

Abstract
Motoneurons are particularly sensitive to mutations in mitofusin-2 (MFN2) that cause the neurological disorder Charcot-Marie-Tooth disease type 2A (CMT2A). MFN2 is a mitochondrial outer membrane protein that, together with its homologue MFN1, fuses mitochondria in most tissues. In this issue of the JCI, Zhou and colleagues show that increasing MFN1 expression in neurons can curtail neurological defects in a CMT2A mouse model. These results show that the ratio of MFN1 to MFN2 can explain the tissue specificity of CMT2A and indicate that augmentation of MFN1 in the nervous system has potential as a possible therapeutic strategy for CMT2A.
The mitofusins: mitochondrial fusion and beyond
The profusion proteins mitofusin-1 and -2 (MFN1 and MFN2) belong to the family of ubiquitous large dynamin-related GTPases, which are found in organisms ranging from yeast to humans (1). Mammalian MFN1 and MFN2 share approximately 80% similarity and contain the same relevant structural motifs (2). Both proteins possess a large cytosolic N-terminal GTPase domain, followed by a spacer, a first coiled-coil heptad-repeat (HR1) domain, a second spacer, two (or one; see ref. 3) very close transmembrane domains (TMs) that cross the outer mitochondrial membrane (OMM), a third spacer, and a second, C-terminal heptad-repeat domain (HR2) (Figure 1A). In the absence of both MFN1 and MFN2, no mitochondrial fusion can occur, leading to severe mitochondrial and cellular dysfunction (4). The exact mechanism of MFN-mediated fusion remains elusive. In a widely accepted model, the C-terminal HR2 domains work in trans to initiate tethering, with subsequent GTPase domain–mediated GTP hydrolysis (refs. 5, 6, and Figure 1B). The recent crystal structure of a cyanobacteria MFN homolog (BDLP) provides a new model in which MFN oligomerization promotes high membrane curvature, which itself is a prerequisite for fusion. GTP hydrolysis then leads to a so-called closed MFN conformation that is possibly detached from the membrane (ref. 7 and Figure 1C). Because both MFN1 and MFN2 can form cis oligomers (8, 9), the model is applicable to not only homo- but also to heterooligomers. Finally, two distinct and dynamic conformational states of mammalian MFNs can exist (ref. 5 and Figure 1D). According to this model, MFNs are tethering nonpermissive in the resting state because of intramolecular, antiparallel HR1-HR2 interactions and because of the relative position of the globular GTPase domain close to the OMM. In contrast, in the tethering permissive state, the destabilization of the intramolecular HR1-HR2 interaction allows the HR2 domain to extend into the cytosol, where it can encounter and bind HR2 domains of MFNs from the opposing membrane. Flexing of the HR2 domains is then responsible for the juxtaposition of tethered mitochondria, thereby reducing the gap between them and allowing the GTPase-dependent fusion of the opposing membranes. Despite their high degree of homology, MFN1 and MFN2 display functional heterogeneity. GTPase hydrolysis is more efficient in MFN1 than MFN2 (8), and MFN1, but not MFN2, is required for mitochondrial fusion driven by the inner membrane profusion protein optic atrophy 1 (OPA1) (10). Conversely, MFN2 tethers mitochondria to the ER (11) and interacts with the Miro/Milton complex, which is required for axonal transport of mitochondria in neurons (12). Finally, MFN2, but not MFN1, is mutated in the neurological disorder Charcot-Marie-Tooth disease type 2A (CMT2A) (13).
Figure 1. The MFN-mediated fusion models.
