

Abstract
Neurotensin is a peptide hormone released from enteroendocrine cells in the small intestine in response to fat ingestion. Although the mechanisms regulating neurotensin secretion are still incompletely understood, our recent findings implicate a role for extracellular signal–regulated kinase 1 and 2 as positive regulators of free fatty acid-stimulated neurotensin secretion. Previous studies have shown that kinase suppressor of Ras 1 acts as a molecular scaffold of the Raf/MEK/extracellular signal–regulated kinase 1 and 2 kinase cascade and regulates intensity and duration of extracellular signal–regulated kinase 1 and 2 signaling. Here, we demonstrate that inhibition of kinase suppressor of Ras 1 attenuates neurotensin secretion and extracellular signal–regulated kinase 1 and 2 signaling in human endocrine cells. Conversely, we show that overexpression of kinase suppressor of Ras 1 enhances neurotensin secretion and extracellular signal–regulated kinase 1 and 2 signaling. We also show that inhibition of extracellular signal–regulated kinase 2 and exocyst complex component 70, a substrate of extracellular signal–regulated kinase 2 and mediator of secretory vesicle exocytosis, potently inhibits basal and docosahexaenoic acid-stimulated neurotensin secretion, whereas overexpression of exocyst complex component 70 enhances basal and docosahexaenoic acid-stimulated neurotensin secretion. Together, our findings demonstrate a role for kinase suppressor of Ras 1 as a positive regulator of neurotensin secretion from human endocrine cells and indicate that this effect is mediated by the extracellular signal–regulated kinase 1 and 2 signaling pathway. Moreover, we reveal a novel role for exocyst complex component 70 in regulation of neurotensin vesicle exocytosis through its interaction with the extracellular signal–regulated kinase 1 and 2 signaling pathway.
Introduction
Obesity in the United States has reached epidemic levels, with approximately 70% of the population overweight and over 30% of overweight individuals classified as obese [1, 2]. Cancers associated with overweight and obesity account for approximately 40% of all diagnosed cancers in the United States [1]. Excess adiposity is thought to contribute to the development of cancer through the release of free fatty acids (FFAs) by adipose tissue, which have been shown to promote tumorigenesis by serving as metabolic fuel for highly proliferating cells [3–6]. FFAs have also been demonstrated to regulate expression and secretion of hormones and neuropeptides, including the gastrointestinal neuropeptide neurotensin (NT), which is implicated in the promotion of obesity and the development of several types of cancer [7–12].
NT is released in the small intestine in response to fat ingestion and has been implicated in the development of metabolic disorders and many types of cancer, including breast, pancreatic, lung, prostate, and colorectal cancer [9, 10, 13–16]. NT facilitates FFA absorption in the intestine and contributes to lipid metabolism and glucose homeostasis [12]. In mice, NT deficiency reduces intestinal fat absorption and is protective against high-fat diet-induced obesity, hepatic steatosis, and insulin resistance [12]. In humans, elevated plasma concentrations of pro-NT (a stable NT precursor fragment produced in equimolar amounts relative to NT) are associated with insulin resistance, obesity-associated metabolic disorders, and visceral adipose tissue (VAT) inflammation in obese patients [13, 17]. Moreover, high plasma pro-NT levels are predictive of the presence and severity of non-alcoholic fatty liver disease (NAFLD) [18]. Interestingly, both NT secretion and cancer growth are stimulated by unsaturated FAs [7, 19]. Pre-clinical and epidemiologic studies indicate that high intake of unsaturated fatty acids increases cancer risk and promotes cancer progression [5, 7, 19–22]. We have recently shown that NT secretion from enteroendocrine (N) cells is enhanced by common unsaturated dietary FAs, including oleic acid, palmitoleic acid, and docosahexaenoic acid (DHA) [11]. Given its intersecting role in metabolic disorders and tumorigenesis, NT may be a potential therapeutic target for metabolic diseases and cancer.
NT is secreted from N cells via the regulated secretory pathway [23]. After proteolytic processing of the NT precursor (pro-NT), active NT peptide is transported from the trans Golgi network and stored in secretory vesicles until NT release is triggered by extracellular stimuli [23–25]. Upon stimulation, NT-containing secretory vesicles are transported to the plasma membrane and released to the cell exterior via exocytosis [24, 25]. While the most potent extracellular stimulus regulating NT release from the small intestine is fat ingestion, the molecular mechanisms regulating FA-stimulated NT secretion are still incompletely understood [23]. We have previously demonstrated that NT gene expression is enhanced by Ras signaling and that extracellular signal–regulated kinase 1 and 2 (ERK1/2) positively regulate NT gene expression and FFA-stimulated NT peptide secretion [11, 12, 26].
ERK1/2 are mitogen activated protein kinases (MAPKs) that play a central role in the regulation of cell proliferation, differentiation, and survival [27]. ERK1/2 signaling is activated upon ligand binding to membrane receptors and subsequent activation of the small GTPase Ras, which phosphorylates and activates the MAP kinase kinase kinase (MAPKKK) Raf [28]. Activated Raf phosphorylates the MAP kinase kinases (MAPKKs) MEK1 and MEK2 (MEK1/2), which phosphorylate and activate ERK1/2 [28]. Activated ERK1/2 phosphorylates a large number of cytosolic substrates to regulate diverse cellular functions and can also be translocated to the nucleus where it activates transcription factors regulating gene expression [27–29]. Deregulation of Ras/MAPK signaling contributes to approximately one-third of human cancers [30, 31].
Coordination of the Raf/MEK/ERK protein complex and subsequent ERK1/2 phosphorylation is regulated by the scaffold protein kinase suppressor of Ras 1 (KSR1) [32–34]. KSR1 coordinates formation of the Raf/MEK/ERK signaling complex, increasing specificity of MEK phosphorylation by Ras and ERK1/2 phosphorylation by MEK [35–37]. Though a recent study presents contrasting evidence suggesting KSR1 allosterically regulates the interaction between Raf/MEK/ERK, the stimulatory effect of KSR1 on ERK1/2 remains well-substantiated [38]. KSR1 is required for Ras-induced transformation of mouse embryonic fibroblasts (MEFs) and increases proliferative potential of mammary epithelial cells when expressed at optimal levels [34]. KSR1-deficient mice exhibit reduced Ras-dependent mammary tumor formation [39, 40]. In the absence of KSR1, high molecular weight complexes containing KSR1, ERK, and MEK are abolished, and KSR1 knockout mice exhibit reduced ERK signaling [40]. Furthermore, reintroduction of KSR1 into KSR1-deficient mice rescues ERK signaling [34]. However, KSR1 is not required for ERK signaling or normal embryonic development. KSR1-deficient mice develop normally, despite attenuated ERK signaling and slightly reduced T-cell activation [35, 40]. Because KSR1 promotes Ras and ERK activation and yet is dispensable for normal development, its potential as a therapeutic target for Ras-driven cancers is being widely investigated [41]. Given the role of NT downstream of Ras and ERK1/2 signaling, we reasoned that KSR1 may also regulate NT secretion.
