

Gut microbiome and metabolic syndrome regulate nanovaccine response and can be immunomodulated using advanced nanomaterials.
Abstract
Biomaterials-based nanovaccines, such as those made of poly(lactic-co-glycolic acid) (PLGA), can induce stronger immunity than soluble antigens in healthy wild-type mouse models. However, whether metabolic syndrome can influence the immunological responses of nanovaccines remains poorly understood. Here, we first show that alteration in the sensing of the gut microbiome through Toll-like receptor 5 (TLR5) and the resulting metabolic syndrome in TLR5−/− mice diminish the germinal center immune response induced by PLGA nanovaccines. The PLGA nanovaccines, unexpectedly, further changed gut microbiota. By chronically treating mice with antibiotics, we show that disrupting gut microbiome leads to poor vaccine response in an obesity-independent manner. We next demonstrate that the low immune response can be rescued by an immunomodulatory Pyr-pHEMA nanogel vaccine, which functions through TLR2 stimulation, enhanced trafficking, and induced stronger germinal center response than alum-supplemented PLGA nanovaccines. The study highlights the potential for immunomodulation under gut-mediated metabolic syndrome conditions using advanced nanomaterials.
INTRODUCTION
Biomaterials-based nanovaccines can mimic pathogens and are readily phagocytosed by dendritic cells, which prime T cells and B cells in lymphoid tissues, such as lymph nodes. These nanovaccines often induce superior and controlled immune response compared to soluble antigens in healthy, preclinical mouse models (1–4). In comparison to soluble antigens, polymeric nanovaccines, such as those made of poly(lactic-co-glycolic acid) (PLGA) (1–4), can protect the vaccine antigen from degradation and rapid clearance and co-deliver immunomodulatory molecules to boost immune recognition. However, previous work has mostly focused on healthy wild-type (WT) mouse models to understand the immune response without considering biological differences that may affect the immunological outcomes of nanovaccines. Here, we are particularly interested in understanding the role of metabolic syndrome as an umbrella disease because of its prevalence in the United States (34 to 39%) (5, 6) and worldwide (~25%) (7). To the best of our knowledge, there are no previous reports exploring the performance of PLGA nanovaccines and other immunomodulatory nanomaterials in metabolic syndrome.
Metabolic syndrome is a cluster of metabolic disorders, such as systemic inflammation, insulin resistance, and obesity (8, 9). The National Cholesterol Education Program Adult Treatment Panel III definition (10, 11) is one of the most widely used criteria of metabolic syndrome and incorporates the key features of hyperglycemia/insulin resistance, visceral obesity, atherogenic dyslipidemia, and hypertension. Metabolic syndrome can originate from a variety of factors such as gut microbiome, diet, and genetic mutations, among others (12). The gut microbiome is particularly interesting because it has been implicated as a cause of metabolic syndrome (8), and multiple lines of evidence link the gut microbiota and metabolic syndrome to the immune system and, more directly, to vaccine response (13–15). In one example, mice deficient in Toll-like receptor 5 (TLR5, a cell surface protein that senses flagella on gut bacteria) spontaneously developed metabolic syndrome (16) and have also been shown to have a poor soluble antigen response (17). In the latter study, soluble influenza vaccine response showed a direct correlation between expression of TLR5 and the magnitude of the antibody response. The study further showed that TLR5-mediated sensing of the microbiota also affected antibody responses to the inactivated polio vaccine. Despite these studies, it remains unclear whether the gut microbiome can regulate the response of materials-based nanovaccines, which have the potential to mount stronger immune response than soluble vaccines, even in the absence of adjuvants. It also remains unclear whether immunomodulatory nanomaterials can overcome any immune regulations imposed by gut microbiome and metabolic syndrome.
Here, we first elucidate how TLR5-mediated sensing of the microbiota and the associated metabolic syndrome modulates the immune response induced by conventional PLGA nanovaccines. We disrupted the gut microbiome sensing using TLR5−/− mice and observed that the resulting metabolic syndrome diminishes the immune response induced by conventional PLGA nanovaccines in the absence of any exogenous adjuvant. The PLGA nanovaccines show reduced particle trafficking to draining lymphoid tissues, and nanovaccines further changed the selective composition of the gut microbiota. By chronically treating WT mice with antibiotics since weaning, we show that disrupting gut signaling leads to poor vaccine response in an obesity-independent manner. We next engineer an immunomodulatory pyridine–poly(hydroxyethyl methacrylate) (Pyr-pHEMA), which self-assembles with protein antigens to form a nanogel vaccine. The Pyr-pHEMA nanogels overcome the diminished response of PLGA vaccines in the metabolic syndrome model by modulating the immune response in immune cells through TLR2 and mount B cell response higher than alum-supplemented PLGA nanovaccines. The results highlight the potential of advanced nanomaterials as immunomodulatory vaccines under gut-mediated metabolic syndrome conditions.
RESULTS AND DISCUSSION
PLGA nanovaccines mount limited immunity under gut-mediated metabolic syndrome conditions in male mice
To determine the efficacy of PLGA nanovaccines under metabolic syndrome conditions, we first validated the pattern of obesity in a dysregulated gut microbiome model (TLR5−/− mice) at 10 and 16 weeks of age as these encompass the typical age range of in vivo vaccination experiments. We confirmed increased fat pad mass and body weight among male mice at 16 weeks of age (Fig. 1A and fig. S1), which is in congruence with studies by Vijay-Kumar et al. (16). In contrast, female mice did not show increased adiposity at these particular ages. We confirmed metabolic syndrome in male mice at 16 weeks of age with elevated levels of obesity hormone leptin (Fig. 1B), diabetes marker insulin (Fig. 1C), and inflammatory cytokines interleukin-6 (IL-6) and IL-8 in the serum of TLR5−/− mice as compared to WT mice (Fig. 1D). We did not see a change in systemic tumor necrosis factor–α (TNF-α), IL-1β, and IL-10. Unlike previous studies that have quantified proinflammatory cytokine levels in adipose tissues of TLR5−/− mice (16), we quantified interleukins and TNF-α in serum because it may have a more direct systemic impact on the vaccine response. Previous work has shown that gut dysbiosis can trigger systemic inflammation through elevated levels of serum IL-6 through inflammatory cells (18). These studies further showed that chronic exposure to inflammation alters the function of phagocytic cells, rendering them less effective at combating pathogens, which forms the premise of the current work. Collectively, our characterization of mice confirms that TLR5−/− mice developed gut-mediated systemic metabolic syndrome at 16 weeks of age, and only male mice showed an increase in obese phenotype.
Fig. 1. PLGA nanovaccines manifest limited humoral immune response in male TLR5−/−mice.
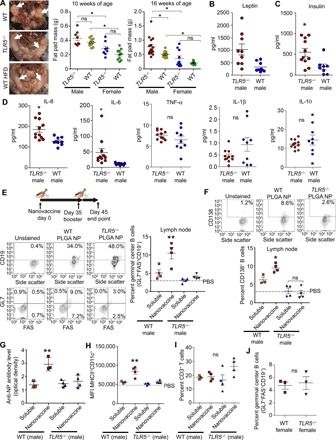
(A) Left: Images of fat pad mass for WT, TLR5−/−, and WT mice on a high fat diet (HFD). Photo credit: J.D.G., Cornell University. Middle and right: Mouse fat pad masses as a function of age and gender. Statistics was performed using a one-way analysis of variance (ANOVA) with Tukey’s post hoc correction (n = 8 to 16). (B and C) Comparison of leptin (B) and insulin (C) levels in TLR5−/− and WT male mice. Statistics was performed using an unpaired, two-tailed t test (n = 8 TLR5−/− and n = 11 WT). (D) Comparison of inflammatory markers in TLR5−/− and WT male mice. Statistics was performed using an unpaired, two-tailed t test (n = 10 each). (E) Timeline for vaccination, germinal center immune response, and gating strategy. The scatterplot presents the percentage of GL7+FAS+CD19+ germinal center B cells in the lymph node of male mice 10 days after booster vaccination with either soluble NP-OVA (4-hydroxy-3-nitrophenylacetyl hapten conjugated to ovalbumin) antigen or PLGA nanovaccines formulated with NP-OVA. PBS, phosphate-buffered saline; FAS, fatty acid synthase. (F) Gating strategy for CD138+ plasma cells. The scatterplot presents the percentage of CD138+ plasma cells in the lymph node after booster vaccination. (G) The scatterplot presents the antigen-specific antibodies in the serum of male mice after immunization. (H and I) Scatterplots present the major histocompatibility complex class II (MHCII) expression on CD11c+ dendritic cells (H) and the percentage of CD3+ T cells (I) in the lymph node after booster vaccination. In (E) to (I), statistics was performed using a one-way ANOVA with Tukey’s post hoc correction (n = 4 except for the soluble NP-OVA WT mice with n = 3). (J) The scatterplot compares the percentage of germinal center B cells in the lymph node of TLR5−/− to WT female mice. Statistics was performed using an unpaired, two-tailed t test (n = 3). In all studies, *P < 0.05 and **P < 0.01. ns denotes nonsignificant differences. MFI, mean fluorescence intensity.
