

Abstract
Septic patients frequently develop cognitive impairment that persists beyond hospital discharge. The impact of sepsis on electrophysiological and molecular determinants of learning is underexplored. We observed that mice that survived sepsis or endotoxemia experienced loss of hippocampal long-term potentiation (LTP), a brain-derived neurotrophic factor–mediated (BDNF-mediated) process responsible for spatial memory formation. Memory impairment occurred despite preserved hippocampal BDNF content and could be reversed by stimulation of BDNF signaling, suggesting the presence of a local BDNF inhibitor. Sepsis is associated with degradation of the endothelial glycocalyx, releasing heparan sulfate fragments (of sufficient size and sulfation to bind BDNF) into the circulation. Heparan sulfate fragments penetrated the hippocampal blood-brain barrier during sepsis and inhibited BDNF-mediated LTP. Glycoarray approaches demonstrated that the avidity of heparan sulfate for BDNF increased with sulfation at the 2-O position of iduronic acid and the N position of glucosamine. Circulating heparan sulfate in endotoxemic mice and septic humans was enriched in 2-O– and N-sulfated disaccharides; furthermore, the presence of these sulfation patterns in the plasma of septic patients at intensive care unit (ICU) admission predicted persistent cognitive impairment 14 days after ICU discharge or at hospital discharge. Our findings indicate that circulating 2-O– and N-sulfated heparan sulfate fragments contribute to septic cognitive impairment.
Keywords: Neuroscience
Keywords: Extracellular matrix, Glycobiology, growth factors
Introduction
Septic patients frequently experience cognitive dysfunction that persists beyond hospital discharge, impairing survivors’ quality of life and ability to return to work (1–3). This cognitive impairment has been largely attributed to brain injury occurring early in sepsis, arising from pathogenic processes including cerebral microvascular thrombosis, metabolic derangements, and IL-1β–dependent neuroinflammation (4–8). Less is known about the long-term functional effects of sepsis on the electrophysiological and molecular neuronal pathways responsible for learning.
Classic neurosurgical studies have identified the hippocampus as the anatomic center of memory. Learning requires synaptic plasticity between key hippocampal neuronal networks, including the strengthening of cornu ammonis region 3 (CA3) to cornu ammonis region 1 (CA1) neuronal connections through a process called long-term potentiation (LTP), the key molecular mechanism driving spatial memory (9). Hippocampal LTP is dependent on the neuronal growth factor brain-derived neurotrophic factor (BDNF), as demonstrated by the induction of cognitive impairment by experimental BDNF sequestration (10). Interestingly, experimental and human sepsis has been associated with hippocampal pathology, including acute dysfunction of the hippocampal blood-brain barrier as well as later loss of hippocampal volumes in sepsis survivors (11–13).
There is increasing appreciation for the importance of the endothelial glycocalyx in sepsis pathophysiology. The glycocalyx is a ubiquitous endovascular layer enriched in heparan sulfate, a linear polysaccharide of repeating glucosamine-hexuronic (iduronic or glucuronic) acid disaccharide units. These disaccharides may be sulfated at specific sites (including the 2-O position of iduronic acid and/or the 6-O or N position of glucosamine), imparting a domain patterning of negative charge. During sepsis, the glycocalyx is fragmented, releasing heparan sulfate hexa- and octasaccharides into the bloodstream (14–16). These fragments have the capacity to interact with soluble proteins (such as growth factors) with remarkable specificity through sulfation-based electrostatic interactions, influencing a variety of homeostatic and/or pathologic signaling pathways (17). Shed fragments circulate for several days after sepsis onset and correlate with clinically significant outcomes including acute kidney and lung injury (15, 18).
Given the importance of the growth factor BDNF to cognition, as well as the ability of circulating heparan sulfate fragments to influence growth factor signaling, we hypothesized that hippocampal penetration of circulating heparan sulfate fragments leads to sequestration of BDNF, impairing LTP and inducing septic cognitive impairment. In this study, we used multimodal ex vivo, in vivo, and human studies to demonstrate that N- and 2-O–sulfated heparan sulfate fragments are released into the circulation during sepsis and inhibit BDNF-mediated hippocampal LTP, leading to cognitive dysfunction in both mice and humans.
