

SIRT7 plays a critical role in dissembling and deactivating ATM when DNA damage repair is completed.
Abstract
The activation of ataxia-telangiectasia mutated (ATM) upon DNA damage involves a cascade of reactions, including acetylation by TIP60 and autophosphorylation. However, how ATM is progressively deactivated after completing DNA damage repair remains obscure. Here, we report that sirtuin 7 (SIRT7)–mediated deacetylation is essential for dephosphorylation and deactivation of ATM. We show that SIRT7, a class III histone deacetylase, interacts with and deacetylates ATM in vitro and in vivo. In response to DNA damage, SIRT7 is mobilized onto chromatin and deacetylates ATM during the late stages of DNA damage response, when ATM is being gradually deactivated. Deacetylation of ATM by SIRT7 is prerequisite for its dephosphorylation by its phosphatase WIP1. Consequently, depletion of SIRT7 or acetylation-mimic mutation of ATM induces persistent ATM phosphorylation and activation, thus leading to impaired DNA damage repair. Together, our findings reveal a previously unidentified role of SIRT7 in regulating ATM activity and DNA damage repair.
INTRODUCTION
DNA damage response and repair involve hierarchical cellular events that integrate hundreds of signal-transducing proteins or pathways. Among the numerous DNA damage-responding factors, ataxia-telangiectasia mutated (ATM) has been demonstrated to be an apical kinase in regulating cellular response to DNA double-strand breaks (DSBs). In undamaged cells, ATM remains inactive as a dimer or higher-order multimer (1). Upon DNA damage, ATM is activated via a series of highly organized machineries, including acetylation by the histone acetyltransferase TIP60 at lysine 3016, intermolecular autophosphorylation at multiple sites, destabilization of linker histone H1.2, and stimulation by the MRE11-RAD50-NBS1 (MRN) complex (1–7). Activated ATM can then phosphorylate various factors to regulate checkpoint activation; DNA damage response and repair, including Nijmegen breakage syndrome 1 (NBS1); H2A histone family member X (H2A); and checkpoint kinase 2 (CHK2) (8–10). For example, H2AX phosphorylation at Ser139 (namely, γ-H2AX), which is one of the earliest events to occur in response to DNA damage, is enriched at DNA damage sites and forms a platform with a mediator of DNA damage checkpoint protein 1 to facilitate the recruitment of downstream repair factors (11–13). During DNA damage response and repair, ATM stays at the damage sites for a varying amount of time, from minutes to hours, depending on the nature of the DNA damage lesions. ATM is then gradually displaced and inactivated, which is reportedly associated with its dephosphorylation that is regulated by protein phosphatases, such as protein phosphatase 1 (PP1), PP2A, and wild-type p53-induced phosphatase 1 (WIP1) (14–16). Persistent ATM activation leads to impaired DNA repair and cell survival (15). Although much attention has been paid to the mechanisms of ATM activation in response to DNA damage, how ATM is disassembled and deactivated when DNA repair is completed remains poorly understood.
Acetylation of ATM has been known as an essential regulator of its activation and DNA damage repair. Upon DNA damage, the local chromatin structure is transiently condensed and H3K9me3 signals are rapidly increased, which recognize and activate TIP60 (17, 18). TIP60 is also phosphorylated and regulated by protein tyrosine kinase c-ABL, which promotes its binding to H3K9me3 and triggers ATM activation (19). Activated TIP60 then acetylates ATM, which is prerequisite for its autophosphorylation and subsequent activation (2, 20). Other factors, including activating transcription factor 2 and NOTCH1, also modulate ATM activation through TIP60 (21–23). Nevertheless, although the role of TIP60-dependent acetylation in regulating ATM activity has been well established, whether and how ATM acetylation is dynamically regulated in response to DNA damage are largely unexplored.
Sirtuins, as mammalian homologs of the yeast protein Sir2, are nicotinamide adenine dinucleotide (NAD+)–dependent histone deacetylases (HDACs), which regulate diverse cellular and biological functions, including aging, metabolism, genomic stability, and tumorigenesis (24, 25). Among the seven members of the sirtuin family proteins [sirtuin 1 (SIRT1) to SIRT7], SIRT1, SIRT6, and SIRT7 are primarily localized to the nucleus and have been reported to regulate DNA damage response and repair. For example, SIRT1 regulates nucleotide excision repair pathway by deacetylating Xeroderma pigmentosum group A (XPA) and increasing Xeroderma pigmentosum group C (XPC) expression upon ultraviolet irradiation (26, 27). SIRT1 is also involved in the repair of DNA DSBs by deacetylating NBS1 and Ku70 to regulate homologous recombination (HR) and nonhomologous end joining (NHEJ) repair pathways, respectively (28, 29). In addition, SIRT6 participates in the maintenance of genome integrity and DNA damage repair through modifying various repair factors, including poly(ADP-ribose) polymerase 1 (PARP1) and CtBP-interacting protein (CtIP) (30–32). Recent reports also show that SIRT7 promotes NHEJ through histone H3 lysine 18 (H3K18) deacetylation, which facilitates p53-binding protein 1 (53BP1) recruitment (33). These studies strengthen the importance of nuclear sirtuins in regulating DNA damage response and repair, but how SIRT7 is involved still needs to be further investigated.
DNA damage response must be tightly monitored and regulated to ensure faithful DNA repair and prevent unnecessary cellular alterations (34). Similarly, failure of both robust and persistent ATM activation compromises DNA damage response and repair (15). As sequential acetylation and phosphorylation are essential for ATM activation, it is rational to speculate that deacetylation may be critical for ATM dephosphorylation and deactivation. Given the key roles of nuclear sirtuins in DNA damage response and repair, it is possible that a specific member of the sirtuin family may be responsible for the deacetylation and deactivation of ATM. Here, we found that SIRT7 depletion induces persistent ATM acetylation and activation. We demonstrated that SIRT7, but not other sirtuins, deacetylates ATM in vitro and in vivo. Upon DNA damage, SIRT7 deficiency induces delayed ATM inactivation, which leads to decreased DNA damage repair and increased cell death. Our data fill the missing gap of dynamic regulation of ATM acetylation and suggest that SIRT7-dependent ATM deacetylation is critical for regulating ATM activity and DNA damage repair.
RESULTS
SIRT7 deficiency results in persistent ATM acetylation
Acetylation of ATM has been demonstrated to be essential for its activation upon DNA damage. To explore the dynamic regulation of ATM acetylation, we first examined how ATM acetylation was changed in response to DNA damage. Consistent with previous reports (2, 20), we could detect evident ATM acetylation upon ionizing radiation (IR) in a colon cancer cell line (HCT116), using a pan-acetyl-lysine antibody (Fig. 1A). ATM acetylation was then gradually decreased as DNA repair progressed and returned to nearly basal levels after 6 to 8 hours post-IR at 10 Gy, which was also evidently observed for ATM autophosphorylation, a canonical marker for its activation (Fig. 1A). These findings suggest that ATM may be deacetylated and thus deactivated when DNA damage repair is completed.
