

Abstract
Post-transcriptional gene expression regulation of RNA has emerged as a key factor that controls mammalian protein production. RNA trafficking, translation efficiency, and stability are all controlled at the transcript level. For example, in addition to the commonly known processing steps of capping, splicing, and polyadenylation, RNA can be chemically modified. In eukaryotes, N6-methyladenosine (m 6A) is the most prevalent mRNA modification. While the writers, erasers, and readers for m6A are rapidly being uncovered and studied at the whole-cell level, their competitive interplay to regulate methylated RNA transcripts has yet to be elucidated. To address this limitation, we report the development of programmable dPspCas13b-m6A reader proteins to investigate the regulatory effects of specific readers on single transcripts in live cells. We fused the two most well-characterized m6A reader proteins, YTHDF1 and YTHDF2, to a catalytically inactive PspCas13b protein, which can target the reader to a specific RNA of interest using guide RNA (gRNA) complementarity. We then demonstrate that the fused reader proteins each retain their reported functional role on a reporter construct: YTHDF2 induces degradation and YTHDF1 enhances translation. Finally, we show that the system can target endogenous mRNA transcripts within cells, using YTHDF2 as an exemplar, where we found tethering with YTHDF2 leads to decay of the target transcript. The development of dCas13b-based tools to study the regulation of endogenous RNAs will dramatically enhance our understanding of how RNA regulation occurs at the single RNA level. Additionally, our new tools, which permit transcript-specific mediated decay or enhanced protein production, will find utility in synthetic biology applications aimed at controlling genetic information flow at the RNA level.
Graphical Abstract
■ INTRODUCTION
RNA transcribed from the genome in the nucleus bears little resemblance to the RNA polymer it will ultimately become in the cytoplasm where it is translated into protein. Well-known processes such as capping, splicing, and polyadenylation, as well as the recently discovered and ever-expanding list of diverse chemical modifications and editing, significantly alter the properties and fates of a given RNA during the course of its lifetime.1–6 These alterations, which regulate critical aspects of RNA function such as stability, transport, protein binding, and translation, are commonly mediated by interaction with RNA binding proteins.7 Especially in mammalian systems, regulatory processes at the post-transcriptional level are often a key determinant of genetic information flow.8,9 As the array of known RNA regulatory mechanisms continues to rapidly expand, there is an increasing need for approaches to probe these processes in live cells at the single transcript level.
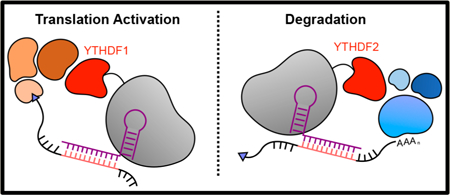
In mammalian cells, N6-methyladenosine (m 6A) is the most prevalent and well-studied mRNA (mRNA) modification.6 m6A has been implicated in various biological processes, such as cell development and differentiation,10–13 viral infections,14–16 and cancer,17–20 by mediating a variety of aspects of RNA processing and regulation such as splicing,21 translation,20,22,23 and stability.7,24 In addition to the discovery of the “writers” and “erasers” that introduce and remove m6A, respectively, several “reader” proteins have been discovered that are able to link site-specific RNA modifications to particular regulatory functions within the cell.25 The YT521-B homology domain family (YTHDF) of proteins is a family of cytoplasmic reader proteins that preferentially bind m6A within a DR(m6A)CH consensus site.26 Early studies mainly focused on two YTHDF family proteins, YTHDF1 and YTHDF2. YTHDF2 has been linked to the degradation of methylated RNA through recruitment of the CCR4-NOT deadenylation complex to the target transcript. In tethering assays on a reporter construct, the presence of YTHDF2 led to a decrease in RNA stability and the initiation of degradation. In co-immunoprecipitation assays, the subdomain region of YTHDF2 that is directly responsible for binding to its interaction partner CCR4-NOT was uncovered.27
YTHDF1 has been linked to the regulation of translation efficiency of a target transcript through interactions with the translation initiation machinery and the ribosome. Tethering assays showed enhanced translation of a reporter gene, and knockdown studies showed YTHDF1 target-wide decreases in translation efficiency and ribosomal occupancy.23 A later study found, however, that YTHDF1 may also induce deadenylation of targeted RNA transcripts in a tethering experiment.27 Studies of viruses concluded that all YTHDF proteins can induce translation upregulation28 or RNA degradation29 when recruited to viral transcripts. However, since many of these experiments have been performed on reporter systems and with all biological regulation machinery present, it is unclear whether YTHDF1 itself initiates deadenylation or it merely has the capability to recruit other proteins, such as YTHDF2, to proceed with deadenylation, or vice versa. If the YTHDF proteins do not have different functions, there must be cellular machinery to regulate their functions on particular transcripts within the cell. Understanding this regulation is important because the ability of the YTHDF proteins to modulate the expression level of individual methylated transcript has also been linked to viral infection14,15 and a variety of cancers.30
While studying cellular readouts as a result of the YTH family proteins with an exogenous reporter plasmid is a critical first step to understanding their biological function, taking these proteins out of their endogenous regulatory network limits these mechanistic studies substantially. More importantly, whole-transcriptome studies using knockdown or overexpression of the reader protein reveals that the same m6A sites on the same transcript can be regulated by both YTHDF1 and YTHDF2 as well as other reader proteins,31,32 suggesting either competition or additional undiscovered regulatory mechanisms are critical for controlling the outcome of the target transcript. To address this and related challenges, tools capable of delivering single reader proteins to single transcripts in a controllable, RNA modification-independent manner, would allow for the interrogation of the reader proteins on target RNA stability and translation efficiency.
