

Summary
Toxin-antitoxin (TA) systems regulate fundamental cellular processes in bacteria and represent potential therapeutic targets. We report a new RES-Xre TA system in multiple human pathogens, including Mycobacterium tuberculosis. The toxin, MbcT, is bactericidal unless neutralized by its antitoxin MbcA. To investigate the mechanism, we solved the 1.8 Å-resolution crystal structure of the MbcTA complex. We found that MbcT resembles secreted NAD+-dependent bacterial exotoxins, such as diphtheria toxin. Indeed, MbcT catalyzes NAD+ degradation in vitro and in vivo. Unexpectedly, the reaction is stimulated by inorganic phosphate, and our data reveal that MbcT is a NAD+ phosphorylase. In the absence of MbcA, MbcT triggers rapid M. tuberculosis cell death, which reduces mycobacterial survival in macrophages and prolongs the survival of infected mice. Our study expands the molecular activities employed by bacterial TA modules and uncovers a new class of enzymes that could be exploited to treat tuberculosis and other infectious diseases.
Keywords: toxin-antitoxin system, NAD, tuberculosis, mycobacterium, bacterial cell death, MbcTA
Graphical Abstract
Highlights
-
•
MbcTA is a RES-Xre toxin-antitoxin system in M. tuberculosis (Mtb)
-
•
MbcT is a NAD+ phosphorylase
-
•
MbcT-catalyzed NAD+ depletion leads to Mtb cell death
-
•
MbcT activity synergizes with antibiotics to reduce Mtb burden in infected mice
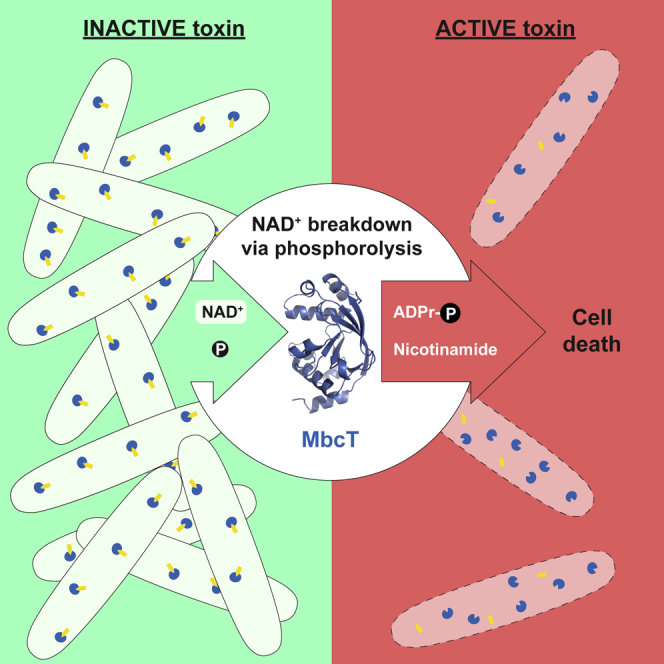
Toxin-antitoxin systems regulate bacterial growth in response to stress through modification of macromolecules, including proteins, RNA, and DNA. Freire et al. show that MbcT, a toxin produced by the tubercle bacillus, induces bacterial cell death through NAD+ phosphorolysis, an unprecedented enzymatic activity.
Introduction
Toxin-antitoxin (TA) systems are widespread in prokaryotes and play a central role in the response and adaptation of bacteria to various stress conditions, including starvation, phage attack, or antibiotic treatment (Hall et al., 2017, Harms et al., 2018, Lobato-Márquez et al., 2016, Page and Peti, 2016). TA systems encode a toxic protein, which targets an essential physiological process in the bacterial cell, together with a toxin-neutralizing “antidote” or antitoxin. Under favorable growth conditions, toxin activity is blocked by the presence of the antitoxin. When faced with antibiotic or environmental stress, the antitoxin is rapidly degraded, which allows the toxin to become activated, thereby reducing the bacterial growth rate (Deter et al., 2017, Hall et al., 2017). TA systems are classified in four families (I–IV) based on the nature of the antitoxin and the associated mechanism of toxin inhibition (Harms et al., 2018). Most studies have focused on type II TA systems, which are composed of a protein antitoxin and toxin pair. Strikingly, type II TA systems are highly abundant in the tuberculosis (TB) bacillus, Mycobacterium tuberculosis (Mtb), in which they are thought to contribute to pathogenicity and persistence (Keren et al., 2011, Ramage et al., 2009, Sala et al., 2014, Slayden et al., 2018). Among the ∼80 TA system-encoding operons identified in the Mtb genome, three antitoxin-encoding genes are essential for viability, as evidenced by saturating transposon mutagenesis studies (DeJesus et al., 2017). This suggests that the cognate toxins of these essential antitoxins are lethal to Mtb, and such TA systems could be exploited for the development of novel anti-TB therapies.
Here, we focus on the Mtb type II TA module Rv1989c-Rv1990c, in which the antitoxin-encoding gene (Rv1990c) is essential, whereas the cognate toxin-encoding gene (Rv1989c) is dispensable for bacterial growth (DeJesus et al., 2017) (Figure S1A). This TA pair was previously identified by in silico genomic analysis of prokaryotic TA loci and classified as a so-called COG5654-COG5642 TA system (Makarova et al., 2009). It was predicted to encode a RES domain-containing toxin and a cognate antitoxin with a XRE-like HTH domain, typically found in phage repressor proteins (Wood et al., 1990) (Figure S1A). According to a SMART search for analysis of protein domain architectures, the three conserved polar groups (R-E-S) that are predicted to form an active site in Rv1989c are Arg47, Glu69, and Ser126 (Letunic and Bork, 2018). COG5654 or RES domains are widely spread in bacteria and often found in conjunction with various other conserved domains. Interestingly, a plasmid-encoded RES-Xre locus from the legume symbiont Sinorhizobium meliloti was reported to function as an active TA system (Milunovic et al., 2014). The Rv1989c-Rv1990c TA system is particularly interesting because it is significantly upregulated in a variety of stress conditions, including in Mtb persister cells (Keren et al., 2011), during hypoxic stress (Rustad et al., 2008), under starvation (Gupta et al., 2017), and within host macrophages (Homolka et al., 2010). A BLASTp search predicts Rv1989c-Rv1990c-like TA systems in multiple mycobacterial species of the M. tuberculosis complex (Tortoli et al., 2017), with orthologs detected in a limited number of strains of opportunistic non-tuberculous mycobacteria (e.g., M. avium). Homologs of this TA system are also present in environmental prokaryotes, such as Gordonia spp (Figure S1B). This is in line with our previous suggestion that the Rv1989c-Rv1990c TA pair was most likely acquired through horizontal gene transfer with environmental bacteria (Becq et al., 2007). To uncover the mechanism of action of the Rv1989c toxin, we used a combination of biochemical, structural biology, and microbiological methods. We show that Rv1989c encodes a novel NAD+ phosphorylase, an enzymatic activity that has never been described thus far, and reveal a synergistic protective effect of toxin activity and antibiotic treatment in a mouse model of Mtb infection.
Results and Discussion
We first expressed Rv1989c and Rv1990c from different inducible promoters in Escherichia coli. Induction of Rv1989c inhibited E. coli growth on agar plates, unless Rv1990c was co-expressed (Figure S2A). In contrast, wild-type (WT) Mtb expressing Rv1989c from a tetracycline-inducible promoter on an integrated plasmid (Ehrt et al., 2005) did not show impaired growth (Figures 1A and 1B). We hypothesized that the quantity of antitoxin protein expressed from the chromosomally encoded Rv1990c gene was sufficient to neutralize the amount of toxin expressed from both the chromosomal Rv1989c gene and the plasmid-encoded copy of Rv1989c. To test our hypothesis, we constructed a Mtb knockout (KO) mutant with a deletion of the entire Rv1989c-Rv1990c operon (MtbΔTA) by homologous recombination, as outlined in Figures S2B–S2E. Indeed, induction of an ectopic copy of the toxin gene in the MtbΔTA strain completely abolished mycobacterial growth, both on agar medium and in liquid culture (Figures 1A and 1B). Further, MtbΔTA displayed a substantial decrease in colony-forming units (CFUs) after induction of the toxin gene, with a loss of more than 3-Log10 in CFUs over only 4 days, suggesting bactericidal activity of the toxin (Figure 1C). We then tested the viability of MtbΔTA cells following ATc-induced expression of Rv1989c by flow cytometry analysis (Figures 1D and 1E) and fluorescence microscopy (Figure 1F) of bacteria labeled with LIVE/DEAD BacLight stains. Addition of ATc to a culture of MtbΔTA carrying an empty vector had no effect on the proportion of cells permeable to propidium iodine (PI). In contrast, for MtbΔTA cells expressing Rv1989c, the proportion of PI-permeable cells increased from 15% in the absence ATc to 57% in the presence of ATc after 4 days incubation, indicative of the bactericidal activity of Rv1989c. To assess the expression level of the Rv1989c gene in our experimental setup, we compared Rv1989c mRNA levels in WT Mtb versus in the Rv1989c-inducible MtbΔTA strain by real-time qPCR (Figure S2F). Rv1989c mRNA level in the Rv1989c-induced MtbΔTA strain was ≈2-fold higher than that in WT Mtb during exponential growth, and ≈2-fold lower than that in Mtb grown under starvation, a natural stress condition known to induce the TA system (Gupta et al., 2017). Thus, the absence of toxicity in an Mtb WT strain and the real-time qPCR analysis shows that in our experimental setup the Mtb cell death we observed by CFU counting and viability analysis in combination with flow cytometry is not due to an overwhelming production of the toxin. Taken together, these results establish that Rv1989c-Rv1990c can function as a bactericidal TA system in Mtb. Hence, we named the Rv1989c-Rv1990c system mycobacterial cidal toxin (MbcT) and antitoxin (MbcA).
Figure 1.

Rv1989c-Rv1990c Is a Bactericidal TA System in Mtb
(A) WT Mtb or MtbΔTA transformed with pGMC derivatives carrying Rv1990c, Rv1989c, or Rv1989c-Rv1990c as indicated were grown in 7H9 ADC Tween medium. Growth on solid medium was tested by spotting serial 10-fold dilutions on 7H11 OADC agar (−ATc) or the same medium supplemented with anhydrotetracycline (+ATc) to induce transcription from the P1 promoter. Plates were observed after 20 days at 37°C. Shown data are representative of three independent experiments.
(B) Growth of WT Mtb or MtbΔTA transformed with pGMC derivatives carrying Rv1990c, Rv1989c, or Rv1989c-Rv1990c in liquid medium as monitored by turbidity measurements (McFarland units). ATc was added at day 0 to induce ectopic gene expression when indicated. Data are represented as mean of three technical replicates ± SD. Shown data are representative of three independent experiments.
(C) Survival of MtbΔTA strains carrying pGMC-TetR-P1-Rv1989c as measured by CFU scoring of liquid cultures. ATc was added at time 0 to induce expression of Rv1989c. Samples were collected at the seven different time points, cells were washed with growth medium to remove ATc and 10-fold serial dilutions were spotted on agar plates for CFU counting. Data are represented as mean of three independent replicates ± SD.