Fig. 1. SIRT7 deficiency results in persistent ATM acetylation.
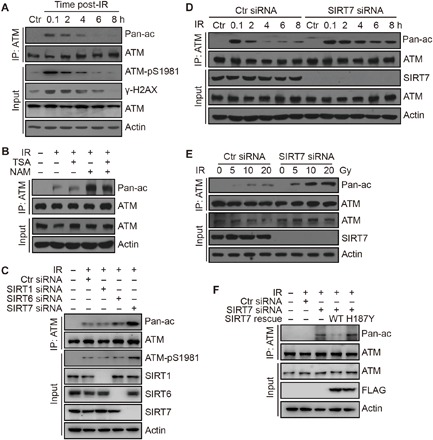
(A) HCT116 cells were irradiated at 10 Gy and released for the indicated time periods. Cell lysates were subjected to immunoprecipitation (IP) analysis. Pan-ac antibody, Pan-acetyl-lysine antibody. (B) HCT116 cells were treated with or without NAM at 10 mM or TSA at 1 μM and irradiated at 10 Gy and released for 8 hours. Cell lysates were subjected to IP analysis. (C) HCT116 cells were transfected with the indicated siRNAs and radiated at 10 Gy and released for 8 hours. Cell lysates were subjected to IP analysis. (D) HCT116 cells were transfected with the indicated siRNAs and radiated at 10 Gy and released for the indicated time periods. Cell lysates were subjected to IP analysis. (E) HCT116 cells were transfected with the indicated siRNAs and irradiated at 5, 10, or 20 Gy and released for 8 hours. Cell lysates were subjected to IP analysis. (F) HCT116 cells were transfected with the indicated siRNAs or plasmids and irradiated at 10 Gy and released for 8 hours. Cell lysates were subjected to IP analysis.
With TIP60 well characterized as the ATM acetyltransferase, we aimed to search for the ATM deacetylase. Cellular exposure to nicotinamide (NAM), a specific sirtuin inhibitor, but not trichostatin A (TSA), a class I and II HDAC inhibitor, could markedly increase ATM acetylation levels upon DNA damage (Fig. 1B). This observation suggests that sirtuin family deacetylases are responsible for ATM deacetylation. To test this hypothesis, we performed a series of knockdown experiments using specific small interfering RNA (siRNA) to target the nuclear sirtuins, SIRT1, SIRT6, and SIRT7. Notably, SIRT1 and SIRT6 depletion only resulted in a slight increase of ATM acetylation, whereas knockdown of SIRT7 led to a marked increase in ATM acetylation levels upon IR (Fig. 1C). In addition, we could also detect specific acetylation on K3016 of ATM after SIRT7 depletion using a specific anti-K3016 acetylation antibody (fig. S1A), suggesting that SIRT7 specifically regulates DNA damage–induced ATM acetylation.
Next, we went on to investigate the role of SIRT7 in the dynamics of DNA damage–induced ATM acetylation. When SIRT7 was depleted by siRNA-mediated knockdown, induction of ATM acetylation was not altered (Fig. 1D), suggesting that ATM could be normally activated. However, acetylation of ATM showed delayed decrease, which persisted through the DNA damage response and repair process (Fig. 1D), suggesting that SIRT7 is responsible for the deacetylation of ATM at late stages of DNA damage repair. Similarly, ATM acetylation was notably increased in SIRT7 knockdown cells when cells were treated with different doses of IR and collected at 8 hours post-IR (Fig. 1E). To further confirm that SIRT7 is responsible for the deacetylation of ATM in cells, we performed a rescue experiment by transfecting wild-type (WT) or enzymatic-dead (H187Y) SIRT7 plasmids into SIRT7 knockdown cells. SIRT7 knockdown led to increased ATM acetylation after 8 hours post-IR, which was repressed by reintroduction of WT but not H187Y SIRT7 (Fig. 1F), suggesting that SIRT7-regulated ATM acetylation is dependent on its enzymatic activity. Together, these data indicate that SIRT7 is responsible for deacetylating ATM, which may be associated with ATM deactivation at late stages of DNA damage repair.
SIRT7 interacts with ATM and directly deacetylates ATM
To gain further insights as to how SIRT7 regulates ATM acetylation, we first investigated the interactions between SIRT7 and ATM. Coimmunoprecipitation (Co-IP) assays using endogenous ATM or SIRT7 antibodies could detect a clear interaction between SIRT7 and ATM in HCT116 cells (Fig. 2, A and B). This interaction was obviously promoted upon DNA damage, especially after 4 to 8 hours post-IR, as measured by Co-IP assays in different cell lines (Fig. 2C and fig. S1B). As the expression of SIRT7 was not altered upon IR-induced DNA damage (fig. S1C), we suspected that SIRT7 may be recruited to chromatin. The enrichment of SIRT7 onto chromatin was markedly increased upon IR treatment, as shown by chromatin fractionation assays in different cell lines (Fig. 2D and fig. S1D). We also showed that SIRT7 threonine phosphorylation was induced upon DNA damage, which is not influenced by ATM inhibitor Ku55933, suggesting that phosphorylation is responsible for SIRT7 recruitment (fig. S1E). These observations further support our proposal that SIRT7 is responsible for ATM deacetylation during the late stages of DNA damage repair.
Fig. 2. SIRT7 interacts with ATM and regulates ATM activity.
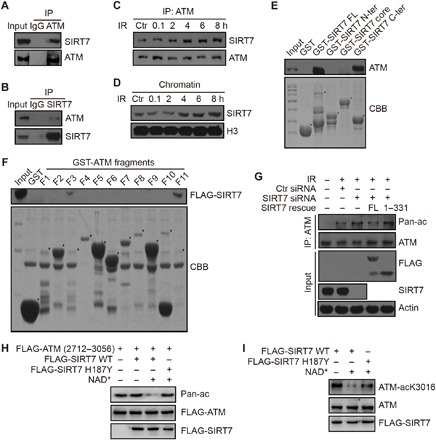
(A and B) Chromatin proteins were immunoprecipitated with an anti-ATM antibody or anti-SIRT7 antibody for IP analysis. IgG, immunoglobulin G. (C) HCT116 cells were treated with IR at 10 Gy and released for the indicated time periods. Chromatin proteins were subjected to IP analysis. (D) HCT116 cells were treated with IR at 10 Gy and released for the indicated time periods. Chromatin proteins were subjected to immunoblotting analysis. (E) GST alone, FL GST-SIRT7, an N-terminal fragment (1 to 89 aa), a deacetylase core fragment (90 to 331 aa), or a C-terminal fragment (332 to 400 aa) were incubated with HCT116 cell lysates for the GST pulldown assay. CBB, Coomassie brilliant blue. “*” represents specific protein bands. (F) GST alone or GST-tagged ATM fragments were incubated with HCT116 cell lysates with overexpressed FLAG-SIRT7 for the GST pulldown assay. CBB: Coomassie brilliant blue “*” represents specific protein bands. (G) HCT116 cells were transfected with the indicated siRNAs or plasmids and irradiated at 10 Gy and released for 8 hours. Cell extracts were subjected to IP analysis. (H and I) Fragment (2712 to 3056 aa) or FL FLAG-ATM proteins were purified from irradiated cells (10 Gy, 1 hour post-IR) and incubated with purified WT or mutant (H187Y) FLAG-SIRT7 with or without NAD+. The acetylation levels were determined by immunoblotting using an anti–pan-lysine-acetylation antibody or anti–ATM-acK3016 antibody.
