

Abstract
Specific host pathways that may be targeted therapeutically to inhibit the replication of Ebola virus (EBOV) and other emerging viruses remain incompletely defined. A screen of 200,000 compounds for inhibition of an EBOV minigenome (MG) assay that measures the function of the viral polymerase complex identified as hits several compounds with an amino-tetrahydrocarbazole scaffold. This scaffold was structurally similar to GSK983, a compound previously described as having broad-spectrum antiviral activity due to its impairing de novo pyrimidine biosynthesis through inhibition of dihydroorotate dehydrogenase (DHODH). We generated compound SW835, the racemic version of GSK983 and demonstrated that SW835 and brequinar, another DHODH inhibitor, potently inhibit the MG assay and the replication of EBOV, vesicular stomatitis virus (VSV) and Zika (ZIKV) in vitro. Nucleoside and deoxynucleoside supplementation studies demonstrated that depletion of pyrimidine pools contributes to antiviral activity of these compounds. As reported for other DHODH inhibitors, SW835 and brequinar also induced expression of interferon stimulated genes (ISGs). ISG induction was demonstrated to occur without production of IFNα/β and independently of the IFNα receptor and was not blocked by EBOV-encoded suppressors of IFN signaling pathways. Furthermore, we demonstrated that transcription factor IRF1 is required for this ISG induction, and that IRF1 induction requires the DNA damage response kinase ATM. Therefore, de novo pyrimidine biosynthesis is critical for the replication of EBOV and other RNA viruses and inhibition of this pathway activates an ATM and IRF1-dependent innate immune response that subverts EBOV immune evasion functions.
Introduction
Filoviruses are filamentous, enveloped viruses with non-segmented, negative-sense RNA genomes (Messaoudi et al., 2015). The filovirus family consists of the genus Ebolavirus, which is comprised of five different species including Zaire ebolavirus (Ebola virus, EBOV), the genus Marburgvirus, which includes Marburg virus (MARV), and the genus Cuevavirus (Afonso et al., 2016). Members of the Ebolavirus and Marburgvirus genera are zoonotic pathogens that have caused repeated outbreaks with substantial lethality in humans (Rougeron et al., 2015). The largest such outbreak on record was caused by EBOV and occurred in West Africa between 2013-2016, resulting in more than 28,000 infections, more than 11,000 deaths and the export of infected cases to the United States and Europe (Spengler et al., 2016). In pregnant women, the fatality rate during the West Africa epidemic was estimated to be 70% (Hayden et al., 2017). The only treatments available for infected individuals were supportive care and experimental therapies, hampering patient treatment and leaving healthcare workers at severe risk. Survivors are known to exhibit persistent infections with virus residing in immune privileged sites, including the eye and testes (Jacobs et al., 2016; Uyeki et al., 2016; Yeh et al., 2015; Zeng et al., 2017). These facts highlight the need for effective anti-filovirus therapies.
The RNA synthesis reactions that replicate the viral genomic RNA and transcribe the viral genes into mRNAs are essential for replication (Muhlberger, 2007). These are therefore potential antiviral targets. These viral RNA synthesis reactions are carried out by a complex of four viral proteins, nucleoprotein (NP), viral protein of 35 kilodaltons (VP35), VP30 and the large (L) protein (Muhlberger et al., 1999). Replication of the viral genomic RNA requires NP, which associates with the viral genomic and antigenomic RNAs throughout the course of infection; VP35, a non-enzymatic cofactor and L. L possesses all the enzymatic activities required for viral transcription and genome replication, including RNA-dependent RNA polymerase activity, guanyltransferase and methyltransferase activities (Muhlberger, 2007). Viral transcription (mRNA synthesis) involves the synthesis of distinct 5’-capped, 3’polyadenylated mRNAs from each of the viral genes and requires, in addition to NP, VP35 and L, the VP30 protein (Muhlberger, 2007). In addition to the required viral proteins, host factors also modulate viral RNA synthesis through interaction with viral factors (Luthra et al., 2015; Luthra et al., 2013; Smith et al., 2010). However, a complete understanding as to how host factors contribute to viral RNA synthesis remains elusive.
Another feature of filovirus replication that is a potential target for therapeutic intervention is viral suppression of innate antiviral defenses. EBOV and MARV have been demonstrated to inhibit interferon-α/β (IFN) responses by several mechanisms (Basler et al., 2003; Basler et al., 2000; Kaletsky et al., 2009; Leung et al., 2010; Mateo et al., 2010; Prins et al., 2010; Reid et al., 2006; Reid et al., 2007; Valmas and Basler, 2011; Xu et al., 2014). These include inhibition of the RIG-I-like receptor (RLR) signaling pathways by VP35 proteins which results in inhibition of IFN production, a block to induction of interferon stimulated gene (ISG) expression and impaired maturation of dendritic cells (Cardenas et al., 2006; Lubaki et al., 2016; Yen et al., 2014; Yen and Basler, 2016). Further, EBOV VP24 and MARV VP40 inhibit IFN-triggered signaling such that IFN-induced ISG expression is blocked (Reid et al., 2006; Reid et al., 2007; Valmas and Basler, 2011; Xu et al., 2014). The importance of these functions for filovirus disease is demonstrated by the severe attenuation of recombinant EBOVs engineered to lack VP35 IFN-antagonist activity (Hartman et al., 2008; Prins et al., 2010).
Dihydroorotate dehydrogenase (DHODH) is a key enzyme in de novo pyrimidine biosynthesis (Reis et al., 2017). DHODH inhibitors exhibit antiviral activity against a range of different viruses with an important component of their antiviral effect attributable to the depletion of the nucleosides necessary for replication of the viral genome (Hoffmann et al., 2011; Ortiz-Riano et al., 2014; Wang et al., 2011; Wang et al., 2016). Such compounds show potent antiviral activities against viruses in cell culture but also have cytostatic effects on rapidly dividing cells. For instance, the DHODH inhibitor brequinar inhibits dengue virus (DENV) replication through depletion of the intracellular pyrimidine levels but was originally developed as a potential anti-cancer agent and was subsequently demonstrated to exhibit immunosuppressive activity (Chen et al., 1992; Cramer et al., 1992; Wang et al., 2011). However, the potent antiviral activity of another DHODH inhibitor, GSK983, against DENV and Venezuelan equine encephalitis virus (VEEV) can be separated from its cytostatic effects if exogenous deoxycytidine is provided, as this facilitates cellular DNA synthesis without restoration of viral RNA synthesis (Deans et al., 2016). Beyond suppression of viral RNA synthesis, DHODH inhibitors have been demonstrated to trigger expression of ISGs, an activity that may also contribute to the antiviral effects of these compounds (Chung et al., 2016; Lucas-Hourani et al., 2013; Wang et al., 2016). Mechanistically, these compounds may not require IFN production for ISG induction but may induce ISGs through activation of the transcription factor Interferon Regulatory Factor 1 (IRF1) (Chung et al., 2016; Lucas-Hourani et al., 2017).
A previous report described small molecule inhibitors of EBOV RNA synthesis identified by high throughput screening (HTS) of an EBOV minigenome assay (Luthra et al., 2017b). This assay measures the function of an EBOV RNA synthesis complex that is reconstituted by transfection into mammalian cells. Among the hits were compounds with an amino-tetrahydrocarbazole scaffold. This scaffold resembled GSK983 which had been previously described as having broad-spectrum antiviral activity (Gudmundsson et al., 2009; Harvey et al., 2009). More recently, GSK983 was demonstrated to cause cell cycle arrest and to suppress virus replication by inhibition of DHODH (Deans et al., 2016). Based on these similarities, we synthesized SW835, the racemate of GSK983. SW835 and another DHODH inhibitor, brequinar, were then tested in the EBOV minigenome assay, and for activity towards infectious EBOV, vesicular stomatitis virus (VSV) and Zika virus (ZIKV). We demonstrate antiviral activities for SW835 and brequinar and find that these activities depend on pyrimidine depletion. Furthermore, these compounds stimulate ISG expression which contributes to antiviral activity. ISG expression is demonstrated to be induced by a mechanism that is not blocked by filovirus innate immune antagonist proteins and is dependent on expression of IRF1 and the cellular kinase ATM. Together, these data demonstrate the critical role of pyrimidine biosynthesis for EBOV replication and clarify the mechanisms by which inhibition of pyrimidine biosynthesis induces innate antiviral responses.
Results
Identification of amino-tetrahydrocarbazole compounds that inhibit EBOV RNA synthesis.
