

Abstract
Aquaporin 0 (AQP0) is essential for eye lens homeostasis as is regulation of its water permeability by Ca2+, which occurs through interactions with Calmodulin (CaM), but the underlying molecular mechanisms are not well understood. Here, we use molecular dynamics (MD) simulations on the microsecond time scale under an osmotic gradient to explicitly model water permeation through the AQP0 channel. To identify any structural features that are specific to water permeation through AQP0, we also performed simulations of aquaporin 1 (AQP1) and a pure mixed lipid bilayer under the same conditions. The relative single-channel water osmotic permeability coefficients (pf) calculated from all of our simulations are in reasonable agreement with experiment. Our simulations allowed us to characterize the dynamics of the key structural elements that modulate the diffusion of water single-files through the AQP0 and AQP1 pores. We find that CaM binding influences the collective dynamics of the whole AQP0 tetramer, promoting the closing of both the extracellular and intracellular gates by inducing cooperativity between neighboring subunits.
Keywords: aquaporins, osmotic transport, water permeability, membrane protein, molecular dynamics, calmodulin
Graphical abstract
1. Introduction
The aquaporins (AQPs) are a ubiquitous transmembrane protein family that selectively transport water across membranes. AQPs assemble in tetramers with each monomer forming an independent water pore (see Fig. 1). Water selectivity is provided by a narrow constriction site (CS-I), located on the extracellular side of the protein formed by conserved Arg, His, and Phe side chains that allows single-file water molecules through the pore and provides a steric barrier to larger molecules, as well as an exclusion site for metal ions [1]. Roughly at the center of the channel, two highly conserved Asn-Pro-Ala (NPA) motifs form a proton-exclusion site [1, 2]. Water molecules also interact with pore-lining backbone carbonyls on either side of the NPA motifs. The pore opens to a vestibule on the cytoplasmic side.
Fig. 1.

Detailed view of a single AQP0 subunit embedded in a lipid bilayer highlighting the key amino acid residue side chains involved in the regulation of water permeation. R187 and H172 form the constriction site I (CS-I) on the extracellular side. Y23, close to the center of the pore, may interact with the water single-file. H66 and Y149 together with F75 (not shown) form the constriction site II (CS-II) on the intracellular side. Water molecules within the pore are also shown. Amino acid residue side chains are shown in licorice representation and colored by atom (C, silver; O, red; N, blue; H, white). For reference, lipid carbonyls are shown in yellow as translucent filled-spheres. The transmembrane direction is along the vertical.
AQP0 is expressed almost exclusively in the eye lens where it plays a key role in lens homeostasis. AQP0 transports water at a rate at least 10-fold lower than the prototypical AQP1 [3, 4]. Two Tyr side chains, Y23 and Y149, have been proposed to play a role in the AQP0 slow water conduction rate (see Fig. 1) [5–8]. Y23 is located between CS-I and the NPA motif. Y149 forms a second constriction site (CS-II), with H66 and F75, on the intracellular end of the channel [5, 6].
AQP0 water permeability is modulated by calmodulin (CaM) in a Ca2+-dependent manner [9, 10]. A CaM molecule interacts simultaneously with two AQP0 subunits through binding to their C-terminal helices (calmodulin binding domains, or CBDs), giving rise to a 2:1 CaM:AQP0 tetramer complex [11, 12]. MD simulations suggest that the binding of CaM allosterically affects the dynamics of CS-II, via Y149, leading to the closure of the channel on the intracellular side [11, 13]. Results from Xenopus oocyte swelling assays show that Ca2+ sensitivity is lost in AQP0 variants lacking Y149 [11], and modulation of water permeability in AQP0 by an increase in Ca2+ is cooperative [14]. However, the underlying molecular mechanisms associated with AQP0 regulation by Ca2+-CaM remain poorly understood.
Here we report MD simulations of the AQP0-CaM complex, AQP0, and AQP1 under an osmotic gradient on the microsecond time scale. Our simulations allow for the sampling of extended permeation regimes and the direct characterization of the dynamics of the single-file water chains. Relative water permeability coefficients computed directly from the net number of waters translocated across the membrane are in good agreement with experimental values. We find that the differences in water permeation rates between the systems can be accounted for by changes in the exchange rates between closed and open state configurations. Notably, we find that the CaM allosteric effect appears to propagate throughout the whole channel in a manner that favors the closing of both CS-I and CS-II. The results strongly indicate that CaM complexation with AQP0 may promote cooperativity between neighboring protein subunits, thereby constraining the overall protein conformational dynamics.