We examined whether altered sensing of the gut microbiome and the resulting metabolic syndrome regulate the success of a model PLGA nanovaccine, which is among the most widely used nanovaccine platforms (1, 19). Mice were immunized subcutaneously either with PLGA nanovaccines formulated with 50 μg of 4-hydroxy-3-nitrophenyl acetyl hapten conjugated to ovalbumin (NP-OVA) or as a soluble NP-OVA antigen. The average diameter of the PLGA nanovaccine was 194 nm (fig. S2). Previous studies have shown that mice immunized with soluble NP-OVA antigen induce NP-specific immune response (20); therefore, our experimental design did not initially include additional adjuvants to better understand the efficacy of PLGA nanovaccines. Thirty-five days later (1, 21), a booster dose was injected, and after another 10 days, cells were harvested from the spleen and the draining lymph node (Fig. 1E). Nanovaccines induced significantly higher GL7+FAS+CD19+ germinal center B cells in the lymph node after immunization as compared to soluble NP-OVA (Fig. 1E). In contrast, there was no difference in the vaccine response between soluble NP-OVA–injected TLR5−/− and nanovaccine-injected TLR5−/− mice. Similar observations with germinal center response were made 6 days after booster immunization (fig. S3). We quantified CD138+ plasma cells in draining lymph nodes and observed a trend similar to that of the germinal center response (Fig. 1F). The antigen-specific immunoglobulin G (IgG) antibodies in the blood serum increased 2.5- to 3.5-fold in WT mice but not in TLR5−/− mice, further confirming that only the WT mice gained the benefits associated with PLGA vaccination, when compared to the soluble antigen (Fig. 1G).
In the WT mice, the PLGA nanovaccines increased the expression of major histocompatibility complex class II (MHCII) on CD11c+ dendritic cells by threefold in the lymph node compared to soluble NP-OVA (Fig. 1H). In contrast, there was no difference in the expression of MHCII marker on dendritic cells in TLR5−/− mice immunized with the nanovaccine or soluble antigen. There were no significant changes in CD3+ T cells across any group (Fig. 1I). Together, these results highlight that engineered nanovaccines underperform in TLR5−/− mice, a model of the altered gut microbiome and metabolic syndrome, as compared to age-matched WT mice.
Since female mice do not develop obese phenotype, we hypothesized that the lack of the metabolic syndrome phenotype would lead to a similar germinal center response in WT versus TLR5−/− female mice. We confirmed the absence of detectable differences in IL-6, IL-8, TNF-α, IL-1β, and IL-10 inflammation in female TLR5−/− and WT mice (fig. S1), suggesting the absence of metabolic syndrome based on the lack of obesity and an inflammation marker. As expected, both WT and TLR5−/− female mice manifested similar germinal center B cell response with PLGA nanovaccines (Fig. 1J). Collectively, studies in female mice support our hypothesis and suggest that the absence of TLR5 alone is not sufficient to modulate vaccine response, implicating aspects of the metabolic syndrome.
TLR5−/− male mice have reduced PLGA nanovaccine trafficking to lymphoid tissues
Previous studies have demonstrated that nanovaccine immunity often depends on the efficient trafficking of particulate vaccines by immune cells to the draining lymph nodes or through direct drainage (22–25). Therefore, we hypothesized that the poor immune response may correlate to the limited trafficking of nanovaccines in TLR5−/− male mice to the draining lymph node as compared to WT mice. To test this hypothesis, we subcutaneously injected PLGA nanovaccine with a model protein fluorescein isothiocyanate (FITC)–bovine serum albumin (BSA) (50 μg) into the left flank and analyzed the site of injection using an IVIS imager followed by flow cytometry. After immunization, TLR5−/− male mice had higher retention of nanovaccine at the injection site relative to WT mice, a pattern that continued through day 4 and diminished by day 6 (Fig. 2, A and B, and fig. S4). We questioned whether the retention of nanovaccine at the injection site could limit the localization of nanovaccines in the draining lymph node, spleen, and other organs. The draining inguinal lymph node of the TLR5−/− mice exhibited a lower accumulation of nanovaccines in TLR5−/− mice than in the WT mice on day 2 (Fig. 2, C and D), an effect that diminished by day 6. While the liver and kidney showed nanovaccine accumulation on days 2 and 6, there were no observed differences between the TLR5−/− and WT mice (fig. S4). Flow cytometry analysis revealed that in the draining lymph node at day 2, there was a twofold reduction of trafficking of F4/80+ macrophages with FITC-PLGA and CD169+ macrophages with FITC-PLGA+ signal among the TLR5−/− mice relative to the WT mice (Fig. 2E). Similarly, we observed a reduced response among CD11c+ dendritic cells in TLR5−/− mice (25% FITC-PLGA+CD11c+ cells) compared to 40% of the cells in WT mice (Fig. 2E), which correlated with the reduced expression of CD86 activation marker in CD11c+ cells in TLR5−/− mice (fig. S5).
Fig. 2. TLR5−/−phenotype in male mice leads to reduced nanoparticle trafficking from the injection site to lymphoid tissue.
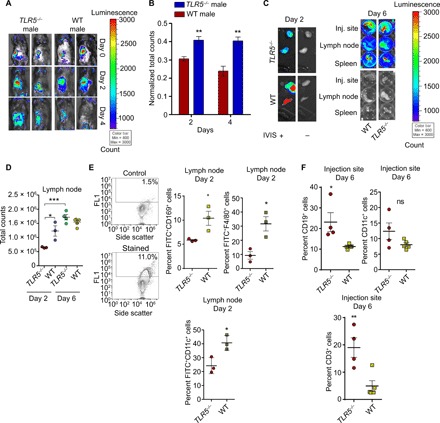
(A) IVIS images demonstrating distribution of nanoparticles at day 0, day 2, and day 4 after injection for TLR5−/− and WT male mice. Images are representative of a cohort of four mice. (B) Quantification of signal at the injection site on days 2 and 4, normalized to day 0 (mean ± SEM). Comparisons at each day were performed using an unpaired, two-tailed t test. (C) Fluorescent images of organs at day 2 and day 6 after injection in male mice. Total counts represent the sum of all counts for all pixels inside the region of interest. (D) Quantification of IVIS signal in lymph node at day 2 and day 6 (n = 4). Statistics was performed using an unpaired, two-tailed t test. (E) Left: Gating strategy for flow cytometry. Top left is an unstained control; bottom left represents a stained sample. Right: Scatterplots represent percent FITC+CD169+ macrophage, FITC+CD11c+ dendritic cells, and FITC+F4/80 macrophages in the lymph node on day 2. Comparisons were made with an unpaired, two-tailed t test. (F) Percent CD19+ B cells, CD11c+ dendritic cells, and CD3+ T cells at the injection site on day 6 after immunization. Comparisons were performed using an unpaired, two-tailed t test. For all flow cytometry data, readouts are presented as means ± SEM. For trafficking studies, n = 3 for both groups on day 2. n = 4 for TLR5−/− group on day 6 and n = 5 for WT group on day 6. In all studies, *P < 0.05, **P < 0.01, and ***P < 0.001. ns denotes nonsignificant differences.
We questioned whether the differential accumulation of FITC-PLGA+ signal in lymph node cells is attributable to the recruitment of antigen-presenting cells at the injection site that might traffic to the draining lymph node. We harvested the injection site, and flow cytometry analysis showed no differences in CD11c+ dendritic cells, CD169+ macrophages, and GR1+ neutrophils at the injection site (Fig. 2F and fig. S6). In contrast to the antigen-presenting phagocytic cells, a twofold elevation of CD19+ B cells and a fourfold increase in the percentage of CD3+ T cells were observed at the injection site in TLR5−/− mice relative to WT mice (Fig. 2F). Since vaccinated TLR5−/− mice do not mount a robust germinal center response (see Fig. 1E) or show T cell immunity, we do not think that these recruited cells contribute to the vaccine response in the draining lymph node in the tested mice. No differences in studied cell populations were seen on day 2 at the injection site (fig. S6).