To investigate whether SIRT7 and ATM directly interact with each other, we performed glutathione S-transferase (GST) pulldown experiments using GST-tagged ATM or SIRT7 proteins. As shown in fig. S2A, we fragmented SIRT7 according to its functional domains. Both full-length (FL) and the C-terminal domain [332 to 400 amino acids (aa)] of SIRT7 interacted with ATM (Fig. 2E). Similarly, we also fragmented ATM and confirmed that fragments 3 and 11 (F3, 523 to 769 aa; F11, FRAP-ATM-TRRAP-C-terminal (FATC) domain, 2842 to 3056 aa) of ATM interacted with SIRT7 (Fig. 2F and fig. S2B). In addition, we performed rescue experiments by reintroducing FL or C terminal–truncated (1 to 331 aa) SIRT7 into SIRT7 knockdown cells. Here, re-overexpression of FL decreased SIRT7 depletion–induced ATM acetylation, but the truncated mutant lacking the C terminus failed to do so (Fig. 2G). As the truncated mutant of SIRT7 was still enzymatically active (fig. S2C), but unable to interact with ATM, this result demonstrates that the interaction between SIRT7 and ATM is essential for ATM deacetylation.
To demonstrate a direct role of SIRT7 in deacetylating ATM, we set up an in vitro SIRT7 deacetylation assay. We transfected cells with a FLAG-tagged ATM fragment (2712 to 3056 aa) and exposed to IR to increase its basal levels of acetylation. We then precipitated the FLAG-ATM fragment as the substrate. Here, WT SIRT7 potently deacetylated ATM in the presence of NAD+, but a catalytically dead mutant SIRT7 could not (Fig. 2H). It has been previously reported that ATM is acetylated at a single lysine residue (K3016) in response to radiation (20). Therefore, we also performed the in vitro assay using an endogenous antibody against acetylated ATM at K3016 (ATM-acK3016) and found out that WT but not a catalytically dead mutant SIRT7 could deacetylate ATM at its K3016 residue (Fig. 2I). In addition, we ruled out the possibility that SIRT7 may regulate ATM acetylation through TIP60 by showing that SIRT7 did not interact with TIP60 and could not deacetylate it (fig. S2, D and E). Together, these results demonstrate that SIRT7 directly interacts with and deacetylates ATM.
SIRT7-mediated ATM deacetylation is required for its dephosphorylation and deactivation
Next, we went on to dissect how SIRT7-mediated ATM deacetylation is involved in DNA damage response and repair. As acetylation is essential for ATM autophosphorylation and activation, we hypothesized that deacetylation of ATM is required for its dephosphorylation and deactivation. To test this possibility, we first examined the dynamics of DNA damage–induced phosphorylation signals in SIRT7-depleted cells. As shown in Fig. 3A, phosphorylation of ATM and its substrates was rapidly induced, which then gradually decreased and was restored to basal levels after about 6 to 8 hours post-IR. In SIRT7 knockdown cells, ATM was still efficiently activated and phosphorylated upon DNA damage (Fig. 3A). Phosphorylation of ATM and its substrates remained at high levels even after 8 hours post-IR (Fig. 3A), indicating that the ATM was still highly activated. Accordingly, ectopic expression of WT SIRT7 efficiently promoted ATM inactivation, as shown in fig. S3A. This result is consistent with the data that mobilization of SIRT7 onto chromatin and its interaction with ATM occurred during the late stages of DNA damage repair.
Fig. 3. SIRT7 affects ATM autophosphorylation and dimer dissociation.
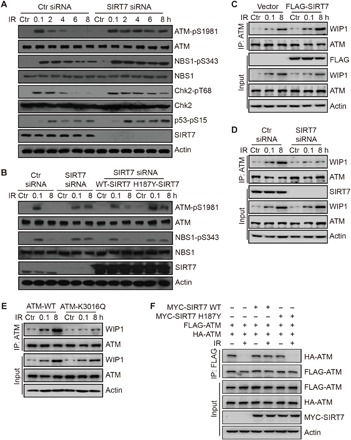
(A) HCT116 cells were transfected with the indicated siRNAs and irradiated at 10 Gy and released for the indicated time periods. Total cell lysates were analyzed by immunoblotting. (B) HCT116 cells were transfected with the indicated siRNAs or plasmids and irradiated at 10 Gy and released for the indicated time periods. Total cell lysates were analyzed by immunoblotting. (C and D) HCT116 cells were transfected with the indicated plasmids or siRNAs and irradiated at 10 Gy and released for the indicated time periods. Whole-cell extracts were subjected to Co-IP analysis. (E) ATM stable knockdown HCT116 cells were transfected with the indicated plasmids and irradiated at 10 Gy and released for the indicated time periods. Whole-cell extracts were subjected to Co-IP analysis. (F) HCT116 cells were transfected with the indicated plasmids and irradiated at 10 Gy and released for 1 hour. Whole-cell extracts were subjected to Co-IP analysis.
To confirm whether this process is dependent on SIRT7 enzymatic activity, we also performed a rescue experiment by reintroducing WT or H187Y SIRT7 into SIRT7 knockdown cells. As shown in Fig. 3B, reintroduction of WT SIRT7 restored the persistent phosphorylation of ATM and its substrate, NBS1, but H187Y mutant SIRT7 failed to do so, suggesting that SIRT7 enzymatic activity is required for the dephosphorylation and deactivation of ATM. In addition, we also tested the phosphorylation of ATM- and RAD3-related (ATR) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which are also critical kinases in DNA damage response and repair. The activities of ATR and DNA-PKcs were not notably altered when SIRT7 was depleted (fig. S3B), as determined by phosphorylation of ATR at T1989 and DNA-PKcs at T2609 (35, 36). Together, these data suggest that SIRT7 is required for dephosphorylation of ATM, and knockdown of SIRT7 leads to persistent ATM activation.
To further explore how SIRT7 regulates ATM dephosphorylation and deactivation, we proposed that SIRT7 may regulate ATM interaction with WIP1, the major phosphatase of ATM (15). To verify this proposal, we performed some Co-IP experiments to detect the interaction between ATM and WIP1 in response to DNA damage. Consistent with previous reports (15), ATM interacted with WIP1 at late stages of DNA damage repair (Fig. 3, C to E), which led to ATM dephosphorylation and deactivation once DNA repair was completed. In SIRT7-overexpressed cells, this interaction was markedly promoted (Fig. 3C). Whereas in SIRT7 knockdown cells, interaction between ATM and WIP1 was attenuated (Fig. 3D). To further demonstrate that SIRT7 regulates ATM interaction with WIP1 through deacetylation, we reintroduced WT or an acetylation-mimic K3016Q mutant ATM in ATM stable knockdown cells. As shown in Fig. 3E, WT ATM efficiently interacted with WIP1, but K3016Q mutant could not, suggesting that SIRT7-mediated deacetylation is required for ATM dephosphorylation and subsequent deactivation.