Results and Discussion
To establish a model of septic cognitive dysfunction, we induced endotoxemia in 8- to 12-week-old male and female C57BL/6 mice through i.p. injection of LPS (10 μg/g BW). Animals injected with LPS had a mortality rate of 18%, with 70% of deaths occurring within 3 days of LPS injection (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI124485DS1). Endotoxemic mice displayed sickness behavior for 3 days, followed by resumption of normal behavior and weight gain by day 7 (Supplemental Figure 1B). To determine whether post-septic mice had memory deficits that persisted despite resumption of normal activity, we performed hippocampus-dependent neurobehavioral testing (contextual fear conditioning) on WT C57BL/6 mice 7 days after i.p. LPS or saline (control) injection. Contextual memory was apparent in saline-treated mice but was impaired in post-LPS–treated mice (Figure 1A). This post-LPS deficit in memory coincided with loss of hippocampal LTP, as measured using 300-μm-thick living hippocampal brain slices isolated from 8- to 12-week-old C57BL/6 mice 7 days after LPS (or saline) treatment (Figure 1B). We observed a similar loss of LTP in mice 7 days after cecal ligation and puncture (CLP) (Figure 1C, CLP or sham), a widely used model of polymicrobial peritonitis–induced sepsis. Intriguingly, we observed that post-LPS hippocampi maintained BDNF content (Figure 1D and Supplemental Figure 2) as well as the ability to induce LTP in response to exogenous BDNF (100 ng/ml, Figure 1E). Treatment of endotoxemic mice with 7,8-dihydroxyflavone (7,8-DHF) (5 μg/g BW administered daily via i.p. injection), a well-tolerated selective agonist of the BDNF receptor tyrosine receptor kinase B (TrkB), prevented loss of memory by day 7, as demonstrated by normalization of contextual fear conditioning (Figure 1F). Taken together, these findings indicate that post-endotoxemic/post-septic mice have impaired memory and loss of LTP, despite the presence of BDNF within the hippocampus and persistence of hippocampal responsiveness to TrkB activation, suggesting induction of a competitive inhibitor of BDNF within the septic hippocampus.
Figure 1. Cognitive impairment in mouse survivors of endotoxemia or sepsis is BDNF and TrkB responsive.

(A) Memory impairment occurred in mice 7 days after i.p. administration of LPS (10 μg/g BW), as demonstrated by loss of freezing behavior (indicating a fearful memory of a previous paw shock) after contextual fear conditioning. (B) Living hippocampal slices isolated from mice 7 days after i.p. LPS administration (as compared with saline) showed impaired LTP, the BDNF-dependent neuronal process responsible for spatial memory. (C) A similar loss of LTP was also seen 7 days after CLP, a model of polymicrobial peritonitis–induced sepsis. Loss of LTP occurred 7 days after LPS, despite (D) maintenance of hippocampal BDNF content and (E) preserved responsiveness to excess (100 ng/ml) exogenous BDNF. (F) Maintenance of the BDNF-responsive molecular machinery of learning after sepsis was further demonstrated by reversal of memory deficits in post-LPS mice treated daily with 7,8-DHF (5 μg/g i.p.), a direct agonist of the BDNF receptor TrkB. *P < 0.05 and **P < 0.01, by t test. For LTP measurements, the left panels represent the mean ± SEM of groups; the right panels represent the average change from baseline over the final 10 minutes of measurement (each data point represents a unique biological replicate). TBS, theta burst stimulation.