(D) MtbΔTA strains carrying pGMC-TetR-P1-Rv1989c or an empty vector control were grown for 4 days in the presence of ATc (+ATc) or left untreated (−ATc). Cells were labeled with the LIVE/DEAD BacLight stains (Syto 9; propidium iodide (PI)) and analyzed by fluorescence-activated cell sorting (FACS). The empty vector control was either left unlabeled (negative control) or heat killed by incubation for 1 h at 100°C (positive control). Quadrants were established using the negative (no stain) and positive (heat-killed Mtb) controls as references. Shown data are representative of two independent experiments.
(E) Bar diagram showing the fraction of MtbΔTA cells permeable to PI and Sypro 9 as determined by FACS analysis (see D). Data are represented as mean of two independent replicates ± SD.
(F) Visualization of Syto9 (green) or PI (red) incorporation in MtbΔTA cells following ATc-induced expression of Rv1989c from pGMC-TetR-P1-Rv1989c by spinning disk confocal microscopy (see D). MtbΔTA cells transformed with empty vector were included as a negative control. PI incorporation is indicative of membrane damage. Representative maximum intensity Z projection images are shown. Scale bar, 5 μm.
See also Figures S1 and S2.
To elucidate the molecular basis of MbcT activity, we solved the high-resolution crystal structure of the MbcTA complex (Figures 2A and S3A; Table 1). The complex adopts a donut-like structure composed of three heterotetrameric MbcTA complexes ([MbcTA]2). The oligomerization state and overall shape of the heterododecameric complex were validated by light scattering and by small-angle X-ray scattering (SAXS) (Figures S3B–S3D; Table 2). MbcA folds into a single structured domain consisting of eight α helices, whereas Mb cT exhibits a β sandwich fold formed by six β strands arranged in two opposing antiparallel β sheets that are flanked and connected by nine α helices (Figure 2B). The lateral side of the substrate-binding pocket of MbcT is formed by a stretch of 11 amino acids arranged in a kinked loop pointing inward (α2-β2 loop) with the side chain of Arg47 extending from the tip of the loop. The main interactions in the MbcTA complex are between residues of the MbcA C terminus and residues, mostly arginines (R27, R33, R43, R47, R72), lining a deep central cleft in MbcT (Figure 2B). To validate the role of the C terminus of MbcA in sterically blocking access to the toxin active site, we designed a truncated MbcA version lacking the last ten C-terminal amino acids (residues 104–113). As expected, this variant was not able to neutralize the toxic effect of MbcT in a MtbΔTA background (Figure S3E).
Figure 2.

Crystal Structure of the MbcTA Complex and Homology of MbcT to ARTs and NADases
(A) Overall structure of the MbcTA heterododecamer consisting of three heterotetramers (3x[MbcT-MbcA]2) arranged around a 3-fold symmetry axis (as indicated by a black triangle). The dashed line box in the front view (left) represents one [MbcT-MbcA]2 heterotetramer formed by two MbcT (blue) and two MbcA (yellow) molecules as indicated in the side view (right).
(B) Cartoon representation of the MbcTA complex and zoom-in of the putative NAD+-binding site. N and C termini and secondary structure elements are labeled (left). Part of the α2-β2 loop (G45GRW48) that lines the putative NAD+-binding site is displayed in red. Zoom (right): interactions based on a distance <3.8 Å as calculated by the PISA server (Krissinel and Henrick, 2004) are indicated by dotted lines. The orientation of the MbcTA complex was modified to optimize visualization of the MbcT/MbcA interaction.
(C) Structure-based alignment of the conserved active-site motifs found in three sequence regions of mono- and poly-ADP-ribosyltransferases (ARTs) and NAD+ glycohydrolases (NADases). The C-alpha atoms of Tyr28 in MbcT do not strictly superimpose with those of Tyr97 and Tyr907 in Dtox and PARP1, respectively (see also D). Numbering of the residues refers to UniProt entries: MbcT (UniProt: Y1989), diphtheria toxin (Dtox) (UniProt: P00588), cholera toxin (Ctoxin) (UniProt: P01555), human poly [ADP-ribose] polymerase 1 (PARP1) (UniProt: P09874), the C-terminal toxin domain (tuberculosis necrotizing NAD+ glycohydrolase toxin TNT) of the Mtb outer membrane channel protein CpnT (UniProt: O05442), P. aeruginosa NAD+ glycohydrolase Tse6 (UniProt: Q9I739), and Streptococcus pyogenes NAD+ glycohydrolase SPN (UniProt: D7S065).
(D) Structural comparison of the active site in MbcT, Dtox in complex with NAD+ (PDB: 1TOX), PARP1 (PDB: 4DQY), and CpnT (PDB: 4QLP). Conserved residues are colored according to their localization in the three distinct regions (cf. C).
See also Figures S3 and S4.
Table 1.
Crystallographic Data Collection, Phasing, and Refinement Statistics
| Native Dataset | S-SAD Dataset | |
|---|---|---|
| Data collection | ||
| Space group | P63 | P63 |
| Cell dimensions (Å,°) | 105.3, 105.3, 108.7 | 105.3, 105.3, 108.7 |
| 90.0, 90.0, 120 | 90.0, 90.0, 120 | |
| Wavelength (Å) | 0.976 | 2.479 |
| Resolution (Å) | 9.99–1.80 (1.86–1.80)a | 91.70–2.51 (2.61–2.51) |
| Rmerge (%) | 0.0562 (1.364) | 0.123 (0.667) |
| I/σ(I) | 21.6 (1.5) | 42.5 (7.3) |
| Completeness (%) | 98.8 (90.2) | 89.3 (70.3) |
| Redundancy | 10.0 (7.5) | 57.6 (49.2) |
| CC1/2 | 0.999 (0.497) | 0.999 (0.920) |
| Total number of reflections | 620,194 (42,332) | 1,225,773 (92,688) |
| Unique reflections | 62,157 (5,639) | 21,269 (1,882) |
| Refinement | ||
| Rwork (%) | 16.23 (27.26) | – |
| Rfree (%) | 21.11 (31.28) | – |
| No. atoms | ||
| Total | 4,865 | – |
| Macromolecules | 4,569 | – |
| Ligands | 18 | – |
| Waters | 278 | – |
| No. protein residues | 589 | – |
| B-factorsb(Å2) | ||
| Macromolecules | 42.4 | – |
| Solvent | 62.8 | – |
| RMSDb | ||
| Bond lengths (Å) | 0.007 | – |
| Bond angles (°) | 0.990 | – |
| Ramachandranc (%) | ||
| Most favored | 99.0 | – |
| Allowed | 1.0 | – |
| Outliers | 0.0 | – |
Values in parentheses indicate the highest-resolution shells and their statistics
Values from PHENIX (Adams et al., 2010)
Values from MOLPROBITY (Chen et al., 2010)
Table 2.
SAXS Data Collection and Derived Parameters
| MbcTA | |
|---|---|
| Data collection | |
| Instrument | P12 at EMBL/DESY, storage ring PETRA III, Germany |
| Beam geometry | 0.2 × 0.12 mm2 |
| Wavelength (Å) | 1.24 |
| q-range (Å−1) | 0.008–0.47 |
| Exposure time (ms) | 20 × 50 |
| Concentration range (mg mL−1) | 0.6–7.1 |
| Temperature (K) | 283 |
| Structural parametersa | |
| I(0) (arbitrary units) (from P(r)) | 31,320 ± 10 |
| Rg (from P(r)) (Å) | 41 ± 1 |
| I(0) (arbitrary units) (from Guinier) | 31,340 ± 30 |
| Rg (Å) (from Guinier) | 41 ± 1 |
| Dmax (Å) | 114 |
| Porod volume (103 Å3) | 262 |
| Molecular mass determinationa | |
| MMPOROD (from Porod volume) (kDa) | 154 ± 15 |
| MMsaxs (from I(0), kDa) | 110 ± 20 |
| MMDAM (from bead model, kDa) | 170 ± 35 |
| Calculated monomeric MM from sequence (kDa) | 197.2 |
| Software employed | |
| Primary data reduction | Automated radial averaging |
| Data processing | PRIMUS |
| Ab initio analysis | DAMMIN |
| Validation and averaging | SASRES, DAMAVER |
| Computation of model intensities | CRYSOL |
| SASBDB entry code | SASDD33 |
Reported for MbcTA at 0.6 mg mL−1
The closest structural relatives to MbcT are ADP-ribosyltransferases (ARTs), in particular bacterial ART toxins and poly (ADP-ribose) polymerases (PARPs) (Aravind et al., 2015, Palazzo et al., 2017, Simon et al., 2014) (Figure S4A). ARTs catalyze the transfer of an ADP-ribose group from an NAD+ donor molecule to a substrate (proteins, DNA, or RNA) and release free nicotinamide (NAA). Bacterial ART toxins are classified into two major groups based on conserved active-site motifs distributed across three regions. The diphtheria toxin (ARTD) group has an H-Y/Y-E motif, also found in PARPs, whereas the cholera toxin (ARTC) group contains an R-S-E motif (Aravind et al., 2015, Simon et al., 2014) (Figure 2C). The structural hallmark of ARTs is a central cleft bearing a conserved NAD+-binding pocket (Aravind et al., 2015, Han and Tainer, 2002). An NAD+-binding pocket is also present in NAD+ glycohydrolases (NADases), such as the bacterial exotoxins TNT (Sun et al., 2015), SPN (Ghosh et al., 2010), and Tse6 (Whitney et al., 2015), but the overall structural homology of MbcT with NADases is less obvious (Figure S4A). Structural superimposition with selected ARTs and NADases suggests that MbcT could consume NAD+ as well, and pinpoints Arg27 in region 1, and Tyr28 and Tyr58 in region 2, as potential NAD+-binding residues (Figures 2C and 2D). Yet, the region-3 residue, which is thought to confer substrate recognition and specificity, is replaced by a glycine (Gly152) in MbcT (Figure 2C). To investigate the functional importance of the putative NAD+-binding site of MbcT and the potential catalytic function of the RES motif (R47-E69-S126), we substituted single residues of MbcT to alanine and assessed the effect on growth inhibition of MtbΔTA. Non-toxic MbcT-R27A, MbcT-R47A, and MbcT-Y58A mutants did not affect the growth of MtbΔTA or E. coli, thus establishing the crucial role of these individual residues for MbcT-catalyzed growth inhibition (Figures S4B and S4C). Surprisingly, Ser126 was not essential for toxicity, whereas MbcT-Y28A and MbcT-E69A retained limited toxin activity. Taken together, these results suggest that MbcT toxicity involves NAD+, but that the catalytic mechanism underlying toxin activity is divergent from that of ART enzymes and NADases.
To identify substrates of MbcT and explore its NAD+-binding activity in vitro, we sought to purify the WT, recombinant MbcT protein. To overcome cell toxicity, we co-expressed full-length WT MbcT with a His-tagged, C-terminal truncation of MbcA (MbcAΔ112–113). WT MbcT is only weakly associated with His-MbcAΔ112–113, allowing for subsequent isolation of WT MbcT by salt-induced dissociation of the His-MbcAΔ112–113-MbcT complex (Figure S5A). In addition to WT MbcT, we also purified the MbcT active-site mutant R27E (MbcT-R27E) from E. coli as a control (Figures S5B and S5C). This variant of MbcT was non-toxic to MtbΔTA cells (Figure S5D). We then incubated recombinant protein with different bacterial cell fractions in the presence of 32P-labeled NAD+ to probe for ADP-ribosylation of cellular protein but did not detect 32P-ADP-ribose-incorporation into the protein fractions (Figure S5E). MbcT also did not modify nucleic acid substrates, in contrast to the mycobacterial DNA-modifying TA toxin DarT (Jankevicius et al., 2016) (Figure S5F).