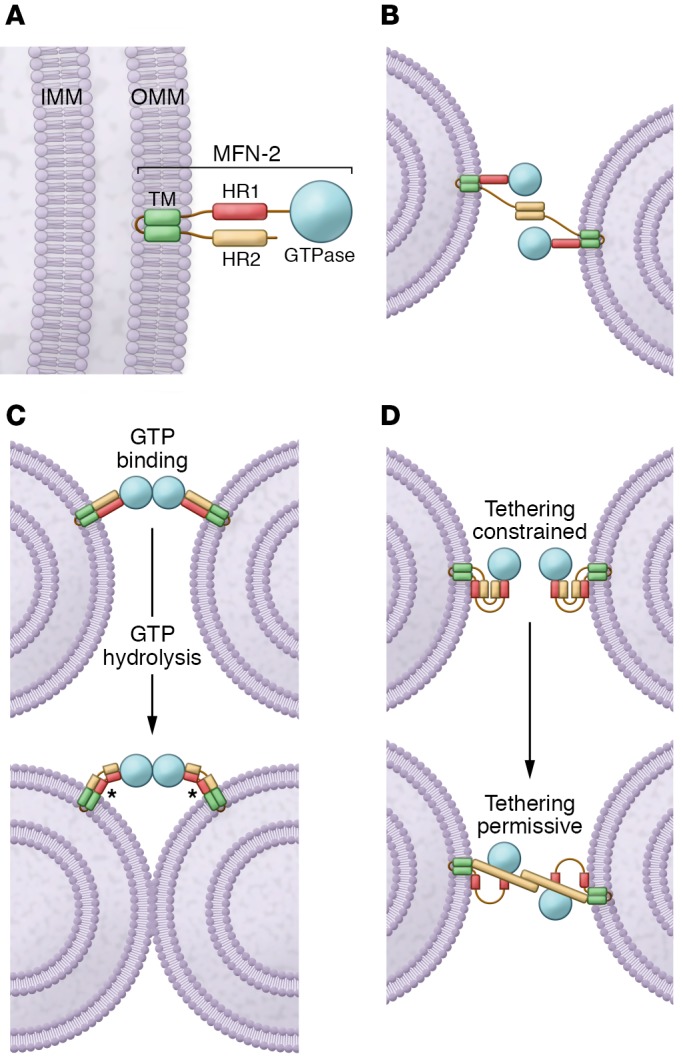
(A) Schematic of the MFN2 protein. MFN2 is embedded in the OMM by a TM and is composed of a GTPase domain and heptad repeat domains (HR1 and HR2). IMM, inner mitochondrial membrane. (B) MFN2 interacts in trans forming either homotypic or heterotypic (with MFN1) dimers to produce the mitochondrial tethering that precedes mitochondrial fusion. (C) MFNs may dimerize in trans upon GTP binding, which leads to long-distance docking of mitochondria. GTP hydrolysis can then induce a large conformational rearrangement of MFNs that brings OMMs into closer proximity. Mitochondrial fusion proceeds as a result of local membrane deformation near the TM domain when MFNs undergo GTP hydrolysis–dependent conformational transition and membrane structure perturbation by the HR1 domain (asterisks). (D) Schematic depiction of the folded (with HR2 constrained; left) and unfolded (with HR2 extended; right) MFN conformations. HR2 unfolding is below.
CMT2A: a disease of mitochondrial fusion, transport, or tethering?
CMT2A is a classic axonal peripheral sensorimotor neuropathy characterized by earlier and more severe involvement of the lower extremities than the upper extremities, distal upper extremity involvement as the neuropathy progresses, and more prominent motor deficits than sensory deficits (14). Most affected individuals develop symptoms in the first or second decade of life (14). MFN2 is the only gene whose pathogenic variants are known to cause CMT2A. More than 100 dominant mutations, the majority of which are missense mutations located in critical protein regions particularly close to or within the GTPase domain and the coil-coiled motifs, in the MFN2 gene have been reported in CMT2A patients (13). However, the molecular mechanism by which MFN2 mutations lead to the disease and, most importantly, how this mechanism can be tackled to modify CMT2A natural history remain obscure. Indeed, because of pleiotropic MFN2 functions, CMT2A pathogenesis might ensue from defects in mitochondrial fusion, transport, or tethering to ER. Moreover, it is unclear why MFN2 mutations preferentially affect the nervous system. The low levels of MFN1 expression in the adult nervous system may explain the neuronal specificity of the defect. Indeed, increased MFN1 expression can curtail the axonal degeneration caused by MFN2 mutants (15). Molecularly, the homooligomeric complexes formed by several MFN2 disease mutants are noncompetent for mitochondrial fusion, while MFN1 can complement mutant MFN2 through the formation of heterooligomeric complexes (16).
In this issue, Zhou and coworkers capitalized on these previous molecular and in vitro discoveries to offer a proof of principle that increased MFN1 activity in the nervous system can ameliorate disease in a mouse model of CMT2A (17). The author used transgenic mice with neuronal-specific expression of CMT2A-associated mutant MFN2 (MFN2R94Q) and demonstrated that mitochondria accumulate in neuronal cell bodies and proximal axons in cortex and spinal cord. The mutant mice recapitulated the neurological features seen in CMT2A patients, including severe early onset sensorimotor deficits, vision loss, and widespread axonal degeneration. Importantly, in vivo and in vitro data support a model in which MFN2R94Q is fusion incompetent and can result in dominant negative suppression of mitochondrial fusion, depending on the level of available fusion-competent MFN1 and MFN2 in the cell. This suggests that, because of the lower levels of endogenous MFN1 in neuronal cells, the promotion of a tethering permissive conformation of HR1-HR2 (Figure 1D) of MFN2R94Q enhances the frequency of failed fusion events and mitochondrial clustering.