Our group and others have shown that highly sulfated heparan sulfate hexa- to octasaccharide fragments are shed into the plasma in animal (19) and human (14) sepsis as a consequence of endothelial glycocalyx degradation. We confirmed these findings using high-sensitivity mass spectrometry multiple reaction monitoring (MRM) (19) analyses of plasma collected after i.p. LPS treatment in mice (Figure 2A) and in a separate cohort of human patients with sepsis, who were enrolled in the Molecular Epidemiology of SepsiS in the Intensive Care Unit (MESSI) study (Figure 2B). Consistent with the known ability of ultra-low-molecular-weight heparins to cross the blood-brain barrier in healthy mice (20), we observed that circulating heparan sulfate fragments also penetrated the blood-brain barrier, as i.v. administered fluorescein-labeled heparan sulfate octasaccharides were observed in the hippocampus (Figure 2D) and cortex (Supplemental Figure 3) in both septic and non-septic mice. Likewise, we observed an increase in hippocampal heparan sulfate content at the point of peak circulating heparan sulfate (1 day after LPS treatment) (Figure 2C). Interestingly, the accumulation of hippocampal heparan sulfate persisted for 7 days after LPS treatment, a time point characterized by impaired cognition (Figure 1A). The persistence of brain heparan sulfate has been recognized in other neurodegenerative disease states, such as Alzheimer’s dementia (21–23). Hippocampal heparan sulfate content eventually normalized in mice 14 days after LPS treatment (compared with mice 14 days after saline treatment), which was coincident with improvement in cognition (Supplemental Figure 4).
Figure 2. Sepsis-associated circulating heparan sulfate fragments penetrate the hippocampus and impede LTP.

Liquid chromatography–tandem mass spectrometry MRM (LC-MS/MS MRM) analyses demonstrated shedding of heparan sulfate (HS) into the plasma of (A) mice 24 hours after i.p. LPS administration (10 μg/g BW vs. saline control) and (B) human patients with sepsis (enrolled in the MESSI cohort and followed longitudinally; control samples represent normal blood donors). (C) Accordingly, an increase in heparan sulfate content was detected in the hippocampus of LPS-injected mice that persisted for 7 days after injection. (D) Fluorescein-labeled, highly sulfated heparan sulfate (heparin) octasaccharides (250 μg) administered i.v. to mice 24 hours after i.p. LPS (10 μg/g) or saline treatment penetrated the hippocampal blood-brain barrier, as observed by confocal microscopy of freshly isolated hippocampal slices. Scale bars: 100 μm. (E) Highly sulfated heparan sulfate (heparin) octasaccharides (degree of polymerization 8 [dp8]) induced loss of LTP when perfused (2.5 μg/ml) over hippocampal slices isolated from healthy (non-septic mice). LTP was rescued by simultaneous perfusion with the TrkB agonist 7,8-DHF (250 nM). *P < 0.05, **P < 0.01, and ***P < 0.001, by ANOVA with Tukey’s correction for multiple comparisons (A, C, and E) or Kruskal-Wallis with Dunn’s test for multiple comparisons (B and D).
We have previously demonstrated that circulating heparan sulfate oligosaccharides can influence growth factor signaling by electrostatically binding positively charged residues of growth factor ligands (19). To determine whether hippocampus-penetrating heparan sulfate fragments interfere with BDNF, the neurotrophin responsible for hippocampal LTP, we isolated hippocampal slices from healthy (non-septic) 8- to 12-week-old C57BL/6 male and female mice and measured LTP in the presence or absence of highly sulfated heparan sulfate (heparin) octasaccharides, approximating the known size and sulfation pattern of circulating heparan sulfate fragments after sepsis (14). Exposure of hippocampal slices to highly sulfated octasaccharides inhibited LTP as compared with matched control slices isolated from the same mice (Figure 2E). Heparan sulfate–mediated inhibition of LTP could be reversed by the downstream activation of TrkB, as octasaccharides were unable to impede LTP when coadministered with 7,8-DHF (Figure 2E). This suggests that the negative effect of heparan sulfate on hippocampal LTP was mediated by inhibition of BDNF/TrkB signaling rather than by any nonspecific injurious effect of the glycosaminoglycan. These findings support the idea that sepsis-induced circulating heparan sulfate fragments penetrate the septic hippocampus and inhibit BDNF-specific molecular mechanisms of learning.