In addition to NAD+ degradation and ADP-ribose (Appr) production, we observed the appearance of an unknown reaction product, dependent on the MbcT concentration (Figure 3A). Interestingly, supplementing the MbcT reaction buffer with sodium phosphate markedly enhanced NAD+ degradation into NAA and the hitherto unknown reaction product (Figure 3B), whereas the MbcT R27E mutant or the MbcTA complex did not trigger NAD+-turnover (Figures 3B and S5F). We performed high resolution mass spectrometry and nuclear magnetic resonance experiments, which identified the additional reaction product as ADP-ribose-1″-phosphate (Appr1p; [M-H]− m/z = 638.0301) (Figure 3C). To our knowledge, MbcT represents the first reported enzyme with NAD+ phosphorylase activity (Figure 3D).
Figure 3.

Enzymatic Activity of MbcT
(A) Representative autoradiograph of a TLC plate showing MbcT-mediated depletion of 32P-NAD+ over time and simultaneous accumulation of 32P-ADP-ribose (Appr) and a secondary reaction product (white arrow). The dotted line indicates the position where samples were applied to the plate. Similar results were obtained in two independent experiments.
(B) Representative HPLC chromatograms of the reaction products of NAD+ (0.5 mM) with MbcT (0.7 μM) in the presence (orange) or absence (black) of sodium phosphate (30 mM). The white arrow indicates the reaction product formed in addition to nicotinamide (NAA) (cf. A). MbcT-R27E does not degrade NAD+ in sodium phosphate (30 mM) buffer (gray line). NAD+, Appr and NAA were identified by retention time comparisons with standards. The observed Appr is an impurity found in the commercial substrate. Similar results were obtained in three independent experiments.
(C) 1H-31P HSQC31 spectra of the reaction products of NAD+ (5 mM) with MbcT (10 mM) and, for reference, of pure Appr (5 mM). Phosphate atoms from the ADP-ribose moiety are colored green, whereas the phosphate atom derived from orthophosphate is highlighted in orange.
(D) Proposed reaction mechanism of MbcT-mediated NAD+ phosphorolysis yielding ADP-ribose-1″-phosphate (Appr1p) and NAA.
(E) Kinetics of NAD+ phosphorolysis by MbcT (50 nM). Km and Vmax values were determined by nonlinear regression analysis with the Michaelis-Menten equation.
(F) Comparison of initial velocity (V0) of NAD+ phosphorolysis of WT MbcT (50 nM) and MbcT-R27E (50 nM) in the presence or absence of sodium phosphate (50 mM). The initial velocities were determined at a substrate concentration of 100 μM. For data in (E) and (F), data are represented as mean of eight and four independent replicates ± SD, respectively.
See also Figures S5 and S6.
A kinetic analysis of MbcT activity, based on NAD+ consumption at saturating orthophosphate conditions, yielded a Km of 110 ± 8 μM (Figure 3E). The turnover number of MbcT for NAD+ phosphorolysis (kcat) was 167 ± 3 s−1 (Figures S6A and S6B). By contrast, MbcT-R27E did not show any detectable NAD+ turnover establishing the essentiality of Arg27 for NAD+ phosphorolysis (Figure 3F). With a catalytic efficiency (kcat/Km) of 1.5 × 106 M−1s−1, MbcT is one of the most effective NAD+-degrading toxins characterized to date, more potent than diphtheria toxin (5 × 105 M−1s−1) (Perikh and Schramm, 2004) and the mycobacterial NADase TNT (8.4 × 104 M−1s−1) (Sun et al., 2015). The high catalytic efficiency of MbcT implies that this enzyme has specifically evolved to carry out NAD+ phosphorolysis.
To determine whether MbcT exerts its toxic effect via NAD+ turnover, we measured the levels of NAD+ in MtbΔTA expressing mbcT. We observed rapid depletion of intracellular NAD+ upon induction of mbcT expression, whereas control strains expressing no toxin or the MbcT-R27E inactive mutant exhibited no decrease in intracellular NAD+ levels (Figure 4A). We also exploited the mbcT-inducible system described above to evaluate MbcT toxicity in vivo. First, we showed that, unlike TNT (Sun et al., 2015), ectopic expression of mbcT in WT Mtb had no deleterious effect on infected human monocyte-derived macrophages (hMDM) (Figure 4B). We then infected hMDM with MtbΔTA transformed with a control vector or a plasmid carrying ATc-inducible mbcT. Induction of mbcT expression 2 days after infection resulted in more than a 10-fold decrease in the intracellular bacterial load (Figure 4C). Next, we infected immune-deficient SCID mice, which are highly sensitive to Mtb infection, with the same MtbΔTA strain. Doxycycline-mediated induction of mbcT after MtbΔTA infection prolonged the survival of infected mice by ∼40% compared to controls without doxycycline (Figure 4D).
Figure 4.

MbcT Activity Can Be Bactericidal In Vivo
(A) Relative intracellular NAD+ levels were measured in MtbΔTA cells transformed with empty vector (Ctrl) or with pGMC derivatives expressing WT MbcT or MbcT-R27E, as indicated. Cultures were grown in the absence (−ATc) or presence of ATc (+ATc) to induce mbcT gene expression from the P1 promoter. Cellular NAD+ was extracted 24 h after induction and measured by a coupled bioluminescence assay. Data are represented as mean of three independent replicates ± SD.
(B) Viability of human macrophages infected with WT Mtb strains carrying a pGMC-TetR-P1-mbcT construct (MbcT), was measured by flow cytometry after labeling with Zombie Aqua Viability Kit. Infected macrophages were cultivated in the presence (+ATc) or absence of ATc (−ATc) to induce mbcT gene expression. MtbΔTA carrying an empty vector was included as control (Ctrl). Data are represented as mean of three independent replicates ± SD.
(C) ATc-induction of mbcT in MtbΔTA results in mycobacterial killing inside human monocyte-derived macrophages. Cells were infected at a multiplicity of infection of 0.1 bacteria/cell with MtbΔTA strains carrying pGMC-TetR-P1-mbcT (mbcT) or empty vector (Ctrl). Toxin expression was induced 2 days of infection by addition of ATc, when indicated.
(D) ATc-induction of mbcT expression in MtbΔTA reduces mycobacterial virulence and improves host survival in immune-deficient SCID mice. Mice were infected with MtbΔTA strains transformed with pGMC-TetR-P1-mbcT (mbcT) or empty vector (Ctrl). Toxin expression was induced by addition of doxycycline (Doxy) in the drinking water of the animals from 7 days onward prior to infection. Mouse survival was followed over time using ten mice per condition. Statistical analysis was performed using the log-rank (Mantel-Cox) test (∗∗∗∗<0.0001).
(E) Number of CFU isolated from the lungs of immune-competent C57BL/6 mice infected with MtbΔTA carrying pGMC-TetR-P1-mbcT (mbcT) or empty vector (Ctrl). At day 21, mice were given isoniazid (INH) or Doxy by daily gavage for 10 days, as indicated. Data are represented as mean of at least four independent replicates ± SD (n = 4–8 mice/group). NS or stars indicate significance as determined by a Student’s t test (∗<0.05; ∗∗<0.01; ∗∗∗<0.001).
In addition, we infected immune-competent C57BL/6 mice with the same bacterial strains and induced mbcT expression with doxycycline 21 days after infection. At this stage, the Mtb load in the lungs reaches a plateau. MbcT induction resulted in the potent killing of Mtb (5-fold reduction in CFUs). Further, MbcT enhanced the therapeutic efficacy of the frontline anti-TB drug isoniazid (INH). Treatment with INH alone led to a 10-fold reduction in CFU relative to untreated mice, whereas INH treatment combined with mbcT expression led to a 100-fold reduction in CFUs, indicative of a synergistic effect (Figure 4E). These results indicate that MbcT is highly toxic to Mtb in vivo when not neutralized by MbcA. As such, small inhibitory molecules able to dislocate the MbcTA complex could be promising candidates for the development of novel therapeutics to control Mtb infection.
The molecular mechanism underpinning MbcT toxicity, NAD+ phosphorolysis, is unprecedented for TA modules. To our knowledge, MbcTA is also the first TA system that degrades an essential cellular metabolite resulting in rapid cell death. Yet, the biological role of the MbcTA system remains elusive. We did not detect any particular phenotype in our MbcTA-KO mutant in a variety of stress conditions in vitro and in vivo (data not shown), so the relevance of the MbcTA system in the Mtb life cycle is difficult to anticipate. This might be because this system would need to be inactivated together with other TA pairs in order to observe a phenotype, as reported for MazEF TA pairs (Tiwari et al., 2015), or because we did not expose the MtbΔTA mutant to the relevant physiological stress.
Strikingly, the Mycobacterium phage Ibhubesi encodes a MbcA homolog, namely PBI_IBHUBESI_52 (Figures S1B and S1C). It is tempting to speculate that the mbcT-mbcA TA system was acquired by Mtb, and possibly other bacteria, to inhibit bacteriophage propagation by triggering self-intoxication, reminiscent of abortive infection TA systems (Dy et al., 2014). In line with this, mycobacteriophage Ibhubesi could have acquired the IBHUBESI_52 gene to neutralize the bacterial defense. The IBHUBESI_52 protein would then be an antidefense protein, mechanistically different but functionally similar to the Gp4.5 protein of bacteriophage T7 (Sberro et al., 2013). Whether TA systems are indeed capable of inducing altruistic killing to prevent phage attack is still under debate (Song and Wood, 2018). Further experiments are needed to test this hypothesis.
Our study identifies MbcT as a highly efficient NAD+ phosphorylase. Further, we show that MbcT activity can be bactericidal in Mtb, in line with previous reports demonstrating that mbcA is an essential gene (DeJesus et al., 2017), and NAD+ depletion is lethal in mycobacteria (Kim et al., 2013, Rodionova et al., 2014, Vilchèze et al., 2010). During the revision process of this paper, Skjerning et al. (2018) reported that plasmid-based expression of three prokaryotic RES-domain containing toxins, including MbcT, resulted in growth arrest of E. coli. Interestingly, the RES toxin from Photorhabdus luminescens (RESPl) triggers depletion of intracellular NAD+ upon expression in E. coli. Although the enzymatic activity of RESPl has not been biochemically validated, it supports our hypothesis that NAD+ degradation is a more general mechanism utilized by prokaryotic TA toxin systems to interfere with bacterial growth. The authors also report the crystal structure of a RES toxin in complex with its cognate Xre antitoxin from Pseudomonas putida (RESPp-XrePp), in which the individual TA components share significant structural similarity with MbcT and MbcA, respectively. The putative NAD+ binding pocket of the RESPp toxin is blocked by the C-terminal region of the Xre antitoxin as observed in the MbcTA complex, further highlighting the functional similarities within the RES-Xre TA systems. However, the toxin and antitoxin proteins assemble into complexes with a different quaternary structure, namely a heterohexameric (RESPp)2-(XrePp)4 complex opposed to the heterododecameric MbcT6-MbcA6 complex.