Acetylation of ATM is also reportedly an essential regulator of its monomerization (20). To investigate whether SIRT7 also regulates ATM monomerization, we engineered a system to monitor this process, in which FLAG-ATM and hemagglutinin (HA)–ATM plasmids were cotransfected. Consistently, we could detect the interaction between FLAG-ATM and HA-ATM, which was almost completely abolished upon DNA damage (Fig. 3F), suggesting that ATM is transited from dimers into monomers. Overexpression of WT SIRT7 evidently blocked DNA damage–stimulated monomerization of ATM, but a catalytically dead mutant SIRT7 failed to do so (Fig. 3F), suggesting that SIRT7 regulates ATM dimer formation through deacetylation. In addition, K3016Q mutant ATM could not dimerize again after 8 hours post-IR, whereas K3016R mutant ATM failed to become monomers upon IR (fig. S3C). These data suggest that SIRT7 prevents ATM dimer dissociation and promotes its dimerization at late stages of DNA repair. Together, these results demonstrate a direct role of SIRT7 in regulating ATM dephosphorylation and deactivation through deacetylation of ATM.
SIRT7 is required for DNA damage repair
ATM is a critical regulator of DNA damage response and repair. To determine whether SIRT7-regulated ATM acetylation also participates in the repair of DSBs, we first examined the role of SIRT7 in DNA damage repair. Consistent with Fig. 3, γ-H2AX, an ATM substrate and a classic marker for DNA repair, was restored to basal levels in control cells (Fig. 4A). In SIRT7 knockdown cells, γ-H2AX remained at high levels even after 8 hours post-IR (Fig. 4A), suggesting that DNA repair was impaired in SIRT7-deficient cells.
Fig. 4. SIRT7 is required for DNA damage repair.
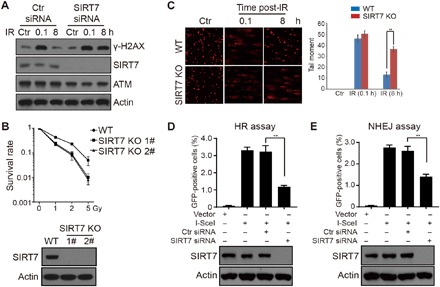
(A) HCT116 cells were transfected with the indicated siRNAs and irradiated at 10 Gy and released for the indicated time periods. Total cell lysates were subjected to immunoblotting analysis. (B) WT or SIRT7 KO (1# and 2#) cells were subjected to colony formation assay. All data represent the means ± SD. (C) WT or SIRT7 KO (1#) cells were subjected to comet assay. The original representative images were shown on the left panel. All data represent the means ± SD. (D and E) DR-U2OS and pEJ5-U2OS cells were transfected with the indicated plasmids or siRNAs and subjected to HR and NHEJ assays, respectively. All data represent the means ± SD.
In addition, we generated SIRT7 knockout (KO) HCT116 cells using CRISPR-Cas9 technology (Fig. 4B, bottom panel). Colony formation analysis showed that SIRT7 KO cells were more sensitive to IR-induced DNA damage (Fig. 4B). Comet assay also demonstrated that repair of DNA breaks was evidently delayed in SIRT7 KO cells (Fig. 4C). Moreover, we used two model cell lines, DR-U2OS and pEJ5-U2OS, to monitor the roles of SIRT7 in HR and NHEJ, respectively. As expected, knockdown of SIRT7 led to decreased efficiencies of both HR- and NHEJ-mediated DSB repair (Fig. 4, D and E), suggesting a more general role of SIRT7 in regulating DNA damage repair. Together, our results confirm that SIRT7 is required for DNA damage repair and cell survival.
SIRT7-mediated ATM deacetylation is required for DNA damage repair
To further explore the biological and functional relevance of SIRT7-dependent ATM deacetylation, we moved on to investigate whether SIRT7 regulates DSB repair through deacetylation of ATM. In comparison to SIRT7 knockdown alone, SIRT7 knockdown cells that were additionally treated with ATM inhibitor (Ku55933) or depleted of ATM showed similar dynamics of γ-H2AX when treated by IR (Fig. 5, A and C). Similarly, in contrast to Fig. 4C, we showed that SIRT7 KO cells that were treated with Ku55933 or depleted of ATM could repair DNA breaks at equivalent rates to WT cells (Fig. 5, B and D), suggesting that SIRT7-regulated DNA repair is dependent on ATM.
Fig. 5. SIRT7-mediated ATM deacetylation is required for DNA damage repair and cell survival.
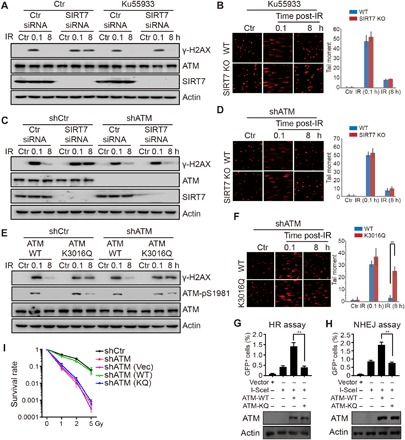
(A) HCT116 cells were transfected with the indicated siRNAs and treated with or without Ku55933 at 10 μM for 12 hours. Cells were irradiated at 10 Gy and released for the indicated time periods. Total cell lysates were subjected to immunoblotting. (B) WT or SIRT7 KO (1#) cells were treated with Ku55933 at 10 μM for 12 hours and then subjected to comet assay. All data represent the means ± SD. (C) ATM stable knockdown (shATM) or control (shCtr) HCT116 cells were treated with Ku55933 at 10 μM for 12 hours. Cells were irradiated at 10 Gy and released for the indicated time periods. Total cell lysates were subjected to immunoblotting. (D) ATM stable knockdown WT or SIRT7 KO cells were subjected to comet assay. All data represent the means ± SD. (E) ATM stable knockdown or control cells were transfected with the indicated plasmids and irradiated at 10 Gy and released for the indicated time periods. Total cell lysates were subjected to immunoblotting. (F) ATM stable knockdown cells were transfected with the indicated plasmids and subjected to comet assay. The bottom panel shows immunoblotting of total cell lysates. All data represent the means ± SD. (G and H) ATM stable knockdown DR-U2OS and pEJ5-U2OS cells transfected with the indicated plasmids and subjected to HR and NHEJ assays, respectively. The bottom panels show immunoblotting of total cell lysates. All data represent the means ± SD. (I) Control knockdown (shCtr), ATM stable knockdown (shATM), or ATM stable knockdown with reintroduction of vector, WT ATM, or K3016Q mutant ATM cells was subjected to colony formation assay. All data represent the means ± SD.