2. Methods
2.1. Simulation Systems
The simulation systems consisted of a single aquaporin tetramer embedded in a mixed POPC:PSM:cholesterol bilayer (0.45:0.35:0.20 molar fractions) separating two aqueous media regions (pure water on the extracellular side and ~1.7 M NaCl on the intracellular side). The separation was accomplished under three-dimensional periodic boundary conditions by the introduction of a fixed barrier parallel to the membrane surface located at the end of the simulation cell and composed of dummy atoms interacting weakly with water molecules and ions via a Lennard-Jones potential (see Fig. 2). A similar simulation set up was recently reported by Wambo et al. [15]. The crystal structures of AQP0 [5] and AQP1 [16] (PDB IDs 1YMG and 1J4N, respectively) were used as initial configurations for the aquaporin-only systems. The model of the AQP0-CaM complex reported by Reichow et al. [11] (PDB ID 3J41) was used for the AQP0-CaM system. An additional POPC:PSM:cholesterol bilayer-only system was generated with the same setup.
Figure 2.

The AQP0-CaM simulation system. The AQP0 tetramer is shown in mauve in secondary structure representation. The CaM proteins are shown in green in molecular surface representation. Lipid carbonyls are shown in yellow as filled-spheres. Waters within the membrane hydrocarbon core region are shown in cyan as filled-spheres. The rest of the waters in the system are shown as cyan dots. Lipid molecules are shown as lines colored by atom (C, silver; O, red; N, blue; H, white). Na+ and Cl− ions are shown in orange and blue filled-spheres, respectively. Lipid molecules, ions, and waters outside the membrane hydrocarbon core region are shown in a cut-away view. The dummy atoms forming the panel wall separating the two solutions are shown in silver as filled-spheres. All the other simulation systems have a similar organization.
2.2. MD Simulations
The μs-timescale simulations were performed on Anton 2, a special-purpose computer for MD simulations of biomolecules [17]. The simulations were first run on a conventional high performance cluster using NAMD 2.11 [18]. In the first portion of the equilibration process, the systems were submitted to 1000 steps of conjugate gradient energy minimization followed by at least 10 ns of MD run at constant pressure (1 bar) and constant temperature (310 K) without an osmotic gradient or a fixed wall. The dummy-atom partition was then introduced and salt ions were added to the intracellular bulk solutions, after which the system was run for at least 2 ns before being transferred to Anton. The CHARMM36m force fields was used for proteins [19], the CHARMM36 force field was used for the lipids [20], and the TIP3P model was used for water [21]. The Anton trajectories were computed using a reversible multiple-timestep algorithm (RESPA) [22] to integrate the equations of motion with a time step of 7.5 fs for the long-range non-bonded forces, and 2.5 fs for short-range non-bonded and bonded forces. All bond lengths involving hydrogen atoms were held fixed using the SHAKE algorithm. The Gaussian-based u-series decomposition method was used to compute long-range electrostatic interactions with a 64 × 64 × 64 grid [17]. Distance cutoffs of 9 Å and 8 Å were used for the Lennard-Jones and short-range electrostatic interactions, respectively.
The simulations were performed at constant temperature (310 K) and constant volume using a Nosé-Hoover chain thermostat [23]. The RESPA algorithm and temperature were implemented using the Multigrator scheme [24]. Analyses and visualizations were performed with VMD 1.9.3 [25] and R [26]. Network representations and analyses were performed with the SNA R package [27], and the principal component analyses were performed with the bio3d R package [28].
Single-channel water permeabilities, pf, in AQP systems were calculated as
where N is the net number of waters translocated during the trajectory, T is the trajectory length, Nb and Tb are the corresponding values for a pure bilayer simulation run under the same conditions, ΔC is the ion bulk concentration difference (estimated from the system density profiles), d is the number of protein subunits, and Vw is the water molar volume.