A screen of 200,000 small molecules identified several compounds sharing an amino-tetrahydrocarbazole scaffold as inhibitors of EBOV RNA synthesis machinery as measured by the EBOV minigenome (MG) assay (Luthra et al., 2017b). In this assay, the plasmids encoding for EBOV polymerase complex proteins, L, NP, VP35 and VP30, are transfected along with a MG plasmid that consists of a Renilla reporter flanked by viral cis-acting regulatory sequences that drive the transcription and translation of the reporter gene. Criteria for hits was inhibition of greater than 70% with less than 20% cell toxicity and a Z-score of greater than 3. The screen identified 320 hits which were confirmed following cherry picking and retesting. Among these were six related compounds with an amino-tetrahydrocarbazole scaffold, of which SW407 was one representative. The structures, percent inhibition and toxicity values of the six amino-tetrahydrocarbazole compounds are included in Table 1. After confirming the activity of SW407 in the MG assay (Figure 1A), we recognized structural similarity to GSK983, a broad-spectrum antiviral agent whose mechanism of action was unknown at the time of our screen. Based on this similarity, we synthesized SW835, the racemic version of GSK983, SW032 (the R-enantiomer) and SW308 (the S-enantiomer) and tested these in the EBOV MG assay to generate a sixteen-point dose response curve alongside corresponding cell viability measurements. SW835 exhibited potent activity with a 50% inhibitory concentration (IC50) and 50% cytotoxic concentration (CC50) similar to the activities of GSK983 and the R-enantiomer SW032 (Figure 1B-D). The S-enantiomer, SW308, was less active in the MG assay consistent with prior studies where this compound was less active than GSK983 against human papilloma virus (HPV) (Gudmundsson et al., 2009) (Figure 1E).
Table1:
Activities and cytotoxicity of amino-tetrahydrocarbazole compounds identified in the original minigenome assay screen when tested at 5 μM.
| Compound name | Structure | % MG inhibition | % Toxicity |
|---|---|---|---|
| SW407 | 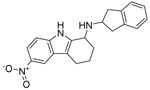 |
93 | 1.4 |
| SW118 |  |
95 | 12 |
| SW743 | 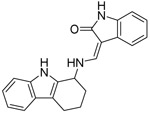 |
81 | 17 |
| SW012 | 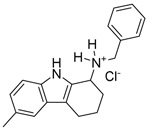 |
78 | 12 |
| SW208 | 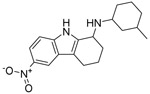 |
78 | 3 |
| SW672 |  |
72 | 14 |
Figure 1. GSK983 and SW835 are potent inhibitors of Ebola minigenome activity.

The Ebola MG assay and cell toxicity in the presence of varying concentrations of (A) SW407, (B) SW835, (C) GSK983, (D) SW032 and (E) SW308. F. The effect of SW835 on the counter screen assay for which the readout is firefly and Renilla luciferase expression from a T7 promoter and an RNA polymerase II promoter, respectively. G. Ebola MG assay activity and cell toxicity of brequinar.
The MG assay relies on T7 RNA polymerase for expression of the minigenome RNA and on RNA polymerase II for expression of the NP, VP35, VP30 and L proteins (Edwards et al., 2015; Jasenosky et al., 2010; Luthra et al., 2017b; Muhlberger et al., 1999). Therefore, we tested for potential inhibition of T7 RNA polymerase or RNA polymerase II by SW835. Cells were transfected with plasmids encoding firefly luciferase under control of a T7 promoter, a T7 RNA polymerase expression plasmid and a plasmid that encodes Renilla luciferase under the control of an RNA polymerase II (polII) promoter. SW835 did not affect the expression of either firefly or Renilla luciferase in this assay (Figure 1F), suggesting that inhibition of minigenome activity is not due to non-specific effects on T7 or RNA polymerase II function or on other requirements for gene expression.
During the course of our study, GSK983 was reported to be an inhibitor of DHODH (Deans et al., 2016). The depletion of pyrimidine pools due to DHODH inhibition was implicated as being responsible for GSK983 antiviral activity against RNA and DNA viruses and for its inhibition of cell growth. Given this, we also examined the effect of a known DHODH inhibitor, brequinar, in the EBOV MG assay (Chen et al., 1992). As with GSK983, brequinar inhibited the EBOV MG assay with an IC50=0.1 μM (Figure 1G).
Next, we assessed in A549 cells the anti-EBOV effects of GSK983; our in-house synthesized compounds SW835, SW032, SW308; and brequinar on replication of a recombinant EBOV that expresses GFP (EBOV-GFP). Following drug pretreatment, cells were infected at a multiplicity of infection (MOI) of 2 in the presence of DMSO or different concentrations of the compounds, ranging from 30 nM to μM, and viral titers in the culture supernatants were assessed two days post-infection. GSK983, SW835 and SW032 reduced viral titers in cell culture supernatants by >200-fold with no effect on cell viability (Figure 2A and B). Interestingly, there was little if any dose-response to these compounds across the range of concentrations tested. The less active enantiomer SW308 that had poor activity in MG assay, was also unable to inhibit EBOV virus replication efficiently (Figure 2A and B). Measured in a separate experiment, brequinar also potently inhibited virus growth (Figure 2C and D). The compounds were further assessed in HeLa cells using an assay that measures GFP expression from EBOV-GFP. GSK983 and SW032 were once again highly active, even at the lowest concentration tested (Figure 2E). However, SW835 reached 50 percent inhibition at ~0.02 μM, and brequinar was determined to have an IC50 value of 0.1 μM. Once again, SW308 was much less active with an IC50 of 1.7 μM (Figure 2E). None of these compounds showed any toxicity against the cells as measured by nuclei staining of the cells (Figure 2F). These data suggest that ongoing DHODH activity is critical target for EBOV replication.
Figure 2. GSK983 and SW835 are potent inhibitors of EBOV replication.

A. Anti-EBOV effects of GSK983 and its enantiomers and SW835. A549 cells were pretreated with compound at the indicated concentrations, infected with EBOV-GFP at an MOI of 2 and two days post-infection culture supernatants were collected, and virus titers were determined by plaque assay. The graphs represent the mean and standard deviation of triplicates. B. Viability of A549 cells treated with indicated compounds 72h post treatment. C. Antiviral activity of brequinar against EBOV-GFP in A549 cells assessed as in panel A. D. Cell viability of brequinar in A549 cells assessed as in panel B. E. The anti-EBOV activity of the compounds in HeLa cells as determined by using EBOV-GFP virus. Percent infectivity was determined by quantification of fluorescence intensities by measuring GFP. The DMSO treated sample to 100%. F. Cell viability was determined in the presence of the indicated compounds in HeLa cells at 24h post-treatment. The cells were fixed, and nuclei were stained with Hoechst stain and were quantified using Cell Profiler software. DMSO treated cells were set at 100%.
As DHODH inhibitors are reported to have broad spectrum antiviral activity, we further assessed the antiviral activity of these compounds for two other RNA viruses, a recombinant vesicular stomatitis virus that expresses GFP (VSV-GFP) and Zika virus (ZIKV). Like EBOV, VSV is a non-segmented negative-sense RNA virus; however, it is from the rhabdovirus family. Conversely, ZIKV is a positive-sense RNA virus from the flavivirus family. As SW835 and GSK983 showed similar activities in the MG and EBOV assays, we focused on SW835 for follow-up experiments. For VSV-GFP, Vero cells were pretreated with compound and infected at an MOI of 0.002 and then fresh compounds were added. The expression of GFP was used as a measure of virus replication. SW835 inhibited VSV-GFP with an IC50 of 0.4 μM (Figure 3A). For ZIKV, immunostaining of viral antigen was used as a measure of virus infectivity in Vero cells infected at an MOI of 1 with compound present both pre- and post-infection. This yielded an IC50 of 0.04 μM with no significant effect on cell viability at 48h post infection (Figure 3B). Brequinar also displayed activity against these viruses with an IC50 of 0.5 μM and 0.3 μM for VSV-GFP and ZIKV, respectively (Figure 3C and D). These data confirmed the broad-spectrum activity of these compounds against RNA viruses.
Figure 3. SW835 and brequinar are potent inhibitors of VSV and ZIKV replication.

A. The antiviral activity of SW835 was measured in Vero cells against VSV-GFP. The cells were pretreated with 3-fold serial dilutions starting at 50 μM for 1h, and then infected with VSV-GFP at an MOI of 0.002. Infectivity was determined by quantifying GFP expression in 96 well plates at 21h post-infection. The y-axis represents percent mean fluorescence activity as determined by normalizing to the mock-treated, VSV-GFP infected samples. B. Antiviral activity of SW385 against ZIKV. Vero cells were treated with compound at four-fold serial dilutions with concentration starting from 1 μM. The cells were infected with ZIKV at MOI=1. ZIKV protein production was quantified using antibody staining (pan-flavivirus antibody, 4G2) via a colorimetric assay at 48h post-infection by measuring absorbance at 650nm. The DMSO treated infected sample is set at 100% activity. C. Antiviral activity of brequinar against VSV-GFP virus. D. Antiviral activity of brequinar against ZIKV. Data represent the mean and standard deviation of triplicates for A-D.
SW835 inhibition of EBOV RNA synthesis involves depletion of pyrimidines.