The recently discovered CRISPR/Cas system has provided an easily programmable way to study nucleic acids in their endogenous environment.33–35 Within the last several years, nuclease-inactived DNA-targeting Cas9 (dCas9) has proven to be a versatile platform for the delivery of effector proteins to single sites in the genome. This has led to new methods to modify the epigenetic properties of cells, including histone methylation and histone acetylation status.36,37 The discovery of RNA-targeting Cas proteins, the Cas13 family of proteins, has opened doors for equivalent studies on endogenous RNA transcripts.38 For example, the RNA-targeting nuclease LwaCas13a can be programmed to target specific RNAs in mammalian systems and can efficiently degrade target transcripts through nuclease activity. Moreover, a nuclease- inactivated “dead” version of LwaCas13a (dLwaCas13a) can be fused to green fluorescent protein for endogenous RNA imaging.39 A nuclease-inactive variant of a Cas13 ortholog, PspCas13b, was fused to the known A-to-I RNA editing enzymes ADAR1 and ADAR2 to show direct RNA editing at endogenous RNA sites.40 Studies with a “dead” Cas13d ortholog furthermore demonstrated the ability of Cas13 to deliver a splicing modulator to individual transcripts.41 Collectively, these studies demonstrate the potential of Cas13-based systems as targeting moieties for tools to study endogenous RNA regulation.
In this present work, we developed dCas13b-m6A reader tools to study the molecular basis of m6A-mediated RNA regulation at specific transcripts (Figure 1A). We engineered dCas13b-YTHDF1 and dCas13b-YTHDF2 effector proteins that can be site-specifically targeted to an RNA of interest. We demonstrate that our constructs maintain the previously reported effects on translation efficiency (YTHDF1, Figure 1B) and degradation (YTHDF2, Figure 1C) using a luciferase reporter system, confirming the functions of these targeted readers in live cells. We go on to show that the fusion proteins can degrade endogenous transcripts in a YTHDF2-dependent manner. Intriguingly, the YTHDF2-mediated decay substantially decreased the expression level of target transcripts, suggesting these tools may find utility for biotechnological applications. Together, this work provides the field with a programmable, versatile set of tools to study endogenous RNA regulation at the molecular level.
Figure 1.
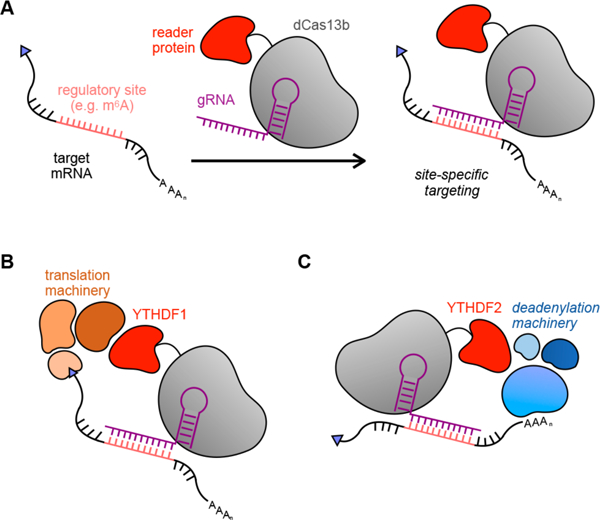
dCas13b fusion proteins can be used to site-specifically deliver regulatory proteins to the transcriptome. (A) General overview of site-specific RNA targeting using dCas13b-guided fusion proteins. (B) dCas13b-YTHDF1 fusion protein can target RNA transcripts to trigger assembly of translation machinery. (C) RNA targeted by YTHDF2 fusion proteins undergo degradation mediated by the CCR4-NOT deadenylation complex.
■ RESULTS AND DISCUSSION
Engineering dCas13b Fusion Proteins To Study the Epitranscriptome.
To provide a simple tool to study RNA modifications, we sought to develop targeted fusion proteins that act in a chemical modification-independent manner. For both YTHDF proteins, the N-terminal domain contains the “effector” region that mediates cellular responses, while the C-terminal domain contains the m6A-binding YTH domain. We therefore chose to omit the C-terminal m6A-binding domain in our fusion proteins to obtain a system that acts independent of RNA methylation. As a result, our constructs can be used to study RNA transcripts in their endogenous context by guiding an effector protein to a known methylation site or to study m6A-initiated downstream effects decoupled from their native regulatory context. Conversely, eliminating the m6A binding component of these proteins abolishes an additional regulatory layer of this system and adds the need to carefully control biological experiments to only capture true biological interactions.
To generate a targeted YTHDF1 reader, we cloned a mammalian expression vector with a catalytically inactive PspCas13b (dPspCas13b) fused to the N-terminal domain of YTHDF1 (NYTHDF1). Truncated versions of the N-terminal domain of YTHDF2 (NYTHDF2) have previously been identified to bind the CCR4-NOT deadenylation machinery,27 maintaining the key functional output of the YTHDF2 reader protein. Therefore, to generate a targeted YTHDF2 reader, we cloned a mammalian expression vector with a dPspCas13b fused to the truncated N-terminal domain of YTHDF2 (YTHDF2(100–200)).
We validated the function of the fusion constructs in live HEK293T cells using a dual luciferase reporter, which allows us to simultaneously analyze relative changes in protein expression by luciferase enzymatic activity and relative changes in luciferase mRNA levels by RT-qPCR (Figure 2A). First, we validated our assay setup using the active Cas13b nuclease, which should be capable of degrading a target RNA through active nuclease activity. We transfected cells with an expression vector for active Cas13b along with a vector that produces either a control off-target gRNA or a gRNA targeting firefly luciferase (Figure 2A). As expected, we observed a substantial decrease of both luciferase RNA as measured by RT-qPCR (Figure 2B, red bar) and protein levels (Figure 2B, blue bar). This control experiment validates that the dual luciferase reporter is capable of measuring both protein and RNA levels produced from the model gene.
Figure 2.