2.3. Graph representations
We used a graph representation to monitor the connectivity of water single files within the AQP subunit pores. A graph is an ordered pair, G = (V,E), comprised of a set of vertices V and a set of edges E. We constructed individual graphs for each protein subunit in every simulation trajectory configuration of each system. The set of vertices included all the water oxygen atoms within a cylindrical region of 12 Å in radius with axis along the transmembrane direction centered between the nitrogen atoms of the two NPA motifs and extended to both membrane surfaces, plus the following pore-lining amino residue atoms in AQP0, all which are within the cylindrical region of interest: polar nitrogen atoms in the R187, H172, and H66 side chains, all heavy atoms in the phenol rings of Y23 and Y149 and in the benzene ring of F75, backbone carboxyl oxygen atoms from G64, A65, H66, G180, A181, and G182, and nitrogen atoms in N68 and N184 (from NPA motifs). A similar set was constructed in AQP1 using the same criteria for corresponding residues. Edges were defined using an interatomic distance cutoff of 3.5 Å between two water oxygens or between water oxygens and protein atoms, which is an estimate of the closest approach. This value was obtained from the average of all the first minimum positions in the corresponding radial distribution functions in each system (3.6 ± 0.1 Å in AQP0 and AQP1, and 3.5 ± 0.1 Å in AQP0-CaM). We reasoned that a single conservative estimate for all three systems would reduce the possibility of generating artifacts when analyzing graph connectivity. Edges between atoms of the same amino acid residue side chain, either bonded or in close proximity, were also added to the graph representation.
2.4. Xenopus oocyte permeability assays
Wild type AQP0 was cloned into a transcription vector (pXbG) containing BglII cloning site flanked by 5’ and 3’ untranslated regions (UTRs) of the Xenopus laevis aeglobin gene driven by the T3 transcription promoter. Oocytes from Xenopus laevis were obtained from Ecocyte (Austin, TX) and injected typically with 10 ng of RNA encoding wild type or R33L AQP0 generated using the mMessage mMachine T3 kit (Ambion/Life Technologies) as described previously [10]. The oocytes were incubated in 100% ND96 (96 mM NaCl 96, 2 mM KCl, 5 mM HEPES, 1.8 mM CaCl2, 1 mM MgCl2, pH 7.5) with the desired test Ca2+ concentration for 5 minutes before the swelling assay. Swelling assays were performed at room temperature (20–21°C) by transferring oocytes from a 200 mOsm to a 70 mOsm (30% (v/v) ND96) solution adjusted to the desired calcium concentration. Water permeability, Pf, was calculated from optical measurements of the increase in cross-sectional area of the oocyte with time in response to diluted ND96 using:
where V is the volume as a function of time, V0 is the initial volume, S0 is the geometric surface area at time 0, Δosm is the osmotic gradient, and Vw is the molar volume of water.
3. Results
We performed multi-μs atomistic MD simulations of AQP0, the AQP0-CaM complex, and AQP1 embedded in a lipid bilayer under an osmotic gradient (hypotonic on the outside, see Fig. 2). The lipid bilayer composition (35% POPC 25% PSM 20% cholesterol) resembles the major components of the oocyte membrane used in the swelling assays [29]. All three AQP systems, as well as a similar pure bilayer system, exhibited net water flow to the hypertonic solution with non-stationary rates (see Fig. S1A). Average permeation rates and total trajectory lengths are reported in Table 1. Analyses were performed over a portion of the trajectory displaying a steady water flow regime for 1 μs in the case of AQP0 and AQP1 and 2 μs in the case of AQP0-CaM, as indicated in Fig. S1B. The single-channel water permeability (pf) values obtained are within the same order of magnitude as reported experimental values [3, 4] (see Table 2), and the corresponding ratios are in good agreement with oocyte swelling assays results (see Fig. 3).
Table 1.
Total simulation trajectory lengths and average permeation rates.
| Simulation System | Total trajectory length (μs) | Average net permeation rate (waters/μs) |
|---|---|---|
| AQP0-CaM | 5.35 | 13 ± 2 |
| AQP0 | 2.63 | 33 ± 2 |
| AQP1 | 2.40 | 122 ± 4 |
| bilayer | 2.40 | 7 ± 1 |
Table 2.
Single-channel osmotic permeability coefficients.
| Simulation System | pf (10−15 cm3/s) | ||
|---|---|---|---|
| This work* | Chandy et al. [3] | Yang and Verkman [4] | |
| AQP0-CaM | 0.7 ± 0.5 | 0.28 | N/A |
| AQP0 | 3.2 ± 0.6 | N/A | 2.5 ± 0.5 |
| AQP1 | 22 ± 1 | 12 | 60 |
Adjusted for temperature differences between experiment and simulation using the activation energies reported by Chandy et al. [3] of 6.9 kcal/mol for AQP0 and 2.4 kcal/mol for AQP1.
Figure 3.