DHODH is required for de novo pyrimidine biosynthesis and its inhibition causes pyrimidine depletion, arresting growth of rapidly dividing cells (Hoffmann et al., 2011; Liu et al., 2000). Previously, the antiviral activity of GSK983 towards the positive-sense RNA viruses DENV and VEEV was shown to be separable from its anti-proliferative activity on rapidly dividing cells by supplementation with pyrimidine salvage metabolites (Deans et al., 2016). Supplementation with exogenous pyrimidine ribonucleosides, either uridine or cytidine, reversed the inhibition of RNA virus replication, while deoxycytidine supplementation decreased cytotoxicity and restored DNA replication but not RNA virus replication. This is due to the inability of 2’-deoxy analogs to be metabolized for ribonucleotide synthesis. We therefore examined the effects of exogenous nucleosides in the EBOV MG assay in presence of SW835 at 24h post compound addition, and in parallel, on cell viability in HEK293T cells, which was assayed at 72h post compound addition to reveal anti-proliferative effects. Addition of either uridine or deoxycytidine reversed the anti-proliferative effects of SW835. However, only uridine reversed the inhibitory activity of SW835 at 1 μM in the MG assay (Figure 4A and B). When tested against infectious EBOV or VSV in A549 cells, the uridine supplementation again reversed the antiviral activity of SW835 but exogenous deoxycytidine did not (Figure 4C and D). In addition, brequinar also showed potent antiviral activity against EBOV and VSV, and these effects were reversed by supplementation of uridine, but not with deoxycytidine (Figure 4E and F). A cell viability assay was performed in parallel, confirming both uridine and deoxycytidine reduced SW835 and brequinar cytotoxicity and rescued A549 cell growth (Figure 4G). Collectively, these results indicate that SW835 activity against EBOV and VSV involves its ability to inhibit DHODH-dependent pyrimidine synthesis.
Figure 4. Deoxycytidine reverses growth inhibition of SW835 without affecting its antiviral activity.

A and B. HEK293T cells were transfected in bulk in a T75 flask with the MG complex plasmids. The next day, cells (1*105) were plated in 96 well plates and treated with DMSO or 1 μM SW835 in the presence of the indicated concentrations of (A) uridine and (B) deoxcytidine. MG activity was measured at 24h post drug treatment. In parallel, cell viability was determined. C. Effect of SW835 (3 μM) on replication of EBOV-GFP (MOI 2) in A549 cells in the presence of 1 mM uridine or 1 mM deoxycytidine at 48 post infection. D. Effect of SW835 on VSV-GFP replication with pyrimidine supplementation. The A549 cells were pretreated with SW835 (10 μM) for 8h with or without exogenously added nucleosides. The cells were infected with VSV-GFP at an MOI of 0.1 and further incubated with drugs for another 16h. The cell supernatants were collected, and viral titers were determined by plaque assay. E. Effect of brequinar (3 μM) on the replication of EBOV-GFP. G. Effect of brequinar (10 μM) on the replication of VSV-GFP. F. Effect of uridine and deoxycytidine on SW835 or brequinar-induced growth inhibition in A549 cells. Cell viability was determined following 72h treatment with no exogenous pyrimidines (control), 1 mM uridine or 1mM deoxycytidine at the indicated concentrations of SW835. Error bars represent the standard deviation for triplicates in A-F.
SW835 activates innate immune genes independent of IFN production and virus infection.
GSK983 and other inhibitors of pyrimidine biosynthesis induce expression of ISGs, the expression of which can be induced by the antiviral cytokines known as IFNs (Harvey et al., 2009; Lucas-Hourani et al., 2013; Lucas-Hourani et al., 2017). Given that ISGs mediate the antiviral effects of IFNs, it was of interest to determine how SW835 and brequinar induce ISG expression and to what extent this contributes to the antiviral effects of these compounds. We first employed firefly reporter gene assays, utilizing HEK293T cell lines stably transfected with firefly luciferase reporter genes regulated either by the IFN-β promoter (IFN-β-FF) or by a promoter consisting of multiple interferon stimulated response elements (ISREs) (ISRE-FF). These cells were treated with SW835 or brequinar at different concentrations, as indicated. Twenty hours post-treatment luciferase activities were determined (Figure 5A and B). Infection with Sendai virus (Cantell strain), an inducer of IFN-β expression, served as a positive control for both cell lines. The cells were also treated with recombinant universal Type I IFN-α (uIFN) as a control for ISRE activation. Neither compound activated the IFN-β promoter (Figure 5A) but both activated the ISRE promoter (Figure 5B), suggesting that ISG induction is independent of IFN production. We also examined expression of ISGs in A549 cells by reverse transcription-quantitative PCR (qRT-PCR) after addition of uIFN, SW835 or brequinar. While all three ISGs examined, ISG54, ISG56, and MX1, responded to uIFN treatment, ISG54 and ISG56 responded to SW835 and brequinar whereas MX1 did not (Figure 5C). This suggests that DHODH inhibitors induce a select set of ISGs and that this occurs by an IFN-independent mechanism.
Figure 5. SW835 induced ISG expression depends on pyrimidine depletion.

Stable HEK293T-IFNβ-FF (A) or ISRE-FF (B) cell lines were plated in 384 well plates. In each case, the cells were allowed to rest for 2h and then treated with DMSO or the indicated amounts of SW835 or brequinar. SeV infection and uIFN (100 units/ml) served as controls. Error bars represent the standard deviation for four replicates. The p-value was determined by one-way ANOVA with Tukey’s test for multiple comparison, *** p< 0.0001. C. The ISG54, ISG56 and MX1 mRNA expression in A549 cells 24h after DMSO, uIFN (100 Units) or SW835 (10μM) addition. D. Stable HEK293T ISRE-FF cells were plated into 96 well plates, treated with compound (10 μM) in the presence or absence of the indicated pyrimidines at 1mM. Luciferase activity was measured at 24h post treatment. E. The endogenous expression of mRNAs for (E) ISG54 and (F) ISG56 in A549 cells after 24h treatment with the DMSO, SW835 or brequinar at 3 μM in the presence or absence of uridine or deoxycytidine at 24h post treatment. Error bars represent the standard deviation for triplicates in A and B-F.
We further evaluated the ability of uridine or deoxycytidine to modulate the response of ISRE-FF reporter cells treated with 10 μM SW835 and brequinar. Recombinant universal IFN (uIFN) was used as positive control. Supplementation with uridine abolished the ISRE activation by SW835 and brequinar but deoxycytidine did not (Figure 5D). In addition to the reporter gene assay, we evaluated the effects of supplementation on SW835 or brequinar-induced endogenous ISG gene expression in A549 cells. Consistent with the reporter gene assay, ISG54 and ISG56 expression was largely reversed by uridine supplementation but not by deoxycytidine supplementation (Figure 5E and F). This suggested that regulation of cellular nucleotide pools is linked to ISG induction.
We further assessed the effect of these compounds on growth of EBOV (MOI=2), VSV (MOI=0.1) and ZIKV (MOI=1) and ISG induction with or without nucleoside supplementation. In these experiments, A549 cells were pre-treated with drugs, 1h for EBOV infection and 8h for VSV and ZIKV infections to achieve optimal antiviral effects. RNA was isolated at 24 or 48h post infection and the expression of viral mRNAs and ISG54 mRNA were evaluated by qRT-PCR (Figure 6A and B). The compounds inhibited EBOV NP mRNA accumulation, which was reversed by uridine but not by deoxycytidine (Figure 6A). Interestingly, uridine supplementation also reversed ISG54 mRNA induction, however in the presence of deoxycytidine ISG induction was maintained, suggesting that ISG induction is associated with pyrimidine nucleotide depletion (Figure 6B). Similar results were obtained with VSV (Figure 6C and D) and ZIKV (Figure 6E and F). Because uridine suppresses both the ISG expression and the antiviral effects of SW835 and brequinar, these data are consistent with a model where ISG expression may augment the antiviral effects of these compounds.
Figure 6. SW835 ISG induction contributes to antiviral activity.

A. A549 were treated with compounds in presence or absence of nucleosides and infected with EBOV-GFP as described in Figure 4C. EBOV NP (eNP) (A) and ISG54 (B) mRNA levels were measured by quantitative real-time RT-PCR (qRT-PCR) 48h post infection. Alternatively, cells were treated with compound and infected with VSV-GFP as described in Figure 4D. The VSV-N (C) and ISG54 (D) mRNA levels were determined at 16h post-infection. Finally, A549 cells were infected with ZIKV (MOI 1) with similar treatment of compounds and pyrimidines as described in Figure 4D. ZIKV RNA or ISG54 mRNA levels were measured at 40h post-infection. Primers specific to the 5’UTR of the viral genome was used to assess viral RNA levels (E) and an ISG54 specific primer was used to assess ISG induction (F). Error bars represent the standard deviation for triplicates in A-F.
SW835 induced ISG activation bypasses the interferon antagonism by filoviral proteins.
