-established despite their importance for understanding orchestrated molecular interactions in pathways in a cell. Because any currently available experimental or computational method has its own strengths and weaknesses, it is effective to employ multiple methods to compensate drawbacks and integrate strengths of each method. The two methods we used in this study, pulse proteolysis coupled with quantitative mass spectrometry and Patch-Surfer2.0, are simple; although they have their own limitations, as discussed above, they can, in principle, be applied for detecting other ligand-binding proteins at a proteomics scale. The method can be applied also for identifying new protein targets for drug molecules.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Justin Arrington and Lenna X. Peterson for proofreading the manuscript.
Funding
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R01GM097528). W.A.T. acknowledges supports by the National Science Foundation (CH1506752) and by the National Institutes of Health (5R01GM088317). D.K. also acknowledges supports from the National Science Foundation (IIS1319551, DBI1262189, IOS1127027, and DMS1614777).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACSPublicationswebsite at DOI: 10.1021/acs.jproteome.6b00624.
Figures showing computational prediction of NAD-binding proteins, pulse-proteolysis condition testing, pulse proteolysis–quantitative mass spectrometry using TMT-duplex labeling, protein identification and quantification with LC–MS, and the number of proteins detected in three replicates using three urea concentrations. A table showing pairwise sequence identity between predicted NAD-binding proteins. (PDF)
A table showing mass spectrometry identification and quantification data. (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Gilson MK; Zhou HX Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 21–42. [DOI] [PubMed] [Google Scholar]
- (2).Sleno L; Emili A Proteomic methods for drug target discovery. Curr. Opin. Chem. Biol 2008, 12 (1), 46–54. [DOI] [PubMed] [Google Scholar]
- (3).Rix U; Superti-Furga G Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol 2009, 5 (9), 616–24. [DOI] [PubMed] [Google Scholar]
- (4).Godl K; Wissing J; Kurtenbach A; Habenberger P; Blencke S; Gutbrod H; Salassidis K; Stein-Gerlach M; Missio A; Cotten M; Daub H An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (26), 15434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hoon S; Smith AM; Wallace IM; Suresh S; Miranda M; Fung E; Proctor M; Shokat KM; Zhang C; Davis RW; Giaever G; St Onge RP; Nislow C An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol 2008, 4 (8), 498–506. [DOI] [PubMed] [Google Scholar]
- (6).Giaever G; Flaherty P; Kumm J; Proctor M; Nislow C; Jaramillo DF; Chu AM; Jordan MI; Arkin AP; Davis RW Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (3), 793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burdine L; Kodadek T Target identification in chemical genetics: the (often) missing link. Chem. Biol 2004, 11 (5), 593–7. [DOI] [PubMed] [Google Scholar]
- (8).Schellman JA Macromolecular binding. Biopolymers 1975, 14 (19), 999. [Google Scholar]
- (9).Pace CN; McGrath T Substrate stabilization of lysozyme to thermal and guanidine hydrochloride denaturation. J. Biol. Chem 1980, 255 (9), 3862–5. [PubMed] [Google Scholar]
- (10).Park C; Marqusee S Quantitative determination of protein stability and ligand binding by pulse proteolysis. Curr. Protoc Protein Sci. 2006, 2011110.1002/0471140864.ps2011s46 [DOI] [PubMed] [Google Scholar]
- (11).Park C; Marqusee S Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2005, 2 (3), 207–12. [DOI] [PubMed] [Google Scholar]
- (12).Dearmond PD; Xu Y; Strickland EC; Daniels KG; Fitzgerald MC Thermodynamic analysis of protein-ligand interactions in complex biological mixtures using a shotgun proteomics approach. J. Proteome Res. 2011, 10 (11), 4948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lomenick B; Hao R; Jonai N; Chin RM; Aghajan M; Warburton S; Wang J; Wu RP; Gomez F; Loo JA; Wohlschlegel JA; Vondriska TM; Pelletier J; Herschman HR; Clardy J; Clarke CF; Huang J Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U. S. A 2009, 106 (51), 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Adhikari J; Fitzgerald MC SILAC-pulse proteolysis: A mass spectrometry-based method for discovery and cross-validation in proteome-wide studies of ligand binding. J. Am. Soc. Mass Spectrom. 2014, 25 (12), 2073–83. [DOI] [PubMed] [Google Scholar]
- (15).Savitski MM; Reinhard FB; Franken H; Werner T; Savitski MF; Eberhard D; Molina DM; Jafari R; Dovega RB; Klaeger S; Kuster B; Nordlund P; Bantscheff M; Drewes G Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346 (6205), 1255784. [DOI] [PubMed] [Google Scholar]
- (16).Huber KVM; Olek KM; Muller AC; Tan CSH; Bennett KL; Colinge J; Superti-Furga G. Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 2015, 12, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liu PF; Kihara D; Park C Energetics-based discovery of protein-ligand interactions on a proteomic scale. J. Mol. Biol 2011, 408 (1), 147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tinberg CE; Khare SD; Dou J; Doyle L; Nelson JW; Schena A; Jankowski W; Kalodimos CG; Johnsson K; Stoddard BL; Baker D Computational design of ligand-binding proteins with high affinity and selectivity. Nature 2013, 501 (7466), 212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stjernschantz E; Oostenbrink C Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys. J 2010, 98 (11), 2682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen YZ; Zhi DG Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins: Struct., Funct., Genet 2001, 43 (2), 217–26. [DOI] [PubMed] [Google Scholar]