Next, to establish a direct role of ATM deacetylation in DNA damage repair, we performed some rescue experiments by retransfecting WT or K3016Q ATM plasmids into ATM stable knockdown cells. As shown in Fig. 5E, reintroduction of WT ATM restored the dynamics of γ-H2AX, whereas reintroduction of K3016Q ATM led to delayed removal of γ-H2AX and ATM phosphorylation. Similarly, K3016Q ATM retransfected cells displayed impaired repair of DNA breaks, as measured by comet assay (Fig. 5F), suggesting that ATM deacetylation is required for efficient DNA repair. In addition, we deleted endogenous ATM in DR-U2OS and pEJ5-U2OS cells using short hairpin RNA. Reintroduction of WT ATM could rescue the impaired efficiencies of HR- and NHEJ-mediated DSB repair, while K3016Q ATM failed to do so (Fig. 5, G and H). To further confirm that SIRT7 regulates DNA repair through ATM deacetylation, we also overexpressed WT or K3016Q ATM in combination with SIRT7 knockdown in DR-U2OS and pEJ5-U2OS cells. Overexpression of K3016Q mutant ATM inhibited HR and NHEJ activities compared to I-SceI alone or WT ATM in control cells but not in SIRT7 knockdown cells (fig. S4, A and B), suggesting that SIRT7 regulates DNA repair through ATM deacetylation.
To explore how ATM deacetylation and persistent ATM activation regulates HR and NHEJ, we performed chromatin fractionation to determine the recruitment of some key DNA repair factors. Chromatin-bound RPA32 and 53BP1, two key factors for HR and NHEJ, returned to nearly basal levels in ATM WT cells after 8 hours post-IR, but not in ATM K3016Q mutant cells (fig. S4C), suggesting that persistent ATM activation impairs the disassembly of DNA repair factors. In addition, cells with re-overexpressed WT ATM displayed comparable survival kinetics to control cells, while re-overexpression of K3016Q ATM showed similar kinetics to ATM-depleted or vector-reintroduced cells (Fig. 5I and fig. S4D). Moreover, SIRT7 depletion–induced sensitivity could be rescued by ATM inhibitor but not deletion of p53 (fig. S4, E and F), suggesting that SIRT7 regulates cell survival through ATM deacetylation. Together, our results demonstrate a direct role of SIRT7 in regulating ATM deacetylation, which is required for ATM deactivation at late stages of DNA damage repair, while SIRT7 deficiency leads to persistent ATM acetylation and compromised DSB repair.
DISCUSSION
In this study, we demonstrate that SIRT7 directly deacetylates ATM at its K3016 residue, which is required for its dephosphorylation and deactivation (Fig. 6). Our data support that SIRT7 depletion or acetylation-mimic mutation of ATM leads to persistent ATM activation and compromised DNA repair. Therefore, we propose a novel model whereby ATM deactivation is regulated by SIRT7-mediated deacetylation and subsequent dephosphorylation by WIP1.
Fig. 6. Schematic model of SIRT7-mediated ATM deacetylation in DNA damage repair.
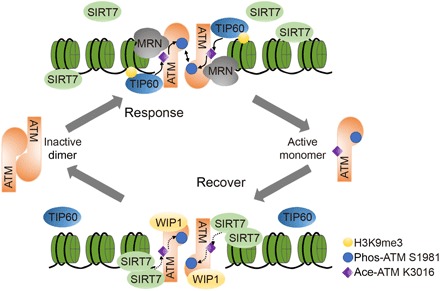
In response to DNA damage, ATM is sequentially modified by TIP60-mediated acetylation and autophosphorylation, which dictates ATM activation in coordination with the MRN complex. At late stages of DNA damage repair, SIRT7 is gradually recruited to DNA damage sites and deacetylates ATM, which is required for its dephosphorylation by phosphatase WIP1. ATM is then dimerized into inactive form. Thus, TIP60- and SIRT7-dependent balance of ATM acetylation controls its activity in response to DNA damage.
As an NAD+-dependent HDAC, SIRT7 controls a wide range of cellular functions through deacetylation of its substrates. For example, SIRT7 regulates cancer cell transformation and NHEJ-mediated DNA repair through deacetylation of H3K18 (33, 37). SIRT7 also targets nonhistone substrate PAF53, which modulates ribosomal DNA occupancy of polymerase I (Pol I) and transcription activation (38). In addition, SIRT7 potentiates the activation of GA binding protein (GABP) via deacetylation of GABPβ1, which is a master regulator of mitochondrial functions (39). Recent studies have also identified a desuccinylase function of SIRT7, which targets H3K122, thus promoting chromatin condensation and DSB repair (40). Here, we show that ATM is a direct substrate of SIRT7-dependent deacetylation. SIRT7 deacetylates ATM at its K3016 both in vivo and in vitro, a major ATM acetylation site in DNA damage (2). Our data provide the missing piece of dynamic regulation of ATM acetylation and suggest that TIP60 and SIRT7 may cooperate to balance ATM activation and deactivation in different stages of DNA damage response and repair through regulating its acetylation status. Moreover, although SIRT7 is reportedly mobilized onto DSB sites in a PARP1-dependent manner (40), we show that phosphorylation of SIRT7 may be responsible for its recruitment, and the exact mechanisms of SIRT7 recruitment remains to be further determined. It will be interesting to explore the possible interactions between SIRT7 and other ATM regulators, which will expand our understanding of the regulatory network of ATM deacetylation.
ATM is a central kinase in the cellular response to DNA damage, which integrates DNA damage signals and cellular responsive machineries by phosphorylating hundreds of downstream factors. Activation of ATM requires a cascade of posttranslational modifications, recruitment, and stimulation by the MRN complex, exposure of DNA breaks, removal of the inhibitory linker histone H1.2, and other mechanisms that are still under investigation (1, 2, 5–7, 41). Activated ATM initiates, amplifies, and supervises the DNA damage signaling to ensure efficient and faithful DNA repair, which is disassembled and deactivated when the DNA repair process is complete. Persistent ATM activation leads to the aberrant activation of downstream proteins, which induces cell death by apoptosis (42), suggesting that timely removal and deactivation of ATM is also critical for DNA repair and cell survival. Nevertheless, how ATM deactivation is regulated remains largely unknown. Previously, it was shown that protein phosphatase, including PP2C- or WIP1-mediated, dephosphorylation is crucial for this process (15). In this study, we reveal that SIRT7-dependent deacetylation of ATM is prerequisite for its dephosphorylation. We show that ATM acetylation and phosphorylation display similar dynamic patterns, which are induced immediately upon DNA damage and then gradually restored to basal levels as DNA repair proceeds. Meanwhile, deletion of SIRT7, which blocks ATM deacetylation, also leads to persistent ATM phosphorylation. We ruled out the possibility that persistent phosphorylation and activation of ATM may result from unrepaired DNA damage because an acetylation-mimic mutation of ATM also abrogated ATM dephosphorylation and deactivation. Moreover, we show that persistent ATM acetylation and activation lead to retention of some key HR and NHEJ repair factors, such as 53BP1 and RPA32. Although the detailed mechanisms still need to be further elucidated, our results demonstrate that prolonged ATM activation impairs DNA damage repair, and a highly coordinated machinery is required to balance ATM acetylation during the repair process.