Filoviruses disable the innate immune responses by interfering with IFN signaling at different steps (Messaoudi et al., 2015). The filovirus VP35 proteins inhibit RIG-I-like receptor signaling to block IFN production, indirectly inhibiting induction of IFN-induced ISGs. EBOV VP24 and MARV VP40 block signaling downstream of IFN receptors to prevent IFN-induced ISG expression. To determine whether ISG induction by SW835 and brequinar can bypass the filoviral blocks to ISG expression, the ISRE luciferase reporter cell line was transfected with expression plasmids for EBOV VP35, EBOV VP24 or MARV VP40. The next day, cells were treated with SW835 or brequinar, with DMSO and uIFN serving as controls. None of the filoviral proteins suppressed ISRE activity induced by these compounds despite inhibition of IFN-induced ISG expression by EBOV VP24 and MARV VP40 (Figure 7A). These data reinforce the view that ISG induction by the compounds is not through the standard IFN-activated Jak-STAT signaling pathway and demonstrate that DHODH inhibitors can bypass filovirus blocks to ISG expression.
Figure 7. SW835 induces ISGs independently of virus infection or IFN production.

A. ISRE activation by SW835 is not suppressed by filovirus interferon antagonists. The stable HEK293T-ISRE-FF cells were transfected with plasmids expressing HA-tagged EBOV VP35 (eVP35), EBOV VP24 (eVP24) or MARV VP40 (mVP40) (50 or 500ng of expression plasmid was transfected, as indicated). The next day cells were treated with DMSO, SW835, brequinar or uIFN. 20h post-treatment luciferase activity was determined. The ‘ns’ indicate the values were not significantly different from vector transfected conditions. The p-value was determined by one-way ANOVA with Tukey’s test for multiple comparison, * p< 0.02. Western blotting was performed to detect the expression of eVP35, eVP24 and mVP40 (at 50 and 500ng) using anti-HA antibody. B. The MEF-wt or IFNAR knockout cells were transfected with ISRE and Renilla reporter plasmids. The next day cells were treated with DMSO, SW835, brequinar or uIFN. 20h later, the luciferase activity was determined. Error bars represent the standard deviation for triplicates in A-B. P-value was determined by one-way ANOVA with Tukey’s test for multiple comparison. ** p< 0.001 and “ns” is not significant.
To further address whether canonical IFN signaling is required for the ISG response, we assessed ISG activation by SW835 in wild type or interferon alpha receptor (IFNAR) knockout mouse embryonic fibroblasts (MEFs) using an ISRE reporter plasmid. SW835 induced activation of the ISRE promoter was not affected in IFNAR knockout cells (Figure 7B), further supporting the conclusion that induction of ISGs is independent of IFN production. Interestingly, ISRE-reporter induction by brequinar in either cell type was not statistically significant compared to DMSO-treated cells, consistent with previous reports that brequinar is less efficient in inducing ISGs in mouse cells as compared to human cells (Chung, 2015).
IRF1 plays a critical role in the antiviral activity of SW835.
As our data suggested that ISG induction by these compounds is independent of interferon production, we further investigated what is driving ISG expression. Some members of the interferon regulatory transcription factor (IRF) family have been shown to activate ISGs in the absence of IFN by binding ISRE promoter sequences. In particular, IRF1 is known to regulate expression of many ISGs and to contribute to induction of an antiviral state (Schoggins and Rice, 2011; Schoggins et al., 2011). Interestingly, over-expression of IRF1 has also been shown to inhibit EBOV replication (Rhein et al., 2015) and many other negative and positive strand RNA viruses (Schoggins and Rice, 2011; Schoggins et al., 2011). Furthermore, IRF1 has also been implicated in promoting the antiviral activity of pyrimidine synthesis inhibitor, brequinar (Chung, 2015; Lucas-Hourani et al., 2013). Therefore, we investigated the role of IRF1 in SW835-induced ISG expression and antiviral activity with VSV-GFP. Knocking down IRF1 using a small interfering RNA (siRNA) abolished the activation of ISRE by SW835 in HEK293T, MEFs-wt, IFNAR knockout MEF and STAT2 knockout MEF cells (Figure 8A and B), suggesting IRF1 expression is critical for ISG induction and that its effect is independent of IFNAR and STAT2. To evaluate whether this effect on ISG induction correlates with the antiviral activity of SW835, we performed VSV-GFP infection studies in A549 cells. Knocking-down IRF1 led to decreased antiviral effects of SW835 (Figure 8C) and this correlated with a decrease in ISG54 mRNA levels (Figure 8D), indicating that innate immune responses can contribute to the antiviral effects of SW835. Cumulatively, these data demonstrate a critical role for IRF1 in the ISG response and antiviral state induced by SW835.
Figure 8. IRF1 contributes to the antiviral activity of SW835.

A. Stable HEK293T-ISRE-FF cells were transfected with an siRNA targeting IRF1 or a scrambled siRNA. 48h later the cells were treated with SW835 or brequinar (10 μM). Luciferase activity was determined at 20h post treatment. Western blot shows the knockdown of IRF1 expression using IRF1 specific antibody. B. The MEF wt, IFNAR knockout or STAT2 knockout cells were transfected with siRNA targeting IRF1 or scrambled siRNA. 48h post transfection cells were additionally transfected with ISRE-firefly and Renilla reporter plasmids. The next day, cells were treated with compounds at 10 μM. 20h post-treatment luciferase activity was measured. The IRF1 knockdown significantly decreased ISRE luciferase activity upon compound treatment. The p-value was determined by one-way ANOVA with Tukey’s test for multiple comparison; *** p< 0.0001. C. Antiviral activity of SW835 against VSV-GFP infected at MOI of 0.01 in IRF1 knockdown A549 cells. The virus titers were determined by plaque assay. IRF1 expression was detected by using anti-IRF1 antibody and anti-GAPDH served as loading control. D. Endogenous expression of ISG54 was determined by qRT-PCR from RNA isolated from cells infected with VSV-GFP described in Panel C.
SW835 upregulates IRF1 in an ATM-dependent manner.
Previously, it was shown that the anti-proliferative effects of DHODH inhibitors such as GSK983 are due to inhibition of cellular DNA synthesis at the S phase of cell cycle (Deans et al., 2016). Such cellular stress responses activate the DNA damage checkpoint kinase, Ataxia Telangiectasia Mutated (ATM), which reduces DNA synthesis, in response to DNA damage (Willis and Rhind, 2009). Interestingly, ATM also plays a role in IFN responses and is involved in regulation of IRF1 in response to DNA damage (Kurz et al., 2004; Pamment et al., 2002; Shiloh, 2006). To determine whether ATM contributes to SW835 or brequinar ISG induction, we performed an ISRE luciferase assay in the absence or presence of ATM kinase inhibitor or DHODH inhibitors (Figure 9A). ISRE activation by both SW835 and brequinar was diminished in the presence of the ATM kinase inhibitor (Figure 9A). To further assess the role of ATM in ISG induction by DHODH inhibitors, knockdown of ATM was performed by using short hairpin RNA constructs (ATM-SH) in the ISRE reporter cells. The knockdown of ATM diminished ISRE activation by SW835 and brequinar, suggesting ATM contributes to ISG expression induced by DHODH inhibitors (Figure 9B). To further correlate ATM with IRF1 regulation, we also examined IRF1 expression by Western blot in 293T cells treated with DHODH inhibitors in the presence or absence of ATM Kinase inhibitor. SW835 and brequinar treatment increased IRF1 expression, which was dampened in the presence of ATM kinase inhibitor (Figure 9C). This suggests that inhibition of pyrimidine biosynthesis induces ISG expression by upregulating IRF1 in an ATM-dependent manner. Together, these data provide evidence that the antiviral activity of SW835 is predominantly governed by DHODH inhibition but also involves activation of ATM and the up-regulation and action of ISGs, including IRF1.
Figure 9. ATM regulates IRF1 levels upon nucleoside depletion by DHODH inhibitors.

A. Stable ISRE-FF cells were treated with ATM Kinase inhibitor (10 μM) 1h prior to addition of DMSO, SW835 (10 μM), brequinar (10 μM) of uIFN. 20h post treatment, luciferase activity was determined. B. Stable ISRE-FF cells were transfected with control or ATM shRNA plasmids and the following day treated with DMSO, SW835 (10 μM), brequinar (10 μM) or universal IFN alpha (uIFN, 100 units/ml). 20h post treatment, luciferase activity was determined. The knockdown of ATM was evaluated by Western blotting using anti-ATM antibody. C. Expression level of IRF1 was determined by Western blotting of HEK293T cells treated with ATM Kinase inhibitor and either SW835, brequinar or uIFN. The ATM Kinase treatment and ATM knockdown significantly decreased ISRE luciferase activity upon compound treatment. The p-value was determined by one-way ANOVA with Tukey’s test for multiple comparison; *** p< 0.0001.
Discussion
In this work, we identified SW835, a racemate of GSK983, as having inhibitory activity towards the EBOV MG assay and demonstrated that these compounds exhibit broad spectrum antiviral activity against EBOV, VSV and ZIKV in cell culture. We further demonstrated that this antiviral activity is related to the capacity of these compounds to inhibit DHODH as supplementation with uridine reverses the antiviral activity. In support of this mechanism of action, we demonstrate that another DHODH inhibitor, brequinar, exhibits similar activities. In addition, based on prior observations that DHODH inhibitors induce ISGs, we demonstrate ISG induction by SW835 and brequinar and link this to depletion of pyrimidine but not deoxypyrimidines. We further link DHODH inhibitor induced ISG expression to activation of signaling by cellular kinase ATM and up-regulation of IRF1. This work establishes a critical role for the pyrimidine biosynthetic pathway for EBOV, VSV and ZIKV growth and clarifies mechanisms by which DHODH inhibitors induce ISGs that may contribute to antiviral activity.