PLGA nanovaccines change the gut microbiota after booster dose
To understand which bacteria correlate with vaccine response, we characterized the changes in gut microbiome with PLGA nanovaccine administration. We performed taxonomic profiling using 16S ribosomal RNA (rRNA) sequencing (26) of the gut microbiome of TLR5−/− and WT mice on stool samples obtained from mice immediately before the booster and 6 days after the booster when the germinal center formation is evident (fig. S3). Overall compositional differences between the gut microbiomes of WT and TLR−/− mice on the day of the booster were subtle (Fig. 3A). However, after the booster, the number of discernable differences between these two groups increased (Fig. 3B). After the booster, there was a significant increase in Proteobacteria and Bacteroidetes in the TLR5−/− mice than WT mice and a concomitant increase in the representation of members of the phyla Firmicutes (P < 0.0001) and Tenericutes (P < 0.01) in WT mice compared to TLR5−/−.
Fig. 3. PLGA nanovaccine immunization alters gut microbiome composition.
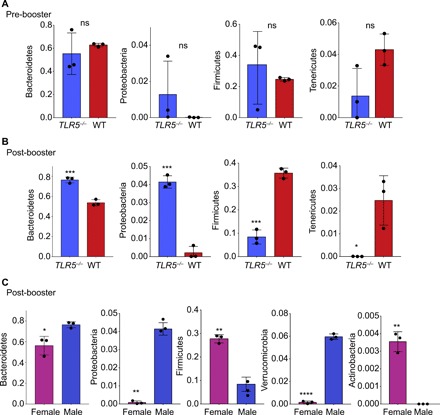
(A and B) Differentially abundant microbial phyla in stool samples from WT (n = 3) versus TLR5−/− (n = 3) male mice, before and after booster doses. (C) Differentially abundant microbial phyla in the stool samples from male (n = 3) versus female (n = 3) TLR5−/− mice, after booster doses. Statistical analysis was performed using paired two-tailed t test. In all studies, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. ns denotes nonsignificant differences.
Since the observed immunological differences between nanovaccine response were only observed in male WT versus TLR5−/− mice and not corresponding age-matched female mice, we next examined the compositional differences in the gut microbiome of TLR5−/− male versus female mice after booster vaccination. Female TLR5−/− mice had more abundance of Actinobacteria (P < 0.001) and Firmicutes (P < 0.005) than male TLR5−/− mice after booster vaccination (Fig. 3C). Male TLR5−/− mice showed an increase in Bacteroidetes, Verrucomicrobia, and Proteobacteria. Although increases in the abundance and instability of Proteobacteria are considered a hallmark of TLR5−/− microbiomes (27), we only observed a notable increase in Proteobacteria after the booster in male mice (P < 0.0001, one-tailed t test). Further examination would be required to determine whether these bacteria play a causative role in vaccination response. Collectively, these results suggest that differential vaccine response in male mice may be attributable to distinct microbiome composition, in addition to reduced metabolic phenotypes, and that these features are not observed in female mice at 16 weeks of age.
Gut microbiome dysbiosis with chronic antibiotics decouples the role of obesity and systemic inflammation in dysregulated vaccine response
We next sought to decouple whether obesity, as a secondary effect of the dysregulated microbiome, was responsible for poor response in TLR5−/− mice. To study these phenomena, we fed WT mice with a cocktail of antibiotics [ampicillin (1.0 g/liter) and neomycin (0.5 g/liter)] in their drinking water, as we have reported earlier (28), starting at 4 weeks of age (Fig. 4A) and continuing through immunization. Unlike TLR5−/− mice, we did not observe an increase in the fat pad mass and body weight of antibiotic-treated mice (Fig. 4B). Microbiome analysis before immunization at 16 weeks suggested that antibiotic-treated WT mice had a significant decrease in Bacteroidetes, Firmicutes, and Actinobacteria and an increase in Proteobacteria (Fig. 4C). Antibiotic-fed mice were immunized at age 16 weeks according to the previously implemented timeline with either NP-OVA PLGA nanovaccine or soluble NP-OVA. We analyzed the immune response and observed no differences in the germinal center B cell (GL7+FAS+CD19+) formation in either the spleen or the draining lymph node (Fig. 4D). Other cell populations, except CD11c+ dendritic cells, remained largely unchanged (fig. S7). Furthermore, blood serum enzyme-linked immunosorbent assay (ELISA) confirmed that there were no differences in antigen-specific antibody levels (Fig. 4E). Before the booster, the antibiotic-treated mice had elevated levels of inflammatory cytokine IL-6 but not IL-8, IL-10, IL-1β, and TNF-α (Fig. 4F). Drawing from these results, we conclude that the differences in the immune response are due to a dysregulation of the intestinal microbiome but not obesity. It is possible that elevated levels of IL-6 may play a crucial role; however, future studies are warranted to decouple the effect of microbiome sensing and IL-6. The antibiotic cocktail delivered to these mice did not entirely ablate the bacterial microbiota but altered toward an increased Proteobacteria composition. We, therefore, conclude that simply perturbing the detection of the microbiota, which leads to increased inflammation, is sufficient to regulate the PLGA nanovaccine responses.
Fig. 4. Antibiotic-mediated dysbiosis of gut microbiome alters adaptive immune response in male WT mice.
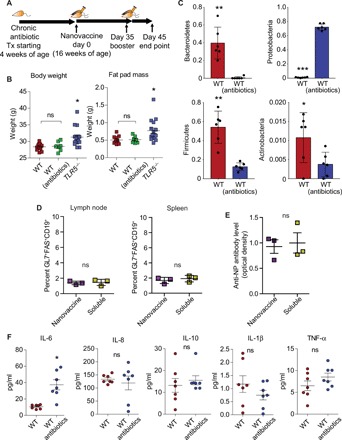
(A) Timeline of antibiotic administration and immunization. Antibiotics were continued from weaning through the end of vaccination. (B) Weight and fat pad mass of mice fed on antibiotics and TLR5−/− mice presented as means ± SEM. Comparisons were made by t test [n = 12 for WT, n = 7 for WT (antibiotics), and n = 16 for TLR5−/−]. (C) Differentially abundant microbial phyla in stool samples from WT (n = 3) versus WT antibiotic-treated (n = 7) male mice before immunization. (D) Percentage of GL7+FAS+CD19+ germinal center B cells in the lymph node and spleen of antibiotic-treated male mice after booster vaccination with either soluble NP-OVA or nanovaccine-formulated NP-OVA. Statistics was performed using an unpaired, two-tailed t test, with populations presented as means ± SEM (n = 3). (E) Scatterplot presents the antigen-specific antibodies in the serum of mice after immunization with nanovaccine and soluble formulation (n = 3). Statistics was performed using an unpaired, two-tailed t test, with populations presented as means ± SEM (n = 3). (F) Inflammation marker levels in the serum of WT and antibiotic-fed WT mice. Statistics was performed using an unpaired, two-tailed t test, with cytokines presented as means ± SEM (n = 7). In all studies, *P < 0.05, **P < 0.01, ***P < 0.001. ns denotes nonsignificant differences.
Pyr-pHEMA nanogel vaccine rescues immune response in the male metabolic syndrome mouse model
We hypothesized that a rational combination of antigen and adjuvant will overcome the poor response imposed by gut or metabolic syndrome by stimulating immune cells. We further hypothesized that a nanovaccine in which the polymer itself has an adjuvant effect may overcome the limitations of dual encapsulation of antigens and adjuvants in nanovaccines. Previously, we had engineered an immunomodulatory nanomaterial where pHEMA was covalently modified with pyridine using 4-dimethylaminopyridine (DMAP) (29). We have previously reported that Pyr-pHEMA nanogels are nontoxic in mammalian cells with increasing doses of nanogels up to 2.0 mg/ml (30). When the modified Pyr-pHEMA polymer was incubated with a protein antigen of interest, the polymer self-assembled to form a nanogel that can activate in vitro cultured dendritic cells and T cells, more than a conventional adjuvant (29). However, it remained unclear whether the immunomodulatory effect of Pyr-pHEMA originated from the polymer itself and whether the Pyr-pHEMA nanogel can overcome the limitations faced by PLGA nanovaccines in the TLR5−/− model.