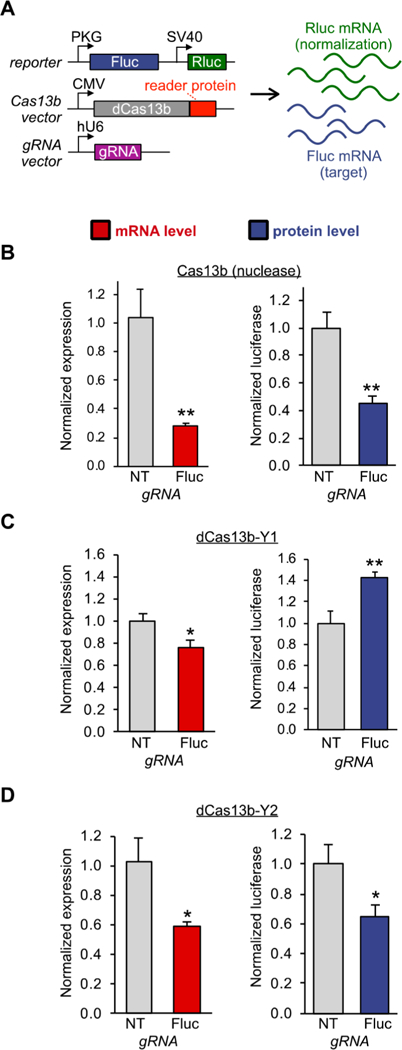
dCas13b-YTHDF fusion proteins can trigger enhanced protein production or RNA knockdown on a reporter transcript. (A) Vector system used to test targeted reader protein tools in a dual luciferase assay. Firefly luciferase (Fluc) is the target of the experiment, while Renilla luciferase (Rluc) is an internal control. RNA levels are monitored by RT-qPCR, and protein production is monitored by luciferase luminescence. Anoff-target gRNA (NT) and a guide RNA targeting Fluc (Fluc) delivery the fusion protein to the transcript. (B) HEK293T cells were co-transfected with the vectors shown in (A), including the active Cas13b nuclease, and 48 h after transfection subjected to RT-qPCR (red bar) and luciferase assay (blue bar). (C) HEK293T cells were co-transfected with the vectors shown in (A), with the dCas13b-YTHDF1 fusion, and assayed by RTqPCR (red bar) and luciferase assay (blue bar). (D) HEK293T cells were co-transfected with the vectors shown in (A), with the dCas13b-YTHDF2 fusion, and assayed by RT-qPCR (red bar) and luciferase assay (blue bar). Student’s t-test; *P < 0.05, **P < 0.01 (n = 3 for RTqPCR, n = 3 biological × 2 technical for Cas13b and dCas13b-Y2 luciferase assays, n = 3 biological × 2 technical + 3 biological replicates for dCas13b-Y1 luciferase assay).
Next, we subjected the new YTHDF-fusion proteins to the same analysis as the active Cas13b nuclease. We transfected HEK293T cells with the dCas13b-NYTHDF1 construct along with either a control gRNA or a gRNA targeting the fusion protein to the firefly luciferase mRNA. Interestingly, delivery of the YTHDF1 protein resulted in a slight decrease in mRNA levels as measured by RT-qPCR (Figure 2C, red bar). Even though the mRNA levels go down, the protein levels consistently increase (Figure 2C, blue bar), as has been observed in previous tethering assays.23 These findings indicate that the targeted YTHDF1 protein maintains function as a fusion construct on a model target gene.
We then assayed whether the YTHDF2 fusion construct also retained its function mediating RNA decay as a targeted protein. We transfected HEK293T cells with an expression vector producing the dCas13b-NYTHDF2 fusion protein, again along with either a control gRNA expression vector or a vector that produces a gRNA targeting firefly luciferase. As expected, targeted YTHDF2 led to a decrease in both mRNA and protein levels (Figure 2D). The level of luciferase decrease observed agrees well with previously obtained data with the full-length N-terminal domain of YTHDF2 in tethering assays.23 As a control, dCas13b lacking a fusion protein showed no measurable change in targeted gene expression or protein production (Figure S1). As some of the previous studies have been conducted in HeLa cells, we also verified the constructs using HeLa cells, observing the same trends as seen for HEK293T cells (Figure S2). Taken together, these findings indicate that both YTHDF proteins retain previously reported functions as part of a targeted fusion construct and can be used to study m6A regulation dynamics in cells.
Determining YTHDF1 Fragments Responsible for Translation Activation.
As previously described, YTHDF2 truncations that maintained downstream function independent of the m6A recognition domain had previously been identified. To study endogenous RNA regulation, we wanted to ensure that the fusion tools are as small and unobtrusive as possible. Unlike YTHDF2, the active subdomain of YTHDF1 that is responsible for binding to the translational machinery has not been identified. We reasoned that our reporter assay would allow us to identify the subdomain responsible for RNA translation activation. We therefore cloned three different truncations of the N-terminal domain of YTHDF1 (aa1–100, aa100–200, aa200–364) fused with dCas13b (Figure 3A). We used the same qPCR and dual luciferase assays as described above to compare the differences in mRNA and protein levels of each truncations with previously obtained data for the full- length N-terminal domain. At the mRNA level, none of the fusions caused dramatic changes, except for the unexpected observation that the YTHDF1(aa1–100) fusion seemed to cause some RNA stabilization (Figure 3B) while inducing less protein production (Figure 3C). More importantly, we found that only the truncation YTHDF1(aa100–200) retains its translation activation activity (Figure 3C). However, we observed some sample variability in the translational activation effect, resulting in data below the statistically significant threshold. We therefore repeated the YTHDF1(aa100–200) experiment with additional replicates, which confirmed the observed translational activation effect (Figure 3D).
Figure 3.