Based on our data from Figure 5 and 6, where uridine supplementation suppressed both ISG expression and antiviral effects of SW835 and brequinar, and data in Figure 8 where knockdown of IRF1 decreases antiviral activity of SW835, we suggest that ISG expression can contribute to the antiviral activity of these compounds. Nonetheless, it is likely that reduction of pyrimidine levels is sufficient to substantially reduce growth of EBOV, VSV or ZIKV. We should also note that data do not exclude a contribution of secondary effects of pyrimidine depletion, other than ISG induction, on replication.
The mechanisms by which DHODH inhibitors stimulate innate immune responses remains incompletely defined but is of interest with regard to their antiviral activity. Our data are consistent with studies indicating that pyrimidine biosynthesis inhibitors do not cause the expression of IFN when applied alone, and virus growth inhibition by these drugs seems to be independent of IFN-α/β synthesis and the canonical Jak-STAT pathway (Chung et al., 2016; Lucas-Hourani et al., 2013; Lucas-Hourani et al., 2017; Wang et al., 2011; Wang et al., 2016). However, some recent reports show a lack of antiviral effect of DHODH inhibitors in Vero cells, which are deleted for the IFN-α/β gene cluster, suggesting a possible role for secreted IFNs in antiviral activity (Cheung et al., 2017). We found that SW835 activates genes involved in the innate immunity, including ISG56 (interferon-induced protein with tetratricopeptide repeats 1; encoded by IFIT1) and ISG54 (interferon-induced protein with tetratricopeptide repeats 2; encoded by IFIT2), independent of type 1 IFNs or exogenous RNA. Our mechanistic studies identified that ISG production did not require the presence of IFNAR or STAT2 suggesting a non-canonical (i.e. IFN-independent) induction of ISGs.
Interestingly, SW835 stimulation of ISGs was regulated by IRF1, consistent with published literature suggesting a role for IRF1 in DHODH induction of such genes (Khiar et al., 2017; Lucas-Hourani et al., 2013). We further investigated how IRF1 is activated in the presence of SW835. We reasoned that because nucleotide deprivation may lead to metabolic stress, the DNA damage repair pathway might be involved. ATM is a cellular kinase involved in DNA damage responses (Kurz et al., 2004; Pamment et al., 2002). Our data demonstrate a role for ATM in the ISG response to either SW835 or brequinar. Furthermore, inactivation of ATM also led to a decrease in IRF1 levels, suggesting that ATM is a critical player regulating IRF1 levels and therefore ISGs upon nucleoside deprivation. It is unclear why ISG induction is reversed by ribonucleoside but not deoxyribonucleoside supplementation, but the observation suggests a role of RNA metabolism in the ISG response. Notably, the mechanism by which DHODH inhibitors induce ISGs subvert filovirus innate immune evasion functions, as ISG induction was not inhibited by filovirus encoded interferon antagonists, including the EBOV VP35 protein, the EBOV VP24 protein or the MARV VP40 protein. Therefore, although ISG responses may not be required for the antiviral activity of these compounds, ISGs could nonetheless contribute to anti-filovirus effects by bypassing viral defenses against host innate immunity.
Figure 10 provides a model consistent with our results, where inhibition of DHODH reduces the de novo production of pyrimidine nucleosides to a level where RNA virus RNA synthesis is impaired. This also inhibits cellular DNA and RNA synthesis. The inhibition of cellular RNA synthesis results in activation of ATM, upregulation of IRF1 expression and induction of ISG expression. While providing deoxycytidine can restore cellular DNA synthesis, this is insufficient to restore RNA virus replication or prevent ISG induction. In contrast, providing uridine relieves inhibition of both virus and the ATM-IRF1-ISG axis. This model highlights the possibility that DHODH inhibitors may have pleiotropic effects on host cell signaling pathways. Better defining these pathways may suggest additional mechanisms that impact antiviral activities.
Figure 10. A proposed model of the antiviral mechanisms of DHODH inhibitors against RNA viruses.

DHODH is a critical enzyme for de novo pyrimidine biosynthesis mediating oxidation of dihydroorotate to orotate. SW835 or other DHODH inhibitors impair the enzymatic activity of DHODH, blocking de novo pyrimidine biosynthesis. This results in the depletion of deoxynucleosides and ribonucleosides, affecting both cell proliferation and RNA virus replication. Depletion of pyrimidine pools also triggers a cellular antiviral response likely through its effect on cellular DNA. This leads to the activation of ATM which induces the expression of IRF1. IRF1 induces ISG expression contributing to inhibition of virus replication.
DHODH inhibitors, including SW835 and brequinar, display potent antiviral activity in vitro but whether DHODH is a viable target in vivo still remains to be fully evaluated. So far attempts to use de novo pyrimidine inhibitors in vivo to control virus infection have met with only partial success (Bonavia et al., 2011; Cheung et al., 2017; Smee et al., 2012; Wang et al., 2011). For example, DHODH inhibitor FA-613 protected 30% of mice from lethal influenza A virus infection (Cheung et al., 2017). A77-1726, the active metabolite of the DHODH inhibitor leflunomide, reduced symptoms in respiratory syncytial virus (RSV)-infected mice, however, DHODH inhibitors administered intranasally to macaques did not suppress RSV infection (Davis et al., 2007; Grandin et al., 2016). Another DHODH inhibitor, NITD-982, active against dengue virus in vitro failed to show efficacy in AG129 mice (Wang et al., 2011). Several factors have been suggested to work against the antiviral activity of DHODH inhibitors in vivo. These include replenishment/maintenance of nucleotide pools in blood plasma which could feed the pyrimidine salvage pathway, thereby compensating for inhibition of the de novo pyrimidine synthesis pathway. In addition, the immunosuppressive effects of DHODH inhibition may also affect the host ability to clear virus infection. Therefore, additional research into in vivo antiviral uses for DHODH inhibitors is warranted. In terms of EBOV, additional studies include optimization of the delivery route, dose and time of delivery of compound in appropriate animal models. In addition, strategies that may improve antiviral efficacy can be explored. Previously, it was shown that deletion of the pyrimidine metabolism enzyme UCK2 (uridine-cytidine kinase), a critical enzyme in the pyrimidine salvage pathway, sensitized cells to GSK983 (Deans et al., 2016). Further investigation is required to evaluate whether UCK2 inhibitors could work synergistically with DHODH inhibitors to suppress RNA virus replication in vivo.
Materials and Methods
Cell culture.
Human embryonic kidney 293T cells (HEK293T), Vero and human A549 cell lines were obtained from ATCC and cultured in Dulbecco's modified Eagle medium (DMEM; Cellgro) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 μg/ml of streptomycin, and 100 units of penicillin (Invitrogen).
We generated stable HEK293T cell lines expressing ISRE and a firefly luciferase reporter gene. To generate the cell lines, the HEK293T were transfected with an ISRE expression plasmid from pGL4.45 (Promega) which consists of five copies of ISRE that drives expression of a firefly reporter gene. The ISGs consist of this regulatory element in their promoter sequences where transcription factors activated by IFN or virus infection can bind. The transient transfections were performed using lipofectamine 2000. Two days post-transfection, culture medium was removed and replaced by fresh media containing hygromycin at 100 μg/ml concentration. Transfected cells were selected under hygromycin. A total of 50 clonal cell populations were screened by treating with IFNβ and screening for luciferase activity. A single ISRE-FF clone was selected for its optimal signal to background ratio when stimulated with recombinant Universal type I interferon-alpha (uIFN) (PBL Assay Science). The stable HEK 293T IFNβ promoter-reporter cell line has been previously described (Luthra et al., 2017b).
Plasmids and Antibodies.
The plasmids for the HTS EBOV minigenome system have been previously described (Edwards et al., 2015). The interferon antagonist plasmids encoding EBOV VP35, EBOV VP24 and MARV VP40 were previously described (Feagins and Basler, 2014, 2015; Luthra et al., 2017b; Xu et al., 2014). The ATM Sh plasmids are previously described (Luthra et al., 2017a). The IRF1 and ATM antibody were purchased from Cell signaling. The GAPDH, β-tubulin, anti-HA and anti-FLAG antibodies were purchased from Sigma- Aldrich.
Chemicals.
Brequinar and GSK983 were purchased from Sigma-Aldrich. SW835, SW032 and SW308 were synthesized in-house. All compounds were >95% pure as assessed by HPLC and hydrogen-1 NMR (1H NMR) analysis. Uridine, cytidine and deoxycytidine were obtained from Sigma-Aldrich. All compounds were diluted to a concentration of 20 mM in DMSO before use. ATM kinase inhibitor, Ku55933, was purchased from Sigma-Aldrich. uIFN and IFN-β were purchased from PBL Assay Science.
