

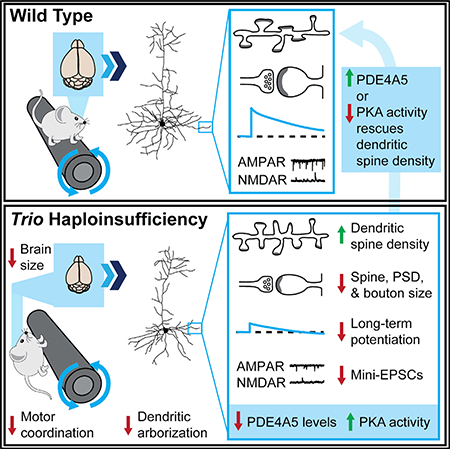
SUMMARY
Heterozygous coding mutations in TRIO are associated with neurodevelopmental disorders, including autism, schizophrenia, bipolar disorder, and epilepsy, and impair TRIO’s biochemical activities. To model mutant alleles, we ablated one or both Trio alleles from excitatory neurons in the cortex and hippocampus of mice. Trio haploinsufficiency increases anxiety and impairs social preference and motor coordination. Trio loss reduces forebrain size and dendritic arborization but increases dendritic spine densities. Cortical synapses in Trio haploinsufficient mice are small, exhibit pre– and postsynaptic deficits, and cannot undergo long–term potentiation. Similar phenotypes are observed in Trio knockout mice. Overall, Trio haploinsufficiency causes severe disease–relevant deficits in behavior and neuronal structure and function. Interestingly, phosphodiesterase 4A5 (PDE4A5) levels are reduced and protein kinase A (PKA) signaling is increased when TRIO levels are reduced. Elevation of PDE4A5 and drug–based attenuation of PKA signaling rescue Trio haploinsufficiency–related dendritic spine defects, suggesting an avenue for therapeutic intervention for TRIO–related neurodevelopmental disorders.
In Brief
Heterozygous TRIO mutations are associated with neurodevelopmental disorders. Katrancha et al. reveal that mouse Trio haploinsufficiency impairs sociability and motor coordination; decreases brain and neuron size; and impairs synaptic anatomy, function, and plasticity. Altering PDE4A5 levels and PKA signaling rescues some synaptic defects, suggesting an avenue to treat TRIO–related neurodevelopmental disorders.
Graphical Abstract
INTRODUCTION
Mutations and rare variants in the coding region of TRIO are enriched in individuals with neurodevelopmental disorders (NDDs), such as autism, schizophrenia, bipolar disorder, intellectual disability, and epilepsy (for review, see Katrancha et al., 2017 and Sadybekov et al., 2017). Analysis of genetic variation in humans indicates that TRIO is highly intolerant to mutation (Genovese et al., 2016; Katrancha et al., 2017; Lek et al., 2016; Samocha et al., 2014), suggesting that mutations in TRIO confer risk for disease. TRIO, also known as triple functional domain protein, interfaces with cell surface receptors and small guanosine triphosphatases (GTPases) to regulate neuronal morphogenesis and synaptic function (Awasaki et al., 2000; Ba et al., 2016; Bateman et al., 2000; Bellanger et al., 2000; van Haren et al., 2014; Iyer et al., 2012; Newsome et al., 2000; Shivalkar and Giniger, 2012; Steven et al., 1998). TRIO9S and TRIO9L, the major isoforms expressed in the mammalian cortex, contain multiple catalytic and accessory domains, including two distinct guanine nucleotide exchange factor (GEF) domains (Bellanger et al., 1998; Blangy et al., 2000; Debant et al., 1996; Katrancha et al., 2017). Many diseaseassociated de novo mutations and rare variants impair TRIO GEF1 domain function but do not interfere with wild–type TRIO function (Katrancha et al., 2017; Sadybekov et al., 2017), suggesting that TRIO haploinsufficiency causes disease.
To study Trio haploinsufficiency in excitatory neurons of the cortex and hippocampus, we crossed mice bearing a conditional “floxed” Trio allele (Zong et al., 2015) with NEX(Neurod6)–Cre mice (Goebbels et al., 2006) to generate heterozygous NEX–Cre, Trio+/flox (NEX–Trio+/−) and homozygous NEX–Cre, Trioflox/flox (NEX–Trio−/−) mice. As modeled by disruption of one Trio allele, Trio haploinsufficiency causes behavioral deficits, including increased anxiety, impaired social preference, and impaired motor coordination. Similar and often more severe phenotypes are observed in NEX–Trio−/− mice. In a gene dosage–dependent manner, Trio loss reduces forebrain size and dendritic arborization but increases dendritic spine densities in the motor cortex. Cortical synapses in Trio haploinsufficient and knockout mice are also smaller than wild–type, exhibit preand postsynaptic deficits, and do not undergo long–term potentiation. Interestingly, phosphodiesterase 4A5 (PDE4A5) levels are reduced and protein kinase A (PKA) signaling is increased when TRIO levels are reduced or eliminated. Elevation of PDE4A5 and drug–based attenuation of PKA signaling rescue Trio haploinsufficiency–related dendritic spine defects. Overall, Trio haploinsufficiency, as observed in patients with NDDs, causes brain– and disease–relevant behavioral, anatomical, functional, and molecular deficits; some of these defects can be rescued by drug–based modulation, suggesting an avenue for therapeutic intervention for TRIO–related NDDs
RESULTS
TRIO Levels Are Reduced in the Cortex and Hippocampus of NEX–Trio+/− and NEX–Trio−/− Mice
NEX–Trio+/− and NEX–Trio−/− mice were generated by crossing mice bearing a conditional Trio allele (Zong et al., 2015), in which exons 22–25 are flanked by LoxP sites, with NEX(Neurod6)–Cre mice (Goebbels et al., 2006). Cre–mediated recombination of Trio creates a premature stop codon at the start of the TRIO GEF1 domain, modeling the protein–truncating R1276X and F1538Intron mutants in schizophrenia and I1329Frameshift mutant in autism (Genovese et al., 2016; Katrancha et al., 2017; De Rubeis et al., 2014). Unlike whole–brain Trio knockout mice (O’Brien et al., 2000; Peng et al., 2010), NEX–Trio+/− and NEX–Trio−/− mice survived into adulthood. At postnatal day 42 (P42), NEX–Trio+/− and NEX–Trio−/− mice had reduced TRIO levels in the cortex (46% and 85%, respectively) and hippocampus (51% and 74%, respectively) relative to wild–type (WT) littermates (Figures 1A–1D). As NEX–Cre is only expressed in excitatory neurons, the remaining TRIO likely reflects residual expression in interneurons or glia (Goebbels et al., 2006). TRIO levels were unaffected in the cerebellum, where NEX–Cre is not expressed (Figures 1A–1D).
Figure 1. NEX–Trio+/− and NEX–Trio−/− Mice Have Smaller Brains and Reduced TRIO Levels in the Cortex and Hippocampus at P42.

(A and B) TRIO levels were reduced in the cortex and hippocampus (Hippo) of NEX–Trio+/− (A) and NEX–Trio−/− (B) mice of both sexes at P42. No change was observed in the cerebellum (Cereb). Repeated–measures (RM) two–way ANOVA of genotype and brain region with post hoc Bonferroni multiple comparisons (Bonf MC) test identified differences for each group (n = 7–8 littermate pairs).
(C and D) Representative immunoblots are shown for male and female NEX–Trio+/− (C) and NEX–Trio−/− (D) mice in the cortex, hippocampus, and cerebellum. The overlapping 268– and 289–kDa bands, representing TRIO9S/L, were quantified (arrows). Other bands represent nonspecific antibody interactions or other isoforms (Katrancha et al., 2017).
(E–G) Total brain weight (E; scale bars represent 0.5 cm) was reduced in NEX–Trio+/− (F) and NEX–Trio−/− (G) mice compared to WT mice of the same sex at P42.
RM two–way ANOVA with post hoc Bonf MC test identified differences (n = 16–21 mice per genotype).
See also Figure S1. Data are represented as mean ± SEM (*p < 0.05; **p < 0.01; ****p < 0.0001).
NEX–Trio+/− and NEX–Trio−/− Mice Have Smaller Brains Than WT Mice
Knockout of both Trio alleles in most cortical and hippocampal neurons with EMX1–Cre resulted in reduced body weight at P17 and reduced brain size at both P17 and P60 (Zong et al., 2015). We found that sex–matched WT, NEX–Trio+/−, and NEX–Trio−/− mice had similar body weights at P42 (Figures S1A and S1C). Both the total brain weight and the combined weight of the cortex and hippocampus were reduced in NEX–Trio+/− and NEX–Trio−/− mice by 4%–7% and 23%–29%, respectively, relative to WT (Figures 1E–1G, S1B, and S1D). The total brain weight and the combined weight of the cortex and hippocampus were also reduced at P21, suggesting a gross anatomical deficit, even during development (Figures S1F and S1G). Body weight at P21 was only reduced in NEX–Trio−/− mice (Figure S1E).
NEX–Trio+/− and NEX–Trio−/− Mice Display Increased Anxiety–like Behavior and Impaired Social Preference
Mutations in TRIO are observed in NDD patients with wide–ranging behavioral deficits. As anxiety and depression are often comorbid with NDDs, we assessed anxiety–like behavior using the open field test (OFT) and elevated plus (E+) maze and behavioral despair using the forced swim test (FST). NEX–Trio+/− mice showed increased anxiety–like behavior in the OFT (Figures 2A and 2B) and E+ maze (Figures 2E and 2F) relative to WT littermates. NEX–Trio−/− mice had variable behavioral phenotypes that were sexually dimorphic: only females spent more time in the center of the OFT, and only males showed reduced open arm time in the E+ maze (Figures 2C, 2D, 2G, and 2H). In the OFT, locomotor activity was decreased in NEX–Trio+/− mice (Figures S2A–S2C), although NEX–Trio−/− mice showed slight hyperactivity (Figures S2D–S2F). Behavioral despair was not observed in NEX–Trio+/− or NEX–Trio−/− mice in the FST (Figures S2G and S2H). Overall, anxiety–like behavior was increased in NEX–Trio+/− mice with no change in behavioral despair, although variable phenotypes were observed in NEX–Trio−/− mice.
Figure 2. NEX–Trio+/− Mice Show Increased Anxiety–like Behavior.

(A and B) NEX–Trio+/− mice of both sexes spent less time in the center of the open field at an absolute level (A) and when controlling for differences in motility (B) in the open field test (OFT).
(C and D) Male NEX–Trio−/− mice showed no change in anxiety, and female NEX–Trio−/− mice showed decreased anxiety relative to WT littermates at an absolute level (C) and when controlling for differences in motility (D) in the OFT.
(E and G) Representative heatmaps show the time (0 s in purple; >10 s in red) that the mouse spent in each location in the elevated plus maze for NEX–Trio+/− (E) and NEX–Trio−/− (G) mice compared to WT littermates. The asterisks (*) indicate the open arms.
(F and H) Male and female NEX–Trio+/− (F) and male NEX–Trio−/− (H) mice spent less time in the open arms of the elevated plus maze than WT littermates. Female NEX–Trio−/− mice (H) showed no change in open arm time. RM two–way ANOVA with post hoc Bonf MC test identified differences (n = 8–16 littermate pairs).
See also Figures S2, S3, S4, and S5. Data are represented as mean ± SEM (*p < 0.05; **p < 0.01).
As social dysfunction is associated with autism, schizophrenia, and other NDDs (Blatt, 2012; Tarbox et al., 2013), we tested how reduced TRIO levels impact social preference. WT mice interacted more with an unfamiliar “stranger” mouse relative to an inanimate object, exhibiting social preference (Figures 3A–3E and S3E). In contrast, NEX–Trio+/− and NEX–Trio−/− mice showed no preference for the stranger mouse relative to the object and reduced social preference relative to WT littermates (Figures 3A–3E), spending equivalent total visit time with the stranger mouse and object (Figures S3A–S3E). These findings reveal that reducing or eliminating TRIO function in excitatory forebrain neurons impairs social function.
Figure 3. NEX–Trio+/− and NEX–Trio−/− Mice Do Not Show a Preference for Social Interaction and Show Decreased Motor Coordination.