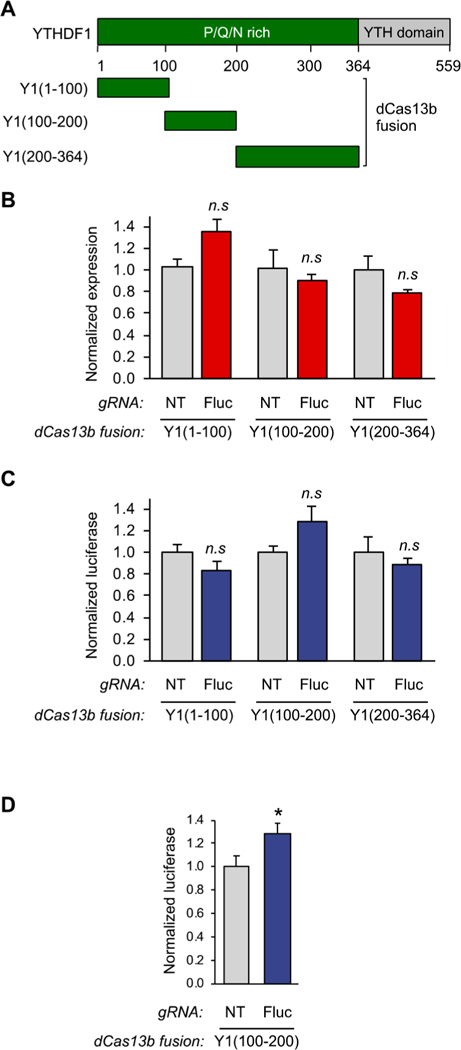
A fragment of the N-terminal domain of YTHDF1 is sufficient to bind to the translation initiation machinery. (A) Schematic diagram of YTHDF1 fragments. (B) qPCR analysis of HEK293T cells transfected with Yl(aa1–100), Yl(aa100–200), or Yl(200–364)-dCas13b fusion proteins and either the NT guide or the Fluc guide using the assay shown in Figure 2A (n = 3). (C) Protein readout analysis of the same Y1(aa1–100), Y1(aa100–200), and Y1(aa200–364) fusion proteins using the dual luciferase reporter system. The Y1(100–200) truncation shows activation of protein production without effecting RNA levels, defining this portion of the protein as the active reader domain (n = 3 biological × 2 technical). (D) Luciferase assay analysis of further replicates of the identified dCas13bY1(100–200) protein. Student’s t-test; *P < 0.03 (n = 9).
Interestingly, unlike with the full-length N-terminal YTHDF1 fusion, we did not observe a decrease in mRNA level for the dCas13b-Y1 (100–200) truncation. The YTHDF1(aa200–364), however, seemed to promote a RNA decrease similar to the level observed with the whole domain. This could be an indication that aa100–200 is responsible for binding to the translation machinery, while aa200–364 binds to other regulatory proteins, such as YTHDF2, in cells. This observation warrants further investigation and further expands the YTHDF-mediated regulatory repertoire. This complexity in reader subdomains could also explain some of the different properties observed for YTHDF1 for specific transcripts under different conditions in the literature and further confirms the need for the tools described here.
Endogenous RNA Targeting with dCas13b-Y2.
Finally, we tested whether endogenous transcripts could also be targeted by the fusion proteins, using dCas13b-YTHDF2 fusion construct as an exemplar. We selected two transcripts: the low-abundant transcript KRAS and the highly abundant PPIB, selected because each are known to be m6A modified26,42,43 and have also been validated for targeting by Cas13b.39 To find an appropriate targeting site, we conducted a small gRNA screen for both targets (Figure S3). To validate our system, we chose to use the gRNAs showing the largest response with our dCas13b-Y2 construct in all following experiments.
After selecting gRNAs targeting either KRAS or PPIB, we transfected HEK293T cells with the dCas13b-YTHDF2 expression vector and a gRNA expression vector. We furthermore transfected cells with the active Cas13b or dCas13b and identical on- and off-target gRNA as positive and negative controls, respectively. For KRAS, we observed a small but reproducible decrease in mRNA levels mediated by YTHDF2 delivery (Figure 4A). Surprisingly, the knockdown efficiency KRAS was similar to what we observed when using the active Cas13b nuclease to actively degrade the transcript (Figure 4B).39 However, an unfused control dCas13b protein had no effect on the transcript (Figure 4C). PPIB was substantially more efficiently knocked down by both dCas13b- YTHDF2 (Figure 4D) and nuclease-active Cas13b (Figure 4E), again with much less effect from a dCas13b control (Figure 4F). These results suggest that the dCas13b-YTHDF2 approach is a new way to trigger a degradation response on endogenous targets. The observed differences in mRNA decay in response to YTHDF2 targeting again showcase the complexity of RNA regulation and the importance of tools to probe transcript-specific responses.
Figure 4.
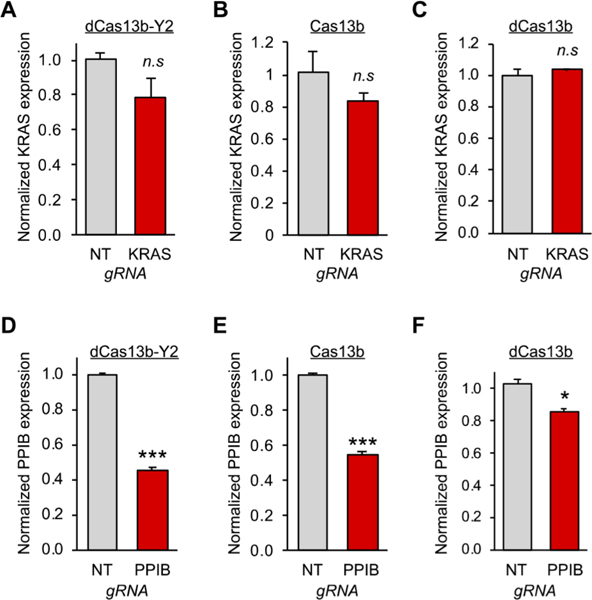
Targeted YTHDF2 readers to study the regulation of endogenous methylated transcripts in live cells. (A) HEK293T cells were transfected with dCas13b-YTHDF2 and either NT gRNA (gray bar) or KRAS gRNA (red bar). After 48 h, mRNA levels were determined by RT-qPCR (B) The same analysis was performed in (A) except the nuclease active Cas13b was delivered. (C) The same analysis was performed in (A) except a dCas13b protein without a fusion was delivered. (D) HEK293T cells were transfected with dCas13bYTHDF2 and either NT gRNA (gray bar) or PPIB gRNA (red bar). After 48 h, mRNA levels were determined by RT-qPCR (E) The same analysis was performed in (D) except the nuclease active Cas13b was delivered. (F) The same analysis was performed in (D) except a dCas13b protein without a fusion was delivered. Student’s t-test; *P < 0.05, ***P < 0.001 (n = 3).