To conclude, our findings pave the way for future exploration of NAD+ phosphorylases in other organisms, and for functional studies of this new class of enzymes in the context of bacterial metabolism. This work also enables the search for small molecule inhibitors that disrupt the MbcTA complex or inactivate the MbcA antitoxin (Williams and Hergenrother, 2012), which could be used in combination with standard drug regimens to combat TB, the most devastating infectious disease globally. More generally, identifying and targeting bactericidal TA systems in bacterial pathogens might illuminate approaches to treat other infectious diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GroEL (E. coli) monoclonal antibody (clone 9A1/2) | Enzo Life Sciences | Cat# ADI-SPS-870-D; RRID: AB_2039163 |
| Bacterial and Virus Strains | ||
| Escherichia coli DH5α | Thermo Fisher Scientific | Cat# 18265017 |
| Escherichia coli BL21(DE3) CodonPlus-RIL | Agilent | Cat# 230240 |
| Escherichia coli W3110 | Genevaux laboratory Hayashi et al., 2006 | N/A |
| Mycobacterium smegmatis mc2155 groEL1ΔC | Noens et al., 2011 | N/A |
| Mycobacterium tuberculosis H37Rv | ATCC | ATCC 27294 |
| Mycobacterium tuberculosis H37Rv Δ(Rv1990c-Rv1989c)::KanR | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nicotinamide adenine dinucleotide [adenylate-32P] | Hartmann Analytic | Cat# ARP0141 |
| Q5 High Fidelity Polymerase | NEB | Cat# M0491 |
| Anhydrotetracycline hydrochloride | Merck | Cat# 37919 |
| Doxycycline hydrochloride | Merck | Cat# D3447 |
| DNase I | Thermo Fisher Scientific | Cat# AM2222 |
| Superscript III Reverse transcriptase | Thermo Fisher Scientific | Cat# 18080044 |
| SYBER Green qPCR Premix ex Taq | Ozyme | Cat# TAKRR420A |
| Thrombin protease | Sigma-Aldrich | Cat# T4648 |
| Protease-Inhibitor Mix HP | Serva | Cat# 39106 |
| Deoxyribonuclease I | Sigma-Aldrich | Cat# DN25 |
| Complete EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat# 11873580001 |
| Recombinant protein: TEV protease | M. Wilmanns lab | N/A |
| Recombinant protein: M. tuberculosis H37Rv MbcA | This study | NP_216506.1 |
| Recombinant protein: M. tuberculosis H37Rv MbcT | This study | NP_216505.1 |
| Recombinant protein: M. tuberculosis H37Rv MbcT-R27E (aa 1–186, ref# NP_216505.1) | This study | N/A |
| Critical Commercial Assays | ||
| NAD/NADH-Glo Detection Reagent | Promega | Cat# G9071 |
| LIVE/DEAD BacLight Bacterial Viability Kits | Thermo Fisher Scientific | Cat# L7007 |
| Zombie Aqua Fixable Viability Kit | BioLegends | Cat# 423101 |
| RNeasy mini kit | QIAGEN | Cat# 74104 |
| RNeasy Protect Bacteria Mini Kit | QIAGEN | Cat# 74524 |
| Penta His HRP conjugate kit | QIAGEN | Cat# 34460 |
| DNeasy Blood & Tissue kit | QIAGEN | Cat# 69504 |
| Deposited Data | ||
| Raw image date | This study; Mendeley Data | https://doi.org/10.17632/y6ynjm5sf3.1 |
| MbcT/MbcA structure | This paper | PDB: 6FKG |
| MbcT/MbcA SAXS data | This paper | SASBDB: SASDD33 |
| Experimental Models: Cell Lines | ||
| Primary Human monocytes from healthy donors | Etablissement Français du Sang | Contract Nr. 121/PVNT/TOU/IPBS01/2009-0052 |
| Experimental Models: Organisms/Strains | ||
| Mouse: SCID CB17/Icr-Prkdcscid/IcrIcoCrl | Charles River | RRID: IMSR_CRL:236 |
| Mouse: C57BL/6J | Charles River | RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| See Table S1 for list of primers used in this study | N/A | N/A |
| Recombinant DNA | ||
| See Table S2 for list of plasmids used in this study | N/A | N/A |
| Software and Algorithms | ||
| SMART | Letunic and Bork, 2018 | RRID: SCR_005026 |
| Jalview 2.10 | Waterhouse et al., 2009 | RRID: SCR_006459 |
| Pymol 2.1.1 | Schrödinger | RRID: SCR_000305 |
| Prism 8 | Prism | https://www.graphpad.com/scientific-software/prism/ |
| OmniSEC | Malvern Panalytical | https://www.malvernpanalytical.com/en/ |
| MassHunter B.07.00 | Agilent | RRID: SCR_015040 |
| OpenLAB CDS ChemStation | Agilent | RRID: SCR_015742 |
| Topspin 3.5 | Bruker | RRID: SCR_014227 |
| 7500 Real-time PCR Software v2.3 | Thermo Fisher Scientific | RRID: SCR_014596 |
| XDS/XSCALE | Kabsch, 2010 | RRID: SCR_015652 |
| CCP4 suite | Winn et al., 2011 | RRID: SCR_007255 |
| SHELX | Sheldrick, 2008 | RRID: SCR_014220 |
| ARP/wARP | Perrakis et al., 1999 | http://www.embl-hamburg.de/ARP/ |
| REFMAC | Vagin et al., 2004 | RRID: SCR_014225 |
| PHENIX | Adams et al., 2010 | RRID: SCR_014224 |
| PDB_REDO | Joosten et al., 2014 | https://pdb-redo.eu/ |
| TLSMD | Painter and Merritt, 2006 | http://skuld.bmsc.washington.edu/∼tlsmd/ |
| Coot | Emsley and Cowtan, 2004 | RRID: SCR_014222 |
| Molprobity | Chen et al., 2010 | RRID: SCR_014226 |
| ATSAS 2.7 | Petoukhov et al., 2012 | RRID: SCR_015648 |
| PDBeFOLD | Krissinel and Henrick, 2004 | http://www.ebi.ac.uk/msd-srv/ssm/ |
| ImageJ | https://imagej.net/Welcome | RRID: SCR_003070 |
| FlowJo | FlowJo | RRID: SCR_008520 |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Annabel Parret (ahaparret@gmail.com).
Experimental Model and Subject Details
Bacterial strains
M. smegmatis mc2155 groEL1ΔC and M. tuberculosis H37Rv (WT) and Mtb mutant strains were routinely grown at 37°C in Middlebrook 7H9 medium (Difco) supplemented with 10% albumin-dextrose-catalase (ADC, Difco) and 0.05% Tween 80 (Sigma-Aldrich) or on Middlebrook 7H11 agar medium (Difco) supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC, Difco). When required, kanamycin (50 μg ml−1), hygromycin (50 μg ml−1), streptomycin (25 μg ml−1) or zeocin (25 μg ml−1) were added to the culture media. E. coli strains DH5α, (DE3) CodonPlus RIL and W3110 (Hayashi et al., 2006) were grown at 37°C in LB (DH5α; W3110) or Terrific Broth medium (Melford) ((DE3) CodonPlus RIL) supplemented with kanamycin (30 μg ml−1), chloramphenicol (34 μg ml−1) or ampicillin (100 μg ml−1) when required. Induction of gene expression is detailed in the Method Details section.
Human Cell Culture
Human monocytes were obtained from healthy blood donors (Etablissement Français du Sang, EFS, Toulouse, France) with written informed consent (under EFS Contract n°121/PVNT/TOU/IPBS01/2009-0052, which was approved by the French Ministry of Science and Technology, agreement nr. AC2009-921, following articles L1243-4 and R1243-61 of the French Public Health Code). Monocytes were prepared following a previously published procedure (Troegeler et al., 2014). Briefly, cells were purified using CD14 microbead positive selection and MACS separation columns (Miltenyi Biotec), according to manufacturer’s instructions. For differentiation of monocyte-derived macrophages, monocytes were allowed to adhere to glass coverslips (VWR international) in 6-well plates (ThermoFischer Scientific), at 1.5x106 cells/well, for 1 h at 37°C in pre-warmed RPMI-1640 medium (GIBCO). The medium was then supplemented with 10% Fetal Bovine Serum (Sigma-Aldrich), 1% sodium pyruvate (GIBCO), 0.1% β-mercaptoethanol (GIBCO) and 20 ng ml−1 human Macrophage Colony-Stimulating Factor (Miltenyi Biotec). Cells were allowed to differentiate for seven days at 37°C under 5% CO2 atmosphere.
Experimental animals
C57BL/6J and SCID CB17/Icr-Prkdcscid/IcrIcoCrl mice were purchased from Charles River and maintained under specific germ-free conditions in the IPBS specific animal facility at 22°C under a 12 h light/dark cycle for at least one week before starting experiments. All animal experiments were performed in animal facilities that meet all legal requirements in France and by qualified personnel in such a way to minimize discomfort for the animals. All procedures including animal studies were conducted in strict accordance with French laws and regulations in compliance with the European community council directive 68/609/EEC guidelines and its implementation in France. All protocols were reviewed and approved by the Comité d’Ethique Midi-Pyrénées (reference MP/03/07/04/09) and the Comité d’Ethique FRBT (APAFIS#1269).
Method Details
Protein homology searches
Rv1989c-Rv1990c-like TA systems were identified in bacterial genomes using NCBI’s standard protein BlastP searches against the non-redundant protein sequence (nr) database. Hits from M. tuberculosis genomes were excluded. From the resulting top 100 hits, only those homologs were withheld for which the hypothetical toxin and antitoxin were encoded by adjacent genes. Selected protein sequences were retrieved from the UniProt database and re-aligned using MAFFT (Katoh and Standley, 2013) within the Jalview software package (Waterhouse et al., 2009). Pairwise protein sequence identities were calculated using the Pairwise Alignment tool in Jalview. Conserved protein were identified with InterPro (Finn et al., 2017). The N-terminal HTH domain of the antitoxin, which is not detected by InterPro, was identified using the HTH motif prediction program available from the NPS@ web server (Combet et al., 2000).
E. coli viability assays
E. coli strain W3110 containing p29SEN or p29SEN-Rv1990c was co-transformed with empty vector (pMPMK6) or pMPMK6-Rv1989c (Table S2), grown to mid-log phase, serially diluted and spotted on agar plates supplemented with appropriate antibiotics. 1% arabinose or 5 μM IPTG or were used to induce expression of Rv1989c and Rv1990c, respectively. Images were taken after overnight incubation at 37°C. Raw images are available on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1).
Construction of M. tuberculosis mutants
Mutant strains of M. tuberculosis H37Rv were constructed by allelic exchange using recombineering (van Kessel et al., 2007). Briefly, two ∼0.5-kb DNA fragments flanking the mbcA-mbcT operon were amplified by PCR from M. tuberculosis H37Rv genomic DNA, using the primers set 1990cAm-Fw/1990cAm-Rv or 1990cAv-Fw/1990cAv-Rv, respectively. These two DNA fragments were inserted into pGem5Z (Promega) flanking a kanamycin-resistance cassette. The recombination substrate was recovered by enzymatic digestion and agarose gel purifications. The recipient strain for recombineering was a derivative of M. tuberculosis H37Rv carrying two plasmids: the integrative plasmid pGMCS-P1-Rv1990c, constitutively expressing mbcA, and pJV53H, a hygromycin-resistant pJV53-derived plasmid expressing recombineering enzymes (van Kessel et al., 2007) (Table S2). This strain was grown in growth medium supplemented with hygromycin until mid-log phase and expression of recombineering enzymes was induced by 0.2% acetamide (Sigma-Aldrich) overnight at 37°C. After induction, electrotransformation was performed with 100 ng of the linear DNA fragment for allelic exchange. After 48 h incubation at 37°C, mycobacteria were plated onto agar plates supplemented with kanamycin. Kanamycin-resistant clones were harvested, cultured in growth medium supplemented with kanamycin and verified to carry the expected allele replacement by colony PCR, using appropriate primers. The pJV53H plasmid was spontaneously lost by serial rounds of culture without hygromycin. Plasmid pGMCS-P1-Rv1990c was removed by transformation with pGMCZ, a similar vector but carrying resistance to zeocin, resulting in the deleted strain WT Mtb Δ(Rv1990c-Rv1989c)::KanR /pGMCZ, further abbreviated as MtbΔTA.