(A) (Middle) In the social preference task, a test mouse (brown) is placed in an open field with a male stranger mouse (gray) and an object (Duplo blocks, similar color and size as the stranger mouse). (Left) Test mice that display social preference spend more time with the stranger mouse relative to the object. (Right) Test mice that display no preference spend equal time with the stranger mouse and object. Target zones are designated by the gray dotted circles.
(B and C) NEX–Trio+/− (B) and NEX–Trio−/− (C) mice did not show preference for the stranger mouse (Str.) relative to the object (Obj.), whereas WT mice displayed normal social preference. A linear regression with post hoc Bonf MC test identified differences (n = 10–18 littermate pairs).
(D and E) The difference in occupancy time between the social and nonsocial zones showed that NEX–Trio+/− (D) and NEX–Trio−/− (E) mice had reduced preference for the social zone compared to sex–matched WT littermates. RM two–way ANOVA with post hoc Bonf MC test identified differences (n = 10–18 littermate pairs).
(F and I) NEX–Trio+/− (F) and NEX–Trio−/− (I) mice of both sexes had a shorter latency to fall off of the accelerating rotarod than WT littermates. RM two–way ANOVA with post hoc Bonf MC test identified differences (n = 11–14 littermate pairs).
(G and H) Male (G) and female (H) NEX–Trio+/− mice had a shorter latency to fall off of the accelerating rotarod than WT littermates in every trial (difference in intercepts), but all groups learned at a rate of 40 s per trial (no difference in slopes).
(J and K) Male (J) and female (K) NEX–Trio−/− mice showed impaired motor learning (significant or suggestive difference in slopes) compared to WT littermates. Male and female WT mice learned at a rate of 32 and 33 s per trial, respectively; male and female NEX–Trio−/− mice learned at a rate of 14 and 18 s per trial, respectively. Linear regressions with post hoc test for differences between slopes and changes in intercepts identified differences (n = 11–14 littermate pairs).
See also Figures S2, S3, S4, and S5. Data are represented as mean ± SEM (*p < 0.05; **p < 0.01; ****p < 0.0001).
We also assessed disease–specific behaviors. Individuals with schizophrenia show deficits in sensorimotor gating (Parwani et al., 2000), so we tested prepulse inhibition of acoustic startle (PPI). NEX–Trio+/− and NEX–Trio−/− mice displayed normal PPI with 30 ms or 100 ms interstimulus intervals (ISIs) for all pre–pulse intensities (6, 12, and 16 dB; Figures S4A–S4D), except male NEX–Trio−/− mice, which exhibited decreased PPI in the most challenging condition (6 dB prepulse intensity with 30 ms ISI). Individuals with autism display repetitive behaviors (Angoa–Pérez et al., 2013), so we assessed nestlet shredding. Male NEX–Trio+/− and NEX–Trio−/− mice exhibited increased nestlet shredding; this effect was driven by a subset of mice, suggesting incomplete penetrance (Figures S4E–S4G). Finally, in a test for recognition memory, NEX–Trio+/− and NEX–Trio−/− mice exhibited normal discrimination between novel and familiar objects and performed similarly to sex–matched WT littermates during object familiarization (Figures S5A–S5E), despite completing the task faster on one or both days (Figures S5F and S5G). Overall, these data suggest that partial and/or full loss of Trio expression does not affect novel object recognition but impacts sensorimotor gating and repetitive behaviors in some male mice.
In summary, NEX–Trio+/− and NEX–Trio−/− mice showed complex behavioral phenotypes that varied in both quality and severity of deficit. Of particular interest, Trio haploinsufficiency resulted in significant impairments in both social function and anxiety, consistent with the prevalence of these symptoms in NDD patients.
NEX–Trio+/− and NEX–Trio−/− Mice Display Gene Dosage–Dependent Deficits in Motor Coordination
Motor abnormalities are early symptoms of autism and schizophrenia (Blatt, 2012; Walker et al., 1994). NEX–Trio+/− and NEX–Trio−/− mice showed a shorter latency to fall off an accelerating rotarod than WT littermates (Figures 3F and 3I). NEX–Trio+/− mice showed no deficit in motor learning, learning at a rate similar to WT littermates (40 s per trial; Figures 3G and 3H). In contrast, NEX–Trio−/− mice (14–18 s per trial) showed an impairment in motor learning rate compared to WT littermates (32 or 33 s per trial; Figures 3J and 3K). No confounding difference in muscle strength was observed using the Kondziela inverted screen test (Figures S3F and S3G). In summary, both NEX–Trio+/− and NEX–Trio−/− mice have deficits in motor coordination, and NEX–Trio−/− mice have additional deficits in motor learning.
NEX–Trio+/− and NEX–Trio−/− Mice Have Decreased Dendritic Arborization, Increased Dendritic Spine Density, and Smaller Synapses in Motor Cortex
As NEX–Cre–mediated recombination is absent in the striatum and cerebellum (Goebbels et al., 2006), we assessed whether the motor deficits are associated with anatomical and functional changes in the motor cortex. We measured dendritic arborization in layer 5 pyramidal neurons (L5 PNs) in motor cortex (regions M1 and M2) of NEX–Trio+/−, NEX–Trio−/−, and WT mice (Figures 4A, 4E, and S6A). Sholl analysis revealed decreased dendritic arbor complexity in NEX–Trio+/− and NEX–Trio−/− mice (Figures 4B and 4F), due to reduced branch point numbers and total dendrite length (Figures 4C, 4D, 4G, and 4H). This phenotype was more pronounced in NEX–Trio−/− mice, indicating a gene dosage dependence.
Figure 4. Dendritic Arborization Is Reduced in the Motor Cortex of NEX–Trio+/− and NEX–Trio−/− Mice.

(A and E) Representative dendritic arbor reconstructions are shown for layer 5 pyramidal neurons (L5 PNs) in NEX–Trio+/− (A) and NEX–Trio−/− (E) mice. (B and F) Sholl analysis revealed decreased dendritic arborization on L5 PNs in NEX–Trio+/− (B) and NEX–Trio−/− (F) mice relative to WT controls; the phenotype was more severe in NEX–Trio−/− mice (F). RM two–way ANOVA with post hoc Bonf MC test identified differences (n = 20 neurons from 4 mice per genotype).
(C and G) Apical and basal dendrite length were reduced on L5 PNs in NEX–Trio+/− (C) and NEX–Trio−/− (G) mice relative to WT controls.
(D and H) The numbers of apical and basal branch points were reduced on L5 PNs in NEX–Trio+/− (D) and NEX–Trio−/− (H) mice relative to WT controls. Unpaired t tests identified differences between groups (n = 20 neurons from 4 mice per genotype).
See also Figure S6. Data are represented as mean ± SEM (***p < 0.001; ****p < 0.0001).
We then assessed dendritic spine density in the motor cortex and hippocampus using the Thy1–GFP–M mouse line (Feng et al., 2000). NEX–Trio+/− and NEX–Trio−/− mice showed increased dendritic spine density on basal dendrites of L5 PNs in motor cortex (Figures 5A–5D), and NEX–Trio−/− mice showed increased dendritic spine density on apical and basal dendrites of hippocampal CA1 neurons (Figures S6D and S6E). No difference in spine density was observed in hippocampal CA1 neurons of NEX–Trio+/− mice (Figures S6B and S6C). Given the increased dendritic spine density in the motor cortex, we examined synaptic ultrastructure using electron microscopy (Figure 5E). Synapse density was significantly or suggestively increased in NEX–Trio−/− and NEX–Trio+/− mice (Figures 5F and 5G). Dendritic spine area, presynaptic density (PSD) length, and presynaptic bouton area were all decreased in NEX–Trio+/− and NEX–Trio−/− mice (Figures 5H–5M). In summary, Trio haploinsufficiency and knockout decreased dendritic arbor length, increased dendritic spine density, and decreased synapse size in the motor cortex.
Figure 5. NEX–Trio+/− and NEX–Trio−/− Mice Have Increased Dendritic Spine Density and Smaller Synapses in the Motor Cortex.

(A and C) Representative basal dendrite segments from L5 PNs in the motor cortex of NEX–Trio+/− (A) and NEX–Trio−/− (C) mice with WT controls. Scale bars represent 5 mm.
(B and D) Dendritic spine density was increased on L5 PNs in the motor cortex of NEX–Trio+/− (B) and NEX–Trio−/− (D) mice relative to WT controls. A linear regression with post hoc Bonf MC test identified differences (n = 17–27 dendrite segments from R3 mice per group).
(E) Representative electron micrographs from L5 motor cortex of NEX–Trio+/−, NEX–Trio−/−, and WT mice. Scale bars represent 0.3 μm.
(F and G) Cortical synapse density (synapses per mm ) trended (p = 0.085) toward an increase in NEX–Trio+/− mice (F) and was increased in NEX–Trio−/− mice (G).
(H and I) PSD length was decreased in L5 motor cortex of both NEX–Trio+/− (H) and NEX–Trio−/− (I) mice.
(J and L) Presynaptic bouton area was decreased in L5 motor cortex of both NEX–Trio+/− (J) and NEX–Trio−/− (L) mice.
(K and M) Dendritic spine area was decreased in both NEX–Trio+/− (K) and NEX–Trio−/− (M) mice. For (F)–(M), a linear regression with post hoc Bonf MC test identified differences (n = 54 fields of view [55 μm2 each] from 3 mice per group).
See also Figure S6. Data are represented as mean ± SEM (**p < 0.01; ***p < 0.001; ****p < 0.0001).
Presynaptic Release Probability, Postsynaptic Currents, andLong–Term Potentiation Are Impaired in NEX–Trio+/− and NEX–Trio−/− Mice
L5 PNs in motor cortex exhibit task–related firing and plastic changes when mice run on an accelerating rotarod (Costa et al., 2004). To measure synaptic transmission, we recorded evoked excitatory postsynaptic currents (eEPSCs) and miniature excitatory postsynaptic currents (mEPSCs) in motor cortex L5 PNs. The NMDA receptor (NMDAR)/AMPA receptor (AMPAR)mediated eEPSC ratio was increased in NEX–Trio+/− mice (Figures 6A and 6B), due to decreased AMPAR–eEPSC amplitudes (Figures S7A and S7B), consistent with decreased PSD length and spine area. In contrast, the NMDAR/AMPAR eEPSC ratio was decreased in NEX–Trio−/− mice relative to WT (Figures 6A and 6B), with both NMDAR– and AMPAR–eEPSC amplitudes decreased (Figures S7A and S7B), a more severe phenotype.
Figure 6. NEX–Trio+/− and NEX–Trio−/− Mice Exhibit Deficits in Pre– and Postsynaptic Function and Synaptic Plasticity.

(A) Representative traces of NMDAR– and AMPAR–eEPSCs from NEX–Trio+/−, NEX–Trio−/−, and WT L5 PNs in the motor cortex (M1).
(B) The NMDAR/AMPAR eEPSC ratio was increased in NEX–Trio+/− mice and decreased in NEX–Trio−/− mice relative to WT controls. Unpaired t tests identified differences between groups (n = 12–14 neurons from 4 mice per group).
(C and D) NEX–Trio+/− and NEX–Trio−/− mice had decreased AMPAR–mEPSC amplitude (C, left) and frequency (D, left). NEX–Trio−/− mice also had decreased NMDAR–mEPSC amplitude (C, right) and frequency (D, right). NEX–Trio+/− mice trended toward decreased NMDAR–mEPSC amplitude (C, right) and had decreased NMDAR–mEPSC frequency (D, right).
(E) Representative traces of AMPAR– and NMDAR–mEPSCs from WT, NEX–Trio+/−, and NEX–Trio−/− L5 PNs in the motor cortex (M1).
(F and G) NEX–Trio+/− mice showed no difference in AMPAR– (F) or NMDAR–mEPSC (G) decay time. NEX–Trio−/− mice showed no difference in AMPAR–mEPSC decay time (F) but increased NMDAR–mEPSC decay time (G). To consistently analyze NEX–Trio+/− and NEX–Trio−/− mice independently, unpaired t tests identified differences between groups (n = 28–36 neurons from 5–7 mice per group).
(H) Representative traces of eEPSCs from WT, NEX–Trio+/, and NEX–Trio−/− L5 PNs in the motor cortex (M1) evoked by paired–pulse stimulation of M1 layer 2/3.