In this study, we examine the role of KSR1 in NT secretion and ERK1/2 signaling in the human endocrine cell lines BON and QGP-1. We demonstrate that inhibition of KSR1 reduces, while its overexpression enhances NT secretion and ERK1/2 signaling. We also show that Exo70, a component of the exocyst complex and direct substrate of ERK2, positively regulates NT secretion. These findings describe a role for KSR1 as a positive regulator of NT secretion through activation of the ERK1/2 signaling pathway and suggest that the interaction between ERK2 and Exo70 contributes to NT secretion in human endocrine cells.
Materials and methods
Reagents
Phospho-ERK1/2 and ERK1/2 antibodies were from Cell Signaling Technology (Danvers, MA). NT antibody for immunofluorescence was from Abcam (Cambridge, UK). Exo70 antibody for western blot analysis was from Santa Cruz Biotechnology (Dallas, TX). KSR1 antibody was from LifeSpan BioSciences (Seattle, WA). GFP antibody was from Clontech (Mountain View, CA). β-actin antibody and docosahexaenoic acid (DHA) were from Sigma-Aldrich (St. Louis, MO). PD0325901 was from Selleck Chemicals (Houston, TX). ON-TARGETplus SMARTpool (KSR1) and ON-TARGETplus Non-targeting Control Pool siRNA were from GE Dharmacon (Lafayette, CO). Non-targeting control shRNA and shRNA targeting KSR1, ERK2, and Exo70 in bacterial glycerol stock were from Sigma-Aldrich. MSCV-IRES-GFP, MSCV-KSR1-IRES-GFP, pEGFP-control and pEGFP-C3-Exo70 plasmids were from Addgene (Cambridge, MA). pECFP-N-KSR1 plasmid was from Dr. Emilia Galperin’s lab (University of Kentucky).
Cell culture, transfection, and lentiviral transduction
The BON cell line was derived from a human pancreatic carcinoid tumor and characterized previously [42, 43]. BON cells were cultured in Dulbecco’s Modified Eagle’s Medium/Nutrient F-12 Ham supplemented with 5% fetal bovine serum (FBS) in 5% CO2 at 37°C. The QGP-1 cell line, derived from a pancreatic somatostatinoma (Japan Health Sciences Foundation, Osaka, Japan), was maintained in RPMI-1640 medium supplemented with 10% FBS [44]. siRNA transfections were performed using RNAiMAX (Life Technologies, Grand Island, NY). Forty-eight h after transfection, BON and QGP-1 cells were treated with 100μM DHA in serum-free medium for 90 min. Media was collected for NT-EIA and cells were lysed for western blot or RNA extraction.
For generation of cell lines expressing KSR1-targeting shRNA, BON or QGP-1 cells were plated in 6-well plates (5 × 105 cells/well) in growth media containing purified non-targeting control (NTC) or KSR1-targeting shRNA lentivirus. Puromycin-selection was performed to select for cells stably expressing KSR1 shRNA and knockdown efficiency was measured via western blot or quantitative real-time PCR (qRT-PCR). For cell lines expressing ERK2 or Exo70 shRNA, lentivirus of NTC, ERK2, or Exo70 shRNAs were co-transfected with an ectopic packaging vector into 293T cells. At 48 to 72 h post-transfection, viral supernatants were collected and filtered through a 0.45-μm Surfactant Free-Cellulose Acetate sterile syringe filter. BON cells were plated in 6-well plates (5 × 105 cells/well) and incubated with the viral supernatant for 24 h, followed by incubation with growth medium for an additional 24 h.
For generation of cells overexpressing KSR1 or Exo70, DNA was isolated from MSCV-IRES-GFP, MSCV-KSR1-IRES-GFP, pEGFP-control, pECFP-N-KSR1, and pEGFP-C3-Exo70 plasmids using the Qiagen Plasmid Miniprep Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Plasmid DNA was used to transfect BON or QGP-1 cells using Lipofectamine LTX with PLUS Reagent. Cells were collected for NT-EIA at 48–72 h post-transfection and overexpression measured via western blot or qRT-PCR.
Immunofluorescence
BON cells were plated on glass coverslips (#1) in 24-well plates at a density of 20×104/cm2 and transfected with pEGFP-control and pECFP-N-KSR1 plasmid DNA using Lipofectamine LTX with PLUS Reagent. 48 h after transfection, cells were fixed in 4% paraformaldehyde/PBS, permeabilized with 0.3% TritonX-100/PBS, and blocked with 0.1% bovine serum albumin/PBS. Cells were incubated with primary antibody for 1 hour, followed by Alexa Fluor-conjugated secondary antibody from Invitrogen for 30 minutes. Images were observed under a Nikon confocal microscope with 40× objective.
Cell treatment and NT enzyme immunoassay (EIA)
Cells were plated in 24-well plates at a density of 15×104/cm2 and grown for 48 h for DHA or drug treatment. Cells transfected with siRNA were treated with 100μM DHA in serum-free growth medium for 90 minutes. shRNA-expressing cell lines were sensitized to FFA treatment after transfection and puromycin selection and were therefore treated with 30μM DHA to reduce toxicity relative to 100μM DHA treatment. Exo70 overexpression experiments were performed with 30μM DHA for consistency with Exo70 shRNA experiments. For treatment with PD compound, cells were pretreated with PD compound for 30 minutes in serum-free growth medium and then treated with 100μM DHA plus PD for an additional 90 minutes. Media were collected for NT secretion measurement using the NT-EIA kit from Phoenix Pharmaceuticals (Belmont, CA) as described previously [45, 46] and cells were lysed for western blotting or RNA isolation. Data obtained from NT-EIA were normalized by protein concentration from corresponding cell lysates.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. cDNA was synthesized from 1μg of total RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). qRT-PCR was performed using a TaqMan Gene Expression Master Mix (#4369016) and TaqMan probes for human KSR1 (ID Hs00300134_m1) and human GAPDH (#4332649) according to manufacturer’s protocol (Applied Biosystems, Austin, TX). Relative mRNA expression was calculated using the comparative ΔΔCt method and represented as fold-change relative to internal controls.
Protein preparation and western blotting
Protein preparation and western blotting were performed as previously described [45, 47]. Briefly, cells were lysed with lysis buffer (Cell Signaling Technology) containing 1mM phenylmethylsulfonyl fluoride (PMSF). Equal amounts of protein were resolved on 4–12% NuPAGE BisTris gels (Invitrogen, Carlsbad, CA) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were incubated with primary antibodies overnight at 4°C followed by incubation with secondary antibodies conjugated with horseradish peroxidase. Membranes were developed using Amersham ECL Western Blotting Detection Reagent (GE Healthcare Life Sciences, Piscataway, NJ) or Immobilon Western Chemiluminescent HRP Substrate (Thermo Scientific, Waltham, MA). Band intensity was measured with ImageJ software using β-actin or total protein as loading controls and expressed as fold-change relative to NTCs.
Statistical analysis
NT secretion values were normalized to protein concentration of corresponding lysates. Descriptive statistics including mean and standard deviation were calculated to summarize NT secretion. Bar graphs represent mean (± SD) fold-change of NT levels in different cell culture conditions. mRNA expression is represented in bar graphs as mean (± SD) fold-change relative to internal controls (human GAPDH). Within each experiment, comparisons across groups were accomplished using one- or two-way analysis of variance models, and pairwise comparisons were performed using contrast statements. Adjustment in p-values due to several pairwise testing within each experiment was performed using the Holm’s procedure. p-values<0.05 were considered statistically significant.