Pyr-pHEMA was synthesized, as reported earlier (29), with a small modification to scale up the production to three times than reported earlier. Briefly, 1.0 mmol of pHEMA polymer was mixed with nicotinoyl chloride hydrochloride (1.2 mmol) in tetrahydrofuran (THF) and DMAP (Fig. 5A). The resulting purified, concentrated Pyr-pHEMA was confirmed by 1H and 13C nuclear magnetic resonance (NMR) spectra acquired in DMSO (dimethyl sulfoxide)–d6 at 25°C, which confirmed that the backbone, linker, and pyridine have experimentally indistinguishable diffusion properties (Fig. 5B). The esterification was confirmed by the following peaks: 7.2 to 9.0 for four pyridine-H peaks, 4.0 to 4.5 for two OCH2 peaks, 1.5 to 2.0 for CH2, and 0.5 to 1.0 for ─CH3. The modification of pHEMA with pyridine decreased the solubility of pHEMA in water (Fig. 5C), and ultraviolet-visible spectroscopy indicated that the absorbance of a pHEMA solution was lower than that of Pyr-pHEMA, thus confirming turbidity of the solution (Fig. 5D). We believe that the self-assembly of nanogels is driven through the displacement of proteins between the aqueous water and less water-soluble Pyr-pHEMA at pH 7.4, with minimized interfacial energy. The encapsulation efficacy of protein was dependent on the initial protein concentration. We observed up to 0.62 ± 0.1 mg of FITC-BSA per milligram of Pyr-pHEMA in nanogels engineered using protein concentrations of 280 μg/ml, compared to 0.2 ± 0.1 mg using protein concentrations of 140 μg/ml (Fig. 5E). A 120-hour release study in phosphate-buffered saline (PBS) indicated 40% protein release in 24 hours with nanogels formed using protein concentrations of 280 μg/ml, as compared to 30% protein release using protein concentrations of 140 μg/ml (Fig. 5F). The release profile saturated after 36 hours at 48 and 38%, respectively. For the remainder of the studies, we continued with nanogels formed using protein concentrations of 280 μg/ml, with an average particle size of 190 nm (fig. S2), comparable to the size of PLGA nanovaccines studied in Fig. 1.
Fig. 5. Pyr-pHEMA nanogels rescue the antigen-specific germinal center response in male TLR5−/−mice and improve particle trafficking.
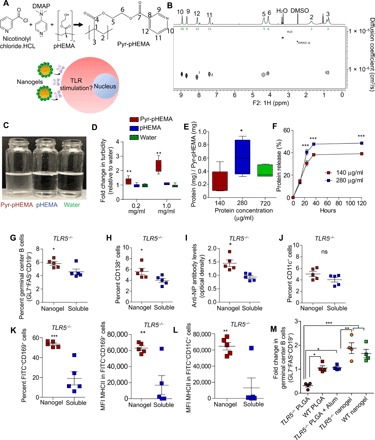
(A) Chemical structure of pyridine functionalized pHEMA and schematic of nanogel interaction with immune cells. (B) Contour plot of diffusion-ordered spectroscopy (DOSY) NMR spectrum of Pyr-pHEMA in DMSO-d6 at 25°C confirming that the backbone, linker, and pyridine have experimentally indistinguishable diffusion properties. ppm, parts per million. (C) Solubility analysis of Pyr-pHEMA and pHEMA as compared to water. Photo credit: M.J.M., Cornell University. (D) Turbidity analysis of Pyr-pHEMA and pHEMA as compared to water using absorbance of solution at 570 nm (n = 5 per formulation). All groups were compared by one-way ANOVA with Tukey’s post hoc test and presented as a box-whisker plot. (E) Loading ratio of Pyr-pHEMA to protein at various soluble protein loading concentrations (n = 5 per formulation, presented as means ± SEM). Comparisons were made by an unpaired, two-tailed t test (*P < 0.05) between protein loading concentrations. (F) Protein release of Pyr-pHEMA in in vitro conditions. All groups were compared by two-way ANOVA with Bonferroni’s correction, and data are presented as means ± SEM (n = 4; ***P < 0.001). (G to J) Pyr-pHEMA nanogel induced germinal center B cell population (G), CD138+ population (H), antigen-specific antibody levels in the serum (I), and CD11c+ dendritic cell population (J) in immunized male TLR5−/− mice. Comparisons were made by an unpaired, two-tailed t test, and all readouts were expressed as means ± SEM (n = 5 WT and n = 5 TLR5−/−). In all studies, *P < 0.05, **P < 0.01, and ***P < 0.001. ns denotes nonsignificant differences. (K and L) Pyr-pHEMA nanogel trafficking and expression of MHCII on CD169 macrophages (K) and CD11c+ dendritic cells (L) in the lymphoid tissues were compared to soluble antigen. Comparisons were made by an unpaired, two-tailed t test, and all data were presented as means ± SEM (n = 5 nanogel and n = 5 soluble formulation). (M) Fold change in GL7+FAS+CD19+ germinal center B cell population between Pyr-pHEMA nanogel, PLGA nanovaccines, and PLGA nanovaccines adjuvanted with alum. All particles were loaded with NP-OVA antigen. All groups were compared by one-way ANOVA with Tukey’s post hoc test, and data are presented as means ± SEM (n = 4). In this study, *P < 0.05, **P < 0.01, and ***P < 0.001.
We immunized male TLR5−/− mice either with Pyr-pHEMA nanogels formulated with 50 μg of NP-OVA or as a soluble antigen to determine whether the nanogel would boost the immune response beyond the soluble formulation. Ten days after the booster dose, Pyr-pHEMA nanogels induced a significant 1.5-fold higher GL7+FAS+CD19+ germinal center B cells in the lymph node as compared to soluble NP-OVA (Fig. 5G). This is in contrast with PLGA vaccines (Fig. 1E), suggesting that Pyr-pHEMA can rescue the poor immune response in the male TLR5−/− mouse model. Analysis of CD138+ cells unveiled a similar trend, and the Pyr-pHEMA response was comparable in TLR5−/− and WT mice (Fig. 5H and fig. S8). The antigen-specific antibodies in the blood serum confirmed that engineered Pyr-pHEMA nanogel rescued the limited immune response shown in PLGA nanovaccines as compared to the soluble antigen (Fig. 5I). However, as shown in Fig. 5J, there was no difference in the CD11c+ dendritic cells in TLR5−/− mice immunized with the Pyr-pHEMA nanovaccine or soluble antigen.
In separate Pyr-pHEMA nanogel trafficking studies, flow cytometry analysis revealed that in the draining lymph node at day 2, there was a 2.5-fold increase in trafficking of CD169+ macrophages with FITC-OVA+ Pyr-pHEMA nanogels (Fig. 5K). The trafficked macrophages showed significantly higher MHCII expression in mice immunized with FITC-OVA–loaded Pyr-pHEMA nanogel than soluble FITC-OVA antigen. Although we did not see a difference in percent CD11c+ dendritic cells between Pyr-pHEMA nanogels and soluble antigen (fig. S8), the expression of MHCII marker on CD11c+ dendritic cells (Fig. 5L) was similar to that on the CD169+ macrophages. In addition, we observed that the Pyr-pHEMA nanogels did not accumulate differentially in the kidney or liver (fig. S9). Collectively, these results led us to conclude that Pyr-pHEMA nanogels can induce an increased immune response in TLR5−/− mice with dysregulated gut sensing and associated metabolic syndrome. Last, in separate immunization studies, we directly compared the germinal center B cell response in PLGA nanovaccine alone, PLGA nanovaccine with gold-standard adjuvant alum, and Pyr-pHEMA nanogel. As indicated in Fig. 5M, addition of alum to PLGA nanovaccines rescued the B cell response in TLR5−/− mice to the level of WT mice immunized with PLGA nanovaccines alone. In contrast, Pyr-pHEMA nanogels without any exogenous adjuvant induced higher (P < 0.001) germinal center response in TLR5−/− mice as compared to alum-supplemented PLGA nanovaccines. Therefore, Pyr-pHEMA nanogels offer an immunomodulatory effect without the combinatorial delivery of adjuvant molecules.