Persistent DNA damage response, especially prolonged activation of ATM and retention of γ-H2AX, is harmful to DNA damage repair and cell survival. For example, tumor suppressor histidine triad nucleotide binding protein 1 (HINT1) is recruited to sites of DNA damage and facilitates DNA damage repair (43). However, HINT1 deficiency leads to persistent γ-H2AX focal expression at DNA damage sites and disturbs the subsequent DNA damage repair process (43). In addition, ATR-dependent γ-H2AX prolongation is also detrimental to DNA damage repair, and cancer cells deficient in Wolf-Hirschhorn syndrome candidate 1 exhibit marked retention of γ-H2AX following replication stress (44). The adenoviral protein E4orf6 is a DNA damage inducer that interrupts DNA damage repair by inhibiting protein phosphatase PP2A, which suppresses γ-H2AX dephosphorylation and leads to significant retention of γ-H2AX (45). Consistent with our findings, these proteins do not affect the formation of γ-H2AX foci but impair the removal of γ-H2AX following DNA damage. Moreover, aberrant activation of CHK2 and p53 are also consequences of persistent ATM activation, which leads to abnormal checkpoint activation and apoptosis (42). Our results are also consistent with ATM-dependent apoptosis, which may explain why SIRT7 deficiency induces cell death and compromises colony formation.
In summary, we present a previously unidentified mechanism of how SIRT7 is involved in regulating ATM activity. Our study provides new insights into the dynamic regulation of ATM acetylation and activation, which opens a new window to target SIRT7, as a dominating factor in ATM deactivation, for cancer therapy. As we have previously shown, SIRT7 levels correlate with the clinical response and prognosis of cancer chemotherapy (46). Further investigations into the translational relevance of SIRT7-regulated ATM deacetylation are now warranted and may ultimately lead to the identification of new promising drugs for cancer treatment.
MATERIALS AND METHODS
Cell culture
HCT116, LoVo, and human embryonic kidney (HEK) 293T cells were obtained from the American Type Culture Collection (USA). These cell lines were cultured in McCoy’s 5A medium or Dulbecco’s modified Eagle’s medium (M&C Gene Technology, China) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% antibiotics (M&C Gene Technology, China). All cell lines were maintained in a humidified incubator at 37°C with 5% CO2 atmosphere. Inhibitors, including NAM, TSA, and Ku55933, were purchased from Selleck (Selleckchem, USA).
In vitro chemoradiation treatment
Cells were irradiated with a biological x-ray irradiator RS2000pro (Rad Source Technologies, USA) with radiation output of 160 kV, 25 mA at a dose rate of 4.125 Gy/min.
Plasmids and siRNA
The human SIRT7 expression constructs (WT SIRT7 and H187Y SIRT7) were gifts from K. F. Chua (Stanford University, USA). The following siRNAs were used to silence target genes by transfecting with Lipofectamine 2000 (Life Technologies–Invitrogen, USA), according to the manufacturer’s instructions:
scrambled siRNA, UUCUCCGAACGUGUCACGU; SIRT7 siRNA1, AGCCATTTGTCCTTGAGGAA; SIRT7 siRNA2, GAACGGAACTCGGGTTATT.
Whole-cell extraction
Equal numbers of harvested cells were washed with phosphate-buffered saline (PBS) by centrifugation at 10,000 rpm (9391g) at 4°C for 30 s, and the cell pellet was resuspended in 30 μl per 106 cells of 2× proteinase inhibitor buffer, consisting of one cocktail proteinase inhibitor pellet (Roche Holding AG, Switzerland) in 3.5 ml PBS. An equal volume of 2× sample buffer (950 μl of Laemmli buffer + 50 μl 2-mercaptoethanol) was added to the resuspended cells. The samples were boiled for 10 min with a pulse vortex every 5 min and then pelleted by centrifugation at 12,000 rpm (13,523g) for 15 min.
SDS-PAGE and Western blotting
Western blotting was used to evaluate protein levels as previously described (47), with minor modifications. Briefly, equal amounts of protein were sized-fractionated on 6 to 15% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels. The antibodies used were anti-SIRT7 (sc-135055), anti–pan-lysine-acetylation (9441S), anti–γ-H2AX (2577, 05-636), anti-ATM (GTX70103), anti–ATM-S1981p (5883S), anti–NBS1-S343p (3001S), anti-NBS1 (sc-8580), anti–CHK2-T68p (2661S), anti-CHK2 (2662P), anti–p53-S15p (9284S), anti–ATR-T1989p (GTX128145), anti-ATR (RTL-0416), anti–DNA-PKcs-T2609p (AP0424), anti–DNA-PKcs (sc-1552), anti-H3 (ab1791), anti-actin (TA-09), and anti-FLAG (F1804). Antibody against ATM-acK3016 was a mixture of self-generated antibodies.
Coimmunoprecipitation
Cells were harvested by centrifugation at 10,000 rpm (9391g) at 4°C for 30 s in PBS. The cell pellet was resuspended in 500 μl of buffer A [10 mM Hepes (pH 7.9), 10 mM KCl, 1.5 mM NaCl, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol (DTT), and 1% protease inhibitor cocktail] with Triton X-100 at a final concentration of 0.1%. The cells were then incubated on ice for 8 min and centrifuged at 3721 rpm (1300g) at 4°C for 5 min. The supernatant was discarded, and the cell pellet was washed once with buffer A and collected by centrifugation at 3721 rpm (1300g) at 4°C for 5 min. The cell pellet was resuspended in 500 μl of buffer [20 mM tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, and 1% protease inhibitor cocktail] and sonicated 10 times on ice using a microtip at 35% amplitude for 1 s. The sonicated samples were then centrifuged at 12,000 rpm (13,523g) at 4°C for 10 min, and the chromatin fraction was collected in the supernatant. Antibody (1 to 2 μg) was added to 1 ml of cell lysate and incubated at 4°C for 8 to 12 hours. After adding protein A/G agarose beads, the incubation was continued for 4 hours. The immunoprecipitants were washed three times with buffer [20 mM tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 0.1% NP-40] and eluted with SDS loading buffer by boiling for 5 min. The supernatant was subjected to Western blotting after centrifugation.
Chromatin fractionation
Cells were harvested and washed in PBS by centrifugation at 10,000 rpm (9391g) at 4°C for 30 s. The cell pellet was resuspended in 500 μl of buffer I [150 mM NaCl, 50 mM Hepes (pH 7.5), 1 mM EDTA, 0.1% Triton X-100, and 1% protease inhibitor cocktail] for 3 min on ice and then centrifuged at 13,000 rpm (15, 871g) at 4°C for 3 min, and the detergent extractable supernatant was collected. The insoluble pellet was washed twice in buffer I without Triton X-100 by centrifugation at 13,000 rpm (15, 871g) at 4°C for 3 min, and the remaining pellet containing the chromatin sample was resuspended in SDS loading buffer and boiled before analyzed by Western blotting.