Water channel permeability rate (pf) ratios from simulations compared to corresponding experimental whole-cell permeability rate (Pf) values (see Methods section). The AQP1:AQP0-CaM experimental ratio is from Németh-Cahalan and Hall [9], while the other two experimental ratios are from this work.
Single-file water transport through protein pores is modulated by the polar interactions between the confined water molecules and pore-lining residues [30]. Thus, in order to reveal key mechanistic differences of the water transport through AQP1, AQP0, and the AQP0-CaM complex, we analyzed the dynamics of the networks formed inside the individual channels by water molecules and pore-lining residues. We describe these networks using a graph representation comprising water oxygen atoms, heavy atoms from flexible side chains involved in permeation regulation, and exposed backbone heavy atoms as vertices. Graph edges are defined by a proximity criterion between water-water and water-protein atom pairs, as well as protein-protein atom pairs within a single amino acid residue side chain (see Methods section for details). In selecting amino acid residues for the vertices sets, we took into account the available structural information from simulations and experiments regarding the water-protein interactions. In AQP0 and AQP0-CaM, we included atoms from the side chains of the CS-I and CS-II residues, the Y23 side chain, as well as the backbone carbonyl oxygens exposed to the pore and the Asp nitrogen atoms in the NPA motifs. A similar set was constructed for AQP1, including atoms from the side chains of T159, H76, and L85, which are the positions equivalent to the AQP0 CS-II residues, and the F24 side chain, which is the equivalent position to Y23 in AQP0. Additional details of the graph construction used to model these networks can be found in the Methods section.
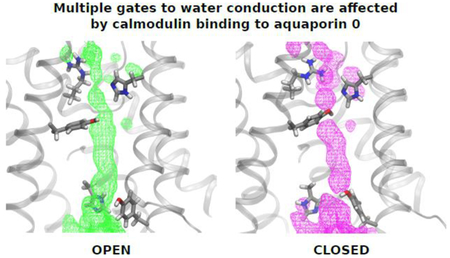
We constructed a graph for each configuration stored in the trajectory (every 240 ps) and determined all the shortest paths between two arbitrary water molecules in the aqueous solutions on either side of the membrane that pass through a single aquaporin pore (see Fig. 4). Configurations exhibiting paths that include only water oxygen atoms or exposed backbone heavy atoms yield uninterrupted water distributions that are associated with fast water translocation and, therefore, can be readily described as open states of the pore (Fig. 5A). Similarly, paths involving heavy atoms from any of the flexible side chains indicated above yield water distributions that are interrupted by these residues (see Figs. 5B–5D).
Figure 4.

Spatial representation of a shortest path in a graph describing a water single file (see Methods section for details on graph construction) connecting the two bulk solutions through an AQP0-CaM pore. In this example, the path involves R187 and H172 nitrogen atoms (CS-I), the A181 carbonyl oxygen, the Y23 hydroxyl oxygen, and the Y149 (CS-II) hydroxyl oxygen. The pore is then considered to be closed at both constriction sites with participation of Y23. Waters oxygen atoms in the set of vertices of the underlying graph are shown as red spheres. The graph edges forming the path are shown as green bonds. Amino acid residue atoms in the graph vertices set that form part of the path are labeled. Amino acid residues without atoms in the path are not shown. Hydrogen atoms were omitted for clarity.
Fig. 5.

Water distributions (as surfaces of equal occupancy) for all the analyzed configurations in the AQP0-CaM trajectory with A) open-state connected paths; B) connected paths involving R187; C) connected paths involving R187 and Y23; D) connected paths involving R187, Y23, and Y149. In all cases the surface corresponding to occupancy of 38% is shown over a configuration snapshot from corresponding set. The R187, H172, Y23, Y149, and H66 are shown in licorice representation and colored by atom (C, silver; O, red; N, blue; H, white).
Most configurations in the AQP0 trajectories included at least one connected path extending from one aqueous medium to the other (see Table 3); a small percentage of configurations without connected paths is to be expected since the diffusive nature of the permeation process through the irregular pore geometry may not always produce instantaneous local configurations that agree with our proximity criterion for the definition of graph edges. In the AQP1 trajectory approximately half of the configurations do not exhibit a fully connected path from one aqueous medium to the other. Visual inspection revealed that the connecting loop between helices H2 and HB (G74-L77) intrudes into the pathway of two subunits blocking the water single-file. This single-channel conformation is not captured by our graph representation since it involves several inter-residue contacts. To our knowledge, this connecting loop has no regulatory role in AQP1. Nevertheless, we find a close correlation between the occurrence of open state configurations, as defined by our graph representation, and the evolution of water translocation in all three systems (see Fig. S1).