Pyr-pHEMA nanogel immunogenicity is mediated by TLR2 on immune cells
It is well established that materials or biologics that mimic bacterial pathogen-associated molecular patterns are capable of stimulating TLRs on the surface of immune cells. In a competing theory by Seong and Matzinger (31), hydrophobic portions of biological molecules and potentially materials can act as universal damage-associated molecular patterns to initiate an immune response, in particular through TLR2 and TLR4 (32–35). New studies have shown that select small-molecule compounds that are aromatic in nature and structurally unrelated to any known TLR2 agonists can function as TLR2 agonists (33). Since Pyr-pHEMA has hydrophobic pyridine attached to pHEMA and pyridine is an aromatic compound, we next investigated whether Pyr-pHEMA nanogel would stimulate immune cells either through TLR2 or TLR4 or, alternatively, through TLR5 or TLR9. We transfected TLR2, TLR4, or TLR9 into human embryonic kidney (HEK) 293 cells (that endogenously express TLR5) and exposed the cells to Pyr-pHEMA nanogels carrying NP-OVA antigen (20 μg/ml), antigen alone (20 μg/ml), or media. Pyr-pHEMA nanogels stimulated TLR2 signal, as indicated by high relative light units of luciferase (Fig. 6A), but had minimal effect on TLR4, TLR5, and TLR9, similar to antigen alone and media. These results suggest that Pyr-pHEMA nanogels function through TLR2 (Fig. 6B) and that the stimulation is driven by polymer and not the complexed protein. We tested this hypothesis by comparing immune response in TLR2−/−, TLR4−/−, and WT mice. Previous studies (36) have shown that the small intestinal TLR5 expression is not regulated through TLR2 or TLR4, as shown by unchanged levels of small intestinal TLR5 transcript in small intestinal tissues of TLR2−/− and TLR4−/− mice. These studies further showed that TLR5−/− mice did not affect TLR2 and TLR4 mRNA levels. Therefore, we do not expect TLR5 to be influenced in TLR2 and TLR4 mice. We first exposed bone marrow–derived primary dendritic cells from TLR2−/−, TLR4−/−, and WT mice to the nanogel formulation (20 μg/ml). We observed that the MHCII and CD80 expression on nanogel-incubated cells was significantly reduced in cells derived from TLR2−/− mice but not in TLR4−/− and WT mice (Fig. 6C). To further validate our hypothesis, we immunized TLR2−/−, TLR4−/−, and WT mice with NP-OVA as a soluble antigen or formulated with Pyr-pHEMA nanogel. We observed that while both WT and TLR4−/−-deficient mice had significantly elevated GL7+FAS+CD19+ germinal center B cell population relative to their soluble counterparts, TLR2−/−-deficient mice did not experience any increase in humoral immunity relative to the soluble antigen (Fig. 6D and fig. 10). These results explain that in vivo humoral immunity is regulated by Pyr-pHEMA nanogel and, more specifically, through TLR2 interactions. Whether hydrophobicity and aromaticity are truly the drivers of Pyr-pHEMA response will be the subject of future investigations.
Fig. 6. Immunomodulatory effects of Pyr-pHEMA are mediated through TLR2.
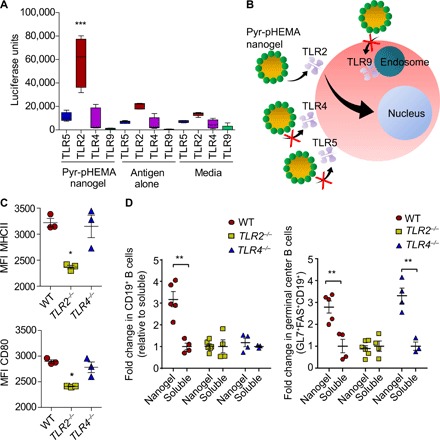
(A) TLR stimulatory activity of Pyr-pHEMA nanogels and controls in HEK293 cells transfected with TLR2, TLR4, and TLR9. HEKs endogenously express TLR5. Luciferase activity indicates activation of a TLR. Statistics was performed using a two-way ANOVA with Bonferroni’s correction (n = 5). (B) Schematic presenting the proposed hypothesis that Pyr-pHEMA nanogels function through TLR2. (C) MFI of MHCII and CD80 on bone marrow–derived dendritic cells that are exposed to Pyr-pHEMA nanogels. Statistics was performed using a one-way ANOVA with Tukey’s post hoc correction (n = 3), with all data presented as means ± SEM (n = 3). (D) Fold change in CD19+ B cell and GL7+FAS+CD19+ germinal center B cell population between nanogel and soluble formulations. All nanogel groups were compared by an unpaired, two-tailed t test with their soluble counterparts, and data are presented as means ± SEM (n = 5 WT nanogel, n = 4 WT soluble, n = 6 TLR2−/− nanogel, n = 5 TLR2−/− soluble, n = 4 TLR4−/− nanogel, and n = 3 TLR4−/− soluble). In all the studies, *P < 0.05, **P < 0.01, and ***P < 0.001. ns denotes nonsignificant differences.
CONCLUSION
While recent work links intestinal microbiome to immunity and soluble antigens (17, 37–39), we demonstrate how the gut microbiome and the resulting metabolic syndrome modulate humoral immunity against biomaterials-based nanovaccines. Collective analyses of male TLR5−/− mice and antibiotic-treated mice provide early indications that gut microbiome–mediated inflammation, and perhaps not obesity, could be the driver of poor PLGA nanovaccine response. The impact of obesity could be further disentangled from that of the microbiota using obese animal models or alternative models, such as fecal transfer experiments and cohabitation studies, and will be investigated in the future. We chose TLR5−/− mice because this model has been linked to both metabolic syndrome and vaccine responses (16, 17, 40). The symptoms developed by TLR5−/− mice are milder than those developed by ob/ob obese mice. Gut inflammation in the TLR5−/− mice is caused directly by microbiota, and other traits of the TLR5−/− phenotype are downstream of increased gut inflammation (16, 41, 42). Pulendran and colleagues showed that antiviral vaccine itself does not directly signal through TLR5 but rather that the intestinal microbiota contribute to TLR5-mediated enhancement of immunity to soluble human influenza vaccine and to the inactivated polio vaccine (17). Several human influenza vaccine formulations in clinical trial have used flagellin as an adjuvant for TLR5 signaling [e.g., VAX102 (M2e-flagellin) universal influenza vaccine; ClinicalTrials.gov identifier: NCT00921947]. Future studies will investigate the effect in other metabolic syndrome mouse models. The studies reported here are at 16 weeks of age when male mice manifest metabolic syndrome phenotype. Although female mice do not manifest metabolic syndrome phenotype at 16 weeks of age, future investigation in aged mice is needed to confirm that it is a gender-specific phenomenon. In the current study, we engineered a TLR2 stimulating Pyr-pHEMA nanogels, which modulated the immunity in a gut microbiome–mediated metabolic syndrome mouse model. The causative effects of nanoparticle-microbiome changes with respect to PLGA and the interaction of Pyr-pHEMA with gut microbiome will be evaluated in future studies. The nano-dimension can span a broad length scale, and the response demonstrated may be different with particles that are different in size from those studied here. Therefore, further studies are needed to elucidate the role of various nanoparticle sizes and material characteristics. We anticipate that these studies may further pave the path to discover how modulations to gut microbiome coupled with immunomodulatory vaccines could improve responses in patients with metabolic syndrome. We also anticipate that these results will lay the foundation for future work in infectious disease vaccines and vaccination in the populations that manifest metabolic syndrome (43, 44) and predict how metabolic diseases, gut pathologies, and other inflammatory conditions may affect nanovaccine response.
MATERIALS AND METHODS
TLR5−/− mouse phenotype characterization
All mice were characterized by weighing the whole mouse at the time of euthanasia, followed by determining the weight of the epididymal fat pads. Blood was collected at euthanasia to quantify the systemic inflammation. Blood serum was stored at −80°C and analyzed at the Duke Molecular Physiology Institute Biomarkers Shared Resource. Serum was assayed using a custom Proinflammatory Panel (Meso Scale Diagnostics) measuring IL-1β, IL-6, IL-8, IL-10, and TNF-α and a Mouse Metabolic Kit (Meso Scale Diagnostics; Rockville, MD). All animal studies were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) at Cornell University.