GST pulldown
GST or GST fusion proteins were expressed in Escherichia coli by isopropyl-β-d-thiogalactopyranoside (0.1 mM) induction overnight at 16° or 28°C and purified using glutathione Sepharose 4B beads (GE Healthcare, USA). Equal amounts of individual fusion protein were incubated with GST fusion proteins (from E. coli) in buffer [10 mM tris-HCl (pH 8.0), 1 mM EDTA, and 100 mM NaCl] for 4 hours at 4°C. After three washes in the same buffer by centrifugation at 1000 rpm (94g) at 4°C for 1 min × 3 times, the precipitated components were analyzed by Western blotting.
In vitro deacetylation assay
HCT116 cells were transfected with truncated (2712 to 3056 aa) or FL FLAG-ATM plasmids using Lipofectamine 2000, and the cells were subjected to radiation to induce ATM acetylation. FLAG-ATM was purified from HCT116 cells by immunoprecipitation with M2 beads (Sigma, USA). The beads were washed in high-salt buffer [50 mM tris-HCl (pH 8.0), 1 M NaCl, 0.2 mM EDTA, and 10% glycerol] before subjected to deacetylation assay. HEK293T cells were transfected with FL FLAG-SIRT7 plasmids using Lipofectamine 2000. FLAG-SIRT7 was purified by immunoprecipitation with M2 beads and eluted by 3× FLAG peptide (0.125 mg/ml). Purified acetylated FLAG-ATM was incubated with eluted FLAG-SIRT7 in deacetylation buffer [10 mM tris-HCl (pH 8.0), 4 mM MgCl2, 0.2 mM DTT, and 10% glycerol] in the presence or absence of 5 mM NAD+ at 30°C for 1 hour. The samples were then analyzed by Western blotting.
CRISPR-Cas9–based gene editing
SIRT7 KO HCT116 cell lines were generated via Lipofectamine 2000 transfection with small guide RNA constructs (sequence 1, GAGCCGCTCCGAGCGCAAAG; sequence 2, CGAGAGCGCGGACCTGGTAA; cloned into a p × 459/Puro vector, a gift from C. Zhou in Peking University). A detailed protocol has been previously reported (48).
Immunofluorescence
HCT116 cells (40 to 50% confluence) were seeded on glass-bottom dishes and incubated overnight in cell culture medium in a humidified incubator at 37°C. The cells were transfected according to the manufacturer’s protocol with indicated plasmids or treated with chemicals. The cells were then fixed in 4% paraformaldehyde at room temperature and permeabilized with methanol for 10 min at −20°C. The cells were then blocked using blocking buffer (0.8% bovine serum albumin in PBS) and then incubated overnight with the indicated primary antibodies (1:100 dilution for all antibodies) at 4°C. After being washed three times with blocking buffer, the cells were exposed to the secondary antibodies (1:100 dilution) conjugated to FITC/TRITC (fluorescein isothiocyanate/tetramethyl rhodamine isothiocyanate) and then embedded with 4′,6-diamidino-2-phenylindole. The immunofluorescence signal was observed under a fluorescence microscope (Olympus, Japan).
Comet assay
Comet assay was performed as previously described (49). Briefly, cells were treated as indicated and then mixed gently with premelted low-temperature–melting agarose at a volume ratio of 1:1 (v/v) and spread on glass slides. The slides were then submerged in precooled lysis buffer at 4°C for 90 min. After rinsing, the slides were electrophoresed in running buffer [1 mM Na2EDTA and 300 mM NaOH (pH 13.0)] at 1.0 V/cm for 20 min and then stained with propidium iodide (5 μg/ml). Fluorescent images of more than 100 nuclei were captured under an Olympus FV1000-IX81 confocal microscope (Tokyo, Japan).
Flow cytometry
Cells were harvested at 48 hours after transfection with DsRed or I-SceI and resuspended in 0.5 ml of PBS for fluorescence-activated cell sorting analysis. Green fluorescent protein (GFP)–based repair efficiency was calculated as the ratio of GFP-positive cells over the DsRed-positive cells.
Colony formation
Equal numbers of HCT116 cells were seeded in cell culture medium containing 10% FBS and 1% antibiotics in 60-mm dishes. Cells were irradiated by x-ray after 12 hours of seeding and continued to culture at 37°C, 5% CO2 for 2 weeks, and then stained with crystal violet.
Statistics
Statistical comparisons were carried out using the Student’s t test. A P < 0.05 was considered statistically significant (not significant, P > 0.05; **P < 0.01, ***P < 0.001). At least three independent experiments were performed in all cases.
Supplementary Material
Acknowledgments
We would like to thank K. F. Chua (Stanford University, USA) for providing the SIRT7-WT and SIRT7-H187Y FLAG-tagged plasmids and for helpful suggestions. F. d’Adda di Fagagna (IFOM, the FIRC Institute of Molecular Oncology, Italy) for providing the GST-tagged ATM fragments. We would also like to thank J. Tamanini (Shenzhen University Health Science Center, ETediting) for proofreading the manuscript before submission. Funding: This study was supported by the National Key R&D Program of China (grant number 2017YFA0503900), the National Natural Science Foundation of China (grant numbers 81802811, 81621063, 81530074, 31570812, and 81720108027), and the Shenzhen Municipal Commission of Science and Technology Innovation (grant numbers JCYJ20160427104855100 and JCYJ20170818092450901). Author contributions: W.-G.Z., M.T., and Z.L. conceived, designed, and performed the experiments and wrote the manuscript. M.T., Z.L., C.Z., X.L., B.T., Z.C., Y.L., Y.C., L.J., Hui Wang, and L.W. analyzed the data and performed material preparation. X.X., J.W., B.L., and Haiying Wang discussed the results and commented on the manuscript. W.-G.Z. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav1118/DC1
Fig. S1. SIRT7 is recruited onto chromatin upon IR treatment.
Fig. S2. SIRT7 interacts and deacetylates ATM.
Fig. S3. Deletion of SIRT7 had no effects on ATR or DNA-PKcs activation.
Fig. S4. SIRT7 regulates DNA repair and cell survival through ATM deacetylation.