Results
KSR1 inhibition reduces NT secretion
KSR1 is expressed in intestinal epithelial cells, where it is protective against cytokine-induced injury [48, 49]. NT is secreted from specialized enteroendocrine (N) cells of the small intestine and is stimulated by ingestion of dietary fats [50–53]. To determine whether KSR1 is expressed in human endocrine cells and characterize its localization relative to NT, we used the BON cell line, which synthesizes and secretes NT in a manner equivalent to that of N cells in the small bowel. BON cells were transfected with CFP-tagged KSR1 plasmid DNA to enable detection of KSR1with fluorescent confocal microscopy at 488nm and subsequently labeled with fluorescent anti-NT antibody [42]. Consistent with previous evidence, we show that NT is localized to secretory vesicles in human endocrine cells, while KSR1 exhibits diffuse cytosolic staining (Fig 1). Interestingly, distribution of KSR1 and NT is largely mutually exclusive, where KSR1 expressing cells do not contain NT-containing secretory vesicles and cells retaining large amounts of NT do not express high levels of KSR1.
Fig 1. KSR1 expression stimulates NT release from BON cells.
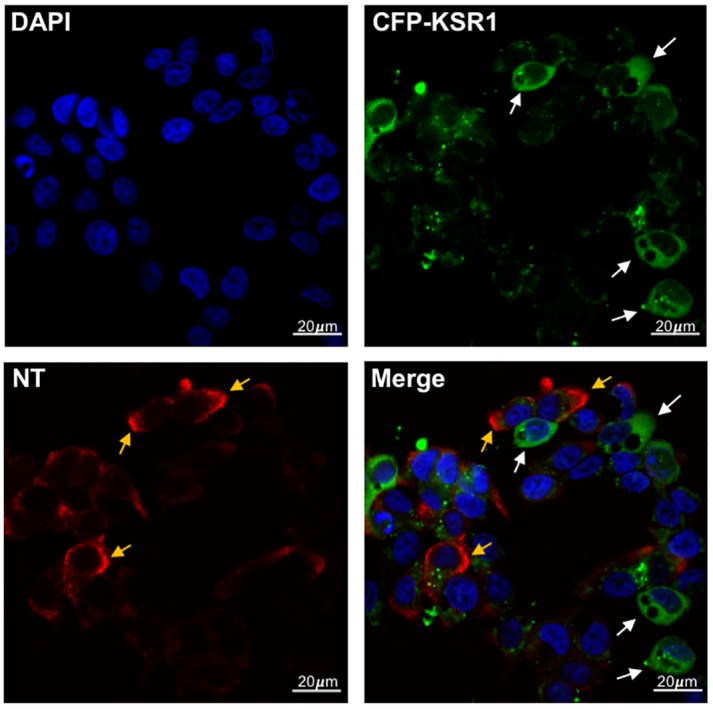
BON cells were transfected with pECFP-N-KSR1 (green) and labeled with immunofluorescent anti-NT antibody (red) and observed via confocal microscopy with 40x objective. White arrows indicate KSR1-positive cells, yellow arrows indicate NT labeling. Scale bar indicates 20μm.
We have previously demonstrated that ERK1/2 plays a stimulatory role in NT secretion [11, 54]. The function of KSR1 as a scaffold of the Raf/MEK/ERK complex has been reported to enhance ERK1/2 signaling [35–37]. To determine whether KSR1 is involved in NT secretion, we transfected BON cells with small interfering RNA (siRNA) against KSR1. Forty-eight h after transfection, cells were treated for 90 min with 100μM docosahexaenoic acid (DHA), a long-chain unsaturated FA that we have previously shown stimulates NT secretion, thereby mimicking the effects of fat-ingestion in the small bowel. KSR1 knockdown was confirmed via qRT-PCR and NT secretion was measured using NT-EIA. Consistently, inhibition of KSR1 attenuates DHA-stimulated NT secretion (Fig 2a).
Fig 2. KSR1 inhibition attenuates FA-stimulated NT secretion.
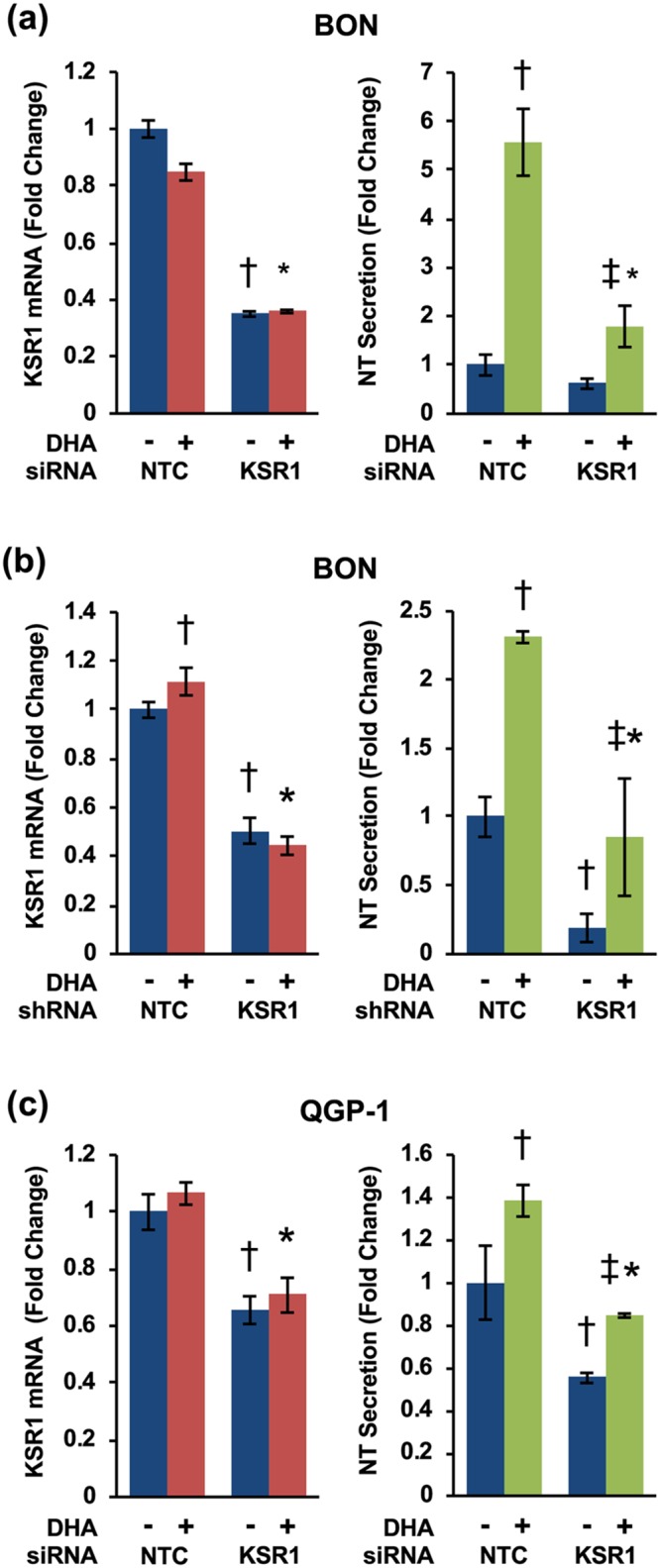
(a) BON cells were transfected with KSR1-targeting siRNA or KSR1-targeting shRNA (b) and treated with or without DHA for 90 min. Total RNA was isolated and qRT-PCR performed targeting KSR1 with GAPDH as the internal control (left) (†p<0.05 vs. untreated NTC; *p<0.05 vs. NTC plus DHA). Media were collected and NT-EIA performed (right) (†p<0.05 vs. untreated NTC; *p<0.05 vs. NTC plus DHA; ‡p<0.05 vs. untreated KSR1-siRNA/shRNA groups.) (c) QGP-1 cells were transfected with KSR1-targeting siRNA and treated with or without DHA for 90 min. Total RNA was isolated and qRT-PCR performed targeting KSR1 with GAPDH as the internal control (left) (†p<0.05 vs. untreated NTC; *p<0.05 vs. NTC plus DHA). Media were collected and NT-EIA performed (right) (†p<0.05 vs. untreated NTC; *p<0.05 vs. NTC plus DHA; ‡p<0.05 vs. untreated KSR1-siRNA group). All data represent mean +/- SD. Data are representative of three independent experiments.