Mouse immunizations
Mice were immunized either subcutaneously or intraperitoneally with 50 μg of NP-OVA encapsulated into PLGA or complexed to Pyr-pHEMA, a dose-matched soluble formulation (50 μg/ml), or PBS. PLGA particles were synthesized using Resomer RG 503 H (Sigma-Aldrich; molecular weight, 24,000 to 38,000) with polyvinyl alcohol (Sigma-Aldrich; 2% w/v; molecular weight, 40,000 to 70,000; 80 to 90% hydrolyzed) using previously published protocols (45). The release profile of biologics from PLGA has been reported earlier by us (19) and shows an initial burst release followed by sustained release phase and a next phase of release observed around day 20. At indicated timelines in figures, a booster injection was applied. Blood was collected via the submandibular vein from the mice, and antigen-specific antibodies were quantified with ELISA. After either 6 or 10 days, animal organs were harvested and lymphoid tissue was prepared for flow cytometry. For Pyr-pHEMA, TLR5−/− mice were intraperitoneally immunized either with nanovaccines formulated with 50 μg of NP-OVA or as a soluble antigen to determine whether the nanogel would boost the response beyond the soluble formulation. Previous work has shown that subcutaneous, intramuscular, and intraperitoneal injections all elicit a strong immune response in engineered vaccines; however, intraperitoneal vaccination has a shorter timeline for induction of immune response (46, 47). Two weeks later, a booster dose was injected, and after another 10 days, cells were harvested from the draining lymph node. Similar studies were performed with TLR2−/−, TLR4−/−, and WT mice with matched age and gender. PLGA nanovaccines with adjuvant Imject Alum (Thermo Scientific), which contains an aqueous solution of aluminum hydroxide (40 mg/ml) and magnesium hydroxide (40 mg/ml) plus inactive stabilizers, were prepared as per the manufacturer’s recommendation. Briefly, Imject Alum was added dropwise with constant mixing to the NP-OVA so that the final volume ratio of Imject Alum to immunogen was 1:1 and then encapsulated inside PLGA nanovaccine and injected intraperitoneally into mice. PLGA nanovaccine without adjuvant and Pyr-pHEMA nanogel were used as comparative groups in the adjuvant study and were injected intraperitoneally. All animal studies were performed in compliance with the IACUC at Cornell University. The following antibodies or their fluorophore variations were used in this study: Peridinin Chlorophyll Protein Complex (PerCP) PerCP-eFluor 710–anti-CD3 (17A2), PerCP-Cyanine5.5–anti-CD8 (53-6.7), Phycoerythrin (PE)–Cyanine7–anti-CD11c (N418), eFluor 660–anti-CD169 (SER-4), and PE–anti-F4/80 (BM8) from eBioscience; PE–anti-MHCII (M5/114.15.2) and PE–anti-CD138 (300506) from Invitrogen; and Allophycocyanin (APC)–anti-CD19 (1D3), FITC–anti-GL7 (GL7), and PE–anti-CD95 (Jo2) from BD Pharmingen.
Microbiome analysis
DNA was isolated from mouse fecal pellets using the MO BIO PowerSoil DNA Isolation Magnetic Kit with the recommended proteinase K step to assist in cell lysis for gut microbiome samples. 16S rRNA libraries were prepared using the Earth Microbiome Project protocol with primers as described by Walters et al. (48). Libraries were sequenced on the Illumina MiSeq platform, and each sample had over 10,000 paired-end 150 ×150 base pair (bp) reads. Sequences were analyzed using mothur (49). Paired sequences were stitched together, and those with ambiguous nucleotides or those longer than 275 bp were removed. Chimeras were identified using UCHIME (50) and removed. Stitched reads were aligned to the SILVA rRNA database v.128 (51) and clustered at 97% sequence identity, and 16S rRNA sequences were assigned taxonomies using the Ribosomal Database Project naïve Bayesian classifier with a confidence threshold value of 80% (52).
Chemical synthesis of Pyr-pHEMA using DMAP
Some of the following methods are similar to those previously published (29). Pyr-pHEMA synthesis was performed through the esterification of the hydroxyl group on pHEMA. Briefly, pHEMA (molecular weight, 20,000 Da; 150 mg, 1.2 mmol OH residues) was dispersed in anhydrous THF (Sigma-Aldrich; 5 ml) at room temperature. DMAP (Sigma-Aldrich; 451 mg; 3.6 mmol) was added, and the mixture was allowed to stir at room temperature until the solution became homogeneous. Nicotinoyl chloride hydrochloride (Alfa Aesar; 411 mg; 2.3 mmol) was dispersed in THF and added dropwise to the pHEMA solution mixture under inert nitrogen atmosphere. The esterification was allowed to proceed at room temperature for 48 hours. A precipitate appeared within 24 hours and was separated from the liquid by centrifugation at the end of the reaction. The precipitate was further washed with THF, and the supernatant was combined and precipitated in cold diethyl ether. The final solid precipitate was rinsed thoroughly with ether three times to remove excess DMAP. Last, the precipitate was collected and dried under vacuum for 3 days at room temperature. 1H and two-dimensional NMR spectra were acquired in DMSO-d6 at 25°C on a 600-MHz Varian Unity INOVA spectrometer operating at 599.50 and 150.76 MHz for 1H observation and 13C decoupling, respectively, using a 5-mm, inverse, triple-resonance probe head. 1D 13C spectrum was acquired in DMSO-d6 at 25°C on a 500-MHz Varian Unity INOVA spectrometer operating at 125.68 MHz for 13C observation, using a 5-mm, direct observe, dual probe head. Spectra were processed and analyzed in MNova 12.0.2 (Mestrelab Research S.L., Santiago de Compostela, Spain). Diffusion-ordered NMR dataset was acquired with the double, bipolar pulse stimulated echo sequence, as supplied in the DOSY package of VnmrJ 3.2 (Dbppste_cc), with 80-ms diffusion delay and 4.4-ms bipolar diffusion gradient pulses.
Fabrication of Pyr-pHEMA self-assembly nanogel
Some of the following methods are similar to those previously published (29). Nanogels were formed by dropwise addition of 0.2 ml of Pyr-pHEMA (2 mg/ml in dimethylformamide) into a 5-ml solution of a protein as a model antigen (NP-OVA and FITC-BSA) in PBS under stirring at 900 rpm at room temperature. The resulting product was separated from unreacted components by centrifugation at 20,000g and washed three times. Characterization of size was performed using Zetasizer Nano ZS (Malvern Instruments). We determined the protein loading efficiency using FITC-BSA nanogels and a BioTek Synergy H1 plate reader (488-nm excitation and 525-nm emission). Protein amount was quantified using a standard curve [FITC-BSA (0 to 60 μg/ml)]. FITC-BSA nanogels were prepared using stock protein solution concentrations of 60,120 and 240 μg/ml with 0.2 ml of Pyr-pHEMA polymer (2 mg/ml). Samples were centrifuged three times and thoroughly washed. To quantify release rate, nanogels were placed in a 96-well plate at a known protein polymer concentration. Every 24 hours, several wells were removed and placed into Eppendorf tubes. These were spun down at 20,000g to separate nanogels from soluble protein, and the fluorescence of the protein nanogels was quantified, as previously discussed. For further studies, nanogels formulated with a protein concentration of 280 μg/ml were used as their diameters most closely matched comparable PLGA nanoparticles.
TLR luciferase assay
The quantification of TLR activity was performed as previously described (53). Briefly, HEK293 cells (2 × 104) were transiently transfected in 96-well plates using TransIT (Mirus) with TLR4-encoding plasmids, 5× nuclear factor κB–luciferase, and empty vector equal to 200 ng per well of total DNA. After overnight culture, the cells were treated with Pyr-pHEMA nanogels with antigen (20 μg/ml), antigen alone (20 μg/ml), and media for 18 hours. Assay validation was performed using Pam3Cysk (1 μg/ml) (TLR2), lipopolysaccharide (100 ng/ml) (TLR4), flagellin (100 ng/ml) (TLR5), or CpG DNA (1 μg/ml) (TLR9) for 18 hours. Cell lysates (5× reporter lysis buffer; Promega) were assayed for luciferase activity using the luciferase substrate [20 mM tricine, 2.67 mM MgSO4⋅7H2O, 33.3 mM dithiothreitol, 100 mM EDTA, 530 mM adenosine 5′-triphosphate, 270 mM acetyl coenzyme A, luciferin (132 mg/ml), 5 mM NaOH, and 265 mM magnesium carbonate hydroxide] on a Veritas luminometer (Turner BioSystems) with an injector.