Heparan sulfate disaccharides may be sulfated at the N and 6-O positions of glucosamine or the 2-O position of iduronic acid (Figure 3A), imparting a domain patterning of negative charge that determines the ability of the heparan sulfate polysaccharide to selectively bind soluble ligands. We performed surface plasmon resonance (SPR), a technique that allows for quantification of glycosaminoglycan-protein interaction, to identify the sulfation sites that participate in heparan sulfate–BDNF binding. We found that heparan sulfate fragments bound BDNF in a length- and sulfation-dependent manner (Supplemental Figure 5). Since SPR does not inform the precise sequence or pattern of sulfation necessary for BDNF binding, we used a glycosaminoglycan microarray (glycoarray) composed of 52 heparan oligosaccharides of varying sulfation patterns (24). This approach revealed that BDNF-avid oligosaccharides commonly contained disaccharides with both N- and 2-O sulfation sequences (Figure 3B and Supplemental Figure 6). Intriguingly, circulating heparan sulfate fragments collected from post-LPS–treated mice (Figure 3C) and septic humans (Figure 3D, Supplemental Figure 7, and Supplemental Table 1) were similarly enriched in N and 2-O sulfation sequences on mass spectrometry MRM analysis. This enrichment in N and O sulfation was observed in the hippocampi of post-septic mice and persisted for 7 days (Figure 3E and Supplemental Table 2), indicating that mice maintained BDNF-avid heparan sulfate fragments within their hippocampi at time points coincident with post-septic cognitive impairment.
Figure 3. Importance of heparan disaccharide sulfation in BDNF binding and septic cognitive impairment.

(A) Heparan sulfate disaccharides may be sulfated at the N (NS) and 6-O (6S) positions of glucosamine and the 2-O (2S) position of iduronic acid. (B) Glycoarray analyses showed that heparan sulfate oligosaccharides, which bind to BDNF (n = 12), were enriched in NS and 2S compared with low-affinity (n = 40) oligosaccharides. 3S sulfation (found only in heparin) was not associated with binding. (C and D) Circulating heparan sulfate fragments detected by mass spectrometry in plasma collected (C) 24 hours after i.p. LPS administration (10 μg/g BW) in mice or (D) at the time of ICU admission in patients (n = 20) with sepsis were enriched in N sulfation (including NS, NS2S, NS6S, and TriS disaccharides) and 2-O sulfation (NS2S, 2S, 2S6S, and TriS). Control patients were normal blood donors (n = 9). (E) Mouse hippocampi after sepsis were enriched in N- and 2-O sulfation on day 7 (n = 3 for all time points and groups). (F) Patients who showed moderate to severe cognitive impairment (MoCA score <21 or cognitive inability to perform the test, n = 6) at the time of hospital discharge (or on day 14 after ICU discharge) were those who previously had (at the time of ICU admission, day = 0) circulating heparan sulfate enriched in NS2S disaccharides or a combination of NS, NS2S, and NS6S disaccharides. n = 14 patients with normal to mild impairment. Horizontal lines in the boxes represent the median, borders represent the 25th and 75th percentiles, and whiskers represent the upper and lower adjacent values (1.5 × interquartile range, Tukey’s method). Outside values were included in the analyses. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, by t test for single comparisons (B–D), ANOVA with Tukey’s correction for multiple comparisons (E), or Wilcoxon rank-sum test (F).
We performed additional glycoarray experiments to define the avidity of heparan sulfate oligosaccharides for pro-BDNF, a proenzyme incapable of promoting LTP (25). We observed no pro-BDNF binding to any heparan sulfate oligosaccharide sequence (Supplemental Figure 8A), suggesting that the cleavage of pro-BDNF to active BDNF exposes a heparan sulfate binding site. Other known heparan sulfate binding proteins (e.g., antithrombin III, platelet factor 4) bound to heparan sulfate sequences distinct from those that bound BDNF. Similarly, the heparan sulfate 10e4 antibody (26) failed to bind BDNF-avid sequences (Supplemental Figure 8, B–D). We leveraged this inability of the 10e4 antibody to bind BDNF-avid sequences to demonstrate that sequestration of non–BDNF-avid heparan sulfate sequences failed to prevent loss of LTP (Supplemental Figure 9), further confirming the sulfation sequence specificity of the observed BDNF-inhibitory effect of heparan sulfate.