To verify these results, we next established BON cell lines stably expressing KSR1-targeting shRNA. shRNA-mediated knockdown of KSR1 reduces basal and DHA-stimulated NT secretion from BON cells (Fig 2b). To further confirm these findings, we established KSR1 knockdown using siRNA in another human endocrine cell line, QGP-1. QGP-1 cells, derived from a pancreatic somatostatinoma, produce and secrete high levels of NT in response to FFA-stimulation, including DHA [11]. Consistent with the effect of KSR1 on NT secretion in BON cells, inhibition of KSR1 in QGP-1 cells reduces basal and DHA-stimulated NT-secretion (Fig 2c). Together, these data demonstrate that KSR1 regulates NT release from human endocrine cells.
KSR1 inhibition reduces ERK1/2 phosphorylation
The scaffolding function of KSR1 on the Raf/MEK/ERK complex has been demonstrated to coordinate and enhance ERK1/2 activation [32–34, 54]. KSR1-deficient mice exhibit attenuated ERK1/2 signaling and a reduction in MEK- and ERK-containing high molecular weight complexes [40]. To determine whether the effect of KSR1 on NT secretion is mediated by the ERK1/2 signaling pathway, BON and QGP-1 cells transfected with KSR1-targeting siRNA were treated with 100μM DHA to stimulate NT secretion and lysed for western blotting. Consistently, inhibition of KSR1 decreased ERK1/2 phosphorylation (p-ERK) (Fig 3). These findings are consistent with studies demonstrating that KSR1 positively regulates ERK1/2 signaling and indicate that the effect of KSR1 on NT secretion in human endocrine cells is mediated by its interaction with the Raf/MEK/ERK signaling complex.
Fig 3. Knockdown of KSR1 attenuates ERK1/2 signaling.

(a) BON and (b) QGP-1 cells were transfected with KSR1-targeting siRNA and treated with or without 100μM DHA for 90 min. Cells were lysed and western blotting analysis performed. Band intensity is indicated below the corresponding band and expressed as fold-change relative to NTC. Each blot is representative of three independent experiments.
KSR1 overexpression stimulates NT secretion and ERK activity
As a scaffold protein, KSR1 can produce both inhibitory and stimulatory effects on ERK1/2 signaling when overexpressed [35]. At optimal expression levels, KSR1 facilitates maximal coordination of Raf/MEK/ERK and enhances ERK1/2 signaling, while at levels above or below this threshold, ERK1/2 signaling is inhibited. As such, the effects of KSR1 overexpression are tissue specific and based on endogenous levels of KSR1 expression [34, 35, 40]. To determine the effects of KSR1 overexpression on ERK1/2 signaling in human endocrine cells, we transfected BON cells with KSR1-overexpressing plasmids and performed western blot to assess ERK1/2 activation. Overexpression of KSR1 enhances ERK1/2 phosphorylation, suggesting that endogenous KSR1 expression in BON cells is below the threshold for coordinating maximal Raf/MEK/ERK signaling (Fig 4a and 4b). To determine whether the effect of KSR1 overexpression on ERK1/2 signaling corresponds to an increase in FFA-stimulated NT secretion, we overexpressed KSR1 in BON and QGP-1 cells and performed NT-EIA. Consistent with the observed increase in ERK1/2 signaling, DHA-stimulated NT secretion is enhanced by KSR1 overexpression (Fig 4c–4d). These data establish KSR1 as a positive regulator of FFA-mediated NT secretion in endocrine cells through activation of the Raf/MEK/ERK signaling pathway.
Fig 4. Overexpression of KSR1 enhances NT secretion and ERK activation.

(a) BON cells were transfected with pEGFP-control and pECFP-N-KSR1 plasmid DNA and QGP-1 cells were transfected with MSCV-IRES-GFP and MSCV-KSR1-IRES-GFP plasmid DNA (b) and treated with or without 100μM DHA for 90 min. Cells were lysed and western blotting analysis performed. Band intensity is indicated below the corresponding band and expressed as fold-change relative to NTC. Each blot is representative of two independent experiments. (c) BON cells were transfected with pEGFP-control and pECFP-N-KSR1 plasmid DNA and QGP-1 cells were transfected with MSCV-IRES-GFP and MSCV-KSR1-IRES-GFP plasmid DNA (d) and treated with or without 100μM DHA for 90 min. Media were collected and NT-EIA performed. Panel (c): †p<0.05 vs. untreated pEGFP; *p<0.05 vs. pEGFP plus DHA; ‡p<0.05 vs. untreated CFP-KSR1; Panel (d): †p<0.05 vs. untreated MSCV-GFP; *p<0.05 vs. MSCV-GFP plus DHA; ‡p<0.05 vs. untreated MSCV-GFP-KSR1. Data represent mean +/- SD and are representative of three independent experiments.
Exo70 positively regulates NT secretion
Upon extracellular stimulation of NT release, NT-containing vesicles are transported to the plasma membrane for exocytosis and extracellular release [24, 25]. Tethering of secretory vesicles to the plasma membrane prior to secretion is mediated by the exocyst, an octameric protein complex comprised of Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70, and Exo84 [55, 56]. Exo70 is a direct substrate of ERK2, which phosphorylates Exo70 at Ser250 and thereby modulates the processes mediating vesicle exocytosis [57]. Whether Exo70 regulates exocytosis of NT secretory vesicles is unknown. To determine whether Exo70 is involved in ERK-mediated NT secretion, we generated BON cell lines stably expressing ERK2 shRNA. Consistent with previous reports, ERK2 inhibition reduces Exo70 expression in BON cells (Fig 5a) [57]. NT secretion is also significantly attenuated by ERK2 knockdown (Fig 5b). To verify these results, we pre-treated BON cells with the MEK inhibitor PD 0325901 to inhibit MEK-induced phosphorylation of ERK1/2 [58]. MEK inhibition abolished ERK1/2 phosphorylation in basal and DHA-stimulated BON cells (Fig 5c) and inhibited DHA-stimulated NT secretion (Fig 5d).