(I) An increase in the paired–pulse ratio was observed in NEX–Trio+/− and NEX–Trio−/− mice, suggesting a deficit in presynaptic release probability. A one–phase decay global fit analysis identified differences between groups. Post hoc Bonf MC test indicated that NEX–Trio+/− was significantly different from WT at35* and 60** ms (p = 0.07 at 300 ms), and NEX–Trio+/− was significantly different from WT at 35****, 60****, 100***, and 300** ms (p = 0.18 at 200 ms; n = 20–26 neurons from 5–7 mice per group).
(J) NEX–Trio+/− and NEX–Trio−/− mice had impaired LTP in the motor cortex, showing only slight induction and no potentiation compared to WT controls. LTP was induced at 0 min. RM two–way ANOVA with post hoc Bonf MC test identified differences (n = 9–11 neurons from ≥6 mice per group).
See also Figures S6 and S7. Data are represented as mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Consistent with the observed eEPSC changes, the amplitudes of AMPAR–mEPSCs were significantly decreased in NEX–Trio+/− and NEX–Trio−/− mice (Figures 6C and 6E). There was a trend toward reduced amplitude of NMDAR–mEPSCs in NEX–Trio+/− mice (p = 0.0517) and a significant reduction in NEX–Trio−/− mice (Figures 6C and 6E). The frequencies of AMPAR– and NMDAR–mEPSCs were also decreased in NEX–Trio+/− and NEX–Trio−/− mice (Figures 6D and 6E). NEX–Trio−/− mice showed an altered mEPSC decay time for NMDARs (Figures 6F and 6G). To assess presynaptic function, we measured paired–pulse ratio (PPR) in L5 PNs in motor cortex at five interstimulus intervals (35, 60, 100, 200, and 300 ms; Figures 6H and 6I). Increased PPR was observed in NEX–Trio+/− and NEX–Trio−/− mice (Figure 6I), suggesting a deficit in presynaptic release probability, which likely contributes to the decreased mEPSC frequency (Figure 6D).
Pre– and postsynaptic functions were also measured in the hippocampus. In NEX–Trio+/− mice, the NMDAR/AMPAR eEPSC ratio trended toward an increase due to decreased AMPAR–eEPSC amplitudes (Figures S6F–S6I). In NEX–Trio−/− mice, the NMDAR/AMPAR eEPSC ratio was not significantly changed (Figures S6F–S6I). Increased hippocampal PPR at a 75–ms interstimulus interval was only observed in NEX–Trio−/− mice (Figures S6J and S6K), suggesting that hippocampal neurons had lower sensitivity to Trio disruption relative to cortical neurons.
Finally, to assess synaptic plasticity in the motor cortex, we analyzed long–term potentiation (LTP), via theta burst stimulation (TBS), in NEX–Trio+/− and NEX–Trio−/− mice, which showed a lack of induction and maintenance of TBS–LTP relative to WT (Figure 6J). Our data suggest that Trio haploinsufficiency and knockout impair both pre– and postsynaptic function and synaptic plasticity.
PDE4A5 Levels Are Reduced in a Trio Gene Dosage–Dependent Manner, and Restoration of PDE4A5 Rescues Trio–Related Increases in Dendritic Spine Density
We used mass–spectrometry–based proteomic and phosphor–proteomic analyses of the motor cortex to identify proteins whose levels or function were altered in NEX–Trio+/− and NEX–Trio−/− mice. Of the 8,379 proteins quantified, 193 and 101 were significantly up– or downregulated, respectively, with Trio loss (Figure S7C). Proteome profiling confirmed TRIO depletion in NEX–Trio+/− and NEX–Trio−/− mice (Figure S7D), along with decreases in other NDD–associated proteins: phosphodiesterase (PDE) 4D, clathrin heavy chain (CHC), PDE4A, and P21 or RAC1 activated kinase 4 (PAK4) (Figures 7A and S7E). Quantitative immunoblotting corroborated these findings in NEX–Trio−/− mice as follows: 11% PDE4D, 8% CHC, 28% PDE4A, and 25% PAK4 (Figures 7B and 7C). Of interest, PDE4A isoform 5 (PDE4A5) was reduced in a Trio dosage–dependent manner, exhibiting 21% and 44% reductions in NEX–Trio+/− and NEX–Trio/ mice, respectively (Figures 7D and 7E). As PDE4A5 is an attenuator of PKA signaling, we queried the phosphoproteomic data for enrichment or depletion of known kinase–substrate pairs (Weidner et al., 2014). Nine pairs were significantly increased in either the NEX–Trio+/− or NEX–Trio−/− samples, with PKA and its substrates most significantly changed (Figure S7F; Weidner et al., 2014). PKA substrate phosphopeptide levels were consistently increased with relatively little change in protein levels, and activity–inducing phosphorylation of the catalytic subunit of PKA (phosphorylation site T198) was increased in NEX–Trio+/− and NEX–Trio−/− mice (Figure 7F). Overall, Trio loss downregulates PDE4A5 protein levels, increases PKA signaling, and increases phosphorylation of PKA substrates in the motor cortex.
Figure 7. NEX–Trio+/− and NEX–Trio−/− Mice Have Decreased PDE4A5 Levels and Increased PKA Signaling, and PDE4A5 Overexpression and Drug–Based Attenuation of PKA Signaling Rescue Trio–Induced Increases in Dendritic Spine Density.

(A) Heatmap illustrating the log2 fold change (FC) of select proteins compared to WT quantified by mass–spectrometry–based proteomics. All selected proteins were reduced in the motor cortex of NEX–Trio−/− and NEX–Trio+/− mice compared to WT.
(B) Representative immunoblots for selected proteins in the cortex are shown for male NEX–Trio−/− and WT mice (P42); bands of interest are marketed by arrows (single band) or brackets (multiple bands).
(C) PDE4D, CHC, PDE4A, and PAK4 protein levels were reduced in the cortex of NEX–Trio−/− mice relative to WT littermates.
(D and E) PDE4A5 protein levels were reduced in the cortex of NEX–Trio−/− (D) and NEX–Trio+/− (E) mice. Representative immunoblots are shown. Ratio paired t test identified differences (n = 6 littermate pairs).
(F) Scatterplot representing the average log2 FC compared to WT for PKA substrates detected in the kinase–substrate pair enrichment analysis. The log2 FCs at phosphorylation sites are plotted against the log2 FCs of the respective protein levels. Each color represents a distinct PKA substrate phosphorylation site; half circles represent NEX–Trio+/− data, and open circles represent NEX–Trio−/− data.
(G) Representative immunofluorescence images of Trio+/flox neurons transfected with just GFP (WT) or GFP–P2A–Cre (Cre–Trio+/−; top) for 6 days. A subset of neurons was treated with PDE4A5 overexpression (middle) or 100 μM Rp–cAMPS (bottom). Scale bar represents 5 mm.
(H) Dendritic spine density was increased in Cre–positive Trio+/flox neurons, and this was rescued with overexpression (o/e) of PDE4A5 for 6 days or treatment with 100 mM Rp–cAMPS for 3 days. Unpaired t tests identified differences between groups (n = 15–21 dendrite segments from R3 cells per group).
See also Figure S7. Data are represented as mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
To assess the contribution of PDE4A5 to Trio–related phenotypes, we modeled Trio haploinsufficiency in cultured cortical neurons. First, we verified that Cre expression yielded significant TRIO loss in Trioflox/flox mouse fibroblasts (Figures S7G and S7H). Then, we transfected Trio+/flox neurons with GFP–P2A–Cre to inactivate one Trio allele (Cre–Trio+/−) and compared this to GFP–transfected Trio+/flox neurons (WT). Cre–Trio+/− neurons showed increased spine density and decreased branch tip number (Figures 7G, 7H, and S7I), mimicking the phenotypes observed in NEX–Trio+/− mouse brain (Figures 4 and 5). PDE4A5 overexpression rescued the increased spine density in Cre–Trio+/− neurons, restoring them to WT levels, but did not impact WT spine density (Figures 7G and 7H). PDE4A5 overexpression did not rescue branching deficits in Cre–Trio+/− neurons, indicating that other mechanisms support dendrite branches (Figure S7I). As PDE4A5 levels are reduced and PKA signaling is elevated in NEX–Trio+/− mouse cortex (Figures 7E and 7F), we treated Cre–Trio+/− neurons with Rp–cAMPS, a competitive PKA antagonist (Taylor et al., 1999). Rp–cAMPS treatment rescued the increased dendritic spine density in Cre–Trio+/− neurons (Figures 7G and 7H). These findings suggest that drug–based modulation of the TRIO/PDE4A5–PKA pathway may be a promising treatment for NDDs.
DISCUSSION
NDDs are associated with de novo mutations and rare variants in TRIO’s coding region, many of which impair TRIO GEF1 domain function (Katrancha et al., 2017; Sadybekov et al., 2017). These disorders are also associated with impaired structure and function of excitatory forebrain neurons (Glantz and Lewis, 2000; Goebbels et al., 2006; Hutsler and Zhang, 2010; Konopaske et al., 2014). As only one allele is typically damaged in these patients, we hypothesized that the disease–associated mutations result in haploinsufficiency of TRIO, impairing the function of excitatory forebrain neurons. We discovered that NEX–Trio+/− mice have smaller brains and exhibit profound deficits in dendrite and synapse structure and function; synaptic transmission and plasticity; and motor, social, and anxiety–like behaviors, all hallmarks of NDDs. Similar and often more severe phenotypes are observed in NEX–Trio−/− mice. Overall, this study reveals that Trio haploinsufficiency, as observed in patients with NDDs, causes disease–relevant deficits, suggesting that upregulating TRIO function may be a promising treatment for NDDs.
Trio Disruption Causes NDD–Associated Deficits in Cortical Function and Anatomy
Motor abnormalities are among the earliest symptoms in autism and schizophrenia (Blatt, 2012; Walker et al., 1994). Our data suggest that TRIO plays a role in motor function, as reduced Trio dosage causes motor coordination deficits. Furthermore, we observed altered dendritic spine density, dendritic arborization, and forebrain size in NEX–Trio+/− mice. These phenotypes are consistent with the observations of increased cortical dendritic spine density in autism (Hutsler and Zhang, 2010) and in primary neuronal culture following overexpression of the intellectual disability–associated TRIO D1368V mutation (Sadybekov et al., 2017), decreased dendritic arborization in various NDDs (Kulkarni and Firestein, 2012), and decreased cortical thickness in schizophrenia (van Erp et al., 2018). Our data suggest that TRIO loss may play an important role in NDD–associated deficits in motor coordination and cortical anatomy.
TRIO Promotes PDE4A5–PKA Signaling to Regulate Dendritic Spine Density
Our study links TRIO to PDE4A and downstream PKA–cyclic AMP (cAMP) signaling in a NDD–relevant pathway. We provide evidence of a functional interaction between TRIO and PDE4A5 and their joint impact on dendritic spine density. Our data are supported by previous evidence that both TRIO and PDE4A bind DISC1 (disrupted in schizophrenia 1), which is believed to anchor these proteins within the dendritic spine (Chen et al., 2011; Murdoch et al., 2007). Our data suggest that TRIO and PDE4A5 interact with PKA and cAMP signaling to control dendritic spine density in a shared molecular pathway, a possible therapeutic target for NDDs.
Trio Loss Impairs Both Pre–and Postsynaptic Functionin Mammals
Previous reports suggest that Trio knockdown or reduced TRIO function impairs AMPAR function in the hippocampus (Ba et al., 2016; Herring and Nicoll, 2016; Sadybekov et al., 2017). We further show that Trio disruption impairs AMPAR function in both the motor cortex and hippocampus, even if only one allele is damaged, as in patients with NDDs. We also reveal that reduced TRIO function disrupts normal presynaptic release and bouton area in mammals, possibly restricting glutamate availability in the motor cortex. Similarly, Trio mutant Drosophila exhibit impaired axonal formation and pathfinding as well as decreased boutons per area at the neuromuscular junction (Awasaki et al., 2000; Ball et al., 2010; Bateman et al., 2000; Hakeda–Suzuki et al., 2002; Miller et al., 2013; Newsome et al., 2000; Pawson et al., 2008). Furthermore, LTP in the motor cortex may mediate motor function and skill learning (Li et al., 2017; Rioult–Pedotti et al., 2000; Yu and Zuo, 2011). Our observation that LTP is impaired with reduced TRIO function suggests that defects in plasticity likely contribute to the observed motor coordination and learning deficits. Overall, our data suggest that TRIO plays important roles in both synaptic transmission and plasticity. Furthermore, the observed increase in dendritic spine density, decrease in PSD length, decrease in mEPSC frequency, and impairment in LTP together suggest that there are more immature dendritic spines in the cortex, an anatomical hallmark of many NDDs (Martínez–Cerdeño, 2017).