In addition to RT-qPCR, we next verified changes in the expression level of the protein product of the gene PPIB (cyclophilin B: CypB) by Western blot. As anticipated from the qPCR results, we saw the protein level decreased when we transfected cells with our dCas13b-Y2 construct and a PPIB- targeting gRNA (Figure S4).
Off-target effects are one of the main concerns when using Cas-based systems. To assess how much we perturb the overall transcriptome in cells, we conducted a whole-transcriptome analysis by RNA sequencing. We transfected cells with dCas13b-Y2 or active Cas13b and off-target gRNA or PPIB gRNA, prepared mRNAs libraries, and then analyzed each transcriptome by high-throughput sequencing (HTS). We determined that for dCas13b-Y2 there were 26 differentially expressed transcripts between the NT gRNA and PPIB gRNA triplicates (Figure S5B). We observed changes indicating higher as well as lower expressed transcripts, which could be due to biological on-target effects or mis-targeting of dCas13b. Differential expression could furthermore be a direct result of overexpressing YTHDF2. YTHDF2 is known to recruit cellular machinery that can lead to changes in transcript expression, and we cannot rule out that this happens when we overexpress it. Nonetheless, the off-target effects were comparable to those observed we observed for nuclease-mediate Cas13b targeting (23 differentially expressed transcripts, Figure S5D) and are substantially less than those observed from other technologies like shRNA.41
The transcript we targeted with our dCas13b-Y2 construct was not found to be differentially expressed by HTS (Figure S5). To examine this discrepancy, we conducted further RT-qPCR analyses on the extracted total RNA that was used for HTS. We analyzed the samples prior to mRNA purification and library preparation to rule-out biases and artifacts that may emerge during these steps and also performed comparative analysis with other housekeeping genes to confirm the validity of the on-target effect. Indeed, these additional RT-qPCR experiments reproduced our previous observations, revealing similar decreases in PPIB levels when referencing to either GAPDH or ACTB (Figure S5 A,C). Taken together with our Western blot analysis, we conclude that the relatively modest but significant extent of transcript knockdown is lost during the purification and library preparation steps and was not substantial enough to observe by HTS.
To confirm our HTS experimental and analysis pipeline was functioning properly, we also performed transcriptome analysis with PPIB targeted by the active Cas13b nuclease. We found that while the targeted PPIB transcript is significantly decreased as measured by RT-qPCR, it falls barely into our significance cutoff by HTS. As nuclease-mediated Cas13b outperforms the dCas13b-Y2 knockdown efficiency, we suspect that the overall difference in RNA level must be more substantial to be seen after library preparation and sequencing. It has been previously reported that library preparation and handling can introduce large differences in the outcome.44 We believe that if we optimized our dCas13b-Y2 conditions further to yield a larger decrease in RNA level, we could observe these by RNA-seq. At this point, we can conclude from our RNA-seq results that expressing dCas13b-Y2 in cells does not cause gross changes to the transcriptome. Any potential Casmediated off-target effects will only be uncovered once the system performs well enough for more indepth sequencing analysis.
■ CONCLUSION
We developed and validated dCas13-targeted RNA reader proteins as a new platform to study RNA regulation, focusing here on YTHDF reader proteins of m6A-modified RNA transcripts. Our system is easily programmable by simply changing the gRNA for Cas13b and can therefore be used to study the regulation of any methylated RNA of interest. Furthermore, due to Cas13’s tolerance to fusion proteins, it can also be adapted to study the regulation of any known effector protein of RNA by switching the fusion protein. To our knowledge, this presents the first tool to study RNA regulation dynamics and effects of RNA modifications on single transcripts in an endogenous context.
We noticed a high degree of variability in our cell-based assays that we attribute to differences in transfection and cell state. Further optimization is required to ensure more consistent cellular assays. The difference in response between different endogenous KRAS and PPIB transcripts could indicate that there are regulatory differences between genes. These data also suggest Cas accessibility of a target transcript may be a determinant of overall effectiveness on the transcript, suggesting better RNA-targeting Cas systems are still needed. Overall, these tools provide the ability to investigate such differences on endogenous transcripts and allow for new mechanistic studies regarding RNA regulation of individual transcripts. However, the relatively large size of the currently available RNA-targeting Cas systems could also provide additional complexity to interpreting the results when delivered to transcripts, which could be alleviated by smaller delivery systems.
In this work, we chose to develop and validate our new targeted reader protein tools in the context of transcripts with annotated m6A sites, because these RNAs are already prone to chemical modification-mediated post-transcriptional gene regulation. However, we found that the system also works on the firefly luciferase reporter RNA. In their work, Liu et al.45 show that in a similar reporter construct with the same firefly luciferase coding region as in our luciferase reporter construct, there was no substantial methylation of Fluc by m6A-pulldown followed by RT-qPCR. They furthermore saw the same methylation level with wild-type (GGACU) and a “dead” mutated m6A (GGAUU) consensus site. Since m6A is known to exist predominantly at DRACH consensus sites, this suggests there is no substantial methylation of the firefly luciferase reporter system. Therefore, it is reasonable to assume that our dCas13b-Y2 system is acting independent of the methylation level at its targeting site. However, future work, possibly deploying targeted methyltransferases and demethy- lases, will further clarify these mechanistic questions.