Fig 5. ERK1/2 inhibition attenuates Exo70 expression.

(a) BON cells stably expressing NTC or ERK2 shRNAs were treated with or without 30μM DHA for 90 min. Cells were lysed and western blotting analysis performed. Band intensity is indicated below the corresponding band and expressed as fold-change relative to NTC. (b) Media were collected and NT-EIA performed (†p<0.05 vs. untreated NTC; *p<0.05 vs. NTC plus DHA; data represent mean +/- SD). (c) BON cells pre-treated with DMSO or PD 0325901 for 30 minutes were treated with or without 100μM DHA for 90 min. Cells were lysed and western blotting analysis performed. Band intensity is indicated below the corresponding band and expressed as fold-change relative to NTC. (d) Media were collected and NT-EIA performed (†p<0.05 vs. DMSO without DHA; *p<0.05 vs. DMSO plus DHA; ‡p<0.05 vs. PD without DHA; data represent mean +/- SD). All data are representative of three independent experiments.
To further confirm the role of Exo70 in NT secretion, we generated BON cell lines stably expressing Exo70 shRNA and measured constitutive and FFA-stimulated NT secretion. Inhibition of Exo70 potently inhibits NT secretion from both untreated and DHA-stimulated BON cells (Fig 6a).
Fig 6. Exo70 positively regulates NT secretion.

(a) BON cells stably expressing NTC or Exo70 shRNAs were treated with or without 30μM DHA for 90 min. Media were collected and NT-EIA performed (†p<0.05 vs. untreated NTC; *p<0.05 vs. NTC plus DHA; data represent mean +/- SD). (b) BON cells were transfected with pEGFP-control or pEGFP-C3-Exo70 plasmid DNA and treated with or without 30μM DHA for 90 min. Media were collected and NT-EIA performed (†p<0.05 vs. untreated pEGFP; *p<0.05 vs. pEGFP plus DHA; ‡p<0.05 vs. untreated pEGFP-Exo70; data represent mean +/- SD). (c) BON cells stably expressing NTC or Exo70 shRNAs were treated with or without 30μM DHA for 90 min. Cells were lysed and western blotting analysis performed. Band intensity is indicated below the corresponding band and expressed as fold-change relative to NTC. All data are representative of three independent experiments. (d) Proposed model of KSR1/ERK/Exo70 signaling in the control of NT secretion.
Conversely, overexpression of Exo70 enhances both basal and DHA-stimulated NT secretion (Fig 6b). Moreover, western blot analysis demonstrates that ERK1/2 phosphorylation is attenuated by Exo70 inhibition (6c). Collectively, these data implicate Exo70 as a positive regulator of NT secretion and suggest that the interaction between ERK1/2 and the exocyst complex may mediate exocytosis of NT vesicles from enteroendocrine cells.
Discussion
FAs and NT play stimulatory and intersecting roles in the development of obesity and cancer. Our group recently demonstrated that stimulation of NT secretion by the unsaturated FA, DHA, is mediated by ERK1/2 signaling [11, 54]. Here, we examine the role of KSR1 on DHA-stimulated NT secretion. We show that KSR1 promotes DHA-stimulated NT secretion through the ERK1/2 signaling pathway. We also show that Exo70 inhibition reduces, while its overexpression enhances NT secretion and ERK1/2 phosphorylation. Collectively, the data presented in our current study implicate KSR1 and Exo70 as positive regulators of NT secretion that are integrated through the ERK1/2 signaling pathway.
KSR1 serves as a scaffold of the Raf/MEK/ERK complex, coordinating the spatial interactions between RAF, MEK, and ERK and enhancing specificity of their sequential phosphorylation [37, 40]. These interactions make KSR1 a potent regulator of ERK1/2 signaling [34–37]. We show that KSR1 inhibition reduces DHA-stimulated NT secretion and correspondingly inhibits ERK1/2 phosphorylation in human endocrine cells. In contrast, overexpression of KSR1 enhances NT secretion and ERK1/2 phosphorylation, and immunofluorescent labeling suggests that KSR1 expression promotes release of NT vesicles from N cells. Given the role of KSR1 as a positive regulator of ERK1/2 signaling and our previous studies showing that ERK1/2 enhances NT mRNA expression and peptide secretion, these data suggest that KSR1 positively regulates NT secretion through the Raf/MEK/ERK signaling cascade [11, 54]. KSR1 has been similarly shown to regulate secretion of insulin and inflammatory cytokines, though whether its role in secretion is predominantly positive or negative is unclear [59, 60]. KSR1-deficient mice have higher levels of basal insulin release relative to wild-type mice, suggesting that KSR1 negatively regulates insulin secretion [60]. In T-lymphocytes and splenocytes from KSR1-deficient mice, interferon gamma (IFN-γ) secretion is enhanced relative to wild-type cells, suggesting a negative role for KSR1 in IFN-γ secretion [59]. However, KSR1-deficient T cells exhibit reduced interleukin 17A (IL17A) secretion, suggesting that KSR1 enhances IL17A secretion [59].
These contrasting findings may be attributable to the role of KSR1 as a scaffold protein, which can play negative or positive roles in signal transduction depending on their expression level [35, 61–63]. Scaffold proteins facilitate maximal signaling when expressed in approximate stoichiometry with their ligands [61]. Scaffold inhibition reduces signaling due to depletion of the scaffold, yet scaffold overexpression can also inhibit signaling due to sequestration of the ligands, unless the ligands themselves are also overexpressed [61]. However, in cells endogenously expressing high levels of scaffold, inhibition may bring the scaffold and its ligands to near equivalent amounts, thereby increasing signaling [61]. The conflicting evidence regarding the role of KSR1 in secretory processes may be attributable to variation in endogenous expression levels of KSR1 and its ligands between cell types.
In mouse embryonic fibroblasts (MEFs), expression of KSR1 at 14 times endogenous levels produces maximal ERK1/2 signaling, while levels exceeding this reduce ERK1/2 signaling to levels comparable to that of KSR1-deficient cells [34]. In contrast, ectopic expression of KSR1 in 293T cells inhibits insulin- and phorbol myristate acetate (PMA)-stimulated ERK1/2 signaling [64]. We show that overexpression of KSR1 enhances ERK1/2 phosphorylation and DHA-stimulated NT secretion in neuroendocrine cells. These data suggest that, like MEFs, endogenous KSR1 expression in neuroendocrine cells is below that of the threshold for maximal ERK activation. Alternatively, components of the Raf/MEK/ERK complex may also be overexpressed in response to the downstream effects of ectopic KSR1 expression, thereby restoring stoichiometry of the complex. NT has been shown to activate ERK signaling through Ras activation [65]. Stimulation of NT by overexpression of KSR1 may therefore feedback positively on ERK1/2, optimizing signaling of the Raf/MEK/ERK complex.