Table 3.
Proportion of trajectory configurations exhibiting connected paths
| Simulation System | Proportion of trajectory configurations (%)* |
|---|---|
| AQP0-CaM | 95 |
| AQP0 | 89 |
| AQP1 | 51 |
The number of configurations exhibiting at least one connected path between the opposite aqueous media in the sampled trajectory over the total sampled trajectory length (i.e. 1 μs in AQP0 and AQP1, 2 μs in AQP0-CaM)
Because there are six different amino acid side chains included in our graph representation, the paths can be classified in 64 major categories, or “connected states”, by identifying whether or not they include atoms from any of the residues in the set formed in AQP0 by R187 (CS-I), H172 (CS-I), Y23, F75 (CS-II), H66 (CS-II), and Y149 (CS-II), or the equivalent positions in AQP1. The states observed in each simulation system and their corresponding frequencies expressed as a proportion of the analyzed trajectory, on a per channel basis, are shown in Tables S1 through S3 and are summarized in Fig. 6. Only those connected states with trajectory proportions whose relative errors were less than 0.5 are included. We analyzed all three systems on a per channel basis even though the original structural model of the AQP0-CaM complex is not strictly a homotetramer. As illustrated in Fig. S2, the connected state frequencies of individual subunits of AQP0 are not statistically equivalent in the microsecond timescale of our simulation trajectories. Therefore, when comparing to similar results in the AQP0-CaM system, we cannot assert whether the two different structural environments of the AQP0 subunits induced by CaM result in two distinct functional entities.
Fig. 6.

Trajectory proportions estimates per channel exhibiting a connected path between opposite aqueous media involving side chains heavy atoms in CS-I residues (blue), CS-I and Y23 (cyan, AQP0 only), or CS-I plus CS-II residues with and without Y23 (magenta, AQP0 only). Proportion estimates for water-only single-files, which may include interactions with pore-exposed backbone polar atoms, are shown in green. Connected paths in AQP0 involving CS-II residues only (or equivalent positions in AQP1) or paths involving F24 in AQP1 (equivalent to Y23 in AQP0) had proportion estimates with magnitude comparable to the estimated errors and were not included in the plot.
The most frequent connected states observed in the AQP0 systems can be parsed into four different classes (see Figs. 5 and 6): open states, CS-I closed, CS-I closed plus Y23 block, and fully closed (closed at both CS-I and CS-II), which may include blocking by Y23. In AQP1, the dominant classes are open states and CS-I closed, both of which occur in similar proportions (see Fig. 6). States involving closing at CS-II only in AQP0, or blocking by F24 in AQP1 were observed but with trajectory proportions similar in magnitude to the corresponding error estimates.
We find consistency between the permeability differences between AQP1 and AQP0 (Fig. 3) and our open state trajectory proportion estimates (13.5% for AQP1 and less than 1% in AQP0 systems, see Fig. 6). According to our analysis, the reduction in open state configurations in AQP0 relative to AQP1 can be directly attributed to the interactions with Y23 and CS-II residues, since both AQP1 and AQP0 have similar trajectory proportion estimates for closed states involving only the conserved CS-I. Upon complexation with CaM, there is an increase in the proportion of configurations involving interactions with both CS-I and CS-II. However, the most striking outcome of this analysis is a larger increase in the proportion of configurations involving interactions between the water single-file and CS-I residues over interactions involving CS-II residues. Thus, the presence of CaM affects the dynamics of both CS-II and CS-I, a result consistent with the observed reduction in permeability upon complexation with CaM. In a previous study [11], we identified the effect of CaM binding on CS-II; the effect on CS-I is a novel finding of the present study.