Surprisingly, targeting YTHDF2-mediated decay resulted in dramatic knockdown of certain target genes in our hands, not too different from the knockdown efficiency of nuclease active Cas13b. For example, on the reporter construct, active nuclease-mediated degradation resulted in a ~75% decrease in RNA levels and 50% decrease in protein levels, while YTHDF2-mediated decay lead to a ~40% decrease in RNA levels and a 40% decrease in protein levels (Figure 2B,C). The YTHDF2 knockdown of the endogenous genes tested was also quite comparable to active nuclease-mediated degradation. For example, YTHDF2-mediated decay knocked down PPIB by 60% (Figure 4D), while active Cas13b-mediated decay knocked the gene down by ~55% (Figure 4E). This efficiency in gene expression control suggests that reader proteinmediated decay may be a useful strategy for both synthetic biology and bioengineering applications.33,46 Collectively, these experiments indicate that dCas13b-fusion proteins can be used to study single transcripts in a native biological context and demonstrates there is still much to be learned about RNA regulation.
■ EXPERIMENTAL DETAILS
Cloning.
All plasmids were generated by Gibson Assembly cloning using PCR products amplified with Q5 DNA Polymerase (NEB). The plasmids were sequenced by the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility. All plasmids used in this study are listed in Table S1, which includes free links to fully annotated sequence maps. Vector maps are available online as indicated and are physically available upon request. The original dPspCas13b plasmid (pC0050-CMV-dPspCas13b-longlinker-ADAR2DD(wt)) was a gift from Feng Zhang (Addgene plasmid no. 103866).40
Mammalian Cell Culture and Plasmid Transfection.
HEK293T (ATCC) and HeLa (ATCC) cells were maintained using DMEM (L-glutamine, high glucose, sodium pyruvate, phenol red; obtained from Corning) supplemented with 10% fetal bovine serum (FBS, Gemini Benchmark), and 1% penicillin/streptomycin (P/S, Gibco/Life Technologies). For transfections, cells were cultured in DMEM (L-glutamine, high glucose, sodium pyruvate, phenol red; obtained from Corning) supplemented with 10% FBS. Plasmid transfections were achieved using lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol.
Mammalian Luciferase Assay.
To test changes in protein levels, HEK293T cells were transfected with 150 ng Cas13b/dCas13b-YTHDF1/dCas13b-YTHDF2, 100 ng gRNA, and 10 ng Dual-Glo luciferase reporter (Promega) per well, unless otherwise noted. Due to their lower tolerance of DNA transfection amounts, HeLa cells were transfected with 75 ng Cas13b/dCas13bYTHDF1/dCas13b-YTHDF2, 50 ng gRNA, and 10 ng Dual-Glo luciferase reporter (Promega). For Figure S1B, cells transfected with 80 ng dCas13b unfused construct and 80 ng scFv construct in addition to the luciferase reporter. Cells were plated on a 96-well plate (CellVis) 16 h before transfection and transfected at 80% confluency. A total of 20 μL Opti-MEM I Reduced Serum Medium (ThermoFisher Scientific) per well was used after combining 10 μL Opti-MEM containing 0.5 μL Lipofectamine 2000 and 10 μL Opti-MEM containing the plasmids. Combined solutions were incubated for 15 min before they were slowly pipetted onto cells. After 48 h, luciferase activity was assayed using the DualGlo Luciferase Assay System (Promega) on a Biotek Synergy plate reader according to the manufacturer’s instructions. All experiments were performed in at least three biological and two technical replicates. Firefly luciferase readouts were normalized to the corresponding Renilla luciferase readout to account for differences in transfection efficiency.
Total RNA Isolation and Quantitative PCR.
To determine gene expression levels in HEK293T cells, RNA was isolated, and changes in RNA levels were quantified using RT-qPCR Cells were plated on a 48-well plate (Corning) and transfected with 500 ng Cas13b/ dCas13b-fusion and 300 ng gRNA plasmid. Where applicable, 30 ng DualGlo luciferase reporter (Promega) was added. Total RNA was harvested 48 h after transfection using the RNeasy Mini Kit (Qiagen). Following RNA isolation, RNA was reverse transcribed to cDNA using the PrimeScript RT Reagent Kit (TaKaRa). All qPCR reactions were performed as 20 μL reactions using FastStart Essential DNA Green Master (Roche) and amplified on a LightCycler 96 Instrument (Roche). Expression levels were obtained by subtraction the housekeeping gene (GAPDH or ACTB) Ct value from target Q value and normalizing to the nontargeting (NT) gRNA. Relative abundance was determined using 2-ΔCt. All assays were performed with three biological replicates. The qPCR primers used in this study are listed in Table S2.
Western Blotting.
For Western blotting, HEK293T cells were plated in 12-well plates (Corning) and transfected with 2 μg Cas13b/ dCas13b-YTHDF2 and 1.2 μg gRNA plasmids. After 48 h, cells were washed with ice-cold PBS and lysed in 50 μL RIPA buffer (50 mM Tris, 150 mM NaCl, 0.5% deoxycholate, 2% SDS, pH 7.4). After a 30 min incubation at room temperature, the protein concentration was measured by BCA assay (Thermo Scientific). The appropriate amount of protein (35 μg protein when detecting CypB) was boiled with loading dye for 5 min at 95 °C and loaded onto a 15% SDS PAGE gel. The proteins were transferred onto a PVDF membrane (Millipore) and blocked in 5% milk in TBST. Proteins were detected using a 1:1000 dilution CypB antibody (Santa Cruz) and 1:1000 antimouse HRP-conjugated antibody (Santa Cruz). Membranes were imaged on Fluor Chem R (Protein Simple) after incubation with Super Signal West Pico Plus (Thermo Scientific).