As a secreted peptide, NT is subject to regulation by exocytotic processes, including vesicle trafficking, docking, and fusion with the plasma membrane [24, 25]. Exo70, a component of the exocyst complex that regulates exocytosis, is localized to lipid rafts on the plasma membrane and facilitates docking of secretory vesicles to the membrane prior to secretion [55, 56]. Exo70 is a direct substrate of ERK2, which phosphorylates Exo70 at Ser250 upon EGF stimulation [13]. We have recently demonstrated that DHA-stimulated NT secretion is mediated by an ERK1/2-dependent mechanism in neuroendocrine cells and C57/BL6 mice [11]. In our current study, we show that ERK2 knockdown reduces expression of total Exo70, though no antibodies are currently available for detection of phosphorylated Exo70. Ablation of ERK1/2 phosphorylation with the MEK inhibitor PD 0325901 also abrogated Exo70 expression and NT-secretion. These findings support other studies demonstrating that ERK2 positively regulates Exo70 and promotes its interaction with other components of the exocyst complex [57]. Activation of Exo70 by ERK1/2 signaling also mediates vesicular trafficking and matrix metalloproteinase (MMP) secretion in MDA-MB-231 human breast cancer cells [57]. Our results corroborate these findings and provide further evidence to support the role of ERK1/2 signaling as a regulator of Exo70-mediated secretory processes.
We also find that Exo70 knockdown potently inhibits both basal and DHA-stimulated NT secretion. In contrast, Exo70 overexpression enhances basal and DHA-stimulated NT secretion, suggesting a positive role for Exo70 in regulating NT secretion. Consistent with our findings, Lopez et. al [66] showed that Exo70 regulates insulin secretion. Insulin granules associate with Exo70 at the plasma membrane of β-cells, suggesting that Exo70 facilitates docking of insulin granules with the membrane [66]. Exo70 is also required for proper docking of Glut4-containing vesicles with the plasma membrane during insulin-stimulated Glut4 secretion [67]. Additionally, we show that inhibition of Exo70 attenuates ERK1/2 phosphorylation, suggesting a role for Exo70 in feedback activation of ERK1/2 signaling. Notably, Exo70 mRNA and protein is overexpressed in colon cancer, in which both ERK1/2 and NT are also heavily implicated [68]. The exocyst complex has also been implicated in the epithelial-mesenchymal transition (EMT) in mammary epithelial cells [68, 69]. Studies aimed at further delineating the interaction between Exo70 and ERK1/2 may shed light on their role in processes regulating both secretion and tumorigenesis.
In summary, the present studies demonstrate for the first time that KSR1 positively regulates FFA-stimulated NT secretion and that this effect is mediated by the stimulatory effect of KSR1 on ERK1/2 signaling. Moreover, we reveal a novel role for Exo70 in the regulation of NT secretion (Fig 6d). We also describe a role for ERK1/2 in NT secretion independent of its previously described role on NT mRNA and protein expression by demonstrating its stimulatory effect on the secretory processes mediating NT release. Collectively, these data indicate that KSR1 modulation may have therapeutic potential for diseases driven by NT gene expression and peptide secretion, such as obesity-associated metabolic disorders and Ras-driven cancers.
Acknowledgments
We thank the University of Kentucky Markey Cancer Center’s Research Communications Office for manuscript preparation and the Biostatistics and Bioinformatics Shared Resource Facility for statistical analyses.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by the National Institutes of Health Grants R01 DK112034 (to B.M.E.), R01 DK048498 (to B.M.E.) and NCI P30 CA177558. S.R. is supported by NIH Training Grant T32 DK007778.
References
- 1.Steele CB, Thomas CC, Henley SJ, Massetti GM, Galuska DA, Agurs-Collins T, et al. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005–2014. MMWR Morb Mortal Wkly Rep. 2017;66(39):1052–8. Epub 2017/10/06. 10.15585/mmwr.mm6639e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma Y, Yang Y, Wang F, Zhang P, Shi C, Zou Y, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One. 2013;8(1):e53916 Epub 2013/01/26. 10.1371/journal.pone.0053916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obes Rev. 2012;13(12):1083–95. Epub 2012/09/04. 10.1111/j.1467-789X.2012.01024.x . [DOI] [PubMed] [Google Scholar]
- 4.Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. 2008;37(3):635–46, viii–ix. Epub 2008/09/09. 10.1016/j.ecl.2008.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blucher C, Stadler SC. Obesity and Breast Cancer: Current Insights on the Role of Fatty Acids and Lipid Metabolism in Promoting Breast Cancer Growth and Progression. Front Endocrinol (Lausanne). 2017;8:293 Epub 2017/11/23. 10.3389/fendo.2017.00293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18(2):139–43. Epub 2011/02/08. 10.1097/MED.0b013e3283444b09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhathena SJ. Relationship between fatty acids and the endocrine and neuroendocrine system. Nutr Neurosci. 2006;9(1–2):1–10. Epub 2006/08/17. 10.1080/10284150600627128 . [DOI] [PubMed] [Google Scholar]
- 8.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21(3):427–33. Epub 2000/02/26. . [DOI] [PubMed] [Google Scholar]
- 9.Carraway RE, Plona AM. Involvement of neurotensin in cancer growth: evidence, mechanisms and development of diagnostic tools. Peptides. 2006;27(10):2445–60. Epub 2006/08/05. 10.1016/j.peptides.2006.04.030 . [DOI] [PubMed] [Google Scholar]
- 10.Evers BM, Ishizuka J, Chung DH, Townsend CM Jr., Thompson JC. Neurotensin expression and release in human colon cancers. Annals of surgery. 1992;216(4):423–30; discussion 30–1. Epub 1992/10/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jing Li JS, Xian Li, Stephanie B. Rock, Heather F. Sinner, Heidi L. Weiss, Todd Weiss, Courtney M. Townsend Jr., Tuanyan Gao, B. Mark Evers. FFAR4 is involved in regulation of neurotensin release from neuroendocrine cells and male C57BL/6 mice. Endocrinology 2018. [DOI] [PMC free article] [PubMed]
- 12.Li J, Song J, Zaytseva YY, Liu Y, Rychahou P, Jiang K, et al. An obligatory role for neurotensin in high-fat-diet-induced obesity. Nature. 2016;533(7603):411–5. Epub 2016/05/20. 10.1038/nature17662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melander O, Maisel AS, Almgren P, Manjer J, Belting M, Hedblad B, et al. Plasma proneurotensin and incidence of diabetes, cardiovascular disease, breast cancer, and mortality. JAMA: the journal of the American Medical Association. 2012;308(14):1469–75. 10.1001/jama.2012.12998 . [DOI] [PubMed] [Google Scholar]