Dose response for minigenome and toxicity assays.
The minigenome assay in 384 well format has been previously described (Edwards et al., 2015; Luthra et al., 2017b). Briefly, HEK293T cells (7.5*106 cells) were transfected in T75 flasks using Lipofectamine 2000 (Invitrogen) with plasmids encoding NP (3.7 μg), VP30 (1.5 μg), VP35 (1.9 μg), L (7.5 μg), reporter minigenome plasmid (3 μg) along with T7 polymerase plasmid (3 μg). The following day, cells were trypsinzed and plated in 384 well plates (2*104 cells/ 50 μl/well) using a Combi Reagent dispenser (Thermo Scientific). The cells were allowed to rest for two hours, and compounds were added to reach the indicated final concentrations (50 μM and lower in a 3-fold dilution series) in triplicate. Twenty-four hours post-treatment, Renilla-Glo (Promega) substrate was added, and the luciferase signal was read using an EnVision plate reader (PerkinElmer). The 50% inhibitory concentration (IC50) values were calculated with Prism (GaphPad Software, La Jolla, CA) using a four-parameter, nonlinear regression analysis.
For pyrimidine supplementation experiments, pyrimidine metabolite (uridine, deoxycytidine) stocks were generated at 10 mM in distilled water and were further diluted in DMEM to achieve the desired final concentrations.
To assess the cytotoxicity of the compounds, HEK293T, A549 or Vero cells (1*103 cells/well) were plated in 384 well plates (white opaque Culture Plate, Perkin Elmer). One hour after plating, compounds were added to reach the indicated final concentrations (3 or 4-fold dilution series) in triplicate. Twenty-four hours post-treatment CellTiter-Glo (Promega) was added, and ATP content was determined by reading luminescence using an EnVision plate reader (PerkinElmer). The 50% cytotoxicity concentration (CC50) values were calculated with Prism using a four-parameter, nonlinear regression analysis.
The firefly and Renilla luciferase based counterscreen assay was performed by transfecting T7-driven pTM1 firefly plasmid (3 μg), pCAGGS-T7 (3 μg) and pCAGGS-Renilla (3 μg) in 7.5*106 HEK293T cells in T75 flask using Lipofectamine 2000. The next day cells were seeded into 384 well plates using a multi-well dispenser and compounds were added at the indicated final concentrations. 24h later, the luciferase activity was determined using Dual-Glo reagent (Promega).
Effect of compounds on production of infectious EBOV.
A549 cells (2*104 cells/well) were plated in 24 well plates overnight, and the next day compounds were added at different concentrations starting at 3 μM with 3-fold serial dilutions. One-hour post-treatment, in the biosafety level 4 (BSL-4) conditions at the Galveston National Laboratory, EBOV-GFP was added at an MOI of 2 and drugs were added back to the cell culture medium and left on the cells for the course of the experiment. Forty-eight hours later, the cell monolayers were observed using a fluorescent microscope and supernatants were collected to determine virus titers using plaque assay.
In a separate antiviral assay with EBOV-GFP, the HeLa cells were plated (4*103 cells/well) in 25 μl of medium and grown overnight in 384 well tissue culture plates. The next day cells were treated with compounds (starting at 25 μM with 2-fold serial dilutions) in triplicate to yield a 16-point dose curve. Each well was infected in a BSL-4 laboratory at Texas Biomedical Research Institute with EBOV-GFP to achieve a multiplicity of infection of 0.075-0.15. Cells were incubated with virus for 24h. The cells were then fixed by immersing the plates in formalin overnight at 4°C. Fixed plates were decontaminated and brought to BSL-2. Formalin was decanted from the plates and the plates were washed three times with PBS. The plates with GFP encoding EBOV were stained for cell nuclei using Hoechst at 1:50,000 dilutions. Plates were imaged using Nikon Ti Eclipse automated microscope and nuclei and infected cells were counted using Cell Profiler software.
VSV-GFP assay.
The VSV-GFP virus was kindly provided by Benjamin R. tenOever at the Icahn School of Medicine at Mount Sinai. Vero cells (3*104 cells/well) were plated in 96 well plates overnight, and the next day compounds were added at three-fold serial dilutions starting from 50 μM. One-hour post-treatment, VSV-GFP was added at an MOI of 0.002 and compounds were added back to media. Twenty-one hours post-infection the mean fluorescence intensity (MFI) was measured using an EnVision plate reader. The nucleoside supplementation assay was performed in A549 cells (2*105 cells/well) in 24 well plates. The cells were treated with nucleosides (final concentration of 1 mM) with or without the compounds (SW835 or brequinar used at 10 μM) for 8h followed by VSV-GFP infection at MOI 0.1. After 1h of infection, cells were washed and fresh compounds and nucleosides were replaced. The cell supernatants and TRIZOL samples were collected 16h post infection. The virus titers were determined by plaque assay.
Zika virus assay.
Zika virus (ZIKV) African lineage (MR766) was obtained from ATCC. Vero cells (3*104 cells/well) were pretreated with various doses of compound (four-fold serial dilutions) for one hour. The cells were infected with ZIKV at an MOI of 1 in the presence of compounds. After 1h of infection, cells were washed and media with fresh compounds were added. Forty-eight hours post infection, the cells were fixed with ice cold methanol, washed with assay buffer (PBS with 2% nonfat milk and 0.1% Triton X-100) and incubated with anti-flavivirus group antigen antibody (1:4000 dilution, clone D1-4G2-4-15, Sigma) for two hours at room temperature. The cells were then washed three times with assay buffer and incubated with anti-mouse HRP conjugated secondary antibody (1:4000 dilution, in assay buffer) for one hour at room temperature. The cells were further washed three times with assay buffer and then incubated with TMB substrate (100 μl) (Rockland Immunochemicals, PA) for 30 minutes at room temperature. Absorbance was read at 650nm using EnVision (Perkin Elmer) to determine the virus positive cells.
For nucleoside supplementation, A549 cells (2*105 cells/well), were plated in 24 well plates. The cells were treated as described above for VSV assay and were infected with ZIKV at MOI 1. The TRIZOL samples were collected 40h post infection.
IRF1 siRNA assays.
IRF1 silencing was achieved with a siRNA targeting human IRF1 (Santa Cruz Biotechnology, sc7506) or mouse IRF1 (Santa Cruz Biotechnology, sc35707). A scrambled siRNA was used as a control (Santa Cruz Biotechnology, sc37007). All the siRNA stocks were at 10 μM and were diluted to 1 μM for transfections. The siRNA was transfected in 293T-ISRE-FF or MEFs cells using RNAiMax (Invitrogen). The reaction mix for each well of a 96 well plate is described here. However a master mix was generated for transfecting multiple wells. Briefly, 3.6 μl of siRNA (1 μM) were mixed with 26.4 μl of Opti-MEM (Gibco-Invitrogen). The final siRNA concentration was 30 nM in 120 μΚ The transfection reagent Lipofectamine RNAiMAX (0.2 μl was diluted in 29.8 μl of OPTI-MEM. The two solutions were mixed together and were incubated for 20 minutes at room temperature. To each well, 60 μl of the transfection mix was added followed by cells (6*103 cells/well in 60 μl resuspended in DMEM with 10% FBS without penicillin and streptomycin. The cells were incubated for 48h at 37°C and 5% CO2. For studies in MEFs, cells were transfected with reporter plasmids (ISRE-Firefly luciferase and constitutively expressed Renilla luciferase) with Lipofectamine 2000 (Invitrogen) and then stimulated with compounds (10 μM) or recombinant uIFN at 100 Units/ml. For studies in 293T-ISRE-FF cells, the cells were treated with (10 μM) compound. After 24h of culture in the presence of compounds, firefly luciferase activity was determined using the Neolite reagent for 293T-ISRE-FF cells or Dual-Glo (Promega) for assays in MEFs following manufacturer’s recommendations (Promega).
The siRNA transfection in A549 cells was performed in 24 well plates as described above using 5*104 cells/well in 500 μl. The final siRNA concentration was used at 30 nM. The following day, cells were treated with compounds (10 μM) for 8h followed by infection with VSV at MOI 0.01. The supernatants were collected and TRIZOL was added to cells for RNA isolation at 16h post-infection. The virus titers were determined by plaque assay.
Quantitative RT-PCR.
Cells were plated in 24 well plates (2*105 cells/well). Twenty-four hours later, cells were treated with drugs, drugs with nucleotides, or virus infected. The cells were collected at the indicated time points and RNA was isolated with TRIZOL reagent (Sigma) according to manufacturer’s protocol. The isolated RNA was also subjected to DNase treatment using Ambion DNA-free DNase Treatment and Removal Reagents.