RNA Sequencing and analysis.
To determine the specificity of this dCas13b-based system, we performed RNA-seq analysis. HEK293T cells were plated in 12-well plates (Corning) and transfected with 1.8 μg Cas13b/dCas13b-YTHDF2 and 1.2 μg gRNA plasmid. After 48 h, total RNA was extracted using the RNeasy Mini Kit (Qiagen) followed by a 30 min DNaseI (Fisher) treatment and clean up using the RNA clean up and concentrator kit (Zymo). The resulting total RNA was used as the input for library preparation and for qPCR analysis of the same samples (as described earlier). mRNA extraction and RNA-seq libraries were prepared using the mRNA HyperPrep Kit (KAPA biosystems). Libraries were sequenced on an Illumina HiSeq instrument at the University of Chicago Genomic Facility with at least 14 million reads per library. Reads were mapped to the RefSeqGRCh38 transcriptome, quantified, and pseudoaligned using kallisto.47 To find differentially expressed transcripts, we used sleuth.48 Only genes that had a log2FoldChange of at least 0.6 and a FDR < 0.05 were considered to be differentially expressed.
Supplementary Material
■ ACKNOWLEDGMENTS
This work was supported by the University of Chicago, the National Institute of General Medical Sciences (R35 GM119840, B.C.D.) and the National Human Genome Research Institute (RM1 HG008935, C.H. and B.C.D.) of the National Institutes of Health, and the University of Chicago Medicine Comprehensive Cancer Center (P30 CA14599). C.H. is a Howard Hughes Medical Institute Investigator and is a scientific founder and a member of the scientific advisory board of Accent Therapeutics, Inc.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05012.
Supporting tables and figures (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Anderson P; Kedersha N Nat. Rev. Mol. Cell Biol. 2009, 10, 430. [DOI] [PubMed] [Google Scholar]
- (2).Lee Y; Rio DC Annu. Rev. Biochem. 2015, 84 (1), 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chen M; Manley JL Nat. Rev. Mol. Cell Biol. 2009, 10, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Filipowicz W; Bhattacharyya SN; Sonenberg N Nat. Rev. Genet. 2008, 9, 102. [DOI] [PubMed] [Google Scholar]
- (5).Lim C; Allada R Nat. Neurosci. 2013, 16, 1544. [DOI] [PubMed] [Google Scholar]
- (6).Roundtree IA; Evans ME; Pan T; He C Cell 2017, 169 (7), 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Glisovic T; Bachorik JL; Yong J; Dreyfuss G FEBS Lett. 2008, 582 (14), 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Battle A; Khan Z; Wang SH; Mitrano A; Ford MJ; Pritchard JK; Gilad Y Science 2015, 347 (6222), 664–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Maier T; Guell M; Serrano L FEBS Lett. 2009, 583 (24), 3966–3973. [DOI] [PubMed] [Google Scholar]
- (10).Zhao BS; Wang X; Beadell AV; Lu Z; Shi H; Kuuspalu A; Ho RK; He C. Nature 2017, 542, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ivanova I; Much C; Di Giacomo M; Azzi C; Morgan M; Moreira PN; Monahan J; Carrieri C; Enright AJ; O’Carroll D Mol. Cell 2017, 67 (6), 1059–1067.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Aguilo F; Zhang F; Sancho A; Fidalgo M; Di Cecilia S; Vashisht A; Lee D-F; Chen C-H; Rengasamy M; Andino B; Jahouh F; Roman A; Krig SR; Wang R; Zhang W; Wohlschlegel JA; Wang J; Walsh MJ Cell Stem Cell 2015, 17 (6), 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wen J; Lv R; Ma H; Shen H; He C; Wang J; Jiao F; Liu H; Yang P; Tan L; Lan F; Shi YG; He C; Shi Y; Diao J Mol. Cell 2018, 69 (6), 1028–1038.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tirumuru N; Zhao BS; Lu W; Lu Z; He C; Wu L eLife 2016,5, e15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lichinchi G; Zhao BS; Wu Y; Lu Z; Qin Y; He C; Rana TM Cell Host Microbe 2016, 20 (5), 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lichinchi G; Gao S; Saletore Y; Gonzalez GM; Bansal V; Wang Y; Mason CE; Rana TM Nat. Microbiol 2016, 1, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li Z; Weng H; Su R; Weng X; Zuo Z; Li C; Huang H; Nachtergaele S; Dong L; Hu C; Qin X; Tang L; Wang Y; Hong G-M; Huang H; Wang X; Chen P; Gurbuxani S; Arnovitz S; Li Y; Li S; Strong J; Neilly MB; Larson RA; Jiang X; Zhang P; Jin J; He C; Chen J Cancer Cell 2017, 31 (1), 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cui Q; Shi H; Ye P; Li L; Qu Q; Sun G; Sun G; Lu Z; Huang Y; Yang C-G; Riggs AD; He C; Shi Y Cell Rep. 2017, 18 (11), 2622–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang S; Zhao BS; Zhou A; Lin K; Zheng S; Lu Z; Chen Y; Sulman EP; Xie K; Bogler O; Majumder S; He C; Huang S Cancer Cell 2017, 31 (4), 591–606.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lin S; Choe J; Du P; Triboulet R; Gregory RI. Mol. Cell 2016,62 (3), 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiao W; Adhikari S; Dahal U; Chen Y-S; Hao Y-J; Sun B-F; Sun H-Y; Li A; Ping X-L; Lai W-Y; Wang X; Ma H-L; Huang C-M; Yang Y; Huang N; Jiang G-B; Wang H-L; Zhou Q; Wang X-J; Zhao Y-L; Yang Y-G. Mol. Cell 2016, 61 (4), 507–519. [DOI] [PubMed] [Google Scholar]