- 14.Qiu S, Pellino G, Fiorentino F, Rasheed S, Darzi A, Tekkis P, et al. A Review of the Role of Neurotensin and Its Receptors in Colorectal Cancer. Gastroenterol Res Pract. 2017;2017:6456257 Epub 2017/03/21. 10.1155/2017/6456257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Z, Martinez-Fong D, Tredaniel J, Forgez P. Neurotensin and its high affinity receptor 1 as a potential pharmacological target in cancer therapy. Front Endocrinol (Lausanne). 2012;3:184 Epub 2013/01/22. 10.3389/fendo.2012.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshinaga K, Evers BM, Izukura M, Parekh D, Uchida T, Townsend CM Jr., et al. Neurotensin stimulates growth of colon cancer. Surgical oncology. 1992;1(2):127–34. Epub 1992/04/11. . [DOI] [PubMed] [Google Scholar]
- 17.Barchetta I, Cimini FA, Capoccia D, Bertoccini L, Ceccarelli V, Chiappetta C, et al. Neurotensin Is a Lipid-Induced Gastrointestinal Peptide Associated with Visceral Adipose Tissue Inflammation in Obesity. Nutrients. 2018;10(4). Epub 2018/04/25. 10.3390/nu10040526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barchetta I, Cimini FA, Leonetti F, Capoccia D, Di Cristofano C, Silecchia G, et al. Increased Plasma Proneurotensin Levels Identify NAFLD in Adults With and Without Type 2 Diabetes. J Clin Endocrinol Metab. 2018;103(6):2253–60. Epub 2018/03/29. 10.1210/jc.2017-02751 . [DOI] [PubMed] [Google Scholar]
- 19.Bartsch H, Nair J, Owen RW. Dietary polyunsaturated fatty acids and cancers of the breast and colorectum: emerging evidence for their role as risk modifiers. Carcinogenesis. 1999;20(12):2209–18. Epub 1999/12/11. . [DOI] [PubMed] [Google Scholar]
- 20.Azrad M, Turgeon C, Demark-Wahnefried W. Current evidence linking polyunsaturated Fatty acids with cancer risk and progression. Front Oncol. 2013;3:224 Epub 2013/09/13. 10.3389/fonc.2013.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harnack K, Andersen G, Somoza V. Quantitation of alpha-linolenic acid elongation to eicosapentaenoic and docosahexaenoic acid as affected by the ratio of n6/n3 fatty acids. Nutr Metab (Lond). 2009;6:8 Epub 2009/02/21. 10.1186/1743-7075-6-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thiebaut AC, Jiao L, Silverman DT, Cross AJ, Thompson FE, Subar AF, et al. Dietary fatty acids and pancreatic cancer in the NIH-AARP diet and health study. J Natl Cancer Inst. 2009;101(14):1001–11. Epub 2009/06/30. 10.1093/jnci/djp168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drewe J, Mihailovic S, D’Amato M, Beglinger C. Regulation of fat-stimulated neurotensin secretion in healthy subjects. J Clin Endocrinol Metab. 2008;93(5):1964–70. Epub 2008/02/28. 10.1210/jc.2007-2238 . [DOI] [PubMed] [Google Scholar]
- 24.Michael DJ, Cai H, Xiong W, Ouyang J, Chow RH. Mechanisms of peptide hormone secretion. Trends Endocrinol Metab. 2006;17(10):408–15. Epub 2006/11/07. 10.1016/j.tem.2006.10.011 . [DOI] [PubMed] [Google Scholar]
- 25.Park JJ, Loh YP. How peptide hormone vesicles are transported to the secretion site for exocytosis. Molecular endocrinology. 2008;22(12):2583–95. Epub 2008/08/02. 10.1210/me.2008-0209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evers BM, Zhou Z, Celano P, Li J. The neurotensin gene is a downstream target for Ras activation. J Clin Invest. 1995;95(6):2822–30. Epub 1995/06/01. 10.1172/JCI117987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–39. Epub 2007/05/15. 10.1038/sj.onc.1210414 . [DOI] [PubMed] [Google Scholar]
- 28.Wortzel I, Seger R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer. 2011;2(3):195–209. Epub 2011/07/23. 10.1177/1947601911407328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84. Epub 2006/11/28. 10.1016/j.bbamcr.2006.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirsammer G, Strizzi L, Margaryan NV, Gilgur A, Hyser M, Atkinson J, et al. Nodal signaling promotes a tumorigenic phenotype in human breast cancer. Semin Cancer Biol. 2014;29:40–50. Epub 2014/07/30. 10.1016/j.semcancer.2014.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaiswal BS, Durinck S, Stawiski EW, Yin J, Wang W, Lin E, et al. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin Cancer Res. 2018;24(16):4044–55. Epub 2018/05/16. 10.1158/1078-0432.CCR-17-3674 . [DOI] [PubMed] [Google Scholar]
- 32.Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002;16(4):427–38. Epub 2002/02/19. 10.1101/gad.962902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nature reviews Molecular cell biology. 2005;6(11):827–37. 10.1038/nrm1743 . [DOI] [PubMed] [Google Scholar]
- 34.Kortum RL, Lewis RE. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol. 2004;24(10):4407–16. Epub 2004/05/04. 10.1128/MCB.24.10.4407-4416.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frodyma D, Neilsen B, Costanzo-Garvey D, Fisher K, Lewis R. Coordinating ERK signaling via the molecular scaffold Kinase Suppressor of Ras. F1000Res. 2017;6:1621 Epub 2017/10/14. 10.12688/f1000research.11895.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A. 2009;106(27):11022–7. Epub 2009/06/23. 10.1073/pnas.0901590106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Razidlo GL, Kortum RL, Haferbier JL, Lewis RE. Phosphorylation regulates KSR1 stability, ERK activation, and cell proliferation. J Biol Chem. 2004;279(46):47808–14. Epub 2004/09/17. 10.1074/jbc.M406395200 . [DOI] [PubMed] [Google Scholar]
- 38.Lavoie H, Sahmi M, Maisonneuve P, Marullo SA, Thevakumaran N, Jin T, et al. MEK drives BRAF activation through allosteric control of KSR proteins. Nature. 2018;554(7693):549–53. Epub 2018/02/13. 10.1038/nature25478 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lozano J, Xing R, Cai Z, Jensen HL, Trempus C, Mark W, et al. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 2003;63(14):4232–8. Epub 2003/07/23. . [PubMed] [Google Scholar]
- 40.Nguyen A, Burack WR, Stock JL, Kortum R, Chaika OV, Afkarian M, et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002;22(9):3035–45. Epub 2002/04/10. 10.1128/MCB.22.9.3035-3045.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neilsen BK, Frodyma DE, Lewis RE, Fisher KW. KSR as a therapeutic target for Ras-dependent cancers. Expert Opin Ther Targets. 2017;21(5):499–509. Epub 2017/03/24. 10.1080/14728222.2017.1311325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evers BM, Townsend CM Jr., Upp JR, Allen E, Hurlbut SC, Kim SW, et al. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology. 1991;101(2):303–11. Epub 1991/08/11. . [DOI] [PubMed] [Google Scholar]