How does CaM affect both gates? We can address this question by contrasting the dynamics of these connected states in the three simulation systems. We consider all the transitions between connected states with a lag of 10 ns, which in most cases is the order of magnitude exhibiting a steady number of transitions (as a function of lag time). These transitions are represented in Fig. 7 as directed graphs and in Figs. S2 through S4 as matrix plots. The corresponding marginal frequencies for the transition end states are also shown in Figs. S2 through S4. Given a single pore in a connected state, this analysis can be used to estimate the likelihood of observing a specific connected state after 10 ns. We consider first the case of AQP1. Fig. 7A (see also Fig. S2A) indicates that the open state is the most likely state to follow any connected state, with approximately 56% of the observed transitions leading to it (Fig. S2B). The AQP1 CS-I closed states, involving R197, H172, or both, can all be reached from the open state. Given that both open and closed states occur with similar proportions along the analyzed simulation trajectory (Fig. 6 and Table S1) we can conclude that the AQP1 channel is mostly open during the simulation trajectory. Connected states involving F24, T159, or H76 cannot be reached from any other state, which is consistent with the fact that these amino acid residue side chains have no identifiable role in the regulation of water transport through AQP1.
Figure 7.

Transition state diagrams for the water single-file connectivity through any water pore. The vertices color scheme is the same as in Fig. 6 with the addition of connected paths involving CS-II residues only (or equivalent positions in AQP1), which are shown in orange. Directed-graph edges indicate transitions between connected states occurring between configurations separated by ~10 ns in any water pore. The thickness of each edge is proportional to the transition frequency.
In contrast, in AQP0, the most frequent configurations of the water single file involve blockage by either R187 (in CS-I), or by both R187 and Y23 (see Fig. 6 and Table S2), but reaching R187 is twice as likely as reaching R187-Y23 (Fig. S3B). Trajectory configurations exhibiting an open state are roughly as frequent as any of the fully closed states (~1% configurations per channel; see Table S2), and fully closed configurations are ten times more likely to be reached than open state configurations (Fig. S3B). Taken together, these results indicate that, in contrast to AQP1, the AQP0 channel is mostly in a closed state during the simulation trajectory. Closed configurations in AQP0 involve either CS-I alone, or CS-I and CS-II, both of which appear to be promoted by Y23.
Upon complexation with CaM, the most dominant connected states still involve R187 and Y23 but, while in AQP0 blocking by R187 and R187-Y23 was equally likely, in AQP0-CaM the most likely state is blocking by R187, which is twice as likely as R187-Y23 (see Fig. 6 and Table S3). Similarly, there is an increase in the proportion of transitions leading to R187 (51% in AQP0 vs. 60% in AQ0-CaM) while the rate for R187-Y23 remains the same (see Fig. S4). Blocking by H172 with and without Y23 were also observed. Simultaneous blocking by CS-I and Y23 confines a single water molecule between the two side chains (Fig. 5C), suggesting a water-mediated polar interaction between the two side chains. In contrast, blocking by Y23 alone suggests that complexation with CaM may also induce a direct interaction of Y23 with the single-file that is at least partly steric. A second feature of the AQP0-CaM system is an overall increase in likelihood, with respect to AQP0, of a water single-file blocked at both CS-I and CS-II through an increase in the frequency and reachability of configurations in which water interacts with H66 at CS-II (see Table S3 and Fig. S4). Finally, we observed no transitions leading to an open state with a lag of 10 ns.
How can the complexation with CaM affect both gates? Although water single files appear with similar frequency in both AQP0 and AQP0-CaM, the frequency of closed state configurations is roughly a 40% higher in AQP0-CaM (see Fig. 6), this suggests that CaM restricts the overall conformational dynamics of AQP0. We explore this notion by contrasting the distributions of the geometric centers of the R187, Y23, and Y149 side chains in AQP0 and AQP0-CaM over the portion of the trajectory with a steady water flux. As shown in Fig. 8, in AQP0-CaM, both Y149 and R187 are restricted to specific conformers, while Y23 has a broader distribution that includes regions closer to the pore center. Thus, on the extracellular side, the spatial restriction of R187 reduces the water-mediated interaction with Y23, which in turn allows a more effective block of the permeation pathway by Y23 alone. A restriction in the motion of R187 will also influence how water molecules reach the extracellular entrance of the channel, since they do so as part of the solvation shell of R187. On the intracellular side, CaM shifts the population of the Y149 distribution at the level of CS-II to positions closer to the center of the pore, where it can be available for more water-mediated interactions with H66, resulting in additional confinement or block of water molecules at CS-II.
Figure 8.

Position distributions for the R187, Y23, and Y149 side chains. The abscissa and ordinate correspond, respectively, to the position along the transmembrane direction and the distance on the membrane plane. Distances are measured with respect to axes parallel to the transmembrane direction passing through each water pore. The origin is located between the Asn nitrogen atoms of the NPA motifs.