REFERENCES AND NOTES
- 1.Bakkenist C. J., Kastan M. B., DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Sun Y., Jiang X., Chen S., Fernandes N., Price B. D., A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. U.S.A. 102, 13182–13187 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozlov S. V., Graham M. E., Peng C., Chen P., Robinson P. J., Lavin M. F., Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 25, 3504–3514 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozlov S. V., Graham M. E., Jakob B., Tobias F., Kijas A. W., Tanuji M., Chen P., Robinson P. J., Taucher-Scholz G., Suzuki K., So S., Chen D., Lavin M. F., Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 286, 9107–9119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J. H., Paull T. T., Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Lee J. H., Paull T. T., ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Li Z., Li Y., Tang M., Peng B., Lu X., Yang Q., Zhu Q., Hou T., Li M., Liu C., Wang L., Xu X., Zhao Y., Wang H., Yang Y., Zhu W. G., Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Res. 28, 756–770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim D. S., Kim S. T., Xu B., Maser R. S., Lin J., Petrini J. H. J., Kastan M. B., ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 404, 613–617 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J., ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka S., Rotman G., Ogawa A., Shiloh Y., Tamai K., Elledge S. J., Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. U.S.A. 97, 10389–10394 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A., H2AX: The histone guardian of the genome. DNA Repair 3, 959–967 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Stucki M., Clapperton J. A., Mohammad D., Yaffe M. B., Smerdon S. J., Jackson S. P., MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Stucki M., Jackson S. P., gammaH2AX and MDC1: Anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 5, 534–543 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Goodarzi A. A., Jonnalagadda J. C., Douglas P., Young D., Ye R., Moorhead G. B., Lees-Miller S. P., Khanna K. K., Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 23, 4451–4461 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shreeram S., Demidov O. N., Hee W. K., Yamaguchi H., Onishi N., Kek C., Timofeev O. N., Dudgeon C., Fornace A. J., Anderson C. W., Minami Y., Appella E., Bulavin D. V., Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell 23, 757–764 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Peng A., Lewellyn A. L., Schiemann W. P., Maller J. L., Repo-man controls a protein phosphatase 1-dependent threshold for DNA damage checkpoint activation. Curr. Biol. 20, 387–396 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y., Jiang X., Xu Y., Ayrapetov M. K., Moreau L. A., Whetstine J. R., Price B. D., Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 11, 1376–1382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayrapetov M. K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B. D., DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. U.S.A. 111, 9169–9174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaidi A., Jackson S. P., KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 498, 70–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Sun Y., Xu Y., Roy K., Price B. D., DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 27, 8502–8509 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhoumik A., Takahashi S., Breitweiser W., Shiloh Y., Jones N., Ronai Z., ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol. Cell 18, 577–587 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhoumik A., Singha N., O’Connell M. J., Ronai Z. A., Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem. 283, 17605–17614 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamowicz M., Vermezovic J., d’Adda di Fagagna F., NOTCH1 inhibits activation of ATM by impairing the formation of an ATM-FOXO3a-KAT5/Tip60 complex. Cell Rep. 16, 2068–2076 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imai S., Armstrong C. M., Kaeberlein M., Guarente L., Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Finkel T., Deng C. X., Mostoslavsky R., Recent progress in the biology and physiology of sirtuins. Nature 460, 587–591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ming M., Shea C. R., Guo X., Li X., Soltani K., Han W., He Y. Y., Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc. Natl. Acad. Sci. U.S.A. 107, 22623–22628 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan W., Luo J., SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 39, 247–258 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Jeong J., Juhn K., Lee H., Kim S. H., Min B. H., Lee K. M., Cho M. H., Park G. H., Lee K. H., SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 39, 8–13 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Yuan Z., Zhang X., Sengupta N., Lane W. S., Seto E., SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 27, 149–162 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mostoslavsky R., Chua K. F., Lombard D. B., Pang W. W., Fischer M. R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M. M., Mills K. D., Patel P., Hsu J. T., Hong A. L., Ford E., Cheng H. L., Kennedy C., Nunez N., Bronson R., Frendewey D., Auerbach W., Valenzuela D., Karow M., Hottiger M. O., Hursting S., Barrett J. C., Guarente L., Mulligan R., Demple B., Yancopoulos G. D., Alt F. W., Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Kaidi A., Weinert B. T., Choudhary C., Jackson S. P., Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329, 1348–1353 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., Gorbunova V., SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vazquez B. N., Thackray J. K., Simonet N. G., Kane-Goldsmith N., Martinez-Redondo P., Nguyen T., Bunting S., Vaquero A., Tischfield J. A., Serrano L., SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 35, 1488–1503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciccia A., Elledge S. J., The DNA damage response: Making it safe to play with knives. Mol. Cell 40, 179–204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan D. W., Chen B. P., Prithivirajsingh S., Kurimasa A., Story M. D., Qin J., Chen D. J., Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 16, 2333–2338 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S., Shiotani B., Lahiri M., Maréchal A., Tse A., Leung C. C. Y., Glover J. N., Yang X. H., Zou L., ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 43, 192–202 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barber M. F., Michishita-Kioi E., Xi Y., Tasselli L., Kioi M., Moqtaderi Z., Tennen R. I., Paredes S., Young N. L., Chen K., Struhl K., Garcia B. A., Gozani O., Li W., Chua K. F., SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487, 114–118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S., Seiler J., Santiago-Reichelt M., Felbel K., Grummt I., Voit R., Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol. Cell 52, 303–313 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Ryu D., Jo Y. S., Lo Sasso G., Stein S., Zhang H., Perino A., Lee J. U., Zeviani M., Romand R., Hottiger M. O., Schoonjans K., Auwerx J., A SIRT7-dependent acetylation switch of GABPβ1 controls mitochondrial function. Cell Metab. 20, 856–869 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Li L., Shi L., Yang S., Yan R., Zhang D., Yang J., He L., Li W., Yi X., Sun L., Liang J., Cheng Z., Shi L., Shang Y., Yu W., SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 7, 12235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y. C., Gerlitz G., Furusawa T., Catez F., Nussenzweig A., Oh K. S., Kraemer K. H., Shiloh Y., Bustin M., Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat. Cell Biol. 11, 92–96 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roos W. P., Thomas A. D., Kaina B., DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 16, 20–33 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Li H., Balajee A. S., Su T., Cen B., Hei T. K., Weinstein I. B., The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage. J. Cell Biol. 183, 253–265 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hajdu I., Ciccia A., Lewis S. M., Elledge S. J., Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 108, 13130–13134 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart L. S., Ornelles D., Koumenis C., The adenoviral E4orf6 protein induces atypical apoptosis in response to DNA damage. J. Biol. Chem. 282, 6061–6067 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Tang M., Lu X., Zhang C., Du C., Cao L., Hou T., Li Z., Tu B., Cao Z., Li Y., Chen Y., Jiang L., Wang H., Wang L., Liu B., Xu X., Luo J., Wang J., Gu J., Wang H., Zhu W. G., Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer. Theranostics 7, 1346–1359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu W. G., Hileman T., Ke Y., Wang P., Lu S., Duan W., Dai Z., Tong T., Villalona-Calero M. A., Plass C., Otterson G. A., 5-aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J. Biol. Chem. 279, 15161–15166 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao W., McNutt M. A., Zhu W. G., The comet assay: A sensitive method for detecting DNA damage in individual cells. Methods 48, 46–53 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav1118/DC1
Fig. S1. SIRT7 is recruited onto chromatin upon IR treatment.
Fig. S2. SIRT7 interacts and deacetylates ATM.
Fig. S3. Deletion of SIRT7 had no effects on ATR or DNA-PKcs activation.
Fig. S4. SIRT7 regulates DNA repair and cell survival through ATM deacetylation.