The importance of MFN balance in the nervous system
To determine whether the toxic effects of MFN2R94Q can be alleviated by altering the balance between MFN1 and MFN2, Zhou et al. generated CMT2A mutant mice with nervous system–specific overexpression of MFN1 (17). MFN1 overexpression in the nervous system was not only well tolerated, but also provided complete or near-complete phenotypic rescue of defects in locomotor activity, sensorimotor coordination, and vision in CMT2A mutant mice. In addition, mitochondrial aggregation, axon degeneration, and transcriptomic signature were all rescued by MFN1 augmentation. These data indicate that the MFN1/MFN2 ratio is a key determinant of the sensitivity of the nervous system to the dominant negative effects of mutant MFN2 expression and support exploring elevation of MFN1 levels, thereby setting a new MFN1/MFN2 balance, as a novel therapeutic strategy for CMT2A. The concept is consistent with recent reports showing that first-in-class MFN agonists can overcome dominant mitochondrial defects provoked in cultured neurons by MFN2 mutants (18).
Conclusions and future directions
While the curative potential of mitochondrial fusion activation in the context of CMT2A is exciting, the field must address several open questions. For example, how do MFNs promote mitochondrial fusion and how can a dominant negative mutation be overcome by introducing more copies of a functioning homologue in oligomeric complexes? Perhaps MFN1 and MFN2 display some intrinsic difference in the cooperativity of the GTPase activity, such that MFN2 mutations abolish MFN2, but not MFN1, cooperativity. What is the role of the mitochondrial clumping observed in the CMT2A neurons in disease etiology? These clumps might participate in CMT2A pathogenesis, possibly by impeding mitochondrial trafficking in the long motoneuron axons. Because MFN1 expression also corrects the clumping, it is difficult to extrapolate from the data of Zhou and colleagues whether clumping is a primary pathogenetic event or a simple epiphenomenon of the impaired mitochondrial fusion caused by CMT2A-associated MFN2 mutations.
Irrespective of these outstanding questions, the study by Zhou et al. offers a proof of concept that increasing the MFN1/MFN2 ratio in the nervous system is a possible therapeutic approach for CMT2A. Notwithstanding that MFN2R94Q is the most abundant mutation in CMT2A, more than 100 different dominant MFN2 mutations have been reported in CMT2A patients, suggesting that such a therapeutic approach must be comprehensively tested for other MFN2 mutations. Hope, however, exists that such an approach can be generalized: the first-in-class small-molecule MFN2 activator was shown to rescue phenotypes caused by a different (MFN2T105M) CMT2A mutation (18). Finally, this paper is a beautiful example of how a basic science discovery (that MFN2 participates in mitochondrial movement in neurons) can be followed up in years of intense research and brought to a preclinical proof of principle that might lead to a treatment for patients.
Acknowledgments
Work in the Scorrano lab is supported by the Associazione Italiana per la Ricerca sul Cancro (IG19991), the Fondation Leducq TNE (no. 15004), the Fondazione Cariparo Progetti Eccellenza, the Fondazione Cariplo, the Muscular Dystrophy Association (603731 to LS), the Japan Society for the Promotion of Science (S2603), and the Japan Foundation for Pediatric Research (to KI).
Version 1. 03/18/2019
Electronic publication
Version 2. 04/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019 American Society for Clinical Investigation
Reference information: J Clin Invest. 2019;129(4):1533–1535.https://doi.org/10.1172/JCI127820.
See the related article at Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model.
Contributor Information
Keiko Iwata, Email: kiwata@u-fukui.ac.jp.
Luca Scorrano, Email: luca.scorrano@unipd.it.
References
- 1.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell. 2016;61(5):683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 2.Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. 2003;116(pt 13):2763–2774. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- 3.Mattie S, Riemer J, Wideman JG, McBride HM. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J Cell Biol. 2018;217(2):507–515. doi: 10.1083/jcb.201611194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280(28):26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 5.Franco A, et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016;540(7631):74–79. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305(5685):858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 7.Daumke O, Roux A. Mitochondrial homeostasis: how do dimers of mitofusins mediate mitochondrial fusion? Curr Biol. 2017;27(9):R353–R356. doi: 10.1016/j.cub.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 8.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117(pt 26):6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 9.Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115(pt 8):1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 10.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101(45):15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456(7222):605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 12.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30(12):4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Züchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 14.Bombelli F, et al. Charcot-Marie-Tooth disease type 2A: from typical to rare phenotypic and genotypic features. JAMA Neurol. 2014;71(8):1036–1042. doi: 10.1001/jamaneurol.2014.629. [DOI] [PubMed] [Google Scholar]
- 15.Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci. 2012;32(12):4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176(4):405–414. doi: 10.1083/jcb.200611080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y, et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest. 2019;129(4):1756–1771. doi: 10.1172/JCI124194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rocha AG, et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science. 2018;360(6386):336–341. doi: 10.1126/science.aao1785. [DOI] [PMC free article] [PubMed] [Google Scholar]