Human Protein–Truncating Variants in NDDs Are Modeled by Heterozygous Trio Disruption in the Mouse Brain
Disruption of the Trio allele in NEX–Trio+/− miceclosely models the protein–truncating variants (PTVs) that impair TRIO GEF1 activity, including the schizophrenia– or autism–associated R1276X, I1329Frameshift, and F1538Intron mutations (Genovese et al., 2016; Katrancha et al., 2017; De Rubeis et al., 2014). However, it remains unknown whether missense mutations in the TRIO GEF1 domain, which also impair TRIO’s GEF1 activity (Katrancha et al., 2017; Sadybekov et al., 2017), or other PTVs and missense mutations (e.g., in the GEF2 or kinase domains) would yield similar deficits in neuronal structure, synaptic function, and behavior. The study of mice bearing select disease–associated alleles should reveal whether specific disruptions of TRIO function differentially impact Trio–dependent phenotypes, as has been observed in mice bearing different mutant alleles of Shank3 (Zhou et al., 2016).
Conclusions
TRIO has emerged as a risk gene for many NDDs with mutations likely causing genetic insufficiency. A critical need exists to determine TRIO’s role in NDD pathology, so we performed a systematic investigation of the impact of Trio haploinsufficiency on the vertebrate central nervous system. Our data demonstrate that Trio haploinsufficiency is sufficient to impair normal neuron structure and connectivity as well as synaptic transmission and plasticity. We propose that disruptive or hypomorphic TRIO alleles reduce PDE4A5 levels, increase PKA signaling, and contribute to NDDs through similar impairments in structure and function in affected individuals.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anthony J. Koleske (anthony.koleske@yale.edu ). Restrictions may apply to reagents and resources from outside sources, especially those with Material Transfer Agreements (MTA), including (1) NEX–Cre mice – received under MTA from the Klaus–Armin Nave laboratory at the University of Go¨ ttingen, (2) “floxed” Trio mice – received under MTA from the Minsheng Zhu laboratory at Nanjing University, (3) rabbit primary antibody for Clathrin Heavy Chain – received an in–house stock aliquot from Pietro De Camilli laboratory at Yale University, (4) pAAV–mCherry–IRES–Cre DNA plasmid – available at Addgene (Plasmid #55632), (5) pAAV–CAG–“GFP–P2A–Cre” DNA plasmid – received from Jaime Grutzendler at Yale University, and (6) pTriEx–6–PDE4A5 DNA plasmid – received from Becky Carlyle and Angus Nairn at Yale University.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
NEX–Trio+/− and NEX–Trio−/− mice
Mice bearing a conditional “floxed” Trio allele (Zong et al., 2015) and a NEX(Neurod6)–Cre transgene (Goebbels et al., 2006) were crossed to produce heterozygous NEX–Cre, Trio+/flox (NEX–Trio+/) and homozygous NEX–Cre, Trioflox/flox (NEX–Trio−/−) mice. Wild–type (WT) mice lacked NEX–Cre. For dendritic spine visualization, mice were crossed to the Thy1–GFP–M line (Feng et al., 2000) to produce Thy1–GFP–M, NEX–Cre, Trio+/flox (NEX–Trio+/) and Thy1–GFP–M, NEX–Cre, Trioflox/flox (NEX–Trio−/−) mice; WT mice lacked NEX–Cre. All mice were in a mixed C57BL/6 3 129/SvJ background. Animal genotypes were determined by PCR, as described in the Method Details section.
Mice were group housed with siblings of the same sex and kept on a standard 12–hour light/dark cycle. Behavioral analyses were performed with male and female littermates at postnatal day (P)42–P70. Brain and body weight measurements were performed with male and female mice at P21 and P42; male and female mice were analyzed together at P21. To control for potential sex differences and estrus effects in adult mice (Frankfurt and Luine, 2015), only male mice at P37–P45 were used for electrophysiological analyses, and only male mice at P42 were used for anatomical, proteomic, and phosphoproteomic analyses. All immunoblots were performed on male mice at P42; TRIO immunoblots were also performed on female mice at P42. All procedures were compliant with federal regulations and approved by the Yale University Animal Care and Use Committee.
Trioflox/flox mouse fibroblasts
Trioflox/flox mouse fibroblasts were prepared from embryonic day 13.5 mouse embryos (combination of sexes). Fibroblasts were kept at 37C and 5% CO2 and passaged at 3.75−105 per 10 cm plate every three days until immortalized using the 3T3 protocol. Fibroblasts were maintained in DMEM plus 10% fetal bovine serum (FBS), 2 mM L–glutamine, and penicillin/streptomycin.
Trio+/flox mouse primary cortical cultures
Primary neuronal cultures were prepared from embryonic day 15.5 Trio+/flox mouse embryos (combination of sexes). Cortices were dissected in ice–hold HBSS (Invitrogen) with 25 mM HEPES and 0.5% glucose and then digested in the same solution with 0.3 U/ml papain (Worthington, Lakewood, NJ), 0.1% dispase (Roche, Indianapolis, IN), and 0.01% DNaseI (Sigma) at 37°C (twice for 7 minutes). Tissues were triturated 20 times with a glass Pasteur pipette (Moresco et al., 2005). Neurons were resuspended and plated in Neurobasal A (Invitrogen) supplemented with 2% Gem21 (Gemini) and 10% FBS on 12 mm coverslips precoated with 20 μg/ml polyD–lysine (Corning Inc.) overnight at 37°C and 1 μg/ml laminin (Corning) for 2 hours at 37°C. After 4 hours, the media was changed to serum–free Neurobasal A/Gem21 media containing 1% penicillin/streptomycin and 2 μM L–glutamine, and cultures were maintained at 37°C and 5% CO2. Glial growth was suppressed with 1 μM ara–C (Sigma) on day in vitro (DIV) 3 and 0.5 μM ara–C on DIV6.
METHOD DETAILS
Mouse genotyping
Mouse genotypes were determined by PCR using the following primers for “floxed” Trio (5′–TTG TTC CAT TTA CGT CAC CG–3′ and 5′–CCT CCT CAG GGA AGA GAC TA–3′) (Zong et al., 2015), NEX–Cre (5′–AGA ATG TGG AGT AGG GTG AC–3′, 5′–GAG TCC TGG AAT CAG TCT TTT TC–3′, and 5′–CCG CAT AAC CAG TGA AAC AG–3′) (Omar et al., 2017), and GFP (5′–GCA CGA CTT CTT CAA GTC CGC CAT GCC–3′ and 5′–GCG GAT CTT GAA GTT CAC CTT GAT GCC–3′) (Omar et al., 2017). For PCR analyses, tissue was digested overnight at 55°C in a solution containing Gitschier buffer (2 M Tris (pH 8.8), 1 M NH4SO4, 1 M MgCl2, 10% Triton X–100, and water), beta–mercaptoethanol, and proteinese K. Digests were vortexed, heated at 95°C for 10 minutes, vortexed, and centrifuged. PCR analysis was performed on digested tissue with dNTPs, PCR buffer, relevant primers, MgCl2, and DNA polymerase. PCR protocol was as follows: (1) 90°C for 5 minutes, (2) 94°C for 1 minute, (3) 65°C for 1 minute, (4) 72°C for 1 minute, (5) go to step 2 and repeat 34 times, (6) 72°C for 10 minutes, and (7) 4°C forever.
Immunoblot assays
Cortical, hippocampal, and cerebellar extracts from NEX–Trio+/− mice, NEX–Trio−/− mice, and WT littermates at P42 were created by sonicating brain tissue in 1% sodium dodecyl sulfate (SDS), 50 μM Tris, 2 mM ethylenediaminetetraacetic acid (EDTA), protease inhibitors, and phosphatase inhibitors with a small tip probe sonicator (Heat Systems, Ultrasonics, Inc. model W–375) at 4.5 Amps for 15–30 s. The brain homogenates were immediately placed at 95°C for 5 minutes, snap frozen in liquid nitrogen, and stored at 80°C until immunoblotting. Protein concentrations were determined with a bicinchoninic acid (BCA) assay (Pierce) performed in a 96–well plate on a microplate reader (SpectraMax M5, Molecular Devices) against a standard bovine serum albumin (BSA) curve. 15 mg and 30 mg of brain homogenate were diluted with 4x Laemmli sample buffer (LSB) and separated using SDS polyacrylamide gel electrophoresis on a precast 4%–15% gradient gel (Bio–Rad #456–1084). For TRIO immunoblots, proteins were transferred using wet transfer methods to nitrocellulose at 300 μA for 180 minutes on ice. For other immunoblots, proteins were transferred using wet transfer methods to nitrocellulose at 35 V overnight on ice. Membranes were stained with Ponceau S and imaged, blocked with 5% nonfat milk for 1 hour at room temperature, and incubated overnight at 4C with primary antibodies, which included 1:4,000 TRIO Sec14 (Katrancha et al., 2017; McPherson et al., 2005), 1:1,000 PDE4D (Millipore ABS22), 1:1,000 PDE4A (Abcam ab14607), 1:1,000 PDE4A5 (Abcam ab42094), 1:1,000 PAK4 (Proteintech 14685–1–AP), and 1:1,000 CHC (Pietro De Camilli lab, Yale University) primary antibodies. Membranes were washed with Tris Buffered Saline with Tween 20 (TBST), incubated at room temperature for 2 hours in 1:5,000 goat anti–rabbit secondary antibody (Bio–Rad, 170–6515), and washed again with TBST. Signal was detected with an enhanced chemiluminescence substrate using the Genesis software (Syngene, version 1.4.5.0) on a G:Box Chemi XRQ (Syngene). Densitometry values were calculated in ImageJ and normalized to each lane’s total protein as determined by Ponceau S. For TRIO analyses, densitometry values were normalized again to the 30 mg WT sample for each littermate pair.
Brain and body weight measurements
NEX–Trio+/−, NEX–Trio−/−, and WT mice were weighed at P21 and P42. Brains from NEX–Trio+/−, NEX–Trio−/−, and WT mice were also weighed at P21 and P42 (1) after removing external portions of the brain stem and the olfactory bulbs and (2) after removing the cerebellum and the remainder of the brain stem. Olfactory bulbs and brain stem were also removed before imaging.
Dendrite arbor reconstructions
NEX–Trio+/−, NEX–Trio−/−, and WT mice at P42 were deeply anesthetized by pentobarbital sodium injection (Nembutal Sodium Solution) and transcardially perfused/cleared with PBS for 2 minutes. The following Golgi staining protocol was performed using the FD Rapid GolgiStain Kit (FD NeuroTechnologies, Inc) and adapted from their manual. The brain was quickly and carefully removed, rinsed in PBS, and immersed in Golgi impregnation solution. The brain was moved into new Golgi impregnation solution after 12–24 hours and allowed to sit for 2 weeks in the dark at room temperature. The brain was then transferred to Solution C for 24 hours, before being transferred to a new aliquot of Solution C for an additional 48 hours. Brains were embedded in Optimal Cutting Temperature Compound (Tissue–Tek, 4583) inside a cryomold (10 mm × 10 mm × 5 mm, Tissue–Tek, 4565) and snap frozen in a dry ice and 100% ethanol bath. Sagittal sections were cut at 150 μm on a cryostat (Cryostat 2800 Frigocut–E, Reichert–Jung) set at 22°C and placed into room temperature PBS. The PBS was removed and replaced with Solution C before mounting. Sections were mounted onto gelatin–coated coverslips and allowed to dry for 48 hours at room temperature in the dark. Staining was performed in a series of Coplin staining jars: two 4–minute rinses in double distilled water, one 10–minute rinse in Solutions D/E, two 4–minute rinses in double distilled water, one 4–minute rinse in 50% ethanol, one 4–minute rinse in 70% ethanol, two 4–minute rinses in 100% ethanol, and two 4–minute rinses in xylenes. Slides were coverslipped with Permount (Fisher Scientific, SP15–100) and allowed to dry overnight. Dendrite reconstructions were performed using the Neurolucida software (MicroBrightField) at 100x magnification using a light microscope outfitted with a Z drive. Layer 5 pyramidal neurons were identified based upon the neuron’s location relative to other cells within the cortex, the presence of a pyramidal cell body, and the presence of a strong apical arbor that extended to the surface. Motor cortex (M1 and M2) was identified by comparing to “The Mouse Brain in Stereotaxic Coordinates” (Paxinos and Franklin, 2012). Sholl analysis, total dendrite length, and branch point number were analyzed using the NeuroExplorer software (NEX Technologies). Neuron tracings were performed by an experimenter blinded to genotype. Five neurons were sampled from each animal; four animals were used per genotype.