In vitro nanogel response in TLR2−/−, TLR4−/−, and WT mice
Bone marrow monocytes were isolated from TLR2−/−, TLR4−/−, and WT mice. Mice were euthanized by CO2 asphyxiation. All subsequent steps were performed in a laminar flow hood. The femur was carefully removed from the muscle and washed thoroughly with ethanol to sterilize their exteriors. Either side of the femur was cut with dissection scissors, and bone marrow was flushed into sterile Eppendorf tubes with PBS. Immature bone marrow cells were spun down at 300g and lysed in red blood cell lysis buffer. To differentiate the bone marrow cells into dendritic cells, they were cultured in RPMI medium at a seeding density of 5 × 104 cells per well. RPMI was supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) (20 ng/ml) and placed in an incubator at 37°C and 5% CO2. After 3 days, an additional volume of RPMI with GM-CSF (20 ng/ml) was added. After 6 days, media were replaced with RPMI supplemented with GM-CSF (20 ng/ml) and nanogels (20 μg/ml) were added. Cells were kept in culture for 48 hours, and cell surface marker expression levels were quantified by flow cytometry.
Statistical analysis
All experiments were performed with three or more replicates as detailed in the figure legends. Immunization and trafficking experiments were repeated at least two times. Statistical analysis was performed as detailed in the figure legends and text. All gut microbiome analysis used one-tailed t test, whereas other data analysis used an unpaired two-tailed t test or one-way analysis of variance (ANOVA) with Tukey’s post hoc test or two-way ANOVA with Bonferroni’s correction. Quantitative analyses as scatter or bar graphs are presented as means ± SEM. Microbiome results are presented as cladograms. In all studies, *P < 0.05, **P < 0.01, and ***P < 0.001 unless otherwise stated. Nonsignificance is denoted by “ns.”
Supplementary Material
Acknowledgments
We acknowledge D. Putnam at Cornell University for providing TLR2−/− and TLR4−/− mice. We acknowledge F. Apoorva and P. Graney for technical support. We acknowledge assistance from I. Keresztes at Cornell NMR facility for data acquisition and analysis. Funding: The authors acknowledge partial financial support from the NIH and NIAID (1R01AI132738-01A1 to A.S.) and the NSF CAREER Award (award no. 1554275 to A.S.). The authors acknowledge financial support from the Immunoengineering T32 training grant to M.J.M. (NIH and NIBIB, 1T32EB023860-01A1). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the funding agency. Author contributions: M.J.M. conducted all vaccination studies, analyzed data, and wrote the manuscript with A.S. J.D.G. characterized the mouse phenotypes. H.Z. and I.L.B. analyzed 16S sequencing data. T.T.J., P.R., and K.L. assisted in vaccination studies. S.K. synthesized the Pyr-pHEMA polymer. C.A.L. assisted in transfection studies. M.L. and C.J.H. assisted with colony breeding and antibiotics administration. The concept was conceived by A.S., and funding was generated by A.S. All authors reviewed the manuscript and provided feedback. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. 16S rRNA amplicon sequence data have been posted in the Short Read Archive (SRA accession PRJNA515922). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav9788/DC1
Fig. S1. Characterization of WT and TLR5−/− mice (related to Fig. 1).
Fig. S2. Pyr-pHEMA nanogels are equivalent in size to PLGA nanoparticle vaccines (related to Fig. 1 and Fig. 5).
Fig. S3. Knockout of the TLR5 receptor results in lower germinal center formation in mice immunized with a PLGA nanovaccine (related to Fig. 1).
Fig. S4. PLGA nanoparticle trafficking from the injection site to lymphoid tissue on day 6 and accumulation in the liver and kidneys at days 2 and 6 after injection (related to Fig. 2).
Fig. S5. Expression of CD86 activation marker (related to Fig. 2).
Fig. S6. Injection site analysis (related to Fig. 2).
Fig. S7. Cell populations in the spleen and lymph node of immunized antibiotic-fed mice (related to Fig. 4).
Fig. S8. Immunological characterization of Pyr-pHEMA nanogels (related to Fig. 5).
Fig. S9. Pyr-pHEMA nanogels do not differentially accumulate in tissue after 6 days relative to soluble formulation (related to Fig. 5).
Fig. S10. Immunomodulatory effects of Pyr-pHEMA are mediated through TLR2 (related to Fig. 6).
REFERENCES AND NOTES
- 1.Kasturi S. P., Skountzou I., Albrecht R. A., Koutsonanos D., Hua T., Nakaya H. I., Ravindran R., Stewart S., Alam M., Kwissa M., Villinger F., Murthy N., Steel J., Jacob J., Hogan R. J., García-Sastre A., Compans R., Pulendran B., Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas S. N., van der Vlies A. J., O’Neil C. P., Reddy S. T., Yu S. S., Giorgio T. D., Swartz M. A., Hubbell J. A., Engineering complement activation on polypropylene sulfide vaccine nanoparticles. Biomaterials 32, 2194–2203 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Hubbell J. A., Thomas S. N., Swartz M. A., Materials engineering for immunomodulation. Nature 462, 449–460 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Moon J. J., Huang B., Irvine D. J., Engineering nano- and microparticles to tune immunity. Adv. Mater. 24, 3724–3746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguilar M., Bhuket T., Torres S., Liu B., Wong R. J., Prevalence of the metabolic syndrome in the United States, 2003-2012. JAMA 313, 1973–1974 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Mozumdar A., Liguori G., Persistent increase of prevalence of metabolic syndrome among U.S. adults: NHANES III to NHANES 1999–2006. Diabetes Care 34, 216–219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Neill S., O'Driscoll L., Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 16, 1–12 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Pedersen H. K., Gudmundsdottir V., Nielsen H. B., Hyotylainen T., Nielsen T., Jensen B. A. H., Forslund K., Hildebrand F., Prifti E., Falony G., Le Chatelier E., Levenez F., Doré J., Mattila I., Plichta D. R., Pöhö P., Hellgren L. I., Arumugam M., Sunagawa S., Vieira-Silva S., Jørgensen T., Holm J. B., Trošt K., MetaHIT Consortium, Kristiansen K., Brix S., Raes J., Wang J., Hansen T., Bork P., Brunak S., Oresic M., Ehrlich S. D., Pedersen O., Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Lusis A. J., Attie A. D., Reue K., Metabolic syndrome: From epidemiology to systems biology. Nat. Rev. Genet. 9, 819–830 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundy S. M., Cleeman J. I., Daniels S. R., Donato K. A., Eckel R. H., Franklin B. A., Gordon D. J., Krauss R. M., Savage P. J., Smith S. C. Jr., Spertus J. A., Costa F.; American Heart Association, National Heart, Lung, and Blood Institute , Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112, 2735–2752 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Huang P. L., A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2, 231–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ussar S., Griffin N. W., Bezy O., Fujisaka S., Vienberg S., Softic S., Deng L., Bry L., Gordon J. I., Kahn C. R., Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 22, 516–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kau A. L., Ahern P. P., Griffin N. W., Goodman A. L., Gordon J. I., Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Chatelier E., Nielsen T., Qin J., Prifti E., Hildebrand F., Falony G., Almeida M., Arumugam M., Batto J.-M., Kennedy S., Leonard P., Li J., Burgdorf K., Grarup N., Jørgensen T., Brandslund I., Nielsen H. B., Juncker A. S., Bertalan M., Levenez F., Pons N., Rasmussen S., Sunagawa S., Tap J., Tims S., Zoetendal E. G., Brunak S., Clément K., Doré J., Kleerebezem M., Kristiansen K., Renault P., Sicheritz-Ponten T., de Vos W. M., Zucker J.-D., Raes J., Hansen T.; MetaHIT consortium, Bork P., Wang J., Ehrlich S. D., Pedersen O., Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Sheridan P. A., Paich H. A., Handy J., Karlsson E. A., Hudgens M. G., Sammon A. B., Holland L. A., Weir S., Noah T. L., Beck M. A., Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. (Lond) 36, 1072–1077 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vijay-Kumar M., Aitken J. D., Carvalho F. A., Cullender T. C., Mwangi S., Srinivasan S., Sitaraman S. V., Knight R., Ley R. E., Gewirtz A. T., Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh J. Z., Ravindran R., Chassaing B., Carvalho F. A., Maddur M. S., Bower M., Hakimpour P., Gill K. P., Nakaya H. I., Yarovinsky F., Sartor R. B., Gewirtz A. T., Pulendran B., TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 41, 478–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thevaranjan N., Puchta A., Schulz C., Naidoo A., Szamosi J. C., Verschoor C. P., Loukov D., Schenck L. P., Jury J., Foley K. P., Schertzer J. D., Larché M. J., Davidson D. J., Verdú E. F., Surette M. G., Bowdish D. M. E., Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455–466.