Cloning of expression constructs
M. tuberculosis expression constructs
Rv1989c, Rv1990c or both genes were amplified by PCR using Mtb H37Rv genomic DNA as template and primer pairs clo-rv1990-attB2/clo-Rv1990-attB3, clo-rv1989-attB2/clo-Rv1989-attB3 or clo-rv1990-attB2/clo-Rv1989-attB3, respectively (Tables S1 and S2). Plasmids pGMCS-TetR-P1-Rv1990c, pGMCS-TetR-P1-Rv1989c or pGMCS-TetR-P1-Rv1990c-Rv1989c were constructed by multisite gateway recombination (Schnappinger and Ehrt, 2014), using plasmid pDE43-MCS as destination vector. These plasmids are integrative vectors (insertion at the attL5 mycobacteriophage insertion site in the glyV tRNA gene) and express Rv1990c, Rv1989c or Rv1990c-Rv1989c under the control of P1, a tetracycline-inducible promoter (Ehrt et al., 2005) (Table S2). In pGMCS-TetR-P1-Rv1990c and pGMCS-TetR-P1-Rv1990c-Rv1989c, the sequence harboring the natural Shine-Dalgarno sequence of Rv1990c (AGGAAGACAGGCTGCCC) was placed upstream of the AUG codon of Rv1990c. In pGMCS-TetR-P1-Rv1989c, this same sequence was placed upstream of the GUG start codon of the Rv1989c single open reading frames (see sequence of oligonucleotide clo-rv1989c-B2 in Table S1). The empty vector pGMC-TetR-P1 was also constructed by multisite gateway recombination, but with no gene inserted in front of the P1 promoter.
Generation of a construct for expression of Rv1989c and Rv1990c lacking the last ten codons (pGMCS-TetR-P1-ΔRv1990c(104-113)-Rv1989c) was achieved by PCR amplification of two overlapping DNA fragments using pGMCS-TetR-P1-Rv1990c-Rv1989c as template and the primer pairs clo-rv1990-attB2/1990c-del104_113-Rv or 1990c-del104_113-Fw/clo-rv1989-attB3 (Table S2). The two purified fragments were mixed and used as template for a second round of PCR with the oligonucleotide pair clo-rv1990-attB2/clo-Rv1989-attB3. The final construct was generated by multiple gateway cloning using the resulting purified PCR fragment.
Directed mutagenesis of Rv1989c was performed by PCR amplification of two overlapping DNA fragments carrying the required mutation using pGMCS-TetR-P1-Rv1989c as template and the primer pair clo-rv1989-attB2/Rv1989c_XnA_rev or Rv1989c_XnA_for/clo-rv1989-attB3 (Table S2). Purified PCR fragments were mixed and used as templates for a second round of PCR with the primer pair clo-rv1989-attB2/clo-Rv1989-attB3. The resulting fragments were used for multiple gateway cloning to construct pGMCS-TetR-P1-Rv1989c derivatives with the desired mutations.
M. smegmatis expression constructs
The Rv1990c-Rv1989c operon was PCR-amplified using Q5 High Fidelity Polymerase (New England Biolabs) from Mtb H37Rv genomic DNA using the primer set Rv1990c_NcoI and Rv1989c_HindIII (Table S2). DNA fragments were ligated into pMyNT (Table S1) using NcoI/HindIII restriction enzymes, generating pMyNT-MbcTA encoding N-terminally His6-tagged MbcA and untagged MbcT.
E. coli expression constructs
Rv1989c and Rv1990c were PCR-amplified using Phusion High-Fidelity DNA polymerase (New England Biolabs) from M. tuberculosis H37Rv genomic DNA and ligated into expression vectors pnEK and pnEA-His, respectively, using NdeI/BamHI restriction enzymes (Table S2). Resulting constructs, pnEK-MbcT and pnEA-His-MbcAΔ112−113, encoding untagged MbcT and N-terminally His6-tagged MbcAΔ112−113 were cloned using primers DF101/DF102 and SpeI/XbaI ligation of a synthetic fragment consisting of MbcAΔ112−113 (gBlock; Integrated DNA Technologies), respectively. the mbcT gene was first cloned in pET-28a(+) using restriction enzymes NcoI/HindIII. MbcT mutants (pET-28a(+)-MbcT constructs) were generated by site-directed mutagenesis (Table S2). To generate constructs for the E. coli toxicity rescue assays, Rv1989c was PCR-amplified using Phusion High-Fidelity DNA polymerase (New England Biolabs) with primers for1989-2 and rev1989-2. The PCR product was cloned as an EcoRI/HindIII fragment under the control of an arabinose-inducible promoter (pBAD) into pMPMK6 vector (Mayer, 1995) digested with the same enzymes (Table S2). The mbcA gene was PCR-amplified using Phusion High-Fidelity DNA polymerase (New England Biolabs) using primers for1990-2 and rev1990-2, and cloned as an EcoRI/HindIII fragment under the control of an IPTG-inducible promoter into p29SEN vector (Genevaux et al., 2004) digested with the same enzymes. E. coli strain DH5α was used for all cloning experiments.
Viability Staining and Flow Cytometry
Exponentially growing cultures (OD600 between 0.05 and 0.2) of strain MtbΔTA containing plasmid pGMCS-TetR-P1 (empty vector) or pGMCS-TetR-P1-Rv1989c were divided in two: half was left in standard growth medium (uninduced cultures) and the other half was treated with 200 ng ml−1 of anhydrotetracycline (ATc) to induce expression from the P1 promoter. After various times post-induction, samples were harvested and centrifuged to remove residual ATc. Cells were resuspended in PBS buffer and dilutions were plated on 7H11 OADC agar, to measure colony-forming units. For labeling with LIVE/DEAD BacLight (Molecular Probes) dyes, cells were harvested 4 days post-ATc induction. Cells were centrifuged, resuspended in PBS buffer and stained as recommended by the manufacturer. Labeled cells were either observed by confocal microscopy using an Andor/Olympus spinning disk microscope with an Olympus 100x oil immersion objective or by fluorescence-activated cell sorting using a BD FACS Aria Fusion flow cytometer. Image analysis was performed using ImageJ software and flow cytometry data analysis using FlowJo software. Raw images are available on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1).
RT-qPCR
RT-qPCR quantification of mbcT mRNA was performed on total RNA extracted from Mtb WT cultures grown to exponential phase (OD600 of 0.5) at 37°C in 7H9 + 10% ADC + 0.05% Tween-80. For samples from starved cells, cultures were washed and kept for 24h in suspension in PBS buffer before RNA extraction. In addition, total RNA was extracted from MtbΔTA cultures carrying plasmid pGMCS-TetR-P1 (empty vector) or pGMCS-TetR-P1-Rv1989c, 6 or 24 h post-induction with ATc. RNA was prepared using the RNeasy kit (QIAGEN) following manufacturer’s instructions with slight modifications (Levillain et al., 2017). RNA samples were treated for 30 min with 2U of Turbo DNase (Turbo DNA free kit, Ambion). The amount and purity of RNA were quantified using a NanoDrop ND-1000 apparatus (ThermoFischer Scientific) by measuring absorbance at 260/280 nm. Double-stranded cDNA was reverse-transcribed using the superscript III Reverse Transcriptase kit (Invitrogen), according to the manufacturer’s protocol. For real-time qPCR, specific primers were designed and PCR reactions were performed using SYBR Green Premix Ex Taq (Ozyme), according to the manufacturer’s protocol. All real-time qPCR reactions were carried out using a 7500 Real-Time PCR System and data were analyzed using the 7500 Software version 2.3 (Applied Biosystems). PCR array data were calculated by the comparative cycle threshold method, normalized with the rpoB housekeeping gene, and expressed as mean fold change in experimental samples relative to levels in Mtb WT grown in 7H9 ADC tween medium.
Protein Expression and Purification
For expression of the intact MbcT-MbcA complex, pMyNT-MbcTA plasmid DNA was electroporated into M. smegmatis mc2155 groEL1ΔC (Noens et al., 2011) and cultured in Middlebrook 7H9 medium, supplemented with 0.2% glucose, 0.2% glycerol and 0.05% Tween 80. Protein expression was induced with 2% (v/v) acetamide at an OD600 of 1.5. Cells were pelleted by centrifugation after xh incubation and resuspended in lysis buffer C (30 mM Tris (pH 8.0), 100 mM NaCl, 10 mM imidazole, 10% (w/v) glycerol) containing 1/100 protease inhibitor mix HP, 0.01% deoxyribonuclease I (Sigma-Aldrich) and disrupted using an Emulsiflex C3 high-pressure homogenizer (Avestin) by performing 5 cycles of ∼20,000 psi at 4°C. The cell suspension was centrifuged at 43,000 x g for 45 min at 4°C to pellet cell debris. MbcTA was purified from clarified lysate using a 5 mL HisTrap HP column. Following cleavage of the His6-tag with TEV protease, protein was concentrated and injected onto a Superdex 200 16/60 SEC column (GE Healthcare) pre-equilibrated in SEC buffer (30 mM Tris-HCl pH 8.0, 200 mM NaCl, 10% (w/v) glycerol) for removal of aggregated protein. Fractions containing MbcTA were pooled and concentrated to 12 mg ml−1. Samples were immediately used for crystallization or aliquoted and stored at −80°C. Proteins were routinely concentrated using Spin-X UF concentrators (Corning).
For expression of the MbcT-MbcAΔ112−113 complex, pnEK-MbcT and pnEA-His-MbcAΔ112−113 were co-transformed to E. coli BL21(DE3) CodonPlus-RIL. Protein expression was induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at an OD600 of 0.7. After xh incubation at xC, cells were pelleted by centrifugation and resuspended in lysis buffer A (30 mM Tris-HCl pH 8.0, 50 mM NaCl, 10 mM imidazole and 10% (w/v) glycerol) containing 1/100 protease inhibitor mix HP (Serva), 0.01% deoxyribonuclease I (Sigma-Aldrich). Cell disruption was achieved using an Emulsiflex C3 high pressure homogenizer (Avestin) by performing three cycles of ∼15,000 psi at 4°C. The cell suspension was centrifuged at 43,000 x g for 20 min at 4°C to pellet cell debris. MbcTA-containing lysate was loaded onto a 5 mL HisTrap HP (GE Healthcare) to bind the complex, followed by a salt wash with a linear gradient up to 2M NaCl. This salt wash resulted in MbcT-MbcAΔ112−113 dissociation and subsequent elution of MbcT. Fractions containing MbcT were buffer-exchanged to low salt buffer (30 mM Tris-HCl pH 8.0, 20 mM NaCl and 10% (w/v) glycerol), loaded onto a Mono Q 5/50 anion exchange chromatography column (QIAGEN), further concentrated and injected into a Superdex 75 16/60 size-exclusion chromatography (SEC) column (GE Healthcare) pre-equilibrated in SEC buffer for removal of aggregated protein.