Dendritic spine density analyses
Thy1–GFP–M–expressing NEX–Trio+/−, NEX–Trio−/−, and WT mice at P42 were deeply anesthetized by pentobarbital sodium injection (Nembutal Sodium Solution) and transcardially perfused with 4% paraformaldehyde in phosphate buffered saline (PBS). Brains were postfixed at 4°C overnight and rinsed in PBS; 40 mm coronal slices were acquired on a Vibratome Series 1000 Sectioning System (Lancer). Slices containing the hippocampus were immediately mounted onto slides, while slices containing the motor cortex were stained with 4’,6–diamidino–2–phenylindole (DAPI) to identify the basal dendrites of layer 5 pyramidal neurons in motor cortex. Specifically, slices were incubated in 1:10,000 DAPI solution (Thermo Fisher Scientific, 62248) in 3% BSA in PBS by gently shaking for 10 minutes at room temperature and then washed 3 times with PBS. Slices were mounted onto Colorfrost Plus microscope slides (Thermo Scientific Shandon, 9991013), allowed to dry for 30 minutes, and coated with ProLong Diamond Antifade Mountant (Invitrogen, P36970) and a microscope cover glass (Fisherbrand, 12–545–F). The slides were allowed to dry overnight at 4C. The NEXt day, clear nail polish was added to the edges, and the slides were allowed to dry for 24 hours before imaging. An experimenter blinded to genotype acquired images on an UltraVIEW VoX spinning disc confocal (Perkin Elmer) Nikon Ti–E–Eclipse microscope using a 100x CFI Plan Apo VC, NA 1.4, oil objective and a 14 bit Hamamatsu (C9100–50) EM CCD camera. Full z stack images were exported from Volocity (Perkin Elmer) and merged using the NIH ImageJ software. Layer 5 pyramidal neurons were identified at 10x by observing the neuron’s location based upon the DAPI staining and by a pyramidal cell body with the start of a strong apical dendrite. Motor cortex (M1 and M2) was identified by comparing to “The Mouse Brain in Stereotaxic Coordinates” (4th edition, by George Paxinos and Keither B.J. Franklin). An experimenter blinded to genotype analyzed the data, counting dendritic spines on secondary dendritic branches; groups with direct comparisons were analyzed together.
Electron microscopy
Electron microscopy was performed as previously described (Omar et al., 2017) with minor modifications. NEX–Trio+/−, NEX–Trio−/−, and WT mice at P42 were deeply anesthetized by pentobarbital sodium injection (Nembutal Sodium Solution) and transcardially perfused with 4% paraformaldehyde and 2% glutaraldehyde in PBS. Brains were postfixed at 4C overnight and rinsed in PBS; 50 μm coronal slices were acquired on a Vibratome Series 1000 Sectioning System (Lancer). Slices containing the motor cortex (M1 and M2) were identified by comparing them to “The Mouse Brain in Stereotaxic Coordinates” (4th edition, by George Paxinos and Keither B.J. Franklin); chosen slices were near the point of corpus callosum fusion (1.09 mm rostral to Bregma). The slices were processed for 1 hour in 4% OsO4, dehydrated in an ethanol/propylene oxide series, and stained with 1% uranyl acetate in 70% ethanol for 1 hour. NEXt, the slices were stained with lead citrate, embedded in Epon, resectioned at 70 nm using an ultramicrotome, and collected on Formvar–coated slot grids. Images were taken on a JEOL 1200 EX electron microscope at 12,000x magnification. Synapse density, PSD length, spine head area, and presynaptic bouton area were quantified using ImageJ software. The analyses were performed within a 994 ± 4 mm2 area of layer 5 motor cortex (M1) using eighteen 55.2 ± 0.2 mm2 fields of view per mouse. Synapses were identified by: (1) a plasma membrane surrounding and between pre– and postsynaptic compartments, (2) a PSD in the postsynaptic compartment, and (3) vesicles in the presynaptic compartment. Synapse density was the total number of spines divided by the field of view area. PSD length was the cross–sectional length of the PSD within identified synapses. Spine head and presynaptic bouton areas were measured by tracing the plasma membrane of spines and boutons in identified synapses; the tracings included the entire spine or bouton down to the narrowest part of the spine or axon neck.
Electrophysiological recordings
Slices were prepared and recordings were performed as previously described (Xiao et al., 2016). NEX–Trio+/−, NEX–Trio−/−, and WT mice at P37–P45 were deeply anesthetized with pentobarbital sodium injection (Nembutal Sodium Solution) and their brains rapidly removed and cooled in ice–cold sucrose–containing artificial cerebrospinal fluid (sucrose–ACSF, in mM): 2.5 KCl, 7 MgSO4,7H2O, 1.25 NaH2PO4, 0.5 CaCl2,2H2O, 28 NaHCO3, 7 dextrose, 205 sucrose saturated with 95% O2–5% O2 at 290–320 mOsmol/L, pH 7.4. Coronal slices including the area of hippocampus and M1 cortex were cut at 350 mm in ice–cold sucrose–ACSF on a vibratome (Leica), then recovered in a humidified, oxygenated environment at 33.5C for at least 30 minutes. Slices were perfused during recording at 1 ml/min with ACSF (in mM): 3 KCl, 1 MgSO4•7H2O, 1.25 NaH2PO4, 2 CaCl2,2H2O, 26 NaHCO3, 10 dextrose, 118 NaCl, 0.4 ascorbic acid, 4 Na–lactic acid, 2 Na–pyruvic acid saturated with 95% O2–5% O2 at 290–320 mOsmol/L, pH 7.4. 10 mM bicuculline methiodide (BMI) was included in the ACSF. CA1 and M1 pyramidal neurons were identified using DIC and infrared optics with a water–immersion objective (NIR Apo 40x; BX51, Nikon) at room temperature (RT), and whole–cell recordings were obtained with an Axopatch 700B patch–clamp amplifier (Molecular Devices) using recording electrodes pulled from 1.5 mm glass capillaries on a Flaming–Brown micropipette puller (Sutter Instruments) filled with (in mM): 137 cesium gluconate, 10 NaCl, 0.2 EGTA, 4 Na2ATP, 0.3 Na3GTP, 5 Na2–phosphocreatine, 10 HEPES, 5 QX–314 chloride adjusted to 300 mOsmol/L pH 7.4. Under these conditions, the recording electrodes had a resistance of 3–6 MΩ. Recorded signals were low pass filtered at 2 kHz and digitized at 5 kHz with a Digidata 1440 digitizer (Molecular Devices). Cells were excluded if series resistance deviated > 20% or exceeded 20 MU. Data were collected in pClamp 10.3 and analyzed with Clampfit 10.3.
Excitatory postsynaptic currents (EPSCs) were evoked with 0.05 Hz stimulation by a tungsten bipolar stimulating electrode placed 100 mm away from the recording electrode using stimulus intensities that yielded 50%–60% of the maximal response. AMPAR-eEPSC amplitudes were measured at the peak of the EPSC recorded at 70 mV holding potential. NMDAR–eEPSC amplitudes were recorded from the same neurons at +40 mV 40 ms after the stimulus artifact when AMPAR–eEPSCs have decayed. Miniature EPSCs (mEPSCs) were recorded in whole–cell patch mode. AMPAR–mEPSCs were recorded at 70 mV and NMDAR–mEPSCs were recorded at +40 mV with tetrodotoxin and BMI in the external solution. Amplitude, frequency, and decay time of miniature events were detected using Mini–Analysis software (Synaptosoft, Decatur, GA) and inspected visually to exclude overlapping events and events on a noisy baseline. In the paired–pulse ratio (PPR) experiments, the paired–pulse stimulation (interstimulus intervals (ISIs) of 35, 60, 75, 100, 200 or 300 ms) was delivered via a bipolar tungsten stimulating electrode placed in CA3 Schaffer collateral axons in the hippocampus or in layer 2/3 of the motor cortex. The second EPSC (EPSC2), which was evoked by the second of the paired–pulse stimulations, was divided by EPSC1 to obtain a PPR. PPR was assessed at five ISIs (35, 60, 100, 200, and 300 ms) in M1 neurons and at 75ms ISI in hippocampal neurons. Long–term potentiation (LTP) of evoked EPSCs (recorded at 70 mV) was induced by thetaburst stimulation (TBS, five trains of four pulses at 100 Hz in 200 ms intervals, repeated four times with an interval of 10 s) in the presence of 10 03BCM BMI.
Proteomics and phosphoproteomics sample preparation
Mice at P42 were deeply anesthetized. The motor cortex was removed with the assistance of a mouse Brain Matrix and immediately frozen in liquid nitrogen. Dissected mouse cortex slices were transferred to Covaris tissueTUBE TT1 (Part No. 520001, Plug Part No. 520006), kept on dry ice, and flash frozen in liquid nitrogen immediately prior to cryopulverization using a Covaris CP02 cryoPREP Automated Dry Pulverizer (Part No. 500001). The resulting powdered samples were transferred into 1.5 mL microfuge tubes and kept at 80C until lysis. The cryopulverized samples were lysed in 300 μl lysis buffer (8M urea, 150 mM NaCl, 50 mM TRIS, pH 8.0, 1 mM EDTA, containing protease and phosphatase inhibitors) on ice. Lysates were centrifuged for 30 minutes at 4°C at 10,000 rpm. Protein concentrations of supernatants were determined by BCA (Thermo). Cleared tissue lysates were reduced with 10 mM TCEP and cysteine alkylated with 10 mM iodoacetamide for 30 minutes in the dark at room temperature. Samples were diluted to 4 M urea with 200 mM Tris pH 8.0 and digested with LysC (1:50 enzyme:substrate ratio) for 90 minutes at 37°C. Samples were diluted to less than 2 M urea with 200 mM Tris pH 8.0 before adding trypsin (Promega) at the same ratio for overnight digestion at 37°C. Formic acid (FA) was added to a final concentration of 1%, and the samples desalted using 100 mg, 1 cc Sep–Pak (Waters). A 400 mg aliquot of each sample digest was frozen and dried by vacuum centrifugation, then stored at 80°C until tandem mass tag (TMT10, Thermo lot # SE240163, TMT11 channel, Thermo lot # RK245431) peptide labeling according to manufacturer instructions.
Desalted 11–plex TMT labeled peptides (4.4 mg total) were subjected to basic reversed–phase (RP) chromatography. Peptides were separated using a 4.6 mm × 250 mm RP Zorbax 300 A Extend–C18 column (Agilent, 3.5 mm bead size) on an Agilent 1100 Series HPLC. The 96 minute gradient of solvent A (2% acetonitrile [ACN], 20 mM ammonium formate, pH 10) to solvent B (90% acetonitrile, 20 mM ammonium formate, pH 10) held at 0% B for seven minutes; then went from 0% to 16% B over six minutes; from 16% to 40% B over 60 minutes, from 40% to 44% B over four minutes; from 44% to 60% B in over five minutes, ending with a 14 minute hold at 60% B. The flow rate was 1 ml/min. 84 fractions were collected and concatenated down to 25 final fractions. 5% of each final fraction was used for proteome analysis.
The remaining 95% of each of the 25 fractions were concatenated to 13 fractions. These 13 fractions were IMAC enriched using Ni-NTA Agarose Beads (QIAGEN, Lot No. 154027064) for phosphoproteome analysis as previously described (Mertins et al., 2018), except the NTA was charged with 100 mM iron chloride instead of 10 mM.