Since CaM interacts directly with AQP0 only at the intracellular surface and the CBD helices [11, 13], these results suggest that those interactions may produce stronger correlations in the neighboring subunits throughout the whole channel. In order to test this hypothesis, we performed a principal component analysis (PCA) of the Cα displacements in each protein subunit along the sampled portions of the simulation trajectories. The PCA (reviewed in [31]) reveals the dominant collective modes in each protein subunit, which are represented by the eigenvectors of the covariance matrix of the Cα displacements. The protein conformational dynamics is then described by a vector subspace, with dimension smaller than the total number of degrees of freedom, spanned by the principal components (eigenvectors) carrying most of the displacement variance observed along the simulation trajectory. Therefore, if two protein subunits have similar conformational dynamics, their motion will be described by similar vector subspaces. In order to quantify the overlap between subspaces from different protein subunits, we use a single metric termed the root mean-squared inner product (RMSIP) defined by [32]:
where and are the basis vectors corresponding to two principal component subspaces from different protein subunits, and d is the subspace dimension.
We find a persistently higher overlap between the vector subspaces describing the conformational dynamics of neighboring subunits in AQP0-CaM, with respect to AQP0 and AQP1, up to a subspace dimension accounting for ~90% of the total Cα displacement variance (Fig. 9). This result suggests cooperativity between neighboring AQP0 subunits upon complexation with CaM.
Figure 9.

Root Mean-Squared Inner Product (RMSIP) of the Cα displacements principal component subspaces between neighboring channel sub-units. The largest subspace dimension (10) corresponds to ~90% of the Cα displacements variance in each system.
To assess the effect of the change in collective dynamics in AQP0 upon complexation with CaM on water permeation, we consider how the results of our network analysis compare between neighboring pairs of AQP0 subunits. To that end, we computed the Euclidean distance between vectors formed by the connected state frequencies of each pair of subunits, as well as the Euclidean distance between the connected state transition matrices of each pair of subunits. Each CaM molecule interacts with two AQP0, therefore, when considering neighboring AQP0 subunits in AQP0-CaM, there are two classes of non-overlapping neighboring subunit pairs: those where both subunits interact with the same CaM, and those where each subunit interacts with a different CaM. We find that the Euclidean distances between AQP0 subunit pairs in which both subunits interact with the same CaM molecule are equal to the corresponding distances between pairs formed by subunits interacting with different CaM molecules for both connected state frequency vectors and connected state transition matrices (see Tables S4 and S5). In other words, neighboring pairs of the AQP0 subunits in AQP0-CaM exhibit similar water network dynamics irrespectively of whether the pairs involve a single or both CaM molecules. A similar outcome is not found in AQP0. These results are consistent with the differences in collective dynamics in AQP0 and AQP0-CaM as revealed by the principal component analysis (Fig. 9), and suggest that the restraints imposed by CaM on the AQP0 collective dynamics translate to the water permeation function.
4. Discussion
CaM regulates the water permeability of AQP0 in response to changes in the intracellular Ca2+ concentration. Experimentally validated equilibrium MD simulations, based on the structural model of the AQP0-CaM complex, point to allosteric regulation via changes in the conformational dynamics of the cytoplasmic constriction site (CS-II) upon CaM binding [11, 13]. In particular, the presence of CaM facilitates the formation of a salt-bridge between neighboring AQP0 that restricts the motion of the Y149 side chain to a region where interactions with the water single file are favored [13]. Here, we report simulations on the microsecond timescale under an osmotic gradient, which allowed us to monitor the dynamics of the channel gate under a steady water flow. We find that complexation of AQP0 with CaM results in changes to the conformational dynamics of both CS-II and CS-I, as well as Y23. This effect appears to be the result of a reduction in the collective dynamics of the whole AQP0 tetramer via strong coupling between neighboring subunits and suggests a link between allosteric modulation by CaM and subunit cooperativity [11]. Such a structural mechanism is consistent with the results of Németh-Cahalan et al. [14], showing that in response to high calcium concentration the AQP0 tetramer behaves cooperatively, while in low calcium each subunit acts independently and the response of the tetrameter is additive.