MbcT R27E was produced as described for MbcT-MbcAΔ112−113 with following modifications. MbcT R27E-containing lysate in lysis buffer B (30 mM Tris-HCl pH 8.0, 200 mM NaCl, 10 mM imidazole, 10% (w/v) glycerol) was injected onto a 1 mL HisTrap HP column (GE Healthcare) and eluted using a linear gradient up to 300 mM imidazole. Following cleavage of the His6-tag with thrombin protease, the concentrated protein sample was injected onto a Superdex 75 16/60 SEC column pre-equilibrated in SEC buffer for removal of aggregated protein. Raw gel images are available on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1).
Crystallography
Initial crystallization conditions for the MbcTA complex (12 mg ml−1) were identified using the Morpheus screen (Molecular Dimensions) and the PEGs suite (QIAGEN). Optimized rod-like crystals were obtained by the vapor diffusion method in 0.2 M ammonium sulfate, 0.1 M tri-sodium citrate pH 5.6 and 25% PEG 4000. Prior to data collection, crystals were transferred to a solution containing cryoprotectant that was optimized to a ratio of 2:2:1 of SEC buffer, precipitant, and glycerol, respectively, and mounted in a CryoLoop (Hampton).
All diffraction data were collected at EMBL beamline P13 (Cianci et al., 2017) at the PETRA III storage ring (DESY, Hamburg, Germany) using a low-energy set up with a Helium-cone covering the PILATUS 6M pixel-array detector (DECTRIS Ltd., Baden, Switzerland) running with custom low-energy calibration tables. Data for Sulfur Single-wavelength Anomalous Dispersion (S-SAD) phasing were collected with an X-ray beam of 70 μm in diameter at an energy of 5.0 keV (λ = 2.48 Å) on three different positions of a rod-shaped crystal with approximate dimensions of 300 × 70 × 70 μm3. At each position, 3600 frames of 0.1° per 40 ms exposure time were collected.
High-resolution native data were collected at an energy of 12.7 keV (λ = 0.976 Å) on a crystal with approximate dimensions of 500 × 100 × 100 μm3 with a beam of 100 μm in diameter employing a helical scan between two centring points ca. 400 μm apart. 1800 frames of 0.1° per 40 ms were recorded. Data were integrated with XDS and further processed with XSCALE (Kabsch, 2010) and POINTLESS and AIMLESS from the CCP4 suite of programs (Evans, 2011, Winn et al., 2011).
The crystal structure was solved using the SHELX suite (Sheldrick, 2008) of programs via the HKL2MAP user interface (Pape and Schneider, 2004). Unmerged data collected at low and high energy were supplied to SHELXC as SAD and NATIVE datasets respectively. For substructure solution, the anomalous differences determined by SHELXC were truncated at 3.0 Å. In 100 trials, 16 anomalous sites with occupancies higher than 0.6 were identified by SHELXD with a CFOM of 58.8. Phases calculated based on the substructure, after ten alternating cycles of density modification (assuming a solvent content of 44%) and main-chain auto-building as implemented in SHELXE resulted in 550 residues being placed into the experimentally phased electron density with correlation coefficient between the structure factors calculated for the partial structure and the experimental data of 46.7%.
Using the phases obtained from SHELXE, ARP/wARP (Perrakis et al., 1999) was used to automatically build a first model consisting of 578 residues with Rwork- and Rfree-values of 0.30 and 0.25, respectively. The starting model was manually rebuilt with Coot (Emsley and Cowtan, 2004) and refined by iterative cycles using REFMAC (Vagin et al., 2004), PHENIX (Adams et al., 2010), and the PDB_REDO web server (Joosten et al., 2014) using translation, liberation, and screw-rotation (TLS) groups as identified by the TLSMD server (Painter and Merritt, 2006). The quality of the final model was assessed using Coot (Emsley and Cowtan, 2004), the wwPDB validation server (Gore et al., 2012) and the Molprobity server (Chen et al., 2010). Structural figures were generated using PyMol (Schrödinger). Data statistics are presented in Table 1.
Size Exclusion Chromatography Right-Angle Light Scattering
Protein mass measurements were performed on an Agilent HPLC system connected to a Viscotek 305 tri-detector (Malvern) to monitor static light scattering, refractive index, and UV absorbance. 100 μL sample was loaded onto a Superdex 200 HR 10/300 GL column (GE Healthcare) equilibrated in Size Exclusion Chromatography (SEC) buffer at a flow rate of 0.3 mL min−1. Data were recorded and processed using OmniSEC software (Agilent).
Small Angle X-ray Scattering
Small angle X-ray scattering (SAXS) data were collected at EMBL beamline P12 at the PETRA III storage ring (DESY, Hamburg, Germany) (Blanchet et al., 2015) using a 2M Pilatus pixel detector (DECTRIS) detector, a distance of 3.1 m and a wavelength of 1.24 Å (Table 2). MbcTA was measured at several protein concentrations in a range between ∼0.6 to ∼7.1 mg ml−1. Analysis of the scattering data was performed using the programs from the ATSAS 2.7 package (Petoukhov et al., 2012). The data obtained at the lowest and highest concentration were used for further analysis of MbcTA. The forward scattering I(0) and the radius of gyration Rg were calculated from the Guinier approximation calculated using PRIMUS GNOM (Svergun, 1992) was used to evaluate the pair distribution function, P(r), and to calculate the maximum particle dimension (Dmax). Ab initio models for MbcTA were generated with DAMMIN (Svergun, 1999) utilizing a relaxed disconnectivity criterion without symmetry restrictions. Validation, resolution estimation and averaging for the final model building were performed with SASRES (Tuukkanen et al., 2016) and DAMAVER (Volkov and Svergun, 2003). Theoretical scattering curves were calculated using CRYSOL (Svergun et al., 1995). SUPCOMB (Kozin and Svergun, 2001) was used for superimposition of the calculated ab initio model with the atomic structure. Data statistics are presented in Table 2.
Circular Dichroism Spectroscopy
Samples were diluted to ∼0.1 mg ml−1 in buffer containing 250mM NaF and 10mM sodium phosphate (pH 7.5). Circular dichroism (CD) spectra were recorded between 190 and 320 nm at 10°C in a 1 mm QS quartz cuvette on a Chirascan CD Spectrometer upgraded with an Active Nitrogen Management System (Applied Photophysics). Instrument settings were as follows: 1 nm bandwidth, 1 s response and 0.5 nm data pitch. For each dataset 5 spectra have been averaged and sample buffer subtracted as background. Data were recorded with the Pro-Data Chirascan software (version 4.5.1833).
Isolation of genomic DNA and total RNA
For isolation of genomic DNA, E. coli DH5α cells were cultured in LB medium to an OD600 of 0.7, collected and washed with PBS buffer. Genomic DNA was obtained using the DNeasy Blood & Tissue kit (QIAGEN) according to the manufacturer’s instructions for Gram-negative bacteria, including RNaseA digestion. For isolation of total RNA, E. coli DH5α cells were cultured in LB medium to an OD600 of 0.7, mixed with RNAprotect Bacteria Reagent (QIAGEN) by vortexing followed by incubation for 5 min. at room temperature. Cells were pelleted by centrifugation and RNA was extracted according to the RNeasy Protect Bacteria Mini Kit protocol (QIAGEN) with a minor modification, namely DNase digestion was performed on-column for 30 min at 37°C.
ADP ribosylation assays
For production of cell lysates, E. coli strain DH5α was cultured to an OD600 of 0.7. Cells were pelleted, washed with phosphate buffered saline (PBS) and resuspended in 1x BugBuster lysis reagent (Merck Millipore) supplemented with 1 mM dithiothreitol, 1x complete EDTA-free protease inhibitor cocktail (Roche) and 0.01 mg ml−1 deoxyribonuclease I. After 15 min incubation at room temperature, cell lysate was clarified by centrifugation at 20000 g, 4°C for 10 min. The supernatant was desalted using PD10 columns (GE Healthcare) in Tris buffer (20mM; pH 7.5) and protein concentration was measured using the BCA protein assay kit (ThermoFischer Scientific) following the manufacturer’s instructions. M. smegmatis mc2155 groEL1ΔC (Noens et al., 2011) cells were cultured to an OD600 of 1.5 in Middlebrook 7H9 medium, supplemented with 0.2% glucose, 0.2% glycerol and 0.05% Tween-80. M. smegmatis cell lysate was prepared as described for E. coli DH5α cells, with the exception of the cell lysis step. To ensure complete cell lysis, cell pellets were additionally incubated in a sonication bath for 5 min. Reactions were performed in 10 μL reaction buffer (50 mM Tris (pH 7.4), 200 mM NaCl, 2 mM MgCl2 and 1 mM DTT) containing MbcT and ∼1 μg of protein lysate, ∼50 ng denatured dsDNA or ∼1 μg RNA and/or spiked with 32P-NAD+. The final concentration of MbcT was 1 or 10 μM, when mixed with nucleosides or lysates respectively. The reactions were incubated at 37°C for 1 h. Reactions with protein lysate were analyzed by SDS-PAGE, gels were dried and exposed to autoradiography films. Reactions with DNA and RNA or without substrate were analyzed by thin layer chromatography.
Thin Layer Chromatography
2 μL of each ADP-ribosylation reaction was spotted on polyethyleneimine (PEI) cellulose plates (Merck Millipore), which were air-dried prior to development with 0.25 M LiCl and 0.25 M formic acid. After drying, plates were exposed to an image plate (Fujifilm) and analyzed using a Phosphor-Imager (Fujifilm). Raw images are available on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1).
LC-MS
LC-MS analysis was carried out on an Agilent system consisting of a 1290 Infinity II HPLC coupled to a 6230 TOF mass spectrometer with a dual Agilent Jet Stream (AJS) electrospray ionization source in negative mode. Ionization conditions were as follows: Nebuliser pressure 35 psi; N2 drying gas temperature and flow 200°C and 8 l min−1; N2 sheath gas temperature and flow 300°C and 11 l min−1; and capillary, nozzle, fragmentor, and octupole RF voltages 3000, 2000, 400 and 750 V, respectively. Compounds were separated with a Waters XBridge Amide column (3.5 μm; 4.6 mm × 100 mm). Phase A was 5% acetonitrile, 20 mM ammonium hydroxide and 20 mM ammonium acetate. Phase B was 100% acetonitrile (Yuan et al., 2012). Compounds were eluted at a flow rate of 0.4 mL min−1 and a temperature of 40°C with a gradient of 85%–60% B in 5 min, 60% B for 11 min, 60%–2% B in 5 min and 2%–80% B in 5 min. Data were collected and analyzed with MassHunter B 07.00.
HPLC
Reactions were analyzed on an Agilent 1260 Infinity HPLC system using an Agilent Poroshell 120 EC-C18 column (2.7 um 4.6 × 50 mm) and monitoring absorbance at 260 nm. Elution was achieved with an isocratic flow of 2 ml/min of 10 mM ammonium phosphate pH 5.5 with 2.5% acetonitrile (Muller-Steffner et al., 1994). Data were collected and analyzed with OpenLAB CDS ChemStation (Agilent).
NMR spectroscopy
Spectra were acquired in a Bruker Avance III HD spectrometer operating at a 1H frequency of 700 MHz and equipped with a 5 mm 1H/31P/13C/15N resonance PFG cryogenic probe. Data were processed and analyzed with Topspin 3.5. 1D and COSY HMBC NMR spectra are available on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1).