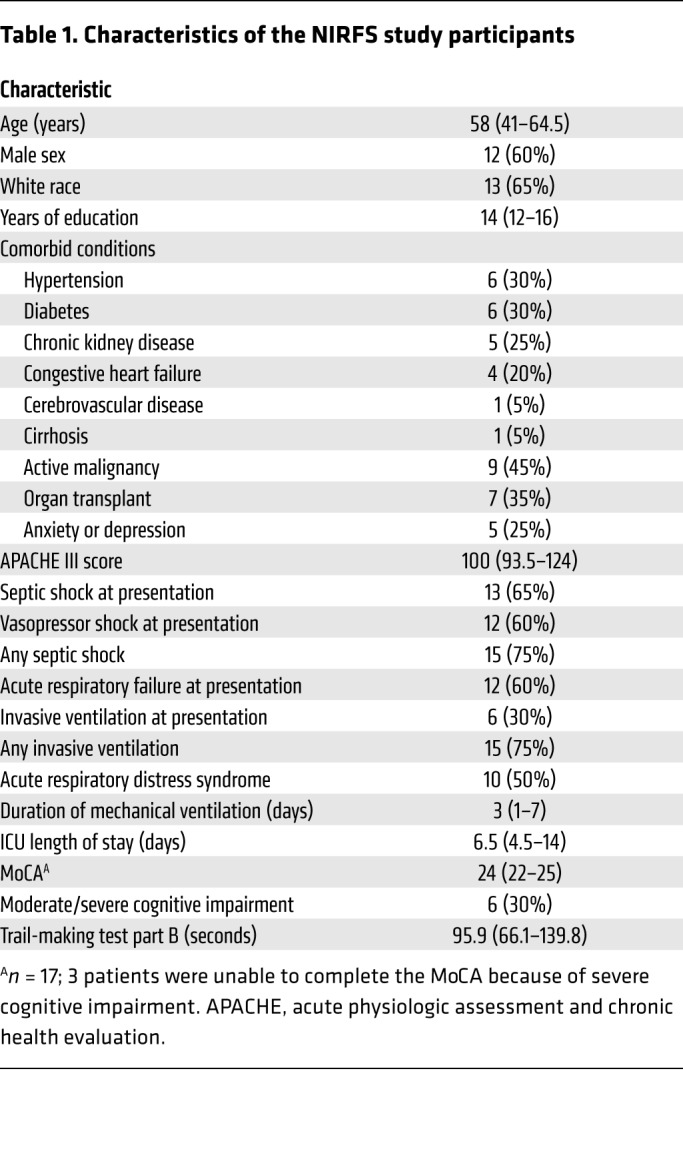
To determine the translational relevance of our findings, we measured heparan sulfate in plasma samples collected on day 0 (i.e., at presentation to the intensive care unit [ICU]) from septic patients enrolled in the Neurocognitive Impairment in Respiratory Failure and Shock (NIRFS) study of the MESSI cohort (Table 1) to determine whether the presence of circulating N- and 2-O–sulfated heparan sulfate predicted cognitive impairment in sepsis. NIRFS patients with persistent cognitive impairment after sepsis (as quantified by a Montreal Cognitive Assessment [MoCA] score <21 or a cognitive inability to perform the test either at hospital discharge or 14 days after ICU discharge) were those who had increased circulating N- and 2-O–sulfated heparan sulfate at the time of ICU admission (Figure 3F and Supplemental Table 3). Taken together, our findings demonstrate that the presence of circulating NS- and/or 2S-enriched, BDNF-avid heparan sulfate fragments at sepsis onset predicts cognitive impairment up to 2 weeks after ICU discharge.
Table 1. Characteristics of the NIRFS study participants.
In summary, our findings demonstrate that septic neurocognitive dysfunction is not only a consequence of inflammatory or ischemic brain injury but may also arise from septic interference with biologic processes (i.e., hippocampal BDNF/TrkB signaling) necessary for memory and cognition. Detection of circulating N- and 2-O–sulfated heparan sulfates may therefore allow the identification of septic patients at high risk of prolonged cognitive impairment, enabling the future development (and personalized implementation) of sulfation sequence–targeted therapeutics to improve memory and other patient-centered outcomes in sepsis survivors.
Methods
Detailed methods are provided in the Supplemental Methods.