A two-step qRT-PCR (Taqman technology, Applied Biosystems) was performed to measure transcription levels for genes of interest. Expression levels of a housekeeping gene, β-actin, was determined and used as an internal reference control. One microgram of total RNA was subjected to cDNA synthesis using oligo(dT) using the SuperScript III first strand cDNA Synthesis Kit following manufacturer’s recommendations (Life Technologies). Quantitative PCR reactions were performed using SYBR green Fast (Applied Biosystems) on a Biorad Real-Time PCR machine. Results were normalized using expression levels of β-actin. For quantitation of gene expression, real-time PCR with the 2−-ΔΔCT method was used in a total of 20 μl per well with 1:10-fold-diluted cDNA.
Interferon and ISRE luciferase assays.
Stable HEK293T cells with IFN-β promoter-firefly luciferase or ISRE promoter-firefly luciferase (2*104 cells/well in 96 well format) were treated with compounds at 10 μM concentration or Sendai virus infected (100 heamagglutinating units) or treated with uIFN (100 Units/ml). Luciferase activity was determined 20h post treatment using Neolite (Perkin Elmer). For the reporter assays in MEFs, the cells (1.5*105 cells/well in 24 well format) were transfected with an ISRE-firefly luciferase reporter plasmid (pGL4.1, 200 ng/well) and a constitutively expressed Renilla luciferase reporter (pRL-TK, 50ng/well) plasmid with Lipofectamine 2000. The next day, the cells were treated with compounds or uIFN (100 units/ml). 20h post treatment, the luciferase activity was determined using the Dual Luciferase kit (Promega). For the ISRE-luciferase assays performed in the presence of ATM kinase inhibitor (Ku55933), 2*105 cells/well in 96 well format were plated and the next day treated with ATM kinase inhibitor (10 μM) for 1h. The cells were then treated with DMSO, SW835 (10 μM), brequinar (10 μM) or uIFN (100 Units/ml). Firefly luciferase activity was determined 20h post compound addition using Neolite (Perkin Elmer).
For ISRE-luciferase assays performed with knockdown of ATM, cells were transfected with 100ng of control shRNA or ATM-specific shRNA plasmids in 96 well luminometer plates with 1*104 cells/well. The next day, the cells were treated with DMSO, SW835 (10 μM), brequinar (10 μM) or uIFN (100 Units/ml). 24h post compound addition, firefly luciferase activity was determined using Neolite reagent (Perkin Elmer). The knockdown of ATM was determined by Western blotting using anti-ATM antibody (Cell Signaling).
Highlights.
An amino-tetrahydrocarbazole compound SW835 Ebola virus, vesicular stomatitis virus and Zika virus replication.
SW835 is the racemic form of GSK983.
SW835 depletes pyrimidines and induces interferon stimulated genes (ISGs).
ISG induction occurs through activation of kinase ATM and transcription factor IRF-1.
SW835 induction of ISGs subverts Ebola virus innate immune evasion mechanisms.
Acknowledgements.
This work was supported by NIH grant U19AI109664 to CFB, AB and JMR, by NIH grants P01AI120943 (Amarasinghe) to CFB and RAD and R01AI125453 to CFB and RAD, by the Welch foundation (I-1612) to JMR. CFB is an Georgia Research Alliance Eminent Scholar in Microbial Pathogenesis. We thank Michelle Meyer for help with the EBOV assays performed at UTMB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonso CL, Amarasinghe GK, Banyai K, Bao Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand FX, Briese T, Bukreyev A, Calisher CH, Chandran K, Cheng J, Clawson AN, Collins PL, Dietzgen RG, Dolnik O, Domier LL, Durrwald R, Dye JM, Easton AJ, Ebihara H, Farkas SL, Freitas-Astua J, Formenty P, Fouchier RA, Fu Y, Ghedin E, Goodin MM, Hewson R, Horie M, Hyndman TH, Jiang D, Kitajima EW, Kobinger GP, Kondo H, Kurath G, Lamb RA, Lenardon S, Leroy EM, Li CX, Lin XD, Liu L, Longdon B, Marton S, Maisner A, Muhlberger E, Netesov SV, Nowotny N, Patterson JL, Payne SL, Paweska JT, Randall RE, Rima BK, Rota P, Rubbenstroth D, Schwemmle M, Shi M, Smither SJ, Stenglein MD, Stone DM, Takada A, Terregino C, Tesh RB, Tian JH, Tomonaga K, Tordo N, Towner JS, Vasilakis N, Verbeek M, Volchkov VE, Wahl-Jensen V, Walsh JA, Walker PJ, Wang D, Wang LF, Wetzel T, Whitfield AE, Xie JT, Yuen KY, Zhang YZ, Kuhn JH, 2016. Taxonomy of the order Mononegavirales: update 2016. Arch Virol 161, 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A, 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77, 7945–7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler CF, Wang X, Muhlberger E, Volchkov V, Paragas J, Klenk HD, Garcia-Sastre A, Palese P, 2000. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci U S A 97, 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonavia A, Franti M, Pusateri Keaney E, Kuhen K, Seepersaud M, Radetich B, Shao J, Honda A, Dewhurst J, Balabanis K, Monroe J, Wolff K, Osborne C, Lanieri L, Hoffmaster K, Amin J, Markovits J, Broome M, Skuba E, Cornella-Taracido I, Joberty G, Bouwmeester T, Hamann L, Tallarico JA, Tommasi R, Compton T, Bushell SM, 2011. Identification of broad-spectrum antiviral compounds and assessment of the druggability of their target for efficacy against respiratory syncytial virus (RSV). Proc Natl Acad Sci U S A 108, 6739–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas WB, Loo YM, Gale M Jr., Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF, 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80, 5168–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SF, Perrella FW, Behrens DL, Papp LM, 1992. Inhibition of dihydroorotate dehydrogenase activity by brequinar sodium. Cancer Res 52, 3521–3527. [PubMed] [Google Scholar]
- Cheung NN, Lai KK, Dai J, Kok KH, Chen H, Chan KH, Yuen KY, Kao RYT, 2017. Broad-spectrum inhibition of common respiratory RNA viruses by a pyrimidine synthesis inhibitor with involvement of the host antiviral response. J Gen Virol 98, 946–954. [DOI] [PubMed] [Google Scholar]
- Chung D, 2015. The Establishment of an Antiviral State by Pyrimidine Synthesis Inhibitor is Cell Type-Specific. J Antimicrob Agents 1. [PMC free article] [PubMed] [Google Scholar]
- Chung DH, Golden JE, Adcock RS, Schroeder CE, Chu YK, Sotsky JB, Cramer DE, Chilton PM, Song C, Anantpadma M, Davey RA, Prodhan AI, Yin X, Zhang X, 2016. Discovery of a Broad-Spectrum Antiviral Compound That Inhibits Pyrimidine Biosynthesis and Establishes a Type 1 Interferon-Independent Antiviral State. Antimicrob Agents Chemother 60, 4552–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer DV, Chapman FA, Jaffee BD, Jones EA, Knoop M, Hreha-Eiras G, Makowka L, 1992. The effect of a new immunosuppressive drug, brequinar sodium, on heart, liver, and kidney allograft rejection in the rat. Transplantation 53, 303–308. [DOI] [PubMed] [Google Scholar]
- Davis IC, Lazarowski ER, Chen FP, Hickman-Davis JM, Sullender WM, Matalon S, 2007. Post-infection A77-1726 blocks pathophysiologic sequelae of respiratory syncytial virus infection. Am J Respir Cell Mol Biol 37, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans RM, Morgens DW, Okesli A, Pillay S, Horlbeck MA, Kampmann M, Gilbert LA, Li A, Mateo R, Smith M, Glenn JS, Carette JE, Khosla C, Bassik MC, 2016. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol 12, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MR, Pietzsch C, Vausselin T, Shaw ML, Bukreyev A, Basler CF, 2015. High-Throughput Minigenome System for Identifying Small-Molecule Inhibitors of Ebola Virus Replication. ACS Infect Dis 1, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagins AR, Basler CF, 2014. The VP40 protein of Marburg virus exhibits impaired budding and increased sensitivity to human tetherin following mouse adaptation. J Virol 88, 14440–14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagins AR, Basler CF, 2015. Amino Acid Residue at Position 79 of Marburg Virus VP40 Confers Interferon Antagonism in Mouse Cells. J Infect Dis 212 Suppl 2, S219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin C, Hourani ML, Janin YL, Dauzonne D, Munier-Lehmann H, Paturet A, Taborik F, Vabret A, Contamin H, Tangy F, Vidalain PO, 2016. Respiratory syncytial virus infection in macaques is not suppressed by intranasal sprays of pyrimidine biosynthesis inhibitors. Antiviral Res 125, 58–62. [DOI] [PubMed] [Google Scholar]
- Gudmundsson KS, Boggs SD, Sebahar PR, Richardson LD, Spaltenstein A, Golden P, Sethna PB, Brown KW, Moniri K, Harvey R, Romines KR, 2009. Tetrahydrocarbazole amides with potent activity against human papillomaviruses. Bioorg Med Chem Lett 19, 4110–4114. [DOI] [PubMed] [Google Scholar]