Enzyme kinetics
All kinetic reactions were performed in 96-well plates. MbcT (50 nM) was incubated with different concentrations of NAD+ sodium salt (Sigma-Aldrich) at 37°C and the reaction time was adjusted in order to assure measurement of the initial rate of the reaction. Reactions were carried out in a final volume of 140 μL in reaction buffer (50 mM sodium-phosphate buffer (pH 7.5), 50 mM NaCl). For each time point, 10 μL of the reaction mixture was added to 300 μL of 5M NaOH and incubated in the dark at room temperature for 50 min to allow for the production of the alkaline-generated fluorescent species of NAD+. Fluorescence was measured at 360/460 nm (excitation/emission filter set) using a TECAN microplate reader. The concentration of NAD+ in each sample was calculated from the relative fluorescence correlated to a standard curve of NAD+. Initial rates of the reaction were determined by the linear regression of the plot of NAD+ consumption versus time, assuming saturating conditions of inorganic phosphate. Each calculated initial rate was plotted versus the corresponding NAD+ concentration. Michaelis Menten kinetics were used to determine KM, Vmax for MbcT under each condition. Kcat was determined from the fit of the plot of Kobs (initial rate divided by enzyme concentration) versus NAD+ concentration. All calculations were performed using GraphPad Prism software.
Western blotting
pET28a(+)-MbcT constructs were transformed to E. coli BL21 (DE3) and cultured in LB at 37°C. Protein expression was induced with IPTG (0.5mM) at an OD600 of 0.7 and cells were harvested 1 h after induction by centrifugation. Cells were resuspended in 1x BugBuster lysis reagent (Merck Millipore) supplemented with 0.13 mg ml−1 protease-inhibitor-mix HP (SERVA Electrophoresis) and 0.01 mg ml−1 deoxyribonuclease I (Sigma-Aldrich) and incubated at RT for 15 min. Protein lysates were separated by SDS-PAGE, transferred to Immuno-Blot PVDF membrane (Bio-Rad) using a Trans-Blot Turbo (Bio-Rad) and blocked overnight using 5% solution of skimmed milk powder (Carl Roth). Membranes were probed with either Penta His HRP conjugate (QIAGEN) or anti-GroEL (E. coli) monoclonal antibody (clone 9A1/2) (Enzo Life Sciences) as the primary antibody. HRP-linked whole Mouse IgG Antibody (GE Healthcare) was used for detection of GroEL. Blots were developed using SuperSignal West Pico Maximum Sensitivity Substrate (ThermoFisher Scientific) and visualized using a ChemiDoc MP (Bio-Rad). Raw blot images are available on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1).
Determination of NAD+ levels in bacterial cells
MtbΔTA strains containing pGMCS-TetR-P1 (empty vector), pGMCS-TetR-P1-Rv1989c or pGMCS-TetR-P1-Rv1989cR27E plasmids were cultured in 7H9 medium (Difco) supplemented with 10% albumin-dextrose-catalase (ADC, Difco), 0.05% Tween-80 (Sigma-Aldrich) at 37°C to OD600 of 0.2 prior to induction of protein expression with 200 ng ml−1 of anhydrotetracycline (ATc). 500 μL of culture was removed 24 h post-induction. Cells were harvested by centrifugation and resuspended in PBS buffer at an OD600 of 0.5. 0.1 μm-diameter glass beads were added to the tubes and cells were lysed by four 60 s pulses at full speed in a bead-beater device. The samples were centrifuged for 1 min at 20,200xg and the lysates were sterilized by filtration. Filtrates were mixed with an equal volume of NAD/NADH-Glo Detection Reagent (Promega) and luciferin bioluminescence was measured after 30 min of incubation using a CLARIOstar plate reader (BMG LABTECH) and normalized to background (PBS-only) signal.
Macrophage infections
Before infection, mycobacterial clumps were disaggregated after at least 20 passages through a 25G needle. Human monocyte-derived macrophages were infected with M. tuberculosis at a multiplicity of infection of 0.3 bacteria/macrophage in complete RMPI medium for 4 h at 37°C. Cells were then washed with RPMI and further incubated at 37°C for 5 days in RPMI supplemented with or without ATc (200 ng ml−1). Measurements of macrophage viability were performed by flow cytometry analysis of cells treated with Zombie Aqua Fixable Viability Kit (BioLegends) as recommended by the manufacturer. Briefly, infected macrophages were recovered from the glass coverslips by treatment with non-enzymatic cell dissociation solution (Sigma-Aldrich). Macrophage pellets were resuspended in 100 μL Zombie Aqua solution in PBS and stained 20 min at 4°C. Macrophages were then washed in PBS, fixed for 2 h at room temperature in 200 μL of PBS containing 4% paraformaldehyde (Polyscience) and analyzed by flow cytometry (LSRII, BD Biosciences).
Mice infections
Six- to eight-week-old female mice (SCID or C57BL/6J, Charles River) were anesthetized in gas chambers containing 0.5% isoflurane. SCID mice were infected by intravenous injection of ∼105 CFUs of Mtb. Groups of 10 mice were provided with drinking water supplemented (or not) with 5% sucrose and 1 mg ml−1 of doxycycline from 7 days onward before infection and during the whole course of the Mtb infection. Survival was followed during time. C57BL/6J mice were infected intranasally with ∼103 CFUs of Mtb in 25 μL of DPBS (GIBCO). At day 21 post-infection, groups of eight mice were fed by daily gavage with either water, isoniazid (25 mg kg−1), doxycycline (1 mg kg−1) or both during 10 days. At day 31 post-infection, mice were sacrificed and lung homogenates were plated onto 7H11 agar plates for CFU scoring.
Quantification and Statistical Analysis
Comparison of survival curves of SCID mice was performed with Log-rank (Mantel-Cox) test in GraphPad Prism software. Significance of variation in CFUs in lungs of infected C57BL/6J mice was performed using unpaired Student’s tests in GraphPad Prism software. No animals were excluded from statistical analysis. p values correlate with symbols as follows: ns = not significant, p > 0.05, ∗ p ≤ 0.05, ∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001.
Data and Software Availability
All raw images as well as the 1D and COSY HMBC NMR spectra are deposited on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1). The accession number for the MbcT-MbcA crystal structure reported in this paper is PDB: 6FKG. The accession number for the MbcT-MbcA SAXS data reported in this paper is SASBDB: SASDD33.
Acknowledgments
We thank K. Aktories and S. Ost for providing CDTa and actin, M. Jeske and K. Temmerman for help with enzyme assays, G. Kelly for NMR data collection analysis, the IPBS imaging (TRI) and zootechnics (Anexplo) core facilities, F. Moreau and C. Berrone for assistance with functional exploration, the SPC facility at EMBL Hamburg and the Proteomics Core Facility at EMBL Heidelberg for technical support, and Y. Rombouts, F. Rauschel, G.R. Stewart, and S.H.E. Kaufmann for comments on the manuscript. NMR data were recorded in the MRC Biomedical NMR Centre at the Francis Crick Institute, which receives core funding from Cancer Research UK (FC001029), the Medical Research Council (FC001029), and the Wellcome Trust (FC001029). This work was supported by the European Union (TBVAC2020), Agence Nationale de la Recherche/Programme d’Investissements d’Avenir (ANR-11-EQUIPEX-0003 and ANR-13-BSV8-0010-01), Fondation pour la Recherche Médicale (DEQ20160334902 and post-doctoral fellowship to A.G.), the Bettencourt-Schueller Foundation, the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001060), the UK Medical Research Council (FC001060), the Wellcome Trust (FC001060), and a Wellcome Trust New Investigator Award (104785/B/14/Z).
Author Contributions
D.M.F., C.G., M.W., A.H.A.P., and O.N. developed the study rationale. A.H.A.P. and D.M.F. designed structural biology, biophysics, and biochemistry experiments. D.M.F. performed structural biology, biophysics, and biochemistry experiments. C.G., A.D.G., and O.N. designed microbiology, macrophage, and mice experiments. C.G., K.A., Y.-M.B., and A.D.G. conducted mycobacterial genetics and macrophage experiments. C.G. and A.C. performed mouse experiments. A.J.S. and P.G. performed initial E. coli experiments. L.B. and D.S. contributed mycobacterial expression vectors. A.P. performed the FACS and microscopy experiments. A.G.-G. and L.P.S.d.C. designed HPLC, NMR, and MS experiments. A.G.-G. performed HPLC, NMR, and MS experiments. D.M.F. and K.S.H.B. performed the isotope experiments. T.P. contributed to construct generation and protein purification. A.T. collected SAXS data. D.F., A.T., and D.I.S. analyzed SAXS data. K.S.H.B. and V.P. provided intellectual input and technical support. D.M.F., T.R.S., and M.C. determined the crystal structure. C.G., A.H.A.P., and O.N. wrote the paper. D.M.F. and M.W. provided critical input. All others revised the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: February 18, 2019
Footnotes
Supplemental Information includes six figures and two tables and can be found with this article online at https://doi.org/10.1016/j.molcel.2019.01.028.
Contributor Information
Annabel H.A. Parret, Email: ahaparret@gmail.com.
Olivier Neyrolles, Email: olivier.neyrolles@ipbs.fr.
Supporting Citations
The following references appear in the Supplemental Information: Botella et al., 2017, Diebold et al., 2011, Gulke et al., 2001.