Proteomics and phosphoproteomics data acquisition
Proteome analysis was performed using a Proxeon EASY–nLC 1200 UHPLC coupled to an Orbitrap Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). The online peptide separation was performed using a 20 cm × 75 μm i.d. silica picofrit capillary column (New Objectives) packed with 1.9 μm ReproSil–Pur C18–AQ beads (Dr. Maisch GmbH) at 50°C. The 110 minute method, 84 minute effective gradient, of solvent A (3% ACN, 0.1% FA) to solvent B (90% ACN, 0.1% FA) started at 2% B and went from 2% to 6% B over one minute; from 6% to 30% B over 84 minutes; from 30% to 60% B over 9 minutes; from 60% to 90% B over 1 minute, holding for five minutes, ramping down to 50% B over one minute and holding for 10 minutes. The flow rate was 200 nl/min for the first 100 minutes and 500 nl/min the last ten minutes. The Q Exactive Plus mass spectrometer performed data–dependent acquisition in positive ion mode. MS1 spectra scanned a range of 300–1800 m/z at a resolution of 70,000, with a maximum injection time (max IT) of 5 ms for a 3e6 AGC target. MS2 spectra scanned a range of 200–2,000 m/z at a resolution of 35,000. The top 12 most abundant peaks, charge state 2 < × < 7, were selected for MS2 with an isolation width of 0.7 m/z and an offset of 0.0 m/z. The AGC target was set to 5e4 with a max IT of 120 ms. An empirically determined normalized collision energy was set to 30. Dynamic exclusion was set to 20 s.
Phosphoproteome analysis was performed as described above except data was acquired on a Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). The Lumos mass spectrometer performed data–dependent acquisition in positive ion mode. MS1 spectra scanned a range of 350–1,800 m/z at a resolution of 60,000, with a maximum injection time (max IT) of 50 ms for a 4e5 AGC target. MS2 spectra scanned at a resolution of 50,000, with a maximum injection time (max IT) of 105 ms for a 5e4 AGC target.
Proteomics and phosphoproteomics data analysis
All .raw files were searched using Spectrum Mill (Agilent Technologies). MS2 spectra were searched against the Uniprot Mouse database (10/17/2014, 41,309 mouse entries, 150 common laboratory contaminants), with a mass tolerance of 20 ppm for both the precursor and product ions. The enzyme specificity was set for LysC/Trypsin and allowing up to three missed cleavages. The fixed modification was carbamidomethylation at cysteine. TMT labeling was required at lysine, but peptide N–termini were allowed to be either labeled or unlabeled. Allowed variable modifications for whole proteome datasets were acetylation of protein N–termini, oxidized methionine, deamidation of asparagine, and pyroglutamic acid at peptide N–terminal glutamine, with a precursor MH+ shift range of 18 to 64 Da. The false discovery rate was determined to be less than 1%. For proteome interpretation, protein identifications were discarded if the protein was only observed by a single peptide. For reporting number of proteins identified protein subgroups, protein gene products (proteoforms) from the same gene, were expanded to account for the observation of different proteoforms. For statistical tests protein subgroups were collapsed to the proteoform with the most evidence.
Primary cortical culture
Cortical cultures were prepared from E15.5 Trio+/flox mouse embryos as described in the Experimental Model and Subject Details section. Cells were transfected with pEGFP–N1 or pAAV–CAG–“GFP–P2A–Cre” and pTriEx–6–PDE4A5 (Becky Carlyle and Angus Nairn, Yale University) DNA by calcium–phosphate precipitation for 6 days starting at DIV9. 100 mM Rp–cAMPS (Tocris 1337, dissolved in water) was applied to a subset of coverslips on DIV12. All coverslips were fixed with 2% PFA at DIV15, then stained with an anti–GFP–booster_Atto488 nanobody. Neurons were imaged and dendritic spine density was analyzed as described in the dendritic spine density analyses section.
On DIV15, cells were fixed at room temperature for 20 minutes with 2% PFA in cytoskeleton buffer (CBS: 10 mM MES pH 6.8, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA and 320 mM sucrose). Fixed cells were permeabilized for 15 minutes with 0.3% TX100/ TBS (150 mM NaCl, 20 mM Tris pH 7.4), washed 3 times with 0.1% TX100/TBS, and blocked for 30 minutes with 0.3% TX100/ 2% BSA/10% Normal donkey Serum/TBS. Cells were then incubated at 4C overnight with anti–GFP–booster_Atto488 nanobody (Chromotek gba–488; 1:500) in 0.1% TX100/2% BSA/TBS. The NEXt morning the cells were washed with 0.1% TX100/TBS, then mounted in AquaMount (Lerner Laboratories 13800) after a final wash with TBS and imaged on a spinning disk confocal as described in the dendritic spine density analyses section. Images were analyzed by an experimenter blind to genotype.
Fibroblast cell immunofluorescence
5×104 Trioflox/flox mouse fibroblast cells were plated the day before transfection and maintained in DMEM plus 10% fetal bovine serum (FBS), 2 mM l–glutamine, and penicillin/streptomycin at 37C and 5% CO2. Cells were transfected with pLL3.7–RFP or pAAV-mCherry–IRES–Cre (Karl Deisseroth, Stanford University, via Addgene Plasmid #55632) using Lipofectamine 3000 (Invitrogen L3000001). Fresh media was put onto cells to remove transfection reagent after 24 hours. Cells were split once, 72 hours after transfection using 0.05% Trypsin–EDTA (GIBCO 25300–054). On Day 4, 12 mm coverslips were coated with 50 μg/ml poly–D–lysine (Corning Inc.) for 20 minutes at room temperature before rinsing 3 times with PBS. Human fibronectin (GIBCO 33016–015) was diluted in PBS to a final concentration of 10 μg/ml before putting on coverslips at 4°C overnight. The following day, coverslips were washed 3 times with PBS before blocking with 1% BSA (American Bio) in PBS for 1 hour at 37°C. Coverslips were washed 3 more times with PBS before plating cells. 1.5−104 transfected fibroblast cells were seeded onto coated coverslips and let spread for 24 hours before fixing and staining.
Cells were immunostained as described in the primary cortical culture section with minor changes. Fixation in 2% PFA in CBS was performed for 5 minutes at room temperature. Cells were incubated with primary antibodies, mouse anti–RFP (Rockland 200–301–379; 1:1,000) and a rabbit antibody generated in–house to the DH portion of the TRIO GEF2 domain (DH2, 1:50) (Katrancha et al., 2017), at 4°C overnight. The NEXt day, the cells were washed 3 times with 0.1% TX100/TBS and sequentially incubated with AlexaFluor568 donkey anti–mouse (Invitrogen A10037; 1:1,000) and AlexaFluor647 donkey anti–rabbit (Invitrogen A31573; 1:1,000), both diluted in 0.1% TX100/2% BSA/TBS, with three 0.1% TX100/TBS washes following each secondary. The coverslips were then mounted and imaged as described in the dendritic spine density analyses section, with identical imaging parameters between conditions. TRIO staining was quantified with NIH ImageJ software by tracing the outline of only transfected cells, using the RFP channel.
Behavioral tests
NEX–Trio+/− mice, NEX–Trio−/− mice, and sex–matched WT littermates were used for all behavioral analyses. Behavioral tests took place between 6 and 10 weeks of age (P42–P70). At P42, mice were habituated to handling for 6 minutes per day for 5 days prior to experimentation.
Novel object recognition task
The novel object recognition test assays recognition memory, a hippocampus–dependent behavior (Broadbent et al., 2009; Cohen et al., 2013). This behavioral task was performed as previously described (Omar et al., 2017; Sfakianos et al., 2007) in a quiet, moderately lit (100 lux) room separate from housing. Mice were habituated to a large polycarbonate cage (45 3 24 3 20 cm) with fitted filtertop covers in the testing room for 1 hour on both days. During object familiarization on day 1, two identical objects were placed on the left and right sides of the testing cage. Mice explored the objects until they accumulated 30 s of tactile exploration time, defined as direct oral or nasal contact. Mice were then returned to their homecage for 48 hours. During the novel object recognition task on day 3, one familiar and one novel object were placed on either side of the testing cage, and mice explored the objects until they accumulated 30 s of tactile exploration time. Mice were excluded from analysis if they failed to explore both objects or accumulate 30 s of tactile exploration time within 6 minutes on either day. Live scoring was performed by an experimenter blinded to genotype, and all sessions were video recorded.
Prepulse inhibition of acoustic startle
The prepulse inhibition of acoustic startle is used to evaluate sensorimotor gating, which is deficient in individuals with schizophrenia (Parwani et al., 2000). This behavioral task was performed as previously described (Baldan Ramsey et al., 2011; Geyer and Dulawa, 2003) in a quiet room separate from housing. Mice were habituated to the testing room for 30 minutes prior to experimentation and then acclimated to the apparatus for 5 minutes. The test session involved four consecutive blocks of test trials, separated by 15 s, with continuous background noise (65 dB). In each block, eight trial types were used: “pulse–alone” trials (baseline startle) involved a 40 ms, 120 dB startle burst; the six independent “prepulse” trials involved a 20 ms prepulse (6, 12, or 16 dB above background) that occurred 30 or 100 ms prior to the 120 dB startle burst; and “no stimulus” trials involved only the background noise to assess spontaneous movement. Blocks 1 and 4 involved six consecutive pulse–alone trials. Blocks 2 and 3 involved six pulse–alone trials, five no stimulus trials, and five of each type of prepulse trial in a pseudo–random order. Overall, ninety–four test trials were performed in a twenty–eight–minute session. Trials with noisy baselines or excessive spontaneous movement prior to the pulse were excluded. Analyses were performed with the SR Lab Startle Response System software (San Diego Instruments) with a 10 ms baseline using an analysis window of 500 ms to 550 ms. All sound levels were measured on the “A” scale in dB (Geyer and Dulawa, 2003).
Nestlet shredding
The nestlet shredding test is a test for stereotyped or repetitive behaviors, often seen in autism spectrum disorders (Angoa–Pérez et al., 2013). This behavioral task was performed in a quiet, moderately lit (100 lux) room separate from housing. Mice were habituated to the testing room for 1 hour prior to experimentation. A standard polycarbonate mouse cage (30 3 19 3 13 cm) was filled with unscented bedding material to a depth of 0.5 cm, the surface was leveled, and a fitted filter–top cover was placed on top. Cotton fiber nestlets (5 cm × 5 cm, 5 mm thick, 2–2.5 g each) were weighed on an analytical balance before being placed on top of the bedding. The test mouse was placed into the test cage and left undisturbed for 30 minutes. The remaining intact nestlet was removed and allowed to dry overnight before being weighed again. The percentage of nestlet shredded was calculated.
Social preference test
The social preference test assays sociability by quantifying preference for a “stranger” mouse relative to an inanimate object (Rapanelli et al., 2017). This behavioral task was performed in a quiet, moderately lit (100 lux) room separate from housing. Mice were habituated to the testing room for 1 hour prior to experimentation. During the habituation phase, mice were placed in a 50 cm × 50 cm plexiglass box with two wire–mesh pencil holders (open side facing downward) for 10 minutes. Then, the mouse was briefly removed while an inanimate object (Duplo blocks of a similar size and color to a mouse) and a male conspecific “stranger” mouse were placed underneath separate wire–mesh pencil holders. The test mouse was then returned to the open field for 10 minutes, tracked using the AnyMaze software (Stoelting Co.), and scored for entries into and time within a social target zone and an equivalent nonsocial target zone. The target zones were defined by an annulus around the wire enclosure.
Open field test
The open field test assays both general locomotor activity and anxiety–like behavior in rodents. This behavioral task was performed in a quiet, moderately lit (100 lux) room separate from housing. Mice were habituated to the testing room for 1 hour prior to experimentation. Individual mice were placed in a large, clear plexiglass box and monitored using the Fusion software connected to a light beam array (Omnitech Electronics, Open Field Test, Version 4.5) for 60 minutes. The following measurements were analyzed based upon beam breaks: total distance traveled, ambulatory time (successive beam breaks), and vertical activity time (z axis beam breaks).
Elevated plus maze
The elevated plus maze assays anxiety–like behavior in rodents. This behavioral task was performed in a quiet, dimly lit (20–30 lux) room separate from housing. The apparatus is raised 36.5 cm above the ground with a 5 cm square center platform and four 30 cm × 5 cm arms; two of the arms are enclosed by 16 cm high walls (Stoelting Co, Wood Dale, IL). Mice were habituated to the testing room for 1 hour prior to experimentation. Mice were tested in 3 trials that were 5 minutes long with at least 10 minutes to rest between trials. Mice were placed directly on the center platform, tracked using the AnyMaze software (Stoelting Co.), and scored for time spent in the open arms.