Statistics
Statistical analyses of data on animal experiments, SPR, glycoarrays, and human subjects heparan sulfate fragment quantification were performed using GraphPad Prism (GraphPad Software). Analysis of all other human subjects data was performed using Stata Software. All replicates refer to studies using biological replicates, which were performed on different days to avoid temporal batch effects. No statistical outliers were excluded. Data represent the mean ± SEM or, for whiskers, were evaluated according to Tukey’s criteria. A two-tailed Student’s t test was performed for single comparisons. A P value of less than 0.05 was considered statistically significant for all analyses.
Study approval
Animal experiments.
All animal experiments were performed with approval of the institutional animal care and use committee of the University of Colorado Office of Laboratory Animal Resources. Experiments were performed in accordance with the Institutional Animal Care and Use Committee Guidebook, published by the NIH Office of Laboratory Animal Welfare (OLAW), as well as the Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines (27). Male and female 8- to 12-week-old C57BL/6 mice, purchased from Charles River Laboratories and The Jackson Laboratory, were used in all studies. At least 3 biologic replicates were used for microscopy and glycosaminoglycan quantification results, and at least 6 biologic replicates were used for all other experiments.
Human subjects.
Human samples and accompanying clinical data were collected from ICU patients enrolled in the NIRFS study, a substudy of the MESSI cohort, which is a single-center, prospective cohort of patients admitted to the ICU at the Hospital of the University of Pennsylvania. This study was approved by the IRB of the University of Pennsylvania. Subjects or their available surrogates provided written informed consent.
Author contributions
JAH, BJA, SAM, JL, NJM, RJL, PSH, and EPS designed the study. JAH, JEO, RMD, and JAF performed animal neuroelectrophysiology and behavioral experiments. JAH and Y. Yang performed microscopy studies. SAM and KO performed Western blot analyses. JAH, GS, Y. Yu, FZ, and XH performed analytical glycobiology experiments. BJA and NJM handled patient enrollment and data acquisition. JAH, BJA, JEO, RMD, GS, FZ, JL, RJL, NJM, PSH, and EPS performed data analysis and prepared the manuscript. EPS was responsible for overall coordination and oversight of the project.
Supplementary Material
Acknowledgments
This work was funded by NIH grants R01 HL125371 (to EPS and RJL); R01 GM125095 (to EPS and PSH); R01 HL137915 (to NJM); K23 HL140482 (to BJA); R01 HL094463 and U01 GM102137 (to JL); and T32 HL007085 (to JAH).
Version 1. 02/05/2019
In-Press Preview
Version 2. 03/18/2019
Electronic publication
Version 3. 04/01/2019
Print issue publication
Funding Statement
To Eric Schmidt and Robert Linhardt
To Eric Schmidt and Paco Herson
To Nuala Meyer
To Brian Anderson
To Jian Liu
To Jian Liu
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019 American Society for Clinical Investigation
Reference information: J Clin Invest. 2019;129(4):1779–1784.https://doi.org/10.1172/JCI124485.
See the related Commentary at The vasculature in sepsis: delivering poison or remedy to the brain?.
Contributor Information
Joseph A. Hippensteel, Email: joseph.hippensteel@ucdenver.edu.
Brian J. Anderson, Email: brian.anderson@uphs.upenn.edu.
James E. Orfila, Email: James.Orfila@ucdenver.edu.
Sarah A. McMurtry, Email: sarah.mcmurtry@ucdenver.edu.
Robert M. Dietz, Email: robert.dietz@ucdenver.edu.
Guowei Su, Email: guoweisu@email.unc.edu.
Joshay A. Ford, Email: joshay.ford@ucdenver.edu.
Kaori Oshima, Email: kaori.oshima@ucdenver.edu.
Yimu Yang, Email: Yimu.Yang@ucdenver.edu.
Fuming Zhang, Email: zhangf2@rpi.edu.
Xiaorui Han, Email: hanx3@rpi.edu.
Yanlei Yu, Email: yuy8@rpi.edu.
Jian Liu, Email: liuj@email.unc.edu.
Robert J. Linhardt, Email: linhar@rpi.edu.
Nuala J. Meyer, Email: nuala.meyer@uphs.upenn.edu.
Paco S. Herson, Email: paco.herson@ucdenver.edu.
Eric P. Schmidt, Email: eric.schmidt@ucdenver.edu.