- (21).Zhu X; Shin WH; Kim H; Kihara D Combined Approach of Patch-Surfer and PL-PatchSurfer for Protein-Ligand Binding Prediction in CSAR 2013 and 2014. J. Chem. Inf. Model 2016, 56 (6), 1088–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Brylinski M; Skolnick J FINDSITE: a threading-based approach to ligand homology modeling. PLoS Comput. Biol. 2009, 5 (6), e1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kinoshita K; Nakamura H Identification of the ligand binding sites on the molecular surface of proteins. Protein Sci. 2005, 14 (3), 711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xie ZR; Hwang MJ Ligand-binding site prediction using ligand-interacting and binding site-enriched protein triangles. Bioinformatics 2012, 28 (12), 1579–85. [DOI] [PubMed] [Google Scholar]
- (25).Sael L; Kihara D Detecting local ligand-binding site similarity in nonhomologous proteins by surface patch comparison. Proteins: Struct., Funct., Genet. 2012, 80 (4), 1177–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhu X; Xiong Y; Kihara D Large-scale binding ligand prediction by improved patch-based method Patch-Surfer2.0. Bioinformatics 2015, 31 (5), 707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chikhi R; Sael L; Kihara D Real-time ligand binding pocket database search using local surface descriptors. Proteins: Struct., Funct., Genet. 2010, 78 (9), 2007–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Meng EC; Shoichet BK; Kuntz ID Automated docking with grid-based energy evaluation. J. Comput. Chem 1992, 13 (4), 505–524. [Google Scholar]
- (29).Morris GM; Goodsell DS; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 19 (14), 1639–1662. [Google Scholar]
- (30).Goldman BB; Wipke WT QSD quadratic shape descriptors. 2. Molecular docking using quadratic shape descriptors (QSDock). Proteins: Struct., Funct., Genet. 2000, 38 (1), 79–94. [PubMed] [Google Scholar]
- (31).Wisniewski JR; Zougman A; Nagaraj N; Mann M Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6 (5), 359–362. [DOI] [PubMed] [Google Scholar]
- (32).Novotni M; Klein R 3D zernike descriptors for content based shape retrieval. In Proceedings of the Eighth ACM Symposium on Solid Modeling and Applications; ACM: Seattle, Washington, 2003. [Google Scholar]
- (33).Baker NA; Sept D; Joseph S; Holst MJ; McCammon JA Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (18), 10037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kyte J; Doolittle RF A simple method for displaying the hydropathic character of a protein. J. Mol. Biol 1982, 157 (1), 105–32. [DOI] [PubMed] [Google Scholar]
- (35).Li B; Turuvekere S; Agrawal M; La D; Ramani K; Kihara D Characterization of local geometry of protein surfaces with the visibility criterion. Proteins: Struct., Funct., Genet. 2008, 71 (2), 670–83. [DOI] [PubMed] [Google Scholar]
- (36).Sael L; Kihara D Binding ligand prediction for proteins using partial matching of local surface patches. Int. J. Mol. Sci 2010, 11 (12), 5009–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hattori M; Okuno Y; Goto S; Kanehisa M Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc 2003, 125 (39), 11853–65. [DOI] [PubMed] [Google Scholar]
- (38).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huang B; Schroeder M LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol. 2006, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2009, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25 (13), 1605–12. [DOI] [PubMed] [Google Scholar]
- (42).Rauniyar N; Gao BB; McClatchy DB; Yates JR Comparison of Protein Expression Ratios Observed by Sixplex and Duplex TMT Labeling Method. J. Proteome Res. 2013, 12 (2), 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).McAlister GC; Nusinow DP; Jedrychowski MP; Wuhr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem 2014, 86 (14), 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Liu PF; Park C Selective stabilization of a partially unfolded protein by a metabolite. J. Mol. Biol 2012, 422 (3), 403–13. [DOI] [PubMed] [Google Scholar]
- (45).Mi H; Thomas P PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tanca A; Biosa G; Pagnozzi D; Addis MF; Uzzau S Comparison of detergent-based sample preparation workflows for LTQ-Orbitrap analysis of the Escherichia coli proteome. Proteomics 2013, 13 (17), 2597–2607. [DOI] [PubMed] [Google Scholar]
- (47).Chen H; Kihara D Effect of using suboptimal alignments in template-based protein structure prediction. Proteins: Struct., Funct., Genet. 2011, 79 (1), 315–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kihara D; Lu H; Kolinski A; Skolnick J TOUCHSTONE: an ab initio protein structure prediction method that uses threadingbased tertiary restraints. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (18), 10125–10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kim H; Kihara D Protein structure prediction using residue-and fragment-environment potentials in CASP11. Proteins: Struct., Funct., Genet. 2016, 84, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Johnson DK; Karanicolas J Ultra-High-Throughput Structure-Based Virtual Screening for Small-Molecule Inhibitors of Protein-Protein Interactions. J. Chem. Inf. Model 2016, 56 (2), 399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.