- (22).Meyer KD; Patil DP; Zhou J; Zinoviev A; Skabkin MA; Elemento O; Pestova TV; Qian S-B; Jaffrey SR Cell 2015, 163 (4), 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang X; Zhao BS; Roundtree IA; Lu Z; Han D; Ma H; Weng X; Chen K; Shi H; He C Cell 2015, 161 (6), 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang X; Lu Z; Gomez A; Hon GC; Yue Y; Han D; Fu Y; Parisien M; Dai Q; Jia G; Ren B; Pan T; He C Nature 2014, 505, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Edupuganti RR; Geiger S; Lindeboom RGH; Shi H; Hsu PJ; Lu Z; Wang SY; Baltissen MPA; Jansen P; Rossa M; Muller M; Stunnenberg HG; He C; Carell T; Vermeulen M. Nat. Struct. Mol. Biol. 2017, 24 (10), 870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dominissini D; Moshitch-Moshkovitz S; Schwartz S; Salmon-Divon M; Ungar L; Osenberg S; Cesarkas K; Jacob-Hirsch J; Amariglio N; Kupiec M; Sorek R; Rechavi G Nature 2012, 485, 201. [DOI] [PubMed] [Google Scholar]
- (27).Du H; Zhao Y; He J; Zhang Y; Xi H; Liu M; Ma J; Wu L Nat. Commun. 2016, 7, 12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kennedy EM; Bogerd HP; Kornepati AVR; Kang D; Ghoshal D; Marshall JB; Poling BC; Tsai K; Gokhale NS; Horner SM; Cullen BR Cell Host Microbe 2016, 19 (5), 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gokhale NS; McIntyre ABR; McFadden MJ; Roder AE; Kennedy EM; Gandara JA; Hopcraft SE; Quicke KM; Vazquez C; Willer J; Ilkayeva OR; Law BA; Holley CL; Garcia-Blanco MA; Evans MJ; Suthar MS; Bradrick SS; Mason CE; Horner SM Cell Host Microbe 2016, 20 (5), 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang S; Sun C; Li J; Zhang E; Ma Z; Xu W; Li H; Qiu M; Xu Y; Xia W; Xu L; Yin R Cancer Leit. 2017, 408, 112–120. [DOI] [PubMed] [Google Scholar]
- (31).Patil DP; Chen C-K; Pickering BF; Chow A; Jackson C; Guttman M; Jaffrey SR Nature 2016, 537, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huang H; Weng H; Sun W; Qin X; Shi H; Wu H; Zhao BS; Mesquita A; Liu C; Yuan CL; Hu Y-C; Huttelmaier S; Skibbe JR; Su R; Deng X; Dong L; Sun M; Li C; Nachtergaele S; Wang Y; Hu C; Ferchen K; Greis KD; Jiang X; Wei M; Qu L; Guan J-L; He C; Yang J; Chen J Nat. Cell Biol. 2018, 20 (3), 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).O’Connell MR; Oakes BL; Sternberg SH; East-Seletsky A;Kaplan M; Doudna JA Nature 2014, 516, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wiedenheft B; Sternberg SH; Doudna JA Nature 2012, 482, 331. [DOI] [PubMed] [Google Scholar]
- (35).Jiang W; Bikard D; Cox D; Zhang F; Marraffini LA Nat. Biotechnol 2013, 31, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hilton IB; D’Ippolito AM; Vockley CM; Thakore PI; Crawford GE; Reddy TE; Gersbach CA Nat. Biotechnol. 2015, 33, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kearns NA; Pham H; Tabak B; Genga RM; Silverstein NJ; Garber M; Maehr R Nat. Methods 2015, 12, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Abudayyeh OO; Gootenberg JS; Konermann S; Joung J; Slaymaker IM; Cox DBT; Shmakov S; Makarova KS; Semenova E; Minakhin L; Severinov K; Regev A; Lander ES; Koonin EV; Zhang F Science 2016, 353 (6299), aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Abudayyeh OO; Gootenberg JS; Essletzbichler P; Han S; Joung J; Belanto JJ; Verdine V; Cox DBT; Kellner MJ; Regev A; Lander ES; Voytas DF; Ting AY; Zhang F Nature 2017,550, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cox DBT; Gootenberg JS; Abudayyeh OO; Franklin B; Kellner MJ; Joung J; Zhang F Science 2017, 358 (6366), 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Konermann S; Lotfy P; Brideau NJ; Oki J; Shokhirev MN; Hsu PD Cell 2018, 173 (3), 665–676.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Linder B; Grozhik AV; Olarerin-George AO; Meydan C; Mason CE; Jaffrey SR Nat. Methods 2015, 12, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Schwartz S; Mumbach MR; Jovanovic M; Wang T; Maciag K; Bushkin GG; Mertins P; Ter-Ovanesyan D; Habib N; Cacchiarelli D; Sanjana NE; Freinkman E; Pacold ME; Satija R; Mikkelsen TS; Hacohen N; Zhang F; Carr SA; Lander ES; Regev A. Cell Rep. 2014, 8 (1), 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lahens NF; Kavakli IH; Zhang R; Hayer K; Black MB; Dueck H; Pizarro A; Kim J; Irizarry R; Thomas RS; Grant GR; Hogenesch JB. Genome Biology 2014, 15 (6), R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yue Y; Liu J; Cui X; Cao J; Luo G; Zhang Z; Cheng T; Gao M; Shu X; Ma H; Wang F; Wang X; Shen B; Wang Y; Feng X; He C; Liu J Cell Discovery 2018, 4 (1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Rauch S; Dickinson BC Biochemistry 2018, 57 (4), 363–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bray NL; Pimentel H; Melsted P; Pachter L Nat. Biotechnol. 2016, 34, 525. [DOI] [PubMed] [Google Scholar]
- (48).Pimentel H; Bray NL; Puente S; Melsted P; Pachter L Nat. Methods 2017, 14, 687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.