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh A., Nie H., Ghosn B., Qin H., Kwak L. W., Roy K., Efficient modulation of T-cell response by dual-mode, single-carrier delivery of cytokine-targeted siRNA and DNA vaccine to antigen-presenting cells. Mol. Ther. 16, 2011–2021 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Gitlin A. D., Mayer C. T., Oliveira T. Y., Shulman Z., Jones M. J. K., Koren A., Nussenzweig M. C., T cell help controls the speed of the cell cycle in germinal center B cells. Science 349, 643–646 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Béguelin W., Rivas M. A., Calvo Fernández M. T., Teater M., Purwada A., Redmond D., Shen H., Challman M. F., Elemento O., Singh A., Melnick A. M., EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat. Commun. 8, 877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Randolph G. J., Angeli V., Swartz M. A., Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5, 617–628 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Mueller S. N., Tian S., DeSimone J. M., Rapid and persistent delivery of antigen by lymph node targeting PRINT nanoparticle vaccine carrier to promote humoral immunity. Mol. Pharm. 12, 1356–1365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H., Moynihan K. D., Zheng Y., Szeto G. L., Li A. V., Huang B., Van Egeren D. S., Park C., Irvine D. J., Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhuang Y., Ma Y., Wang C., Hai L., Yan C., Zhang Y., Liu F., Cai L., PEGylated cationic liposomes robustly augment vaccine-induced immune responses: Role of lymphatic trafficking and biodistribution. J. Control. Release 159, 135–142 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Cleary B., Brito I. L., Huang K., Gevers D., Shea T., Young S., Alm E. J., Detection of low-abundance bacterial strains in metagenomic datasets by eigengenome partitioning. Nat. Biotechnol. 33, 1053–1060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carvalho F. A., Koren O., Goodrich J. K., Johansson M. E. V., Nalbantoglu I., Aitken J. D., Su Y., Chassaing B., Walters W. A., González A., Clemente J. C., Cullender T. C., Barnich N., Darfeuille-Michaud A., Vijay-Kumar M., Knight R., Ley R. E., Gewirtz A. T., Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12, 139–152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guss J. D., Horsfield M. W., Fontenele F. F., Sandoval T. N., Luna M., Apoorva F., Lima S. F., Bicalho R. C., Singh A., Ley R. E., van der Meulen M. C. H., Goldring S. R., Hernandez C. J., Alterations to the gut microbiome impair bone strength and tissue material properties. J. Bone Miner. Res. 32, 1343–1353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purwada A., Tian Y. F., Huang W., Rohrbach K. M., Deol S., August A., Singh A., Self-assembly protein nanogels for safer cancer immunotherapy. Adv. Healthc. Mater. 5, 1413–1419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh A., Agarwal R., Diaz-Ruiz C. A., Willett N. J., Wang P., Lee L. A., Wang Q., Guldberg R. E., García A. J., Nanoengineered particles for enhanced intra-articular retention and delivery of proteins. Adv. Healthc. Mater. 3, 1562–1567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seong S.-Y., Matzinger P., Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 4, 469–478 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Jin M. S., Kim S. E., Heo J. Y., Lee M. E., Kim H. M., Paik S.-G., Lee H., Lee J.-O., Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Guan Y., Omueti-Ayoade K., Mutha S. K., Hergenrother P. J., Tapping R. I., Identification of novel synthetic toll-like receptor 2 agonists by high throughput screening. J. Biol. Chem. 285, 23755–23762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park B. S., Lee J.-O., Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 45, e66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin M. S., Lee J.-O., Structures of the toll-like receptor family and its ligand complexes. Immunity 29, 182–191 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Brandão I., Hörmann N., Jäckel S., Reinhardt C., TLR5 expression in the small intestine depends on the adaptors MyD88 and TRIF, but is independent of the enteric microbiota. Gut Microbes 6, 202–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pulendran B., Systems vaccinology: Probing humanity's diverse immune systems with vaccines. Proc. Natl. Acad. Sci. U.S.A. 111, 12300–12306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villarino N. F., LeCleir G. R., Denny J. E., Dearth S. P., Harding C. L., Sloan S. S., Gribble J. L., Campagna S. R., Wilhelm S. W., Schmidt N. W., Composition of the gut microbiota modulates the severity of malaria. Proc. Natl. Acad. Sci. U.S.A. 113, 2235–2240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ichinohe T., Pang I. K., Kumamoto Y., Peaper D. R., Ho J. H., Murray T. S., Iwasaki A., Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. U.S.A. 108, 5354–5359 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cullender T. C., Chassaing B., Janzon A., Kumar K., Muller C. E., Werner J. J., Angenent L. T., Bell M. E., Hay A. G., Peterson D. A., Walter J., Vijay-Kumar M., Gewirtz A. T., Ley R. E., Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 14, 571–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vijay-Kumar M., Sanders C. J., Taylor R. T., Kumar A., Aitken J. D., Sitaraman S. V., Neish A. S., Uematsu S., Akira S., Williams I. R., Gewirtz A. T., Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Invest. 117, 3909–3921 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvalho F. A., Aitken J. D., Gewirtz A. T., Vijay-Kumar M., TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal Immunol. 4, 102–111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montecino-Rodriguez E., Berent-Maoz B., Dorshkind K., Causes, consequences, and reversal of immune system aging. J. Clin. Invest. 123, 958–965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barzilai N., Huffman D. M., Muzumdar R. H., Bartke A., The critical role of metabolic pathways in aging. Diabetes 61, 1315–1322 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasir J. K., Labhasetwar V., Preparation of biodegradable nanoparticles and their use in transfection. CSH Protoc 2008, pdb.prot4888 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Zhang T.-T., Al-Alwan M., Marshall A. J., The pleckstrin homology domain adaptor protein Bam32/DAPP1 is required for germinal center progression. J. Immunol. 184, 164–172 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Sage P. T., Tan C. L., Freeman G. J., Haigis M., Sharpe A. H., Defective TFH cell function and increased TFR cells contribute to defective antibody production in aging. Cell Rep. 12, 163–171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walters W., Hyde E. R., Berg-Lyons D., Ackermann G., Humphrey G., Parada A., Gilbert J. A., Jansson J. K., Caporaso J. G., Fuhrman J. A., Apprill A., Knight R., Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, e00009-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., Lesniewski R. A., Oakley B. B., Parks D. H., Robinson C. J., Sahl J. W., Stres B., Thallinger G. G., van Horn D. J., Weber C. F., Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R., UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F. O., The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Q., Garrity G. M., Tiedje J. M., Cole J. R., Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinha S. S., Cameron J., Brooks J. C., Leifer C. A., Complex negative regulation of TLR9 by multiple proteolytic cleavage events. J. Immunol. 197, 1343–1352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav9788/DC1
Fig. S1. Characterization of WT and TLR5−/− mice (related to Fig. 1).
Fig. S2. Pyr-pHEMA nanogels are equivalent in size to PLGA nanoparticle vaccines (related to Fig. 1 and Fig. 5).
Fig. S3. Knockout of the TLR5 receptor results in lower germinal center formation in mice immunized with a PLGA nanovaccine (related to Fig. 1).
Fig. S4. PLGA nanoparticle trafficking from the injection site to lymphoid tissue on day 6 and accumulation in the liver and kidneys at days 2 and 6 after injection (related to Fig. 2).
Fig. S5. Expression of CD86 activation marker (related to Fig. 2).
Fig. S6. Injection site analysis (related to Fig. 2).
Fig. S7. Cell populations in the spleen and lymph node of immunized antibiotic-fed mice (related to Fig. 4).
Fig. S8. Immunological characterization of Pyr-pHEMA nanogels (related to Fig. 5).
Fig. S9. Pyr-pHEMA nanogels do not differentially accumulate in tissue after 6 days relative to soluble formulation (related to Fig. 5).
Fig. S10. Immunomodulatory effects of Pyr-pHEMA are mediated through TLR2 (related to Fig. 6).