Supplemental Information
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L., Zhang D., de Souza R.F., Anand S., Iyer L.M. The natural history of ADP-ribosyltransferases and the ADP-ribosylation system. Curr. Top. Microbiol. Immunol. 2015;384:3–32. doi: 10.1007/82_2014_414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becq J., Gutierrez M.C., Rosas-Magallanes V., Rauzier J., Gicquel B., Neyrolles O., Deschavanne P. Contribution of horizontally acquired genomic islands to the evolution of the Tubercle bacilli. Mol. Biol. Evol. 2007;24:1861–1871. doi: 10.1093/molbev/msm111. [DOI] [PubMed] [Google Scholar]
- Blanchet C.E., Spilotros A., Schwemmer F., Graewert M.A., Kikhney A., Jeffries C.M., Franke D., Mark D., Zengerle R., Cipriani F. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY) J. Appl. Cryst. 2015;48:431–443. doi: 10.1107/S160057671500254X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella L., Vaubourgeix J., Livny J., Schnappinger D. Depleting Mycobacterium tuberculosis of the transcription termination factor Rho causes pervasive transcription and rapid death. Nat. Commun. 2017;8:14731. doi: 10.1038/ncomms14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity : all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci M., Bourenkov G., Pompidor G., Karpics I., Kallio J., Bento I., Roessle M., Cipriani F., Fiedler S., Schneider T.R. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. J. Synchrotron Radiat. 2017;24:323–332. doi: 10.1107/S1600577516016465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combet C., Blanchet C., Geourjon C., Deléage G. NPS@: network protein sequence analysis. Trends Biochem. Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- DeJesus M.A., Gerrick E.R., Xu W., Park S.W., Long J.E., Boutte C.C., Rubin E.J., Schnappinger D., Ehrt S., Fortune S.M. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio. 2017;8 doi: 10.1128/mBio.02133-16. e02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deter H.S., Jensen R.V., Mather W.H., Butzin N.C. Mechanisms for differential protein production in toxin – antitoxin systems. Toxins (Basel) 2017;9:1–13. doi: 10.3390/toxins9070211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold M.L., Fribourg S., Koch M., Metzger T., Romier C. Deciphering correct strategies for multiprotein complex assembly by co-expression: application to complexes as large as the histone octamer. J. Struct. Biol. 2011;175:178–188. doi: 10.1016/j.jsb.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Dy R.L., Przybilski R., Semeijn K., Salmond G.P.C., Fineran P.C. A widespread bacteriophage abortive infection system functions through a type IV toxin-antitoxin mechanism. Nucleic Acids Res. 2014;42:4590–4605. doi: 10.1093/nar/gkt1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S., Guo X.V., Hickey C.M., Ryou M., Monteleone M., Riley L.W., Schnappinger D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot : model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans P.R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., Attwood T.K., Babbitt P.C., Bateman A., Bork P., Bridge A.J., Chang H.-Y., Dosztányi Z., El-Gebali S., Fraser M. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45:D190–D199. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genevaux P., Keppel F., Schwager F., Langendijk-Genevaux P.S., Hartl F.U., Georgopoulos C. In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 2004;5:195–200. doi: 10.1038/sj.embor.7400067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh J., Anderson P.J., Chandrasekaran S., Caparon M.G. Characterization of Streptococcus pyogenes beta-NAD+ glycohydrolase: re-evaluation of enzymatic properties associated with pathogenesis. J. Biol. Chem. 2010;285:5683–5694. doi: 10.1074/jbc.M109.070300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore S., Velankar S., Kleywegt G.J. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012;68:478–483. doi: 10.1107/S0907444911050359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulke I., Pfeifer G., Liese J., Fritz M., Hofmann F., Aktories K., Barth H. Characterization of the enzymatic component of the ADP-ribosyltransferase toxin CDTa from Clostridium difficile. Infect. Immun. 2001;69:6004–6011. doi: 10.1128/IAI.69.10.6004-6011.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Venkataraman B., Vasudevan M., Gopinath Bankar K. Co-expression network analysis of toxin-antitoxin loci in Mycobacterium tuberculosis reveals key modulators of cellular stress. Sci. Rep. 2017;7:5868. doi: 10.1038/s41598-017-06003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A.M., Gollan B., Helaine S. Toxin-antitoxin systems: reversible toxicity. Curr. Opin. Microbiol. 2017;36:102–110. doi: 10.1016/j.mib.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Han S., Tainer J.A. The ARTT motif and a unified structural understanding of substrate recognition in ADP-ribosylating bacterial toxins and eukaryotic ADP-ribosyltransferases. Int. J. Med. Microbiol. 2002;291:523–529. doi: 10.1078/1438-4221-00162. [DOI] [PubMed] [Google Scholar]
- Harms A., Brodersen D.E., Mitarai N., Gerdes K. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell. 2018;70:768–784. doi: 10.1016/j.molcel.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Hayashi K., Morooka N., Yamamoto Y., Fujita K., Isono K., Choi S., Ohtsubo E., Baba T., Wanner B.L., Mori H. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100049. 2006.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka S., Niemann S., Russell D.G., Rohde K.H. Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: Delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog. 2010;6:1–17. doi: 10.1371/journal.ppat.1000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankevicius G., Ariza A., Ahel M., Ahel I. The toxin-antitoxin system DarTG catalyzes reversible ADP-ribosylation of DNA. Mol. Cell. 2016;64:1109–1116. doi: 10.1016/j.molcel.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R.P., Long F., Murshudov G.N., Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014;1:213–220. doi: 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren I., Minami S., Rubin E., Lewis K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBiol. 2011;2 doi: 10.1128/mBio.00100-11. e00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-H., O’Brien K.M., Sharma R., Boshoff H.I.M., Rehren G., Chakraborty S., Wallach J.B., Monteleone M., Wilson D.J., Aldrich C.C. A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. USA. 2013;110:19095–19100. doi: 10.1073/pnas.1315860110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin M.B., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Letunic I., Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46:D493–D496. doi: 10.1093/nar/gkx922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levillain F., Poquet Y., Mallet L., Mazères S., Marceau M., Brosch R., Bange F.-C., Supply P., Magalon A., Neyrolles O. Horizontal acquisition of a hypoxia-responsive molybdenum cofactor biosynthesis pathway contributed to Mycobacterium tuberculosis pathoadaptation. PLoS Pathog. 2017;13:e1006752. doi: 10.1371/journal.ppat.1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato-Márquez D., Díaz-Orejas R., García-del Portillo F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016;40:592–609. doi: 10.1093/femsre/fuw022. [DOI] [PubMed] [Google Scholar]
- Makarova K.S., Wolf Y.I., Koonin E.V. Comprehensive comparative-genomic analysis of Type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009;4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.P. A new set of useful cloning and expression vectors derived from pBlueScript. Gene. 1995;163:41–46. doi: 10.1016/0378-1119(95)00389-n. [DOI] [PubMed] [Google Scholar]
- Milunovic B., DiCenzo G.C., Morton R.A., Finan T.M. Cell growth inhibition upon deletion of four toxin-antitoxin loci from the megaplasmids of Sinorhizobium meliloti. J. Bacteriol. 2014;196:811–824. doi: 10.1128/JB.01104-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Steffner H., Muzard M., Oppenheimer N., Schuber F. Mechanistic implications of cyclic ADP-ribose hydrolysis and methanolysis catalyzed by calf spleen NAD+glycohydrolase. Biochem. Biophys. Res. Commun. 1994;204:1279–1285. doi: 10.1006/bbrc.1994.2601. [DOI] [PubMed] [Google Scholar]
- Noens E.E., Williams C., Anandhakrishnan M., Poulsen C., Ehebauer M.T., Wilmanns M. Improved mycobacterial protein production using a Mycobacterium smegmatis groEL1ΔC expression strain. BMC Biotechnol. 2011;11:27. doi: 10.1186/1472-6750-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R., Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016;12:208–214. doi: 10.1038/nchembio.2044. [DOI] [PubMed] [Google Scholar]
- Painter J., Merritt E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Cryst. 2006;39:109–111. [Google Scholar]
- Palazzo L., Mikoč A., Ahel I. ADP-ribosylation: new facets of an ancient modification. FEBS J. 2017;284:2932–2946. doi: 10.1111/febs.14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T., Schneider T.R. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Cryst. 2004;37:843–844. [Google Scholar]
- Perikh S.L., Schramm V.L. Transition state structure for ADP-ribosylation of eukariotic elongation factor 2 catalyzed by diphtheria toxin. Biochemistry. 2004;43:1204–1212. doi: 10.1021/bi035907z. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris R., Lamzin V.S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage H.R., Connolly L.E., Cox J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionova I.A., Schuster B.M., Guinn K.M., Sorci L., Scott D.A., Li X., Kheterpal I., Shoen C., Cynamon M., Locher C. Metabolic and bactericidal effects of targeted suppression of NadD and NadE enzymes in mycobacteria. MBiol. 2014;5:1–9. doi: 10.1128/mBio.00747-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad T.R., Harrell M.I., Liao R., Sherman D.R. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE. 2008;3:1–8. doi: 10.1371/journal.pone.0001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A., Bordes P., Genevaux P. Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins (Basel) 2014;6:1002–1020. doi: 10.3390/toxins6031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sberro H., Leavitt A., Kiro R., Koh E., Peleg Y., Qimron U., Sorek R. Discovery of functional toxin/antitoxin systems in bacteria by shotgun cloning. Mol. Cell. 2013;50:136–148. doi: 10.1016/j.molcel.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D., Ehrt S. Regulated expression systems for mycobacteria and their applications. Microbiol. Spectr. 2014;2 doi: 10.1128/microbiolspec.MGM2-0018-2013. MGM2-0018-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G.M. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Simon N.C., Aktories K., Barbieri J.T. Novel bacterial ADP-ribosylating toxins: structure and function. Nat. Rev. Microbiol. 2014;12:599–611. doi: 10.1038/nrmicro3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjerning R.B., Senissar M., Winther K., Gerdes K., Brodersen D.E. The RES domain toxins of RES-Xre toxin-antitoxin modules induce cell stasis by degrading NAD. Mol. Microbiol. 2018 doi: 10.1111/mmi.14150. Published online October 13, 2018. [DOI] [PubMed] [Google Scholar]
- Slayden R.A., Dawson C.C., Cummings J.E. Toxin antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 2018;76 doi: 10.1093/femspd/fty039. [DOI] [PubMed] [Google Scholar]
- Song S., Wood T.K. Post-segregational killing and phage inhibition are not mediated by cell death through toxin/antitoxin systems. Front. Microbiol. 2018;9:814. doi: 10.3389/fmicb.2018.00814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Siroy A., Lokareddy R.K., Speer A., Doornbos K.S., Cingolani G., Niederweis M. The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 2015;22:672–678. doi: 10.1038/nsmb.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D., Barberato C., Koch M.H.J. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- Tiwari P., Arora G., Singh M., Kidwai S., Narayan O.P., Singh R. MazF ribonucleases promote Mycobacterium tuberculosis drug tolerance and virulence in guinea pigs. Nat. Commun. 2015;6:6059. doi: 10.1038/ncomms7059. [DOI] [PubMed] [Google Scholar]
- Tortoli E., Fedrizzi T., Meehan C.J., Trovato A., Grottola A., Giacobazzi E., Serpini G.F., Tagliazucchi S., Fabio A., Bettua C. The new phylogeny of the genus Mycobacterium : The old and the news. Infect. Genet. Evol. 2017;56:19–25. doi: 10.1016/j.meegid.2017.10.013. [DOI] [PubMed] [Google Scholar]
- Troegeler A., Lastrucci C., Duval C., Tanne A., Cougoule C., Maridonneau-Parini I., Neyrolles O., Lugo-Villarino G. An efficient siRNA-mediated gene silencing in primary human monocytes, dendritic cells and macrophages. Immunol. Cell Biol. 2014;92:699–708. doi: 10.1038/icb.2014.39. [DOI] [PubMed] [Google Scholar]
- Tuukkanen A.T., Kleywegt G.J., Svergun D.I. Resolution of ab initio shapes determined from small-angle scattering. IUCrJ. 2016;3:440–447. doi: 10.1107/S2052252516016018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.A., Steiner R.A., Lebedev A.A., Potterton L., McNicholas S., Long F., Murshudov G.N. REFMAC 5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- van Kessel J.C., Hatfull G.F., Van Kessel J.C., Hatfull G.F. Recombineering in Mycobacterium tuberculosis. Nat. Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- Vilchèze C., Weinrick B., Wong K.W., Chen B., Jacobs W. NAD+ auxotrophy is bacteriocidal for the tubercle bacilli. Mol. Microbiol. 2010;76:365–377. doi: 10.1111/j.1365-2958.2010.07099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V.V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney J.C., Quentin D., Sawai S., LeRoux M., Harding B.N., Ledvina H.E., Tran B.Q., Robinson H., Goo Y.A., Goodlett D.R. An interbacterial NAD(P)+ glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell. 2015;163:607–619. doi: 10.1016/j.cell.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J.J., Hergenrother P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012;20:291–298. doi: 10.1016/j.tim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. Overview of the CCP 4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood H.E., Devine K.M., McConnell D.J. Characterisation of a repressor gene (xre) and a temperature-sensitive allele from the Bacillus subtilis prophage, PBSX. Gene. 1990;96:83–88. doi: 10.1016/0378-1119(90)90344-q. [DOI] [PubMed] [Google Scholar]
- Yuan M., Breitkopf S.B., Yang X., Asara J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw images as well as the 1D and COSY HMBC NMR spectra are deposited on Mendeley Data (https://doi.org/10.17632/y6ynjm5sf3.1). The accession number for the MbcT-MbcA crystal structure reported in this paper is PDB: 6FKG. The accession number for the MbcT-MbcA SAXS data reported in this paper is SASBDB: SASDD33.