Forced swim test
The forced swim test assays behavioral despair in rodents. Mice were habituated to a quiet, moderately lit (100 lux) room separate from housing for 30 minutes prior to experimentation. Mice were then placed in a clear glass cylinder (14 cm in diameter) filled to a depth of 15 cm with water at 25°C and videotaped for 6 minutes. Mobility time was scored during the last 4 minutes of the trial by a blinded experimenter. Mobility was defined as the presence of active or escape directed movement. The total mobility time was subtracted from the total test time (240 s) to determine immobility time.
Accelerating rotarod
The rotarod is used to assess motor coordination and motor learning in rodents. Mice were habituated to a quiet, moderately lit (100 lux) room separate from housing for 30 minutes prior to experimentation. A five–lane rotarod treadmill (Med Associates ENV–577M, 8.75” rod circumference) was used for accelerating analyses. Mice were acclimated to the rotating rod at 4 RPM for 10 s before beginning the test. Then, the rod accelerated from 4 to 40 RPM over the course of 5 minutes (1.2 RPM/10 s) before leveling off at 40 RPM for 2.5 minutes. The maximum trial duration was 7.5 minutes, requiring the mice to travel 1,837.5 inches. Five trials were performed per mouse with a rest time of at least 10 minutes between trials. The latency to fall was recorded per trial.
Kondziela inverted screen test
The inverted screen test assays muscular strength in rodents. This behavioral task was adapted from methods previously described (Deacon, 2013). Briefly, mice were habituated to a quiet, moderately lit (100 lux) room separate from housing for 30 minutes prior to experimentation. The apparatus was a 40 cm square of wire–mesh with 12 mm squares of 1 mm diameter wire; it was surrounded by a square wooden frame (3.5 cm thick). During the training session on day 1, mice were placed in the center of the wire–mesh screen and rotated to an inverted position over 2 s. The screen was placed over a clear open box with the mice 45 cm above a padded surface. Mice were habituated to hanging on the screen until a cumulative hang time of 2 minutes was reached; mice were excluded if they were unable to meet this requirement. During the test session on day 2, mice were placed on the screen as before and given 3 trials to reach a maximum hang time of 8 minutes with 10–minute rest periods between trials. Mice reaching the 8–minute mark were deemed “successful,” and mice failing to reach the mark were “unsuccessful.”
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using either GraphPad Prism 7 or R software. For all assays, NEX–Trio+/− and NEX–Trio−/− mice were analyzed independently. Specific details of ‘n’ and statistical tests are included in the figure legends. Significance was defined by a p value less than 0.05. All pooled data are represented as mean ± SEM.
For tests with two independent variables, a two–way ANOVA was performed prior to direct comparisons with a Bonferroni multiple comparisons post hoc test in Prism. To assay motor learning in the accelerating rotarod task, linear models were created in Prism, and post hoc comparisons were performed on the slopes and elevations/intercepts of the linear models. To assess PPR, a one phase decay global fit analysis was performed with post hoc Bonferroni multiple comparisons. For analyses including more than three independent variables (e.g., genotype, gender, and target zone for the social preference test), a linear regression model (lm function) was created in R. All meaningful interactions were tested, and significant interactions were included in the final model. Post hoc comparisons were performed with a Bonferroni multiple comparisons test in R (pairwise.t.test function with p.adj = “bonf”). Tests involving success rates (proportions) were analyzed using the N–1 Chi–Square test (https://measuringu.com/ab–cal/). Tests involving only one independent variable (genotype) for the comparison of two groups were analyzed using the unpaired Student’s t test. For the proteomics immunoblot follow–up, a ratio paired t test was performed to compare the littermate pairs. The analysis of dendritic spine density was performed using a linear regression model in R (modeling density as a function of the mouse number, neuron number, and genotype) to control for similarities between spine densities acquired from the same neuron or mouse; this was similar for the electron microscopy–based measurements.
For proteomic analyses, the normalized and imputed dataset was subjected to a moderated F–test (Smyth, 2004), followed by Benjamini–Hochberg Procedure correcting for multiple hypothesis testing. We drew an arbitrary cutoff at adjusted p value < 0.05 to identify proteins that significantly differed in at least one group. To consistently analyze NEX–Trio+/− and NEX–Trio−/− mice independently, unpaired t tests identified differences between groups. The heatmap shows the log2 fold change compared to the common reference and was clustered using one minus Pearson correlation metric (https://software.broadinstitute.org/morpheus/). For phosphoproteomic analyses, kinase–substrate pairs enriched in the dataset were identified using the Phoxtrack tool (Weidner et al., 2014). Phoxtrack scores for kinase–substrate sets were reported and plotted if the p value for their enrichment in either the NEX–Trio+/− and NEX–Trio−/− groups was less than 0.05. To visualize how phosphorylation site occupancy changed compared to protein levels, we plotted the mean log2 fold change of the protein level and the phosphosite level for each pair using data from the proteomic and phosphoproteomic analysis.
DATA AND SOFTWARE AVAILABILITY
For proteomic and phosphoproteomic analyses, the accession number for the original mass spectra reported in this paper is MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp): < MSV000083386 >. The data are directly accessible via ftp://massive.ucsd.edu/MSV000083386.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Primary Antibody: TRIO Sec14 domain | Katrancha et al., 2017; McPherson et al., 2005 | N/A |
| Rabbit Primary Antibody: Phosphodiesterase 4D (PDE4D) | Millipore | Cat#ABS22 |
| Rabbit Primary Antibody: Phosphodiesterase 4A (PDE4A) | Abcam | Cat#ab14607 |
| Rabbit Primary Antibody: Phosphodiesterase 4A Isoform 5 (PDE4A5) | Abcam | Cat#ab42094 |
| Rabbit Primary Antibody: P21 (RAC1) Activated Kinase 4 (PAK4) | Proteintech | Cat#14685–1-AP |
| Rabbit Primary Antibody: Clathrin Heavy Chain (CHC) | Pietro De Camilli lab, Yale University | N/A |
| Goat Secondary Antibody: Anti-Rabbit | Bio-Rad | Cat#170–6515 |
| Nanobody: anti-GFP-booster_Atto488 | Chromotek | Cat#gba-488 |
| Mouse Primary Antibody: anti-RFP | Rockland | Cat#200–301-379 |
| Rabbit Primary Antibody: TRIO Guanine Nucleotide Exchange Factor (GEF) Dbl Homology 2 (DH2) domain | Katrancha et al., 2017 | N/A |
| Donkey Secondary Antibody: Anti-Mouse AlexaFluor568 | Invitrogen | Cat#A10037 |
| Donkey Secondary Antibody: Anti-Rabbit AlexaFluor647 | Invitrogen | Cat#A31573 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 3000 | Invitrogen | Cat#L3000001 |
| Permount | Fisher Scientific | Cat#SP15–100 |
| AquaMount | Lerner Laboratories | Cat#13800 |
| ProLong Diamond Antifade Mountant | Invitrogen | Cat#P36970 |
| Rp-cAMPS | Tocris | Cat#1337 |
| Tandem mass tag TMT10 | Thermo | Cat#90406; Lot#SE240163 |
| Tandem mass tag TMT11 channel | Thermo | Cat#1863196B; Lot#RK245431 |
| Poly-D-lysine | Corning | Cat#354210 |
| Human fibronectin | GIBCO | Cat#33016–015 |
| Critical Commercial Assays | ||
| FD Rapid GolgiStain Kit | FD NeuroTechnologies, Inc | Cat#PK401 |
| Deposited Data | ||
| Original mass spectra download under the identifier: < MSV000083386 > | MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) | ftp://massive.ucsd.edu/MSV000083386 |
| Experimental Models: Cell Lines | ||
| Trioflox/flox mouse fibroblasts | This Paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse (C57BL/6 × 129/SvJ): “floxed” Trio allele: Trioflox/flox (Trio−/−) | Zong et al., 2015 | N/A |
| Mouse (C57BL/6 × 129/SvJ): NEX(Neurod6)-Cre transgene: NEX-Cre | Goebbels et al., 2006 | N/A |
| Mouse (C57BL/6 × 129/SvJ): Thy1-GFP-M line: Thy1-GFP | Feng et al., 2000 | N/A |
| Oligonucleotides | ||
| “Floxed” Trio genotyping primer 1: 5′-TTG TTC CAT TTA CGT CAC CG-3′ | Zong et al., 2015 | N/A |
| “Floxed” Trio genotyping primer 2: 5′-CCT CAG GGA AGA GAC TA-3′ | Zong et al., 2015 | N/A |
| GFP genotyping primer 1: 5′-GCA CGA CTT CAA GTC CGC CAT GCC-3′ | Omar et al., 2017 | N/A |
| GFP genotyping primer 2: 5′-GCG GAT CTT GAA GTT CAC CTT GAT GCC-3′ | Omar et al., 2017 | N/A |
| NEX-Cre genotyping primer 1: 5′-AGA ATG TGG AGT AGG GTG AC-3′ | Omar et al., 2017 | N/A |
| NEX-Cre genotyping primer 2: 5′-GAG TCC TGG AAT CAG TCT TTT TC-3′ | Omar et al., 2017 | N/A |
| NEX-Cre genotyping primer 3: 5′-CCG CAT AAC CAG TGA AAC AG-3′ | Omar et al., 2017 | N/A |
| Recombinant DNA | ||
| pEGFP-N1 | Clonetech | N/A |
| pAAV-CAG-“GFP-P2A-Cre” | Jaime Grutzendler, Yale University | N/A |
| pTriEx-6-PDE4A5 | Becky Carlyle and Angus Nairn, Yale University | N/A |
| pLL3.7-RFP | Lin et al., 2013 | N/A |
| pAAV-mCherry-IRES-Cre | Karl Deisseroth, Stanford University (via Addgene) | Addgene Plasmid #55632 |
| Software and Algorithms | ||
| Neurolucida software | MicroBrightField (MBF) Bioscience | https://www.mbfbioscience.com/neurolucida |
| NeuroExplorer software | Nex Technologies | https://www.neuroexplorer.com/ |
| ImageJ software | NIH | https://imagej.nih.gov/ij/ |
| Mini-Analysis software | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis/ |
| Spectrum Mill software | Agilent Technologies | proteomics.broadinstitute.org/ |
| SR Lab Startle Response System software | San Diego Instruments | https://www.sandiegoinstruments.com/product/sr-lab/ |
| AnyMaze software | Stoelting | http://www.anymaze.co.uk/ |
| Fusion software, Version 4.5 | Omnitech Electronics | http://www.omnitech-electronics.com/product/Fusion-Software/510 |
| Morpheus software | Broad Institute of MIT and Harvard | https://software.broadinstitute.org/morpheus/ |
| Phoxtrack tool | Weidner et al., 2014 | N/A |
| pClamp software suite (pClamp and Clampfit) | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/ |
| N-1 Chi-Square test | MeasuringU | https://measuringu.com/ab-cal/ |
| R Studio software | RStudio | https://www.rstudio.com/ |
| Prism 7 software | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Elevated Plus Maze | Stoelting | Cat#60140 |
| Five-lane rotarod treadmill | Med Associates | Cat#ENV-577M |
| Open Field Test | Omnitech Electronics | http://www.omnitech-electronics.com/product/Open-Field-Locomotor-Activity/1010 |
| Optimal Cutting Temperature Compound | Tissue-Tek | Cat#4583 |
| Cryomold (10 mm × 10 mm × 5 mm) | Tissue-Tek | Cat#4565 |
| Colorfrost Plus microscope slides | Thermo Scientific Shandon | Cat#9991013 |
| TissueTUBE TT1 | Covaris | Tube Cat#520001; Plug Cat#520006 |
| CP02 cryoPREP Automated Dry Pulverizer | Covaris | Cat#500001 |
| Axopatch patch-clamp amplifier | Molecular Devices | Cat#700B |
Highlights.