References
- 1.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304(16):1787–1794. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandharipande PP, et al. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369(14):1306–1316. doi: 10.1056/NEJMoa1301372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calsavara AJC, Costa PA, Nobre V, Teixeira AL. Factors associated with short and long term cognitive changes in patients with sepsis. Sci Rep. 2018;8(1):4509. doi: 10.1038/s41598-018-22754-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Annane D, Sharshar T. Cognitive decline after sepsis. Lancet Respir Med. 2015;3(1):61–69. doi: 10.1016/S2213-2600(14)70246-2. [DOI] [PubMed] [Google Scholar]
- 5.Imamura Y, et al. Interleukin-1β causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience. 2011;187:63–69. doi: 10.1016/j.neuroscience.2011.04.063. [DOI] [PubMed] [Google Scholar]
- 6.Mina F, et al. Il1-β involvement in cognitive impairment after sepsis. Mol Neurobiol. 2014;49(2):1069–1076. doi: 10.1007/s12035-013-8581-9. [DOI] [PubMed] [Google Scholar]
- 7.Terrando N, et al. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care. 2010;14(3):R88. doi: 10.1186/cc9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong L, et al. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J Neurosci. 2012;32(49):17714–17724. doi: 10.1523/JNEUROSCI.1253-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84(1):87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 10.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381(6584):706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 11.Hofer S, et al. Injury of the blood brain barrier and up-regulation of icam-1 in polymicrobial sepsis. J Surg Res. 2008;146(2):276–281. doi: 10.1016/j.jss.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Bohatschek M, Werner A, Raivich G. Systemic LPS injection leads to granulocyte influx into normal and injured brain: effects of ICAM-1 deficiency. Exp Neurol. 2001;172(1):137–152. doi: 10.1006/exnr.2001.7764. [DOI] [PubMed] [Google Scholar]
- 13.Semmler A, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84(1):62–69. doi: 10.1136/jnnp-2012-302883. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt EP, et al. The circulating glycosaminoglycan signature of respiratory failure in critically ill adults. J Biol Chem. 2014;289(12):8194–8202. doi: 10.1074/jbc.M113.539452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt EP, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–1223. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Haeger SM, Yang Y, Dailey KL, Ford JA, Schmidt EP. Circulating heparan sulfate fragments attenuate histone-induced lung injury independently of histone binding. Shock. 2017;48(6):666–673. doi: 10.1097/SHK.0000000000000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss RJ, Esko JD, Tor Y. Targeting heparin and heparan sulfate protein interactions. Org Biomol Chem. 2017;15(27):5656–5668. doi: 10.1039/C7OB01058C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt EP, et al. Urinary glycosaminoglycans predict outcomes in septic shock and acute respiratory distress syndrome. Am J Respir Crit Care Med. 2016;194(4):439–449. doi: 10.1164/rccm.201511-2281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, et al. Fibroblast growth factor signaling mediates pulmonary endothelial glycocalyx reconstitution. Am J Respir Cell Mol Biol. 2017;56(6):727–737. doi: 10.1165/rcmb.2016-0338OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Q, et al. The blood-brain barrier accessibility of a heparin-derived oligosaccharides C3. Thromb Res. 2002;105(5):447–453. doi: 10.1016/S0049-3848(02)00050-6. [DOI] [PubMed] [Google Scholar]
- 21.Snow AD, Wight TN. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidoses. Neurobiol Aging. 1989;10(5):481–497. doi: 10.1016/0197-4580(89)90108-5. [DOI] [PubMed] [Google Scholar]
- 22.Snow AD, et al. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am J Pathol. 1990;137(5):1253–1270. [PMC free article] [PubMed] [Google Scholar]
- 23.Snow AD, et al. An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar A beta-amyloid in rat brain. Neuron. 1994;12(1):219–234. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, et al. Construction and characterisation of a heparan sulphate heptasaccharide microarray. Chem Commun (Camb) 2017;53(10):1743–1746. doi: 10.1039/C6CC08204A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang PT, et al. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306(5695):487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- 26.David G, Bai XM, Van der Schueren B, Cassiman JJ, Van den Berghe H. Developmental changes in heparan sulfate expression: in situ detection with mAbs. J Cell Biol. 1992;119(4):961–975. doi: 10.1083/jcb.119.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.