- Hartman AL, Bird BH, Towner JS, Antoniadou ZA, Zaki SR, Nichol ST, 2008. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J Virol 82, 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey R, Brown K, Zhang Q, Gartland M, Walton L, Talarico C, Lawrence W, Selleseth D, Coffield N, Leary J, Moniri K, Singer S, Strum J, Gudmundsson K, Biron K, Romines KR, Sethna P, 2009. GSK983: a novel compound with broad-spectrum antiviral activity. Antiviral Res 82, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG, Friede M, Bausch DG, 2017. Experimental Therapies for Ebola Virus Disease: What Have We Learned? J Infect Dis 215, 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML, 2011. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A 108, 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs M, Rodger A, Bell DJ, Bhagani S, Cropley I, Filipe A, Gifford RJ, Hopkins S, Hughes J, Jabeen F, Johannessen I, Karageorgopoulos D, Lackenby A, Lester R, Liu RS, MacConnachie A, Mahungu T, Martin D, Marshall N, Mepham S, Orton R, Palmarini M, Patel M, Perry C, Peters SE, Porter D, Ritchie D, Ritchie ND, Seaton RA, Sreenu VB, Templeton K, Warren S, Wilkie GS, Zambon M, Gopal R, Thomson EC, 2016. Late Ebola virus relapse causing meningoencephalitis: a case report. Lancet 388, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasenosky LD, Neumann G, Kawaoka Y, 2010. Minigenome-based reporter system suitable for high-throughput screening of compounds able to inhibit Ebolavirus replication and/or transcription. Antimicrob Agents Chemother 54, 3007–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P, 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A 106, 2886–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiar S, Lucas-Hourani M, Nisole S, Smith N, Helynck O, Bourgine M, Ruffie C, Herbeuval JP, Munier-Lehmann H, Tangy F, Vidalain PO, 2017. Identification of a small molecule that primes the type I interferon response to cytosolic DNA. Sci Rep 7, 2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz EU, Douglas P, Lees-Miller SP, 2004. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem 279, 53272–53281. [DOI] [PubMed] [Google Scholar]
- Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, Nix JC, Helgeson LA, Otwinowski Z, Honzatko RB, Basler CF, Amarasinghe GK, 2010. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol 17, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J, 2000. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 8, 25–33. [DOI] [PubMed] [Google Scholar]
- Lubaki NM, Younan P, Santos RI, Meyer M, Iampietro M, Koup RA, Bukreyev A, 2016. The Ebola Interferon Inhibiting Domains Attenuate and Dysregulate Cell-Mediated Immune Responses. PLoS Pathog 12, e1006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Hourani M, Dauzonne D, Jorda P, Cousin G, Lupan A, Helynck O, Caignard G, Janvier G, Andre-Leroux G, Khiar S, Escriou N, Despres P, Jacob Y, Munier-Lehmann H, Tangy F, Vidalain PO, 2013. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog 9, e1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Hourani M, Dauzonne D, Munier-Lehmann H, Khiar S, Nisole S, Dairou J, Helynck O, Afonso PV, Tangy F, Vidalain PO, 2017. Original Chemical Series of Pyrimidine Biosynthesis Inhibitors That Boost the Antiviral Interferon Response. Antimicrob Agents Chemother 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Aguirre S, Yen BC, Pietzsch CA, Sanchez-Aparicio MT, Tigabu B, Morlock LK, Garcia-Sastre A, Leung DW, Williams NS, Fernandez-Sesma A, Bukreyev A, Basler CF, 2017a. Topoisomerase II Inhibitors Induce DNA Damage-Dependent Interferon Responses Circumventing Ebola Virus Immune Evasion. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Jordan DS, Leung DW, Amarasinghe GK, Basler CF, 2015. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J Virol 89, 5148–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Liang J, Pietzsch CA, Khadka S, Edwards MR, Wei S, De S, Posner B, Bukreyev A, Ready JM, Basler CF, 2017b. A high throughput screen identifies benzoquinoline compounds as inhibitors of Ebola virus replication. Antiviral Res 150, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, Amarasinghe GK, Basler CF, 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14, 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo M, Reid SP, Leung LW, Basler CF, Volchkov VE, 2010. Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signaling. J Virol 84, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi I, Amarasinghe GK, Basler CF, 2015. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 13, 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberger E, 2007. Filovirus replication and transcription. Future Virol 2, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S, 1999. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol 73, 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Riano E, Ngo N, Devito S, Eggink D, Munger J, Shaw ML, de la Torre JC, Martinez-Sobrido L, 2014. Inhibition of arenavirus by A3, a pyrimidine biosynthesis inhibitor. J Virol 88, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamment J, Ramsay E, Kelleher M, Dornan D, Ball KL, 2002. Regulation of the IRF-1 tumour modifier during the response to genotoxic stress involves an ATM-dependent signalling pathway. Oncogene 21, 7776–7785. [DOI] [PubMed] [Google Scholar]
- Prins KC, Delpeut S, Leung DW, Reynard O, Volchkova VA, Reid SP, Ramanan P, Cardenas WB, Amarasinghe GK, Volchkov VE, Basler CF, 2010. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J Virol 84, 3004–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF, 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 80, 5156–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF, 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol 81, 13469–13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis RAG, Calil FA, Feliciano PR, Pinheiro MP, Nonato MC, 2017. The dihydroorotate dehydrogenases: Past and present. Arch Biochem Biophys 632, 175–191. [DOI] [PubMed] [Google Scholar]
- Rhein BA, Powers LS, Rogers K, Anantpadma M, Singh BK, Sakurai Y, Bair T, Miller-Hunt C, Sinn P, Davey RA, Monick MM, Maury W, 2015. Interferon-gamma Inhibits Ebola Virus Infection. PLoS Pathog 11, e1005263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeron V, Feldmann H, Grard G, Becker S, Leroy EM, 2015. Ebola and Marburg haemorrhagic fever. J Clin Virol 64, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Rice CM, 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM, 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, 2006. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 31, 402–410. [DOI] [PubMed] [Google Scholar]
- Smee DF, Hurst BL, Day CW, 2012. D282, a non-nucleoside inhibitor of influenza virus infection that interferes with de novo pyrimidine biosynthesis. Antivir Chem Chemother 22, 263–272. [DOI] [PubMed] [Google Scholar]
- Smith DR, McCarthy S, Chrovian A, Olinger G, Stossel A, Geisbert TW, Hensley LE, Connor JH, 2010. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res 87, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler JR, Ervin ED, Towner JS, Rollin PE, Nichol ST, 2016. Perspectives on West Africa Ebola Virus Disease Outbreak, 2013-2016. Emerg Infect Dis 22, 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeki TM, Erickson BR, Brown S, McElroy AK, Cannon D, Gibbons A, Sealy T, Kainulainen MH, Schuh AJ, Kraft CS, Mehta AK, Lyon GM 3rd, Varkey JB, Ribner BS, Ellison RT 3rd, Carmody E, Nau GJ, Spiropoulou C, Nichol ST, Stroher U, 2016. Ebola Virus Persistence in Semen of Male Survivors. Clin Infect Dis 62, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Valmas C, Basler CF, 2011. Marburg virus VP40 antagonizes interferon signaling in a species-specific manner. J Virol 85, 4309–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QY, Bushell S, Qing M, Xu HY, Bonavia A, Nunes S, Zhou J, Poh MK, Florez de Sessions P, Niyomrattanakit P, Dong H, Hoffmaster K, Goh A, Nilar S, Schul W, Jones S, Kramer L, Compton T, Shi PY, 2011. Inhibition of dengue virus through suppression of host pyrimidine biosynthesis. J Virol 85, 6548–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang W, Xu L, Zhou X, Shokrollahi E, Felczak K, van der Laan LJ, Pankiewicz KW, Sprengers D, Raat NJ, Metselaar HJ, Peppelenbosch MP, Pan Q, 2016. Cross Talk between Nucleotide Synthesis Pathways with Cellular Immunity in Constraining Hepatitis E Virus Replication. Antimicrob Agents Chemother 60, 2834–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis N, Rhind N, 2009. Regulation of DNA replication by the S-phase DNA damage checkpoint. Cell Div 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, Pappu RV, Leung DW, Basler CF, Amarasinghe GK, 2014. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 16, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh S, Varkey JB, Crozier I, 2015. Persistent Ebola Virus in the Eye. N Engl J Med 373, 1982–1983. [DOI] [PubMed] [Google Scholar]
- Yen B, Mulder LC, Martinez O, Basler CF, 2014. Molecular basis for ebolavirus VP35 suppression of human dendritic cell maturation. J Virol 88, 12500–12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen BC, Basler CF, 2016. Effects of Filovirus Interferon Antagonists on Responses of Human Monocyte-Derived Dendritic Cells to RNA Virus Infection. J Virol 90, 5108–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Blancett CD, Koistinen KA, Schellhase CW, Bearss JJ, Radoshitzky SR, Honnold SP, Chance TB, Warren TK, Froude JW, Cashman KA, Dye JM, Bavari S, Palacios G, Kuhn JH, Sun MG, 2017. Identification and pathological characterization of persistent asymptomatic Ebola virus infection in rhesus monkeys. Nat Microbiol 2, 17113. [DOI] [PubMed] [Google Scholar]