Trio loss increases anxiety and impairs social preference and motor coordination
Trio loss reduces dendritic arborization but increases dendritic spine densities
Trio loss impairs PDE4A5–PKA signaling, pre– and postsynaptic function, and LTP
Altering PDE4A5 levels and PKA activity rescues Trio–related dendritic spine defects
ACKNOWLEDGMENTS
The authors thank Chris Kaliszewski and Xianyun Ye for assisting with experiments. Thanks to Aaron Levy, Ethan Sarnoski, and Ed Scolnick for helpful comments on the manuscript. Klaus–Armin Nave (University of Göttingen, Max Planck Institute of Experimental Medicine) generously provided the NEX–Cre mouse line. Pietro De Camilli (Yale University) generously provided the CHC primary antibody. Angus Nairn and Becky Carlyle (Yale University) generously provided the pTriEx–6–PDE4A5 DNA plasmid. This work was supported by NIH grants NS089662 (A.J.K.), NS105640 (A.J.K. and Michael J. Higley), MH115939 (A.J.K.), DC013791 (C.A.G.), DC015438 (C.A.G.), DC012441 (C.A.G.), and F31MH116571 (J.E.S.); a pilot award from the Stanley Center/Broad Institute (A.J.K., Dick Mains, and Betty Eipper); a Yale Gruber Science Fellowship (S.M.K.); a National Science Foundation Graduate Research Fellowship (S.M.K.); and NIH training grant T32GM007223 to J.E.S. (Susan J. Baserga).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.022.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Angoa–Pérez M, Kane MJ, Briggs DI, Francescutti DM, and Kuhn DM (2013). Marble burying and nestlet shredding as tests of repetitive, compulsive–like behaviors in mice. J. Vis. Exp, 50978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasaki T, Saito M, Sone M, Suzuki E, Sakai R, Ito K, and Hama C (2000). The Drosophila Trio plays an essential role in patterning of axons by regulating their directional extension. Neuron 26, 119–131. [DOI] [PubMed] [Google Scholar]
- Ba W, Yan Y, Reijnders MRF, Schuurs–Hoeijmakers JHM, Feenstra I, Bongers EMHF, Bosch DGM, De Leeuw N, Pfundt R, Gilissen C, et al. (2016). TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet 25, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldan Ramsey LC, Xu M, Wood N, and Pittenger C (2011). Lesions of the dorsomedial striatum disrupt prepulse inhibition. Neuroscience 180, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball RW, Warren–Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An BS, Wang TT, White JH, and Haghighi AP (2010). Retrograde BMP signaling controls synaptic growth at the NMJ by regulating Trio expression in motor neurons. Neuron 66, 536–549. [DOI] [PubMed] [Google Scholar]
- Bateman J, Shu H, and Van Vactor D (2000). The guanine nucleotide exchange factor Trio mediates axonal development in the Drosophila embryo. Neuron 26, 93–106. [DOI] [PubMed] [Google Scholar]
- Bellanger JM, Lazaro JB, Diriong S, Fernandez A, Lamb N, and Debant A (1998). The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene 16, 147–152. [DOI] [PubMed] [Google Scholar]
- Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, and Debant A (2000). The Rac1– and RhoG–specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol 2, 888–892. [DOI] [PubMed] [Google Scholar]
- Blangy A, Vignal E, Schmidt S, Debant A, Gauthier–Rouvie` re C, and Fort P (2000). TrioGEF1 controls Rac– and Cdc42–dependent cell structures through the direct activation of rhoG. J. Cell Sci 113, 729–739. [DOI] [PubMed] [Google Scholar]
- Blatt GJ (2012). The neuropathology of autism. Scientifica (Cairo) 2012, 703675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Gaskin S, Squire LR, and Clark RE (2009). Object recognition memory and the rodent hippocampus. Learn. Mem 17, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Huang PH, and Cheng HJ (2011). Disrupted–in–schizophrenia 1–mediated axon guidance involves TRIO–RAC–PAK small GTPase pathway signaling. Proc. Natl. Acad. Sci. USA 108, 5861–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Munchow AH, Rios LM, Zhang G, Asgeirsdo´ ttir HN, and Stackman RW Jr. (2013). The rodent hippocampus is essential for nonspatial object memory. Curr. Biol 23, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Cohen D, and Nicolelis MAL (2004). Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr. Biol 14, 1124–1134. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. ; DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RMJ (2013). Measuring the strength of mice. J. Vis. Exp, 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debant A, Serra–Pagès C, Seipel K, O’Brien S, Tang M, Park SH, and Streuli M (1996). The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac–specific and rho–specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 93, 5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller–Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, and Sanes JR (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51. [DOI] [PubMed] [Google Scholar]
- Frankfurt M, and Luine V (2015). The evolving role of dendritic spines and memory: Interaction(s) with estradiol. Horm. Behav 74, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Lande´ n M, Moran JL, Purcell SM, Sklar P, Sullivan PF, et al. (2016). Increased burden of ultra–rare protein–altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci 19, 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer MA, and Dulawa SC (2003). Assessment of murine startle reactivity, prepulse inhibition, and habituation. Curr. Protoc. Neurosci. Chapter 8, Unit 8.17. [DOI] [PubMed] [Google Scholar]
- Glantz LA, and Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave K–A. (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX–Cre mice. Genesis 44, 611–621. [DOI] [PubMed] [Google Scholar]
- Hakeda–Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, and Dickson BJ (2002). Rac function and regulation during Drosophila development. Nature 416, 438–442. [DOI] [PubMed] [Google Scholar]
- Herring BE, and Nicoll RA (2016). Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. USA 113, 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, and Zhang H (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. [DOI] [PubMed] [Google Scholar]
- Iyer SC, Wang D, Iyer EPR, Trunnell SA, Meduri R, Shinwari R, Sulkowski MJ, and Cox DN (2012). The RhoGEF Trio functions in sculpting class specific dendrite morphogenesis in Drosophila sensory neurons. PLoS ONE 7, e33634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrancha SM, Wu Y, Zhu M, Eipper BA, Koleske AJ, and Mains RE (2017). Neurodevelopmental disease–associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum. Mol. Genet 26, 4728–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, and Benes FM (2014). Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 71, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni VA, and Firestein BL (2012). The dendritic tree and brain disorders. Mol. Cell. Neurosci 50, 10–20. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell–Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium (2016). Analysis of protein–coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Ko H, Qian ZM, Yan LYC, Chan DCW, Arbuthnott G, Ke Y, and Yung WH (2017). Refinement of learned skilled movement representation in motor cortex deep output layer. Nat. Commun 8, 15834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y–C, Yeckel MF, and Koleske AJ (2013). Abl2/Arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J. Neurosci 33, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez–Cerdeño V (2017). Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol 77, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson CE, Eipper BA, and Mains RE (2005). Multiple novel isoforms of Trio are expressed in the developing rat brain. Gene 347, 125–135. [DOI] [PubMed] [Google Scholar]
- Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, et al. (2018). Reproducible workflow for multiplexed deep–scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat. Protoc 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Yan Y, Eipper BA, and Mains RE (2013). Neuronal Rho GEFs in synaptic physiology and behavior. Neuroscientist 19, 255–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco EM, Donaldson S, Williamson A, and Koleske AJ (2005). Integrin–mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J. Neurosci 25, 6105–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, Porteous DJ, Millar JK, and Houslay MD (2007). Isoform–selective susceptibility of DISC1/phosphodiesterase–4 complexes to dissociation by elevated intracellular cAMP levels. J. Neurosci 27, 9513–9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Schmidt S, Dietzl G, Keleman K, Å sling B, Debant A, and Dickson BJ (2000). Trio combines with dock to regulate Pak activity during photoreceptor axon pathfinding in Drosophila. Cell 101, 283–294. [DOI] [PubMed] [Google Scholar]
- O’Brien SP, Seipel K, Medley QG, Bronson R, Segal R, and Streuli M (2000). Skeletal muscle deformity and neuronal disorder in Trio exchange factor–deficient mouse embryos. Proc. Natl. Acad. Sci. USA 97, 12074–12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar MH, Kerrisk Campbell M, Xiao X, Zhong Q, Brunken WJ, Miner JH, Greer CA, and Koleske AJ (2017). CNS neurons deposit laminin a5 to stabilize synapses. Cell Rep. 21, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parwani A, Duncan EJ, Bartlett E, Madonick SH, Efferen TR, Rajan R, Sanfilipo M, Chappell PB, Chakravorty S, Gonzenbach S, et al. (2000). Impaired prepulse inhibition of acoustic startle in schizophrenia. Biol. Psychiatry 47, 662–669. [DOI] [PubMed] [Google Scholar]
- Pawson C, Eaton BA, and Davis GW (2008). Formin–dependent synaptic growth: evidence that Dlar signals via Diaphanous to modulate synaptic actin and dynamic pioneer microtubules. J. Neurosci 28, 11111–11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, and Franklin KBJ (2012). The Mouse Brain in Sterotaxic Coordinates, Fourth Edition (Academic Press; ). [Google Scholar]
- Peng YJ, He WQ, Tang J, Tao T, Chen C, Gao YQ, Zhang WC, He XY, Dai YY, Zhu NC, et al. (2010). Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J. Biol. Chem 285, 24834–24844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapanelli M, Frick LR, Xu M, Groman SM, Jindachomthong K, Tamamaki N, Tanahira C, Taylor JR, and Pittenger C (2017). Targeted interneuron depletion in the dorsal striatum produces autism–like behavioral abnormalities in male but not female mice. Biol. Psychiatry 82, 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioult–Pedotti MS, Friedman D, and Donoghue JP (2000). Learninginduced LTP in neocortex. Science 290, 533–536. [DOI] [PubMed] [Google Scholar]
- Sadybekov A, Tian C, Arnesano C, Katritch V, and Herring BE (2017). An autism spectrum disorder–related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun 8, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnström K, Mallick S, Kirby A, et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, and Koleske AJ (2007).Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J. Neurosci 27, 10982–10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivalkar M, and Giniger E (2012). Control of dendritic morphogenesis by Trio in Drosophila melanogaster. PLoS ONE 7, e33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol 3, e3. [DOI] [PubMed] [Google Scholar]
- Steven R, Kubiseski TJ, Zheng H, Kulkarni S, Mancillas J, Ruiz Morales A, Hogue CWV, Pawson T, and Culotti J (1998). UNC–73 activates the Rac GTPase and is required for cell and growth cone migrations in C. elegans. Cell 92, 785–795. [DOI] [PubMed] [Google Scholar]
- Tarbox SI, Addington J, Cadenhead KS, Cannon TD, Cornblatt BA, Perkins DO, Seidman LJ, Tsuang MT, Walker EF, Heinssen R, et al. (2013). Premorbid functional development and conversion to psychosis in clinical high–risk youths. Dev. Psychopathol 25, 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR, Birnbaum S, Ubriani R, and Arnsten AF (1999). Activation of cAMP–dependent protein kinase A in prefrontal cortex impairs working memory performance. J. Neurosci 19, RC23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erp TGM, Walton E, Hibar DP, Schmaal L, Jiang W, Glahn DC, Pearlson GD, Yao N, Fukunaga M, Hashimoto R, et al. ; Karolinska Schizophrenia Project (2018). Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biol. Psychiatry 84, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haren J, Boudeau J, Schmidt S, Basu S, Liu Z, Lammers D, Demmers J, Benhari J, Grosveld F, Debant A, and Galjart N (2014). Dynamic microtubules catalyze formation of navigator–TRIO complexes to regulate neurite extension. Curr. Biol 24, 1778–1785. [DOI] [PubMed] [Google Scholar]
- Walker EF, Savoie T, and Davis D (1994). Neuromotor precursors of schizophrenia. Schizophr. Bull 20, 441–451. [DOI] [PubMed] [Google Scholar]
- Weidner C, Fischer C, and Sauer S (2014). PHOXTRACK–a tool for interpreting comprehensive datasets of post–translational modifications of proteins. Bioinformatics 30, 3410–3411. [DOI] [PubMed] [Google Scholar]
- Xiao X, Levy AD, Rosenberg BJ, Higley MJ, and Koleske AJ (2016). Disruption of coordinated presynaptic and postsynaptic maturation underlies the defects in hippocampal synapse stability and plasticity in Abl2/Arg–deficient mice. J. Neurosci 36, 6778–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, and Zuo Y (2011). Spine plasticity in the motor cortex. Curr. Opin. Neurobiol 21, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, Barak B, Zeng M, Li C, Lu C, et al. (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W, Liu S, Wang X, Zhang J, Zhang T, Liu Z, Wang D, Zhang A, Zhu M, and Gao J (2015). Trio gene is required for mouse learning ability. Brain Res. 1608, 82–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For proteomic and phosphoproteomic analyses, the accession number for the original mass spectra reported in this paper is MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp): < MSV000083386 >. The data are directly accessible via ftp://massive.ucsd.edu/MSV000083386.