- 43.Parekh D, Ishizuka J, Townsend CM Jr., Haber B, Beauchamp RD, Karp G, et al. Characterization of a human pancreatic carcinoid in vitro: morphology, amine and peptide storage, and secretion. Pancreas. 1994;9(1):83–90. Epub 1994/01/01. . [DOI] [PubMed] [Google Scholar]
- 44.Doihara H, Nozawa K, Kojima R, Kawabata-Shoda E, Yokoyama T, Ito H. QGP-1 cells release 5-HT via TRPA1 activation; a model of human enterochromaffin cells. Mol Cell Biochem. 2009;331(1–2):239–45. Epub 2009/06/10. 10.1007/s11010-009-0165-7 . [DOI] [PubMed] [Google Scholar]
- 45.Li J, O’Connor KL, Cheng X, Mei FC, Uchida T, Townsend CM Jr., et al. Cyclic adenosine 5'-monophosphate-stimulated neurotensin secretion is mediated through Rap1 downstream of both Epac and protein kinase A signaling pathways. Molecular endocrinology. 2007;21(1):159–71. 10.1210/me.2006-0340 . [DOI] [PubMed] [Google Scholar]
- 46.Li J, Chen LA, Townsend CM Jr., Evers BM. PKD1, PKD2, and their substrate Kidins220 regulate neurotensin secretion in the BON human endocrine cell line. J Biol Chem. 2008;283(5):2614–21. 10.1074/jbc.M707513200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Hellmich MR, Greeley GH Jr., Townsend CM Jr., Evers BM. Phorbol ester-mediated neurotensin secretion is dependent on the PKC-alpha and -delta isoforms. Am J Physiol Gastrointest Liver Physiol. 2002;283(5):G1197–206. Epub 2002/10/17. 10.1152/ajpgi.00177.2002 . [DOI] [PubMed] [Google Scholar]
- 48.Yan F, John SK, Polk DB. Kinase suppressor of Ras determines survival of intestinal epithelial cells exposed to tumor necrosis factor. Cancer Res. 2001;61(24):8668–75. Epub 2001/12/26. . [PubMed] [Google Scholar]
- 49.Yan F, John SK, Wilson G, Jones DS, Washington MK, Polk DB. Kinase suppressor of Ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J Clin Invest. 2004;114(9):1272–80. Epub 2004/11/03. 10.1172/JCI21022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evers BM, Ehrenfried JA, Wang X, Townsend CM Jr., Thompson JC. Temporal-specific and spatial-specific patterns of neurotensin gene expression in the small bowel. The American journal of physiology. 1994;267(5 Pt 1):G875–82. Epub 1994/11/11. 10.1152/ajpgi.1994.267.5.G875 . [DOI] [PubMed] [Google Scholar]
- 51.Mazella J, Beraud-Dufour S, Devader C, Massa F, Coppola T. Neurotensin and its receptors in the control of glucose homeostasis. Front Endocrinol (Lausanne). 2012;3:143 Epub 2012/12/12. 10.3389/fendo.2012.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mustain WC, Rychahou PG, Evers BM. The role of neurotensin in physiologic and pathologic processes. Curr Opin Endocrinol Diabetes Obes. 2011;18(1):75–82. Epub 2010/12/03. 10.1097/MED.0b013e3283419052 . [DOI] [PubMed] [Google Scholar]
- 53.Vincent JP, Mazella J, Kitabgi P. Neurotensin and neurotensin receptors. Trends in pharmacological sciences. 1999;20(7):302–9. . [DOI] [PubMed] [Google Scholar]
- 54.Li J, Liu J, Song J, Wang X, Weiss HL, Townsend CM Jr., et al. mTORC1 inhibition increases neurotensin secretion and gene expression through activation of the MEK/ERK/c-Jun pathway in the human endocrine cell line BON. Am J Physiol Cell Physiol. 2011;301(1):C213–26. Epub 2011/04/22. 10.1152/ajpcell.00067.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S, Hsu SC. The molecular mechanisms of the mammalian exocyst complex in exocytosis. Biochem Soc Trans. 2006;34(Pt 5):687–90. Epub 2006/10/21. 10.1042/BST0340687 . [DOI] [PubMed] [Google Scholar]
- 56.Wu B, Guo W. The Exocyst at a Glance. J Cell Sci. 2015;128(16):2957–64. Epub 2015/08/05. 10.1242/jcs.156398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ren J, Guo W. ERK1/2 regulate exocytosis through direct phosphorylation of the exocyst component Exo70. Dev Cell. 2012;22(5):967–78. Epub 2012/05/19. 10.1016/j.devcel.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5(4):350–6. Epub 2005/06/16. 10.1016/j.coph.2005.04.007 . [DOI] [PubMed] [Google Scholar]
- 59.Goettel JA, Scott Algood HM, Olivares-Villagomez D, Washington MK, Chaturvedi R, Wilson KT, et al. KSR1 protects from interleukin-10 deficiency-induced colitis in mice by suppressing T-lymphocyte interferon-gamma production. Gastroenterology. 2011;140(1):265–74. Epub 2010/09/30. 10.1053/j.gastro.2010.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klutho PJ, Costanzo-Garvey DL, Lewis RE. Regulation of glucose homeostasis by KSR1 and MARK2. PLoS One. 2011;6(12):e29304 Epub 2011/12/30. 10.1371/journal.pone.0029304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burack WR, Shaw AS. Signal transduction: hanging on a scaffold. Curr Opin Cell Biol. 2000;12(2):211–6. Epub 2000/03/14. . [DOI] [PubMed] [Google Scholar]
- 62.Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332(6030):680–6. Epub 2011/05/10. 10.1126/science.1198701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shaw AS, Filbert EL. Scaffold proteins and immune-cell signalling. Nat Rev Immunol. 2009;9(1):47–56. Epub 2008/12/24. 10.1038/nri2473 . [DOI] [PubMed] [Google Scholar]
- 64.Joneson T, Fulton JA, Volle DJ, Chaika OV, Bar-Sagi D, Lewis RE. Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J Biol Chem. 1998;273(13):7743–8. Epub 1998/04/29. . [DOI] [PubMed] [Google Scholar]
- 65.Zhao D, Keates AC, Kuhnt-Moore S, Moyer MP, Kelly CP, Pothoulakis C. Signal transduction pathways mediating neurotensin-stimulated interleukin-8 expression in human colonocytes. J Biol Chem. 2001;276(48):44464–71. Epub 2001/09/28. 10.1074/jbc.M104942200 . [DOI] [PubMed] [Google Scholar]
- 66.Lopez JP, Turner JR, Philipson LH. Glucose-induced ERM protein activation and translocation regulates insulin secretion. Am J Physiol Endocrinol Metab. 2010;299(5):E772–85. Epub 2010/08/27. 10.1152/ajpendo.00199.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422(6932):629–33. Epub 2003/04/11. 10.1038/nature01533 . [DOI] [PubMed] [Google Scholar]
- 68.Xiao L, Zheng K, Lv X, Hou J, Xu L, Zhao Y, et al. Exo70 is an independent prognostic factor in colon cancer. Sci Rep. 2017;7(1):5039 Epub 2017/07/13. 10.1038/s41598-017-05308-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu H, Liu J, Liu S, Zeng J, Ding D, Carstens RP, et al. Exo70 isoform switching upon epithelial-mesenchymal transition mediates cancer cell invasion. Dev Cell. 2013;27(5):560–73. Epub 2013/12/18. 10.1016/j.devcel.2013.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.