Previous AQP0 simulation studies, all performed on submicrosecond timescales and under hydrostatic equilibrium conditions, used the collective diffusion model of Zhu et al. [33] to compute water permeability coefficients to validate their structural insights [7, 34–37]. The reported values were, overall, in good agreement with the experimental datum of Yang and Verkman [4]. However, the methodology of Zhu et al. [33] relies on computing the diffusion coefficient of a one-dimensional random walk described by a collective variable formed from the displacements of all the waters inside a single channel under equilibrium thermal fluctuations. Because these fluctuations occur on the picosecond timescale, computing this mean-squared displacement in a reliable manner is unfeasible over trajectories that extend over the microsecond timescale. Even if this practical obstacle could be overcome, it is unclear if the underlying model would remain valid as the simulation trajectory samples multiple timescales. We have shown that an osmotic gradient can be modeled using conventional MD simulation methodologies under near-equilibrium conditions over multi-microsecond trajectories, allowing the direct computation of osmotic permeability coefficients that are in good agreement with experimental data.
By contrasting the single-file dynamics between AQP1 and AQP0, we find that the 20- to 40-fold reduction in single-channel water permeability in AQP0 with respect to AQP1 observed in experiments [3, 4] can be accounted for by a complete reversal of the exchange dynamics from a most likely open channel in AQP1 to a most likely closed channel in AQP0.
Equilibrium simulation studies have identified Y23 has a primarily static barrier for permeation [34–36], while Y149 acts as a dynamic gate [11, 34, 36]. In contrast, we observed a more dynamic role for Y23. The most likely connected states in AQP0 single-file configurations involve either R187, or R187 and Y23, and these two connected states interchange with similar rates. Similarly, the closing of CS-II by Y149 is enhanced by the interaction of Y23 with the water single file. Saboe et al. [34] also performed simulations in which the R187, Y23, or Y149 residues side chains were restrained to open or closed configurations. The results of their restrained simulations are in line with our observations, including the fact that, as in AQP1, the key gating element in AQP0 is the arginine side chain in CS-I.
Supplementary Material
Highlights.
Open-closed exchange rates in AQP0 and AQP1 correlate well with permeation rates
CaM binding to AQP0 promotes the closing of the channel at both CS-I and CS-II
AQP0 water permeability reduction upon CaM binding is cooperative
Acknowledgements
This work was supported by National Institutes of Health grant R01 EY005661 (NEI). Anton 2 computer time was provided by the National Center for Multiscale Modeling of Biological Systems at the Pittsburgh Supercomputer Center through grant R01 GM116961 from the National Institutes of Health. The Anton 2 machine at PSC was made available by D.E. Shaw Research. This work also used computing resources at the Texas Advanced Computing Center (TACC) through the Extreme Science and Engineering Discovery Environment (XSEDE) allocation TG-MCB130015. XSEDE is supported by National Science Foundation grant ACI-1548562.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y, Structural determinants of water permeation through aquaporin-1, Nature, 407 (2000) 599–605. [DOI] [PubMed] [Google Scholar]
- [2].de Groot BL, Grubmüller H, The dynamics and energetics of water permeation and proton exclusion in aquaporins, Curr Opin Struct Biol, 15 (2005) 176–183. [DOI] [PubMed] [Google Scholar]
- [3].Chandy G, Zampighi GA, Kreman M, Hall JE, Comparison of the water transporting properties of MIP and AQP1, J Membr Biol, 159 (1997) 29–39. [DOI] [PubMed] [Google Scholar]
- [4].Yang BX, Ma TH, Verkman AS, Water and glycerol permeability of aquaporins 1–5 and MIP determined quantitatively by expression of epitope-tagged constructs in Xenopus oocytes., J Am Soc Nephrol, 7 (1996) A0135–A0135. [DOI] [PubMed] [Google Scholar]
- [5].Harries WE, Akhavan D, Miercke LJ, Khademi S, Stroud RM, The channel architecture of aquaporin 0 at a 2.2-A resolution, Proc Natl Acad Sci U S A, 101 (2004) 14045–14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gonen T, Sliz P, Kistler J, Cheng Y, Walz T, Aquaporin-0 membrane junctions reveal the structure of a closed water pore, Nature, 429 (2004) 193–197. [DOI] [PubMed] [Google Scholar]
- [7].Hashido M, Ikeguchi M, Kidera A, Comparative simulations of aquaporin family: AQP1, AQPZ, AQP0 and GlpF, FEBS Lett, 579 (2005) 5549–5552. [DOI] [PubMed] [Google Scholar]
- [8].Han BG, Guliaev AB, Walian PJ, Jap BK, Water transport in AQP0 aquaporin: molecular dynamics studies, J.Mol.Biol, 360 (2006) 285–296. [DOI] [PubMed] [Google Scholar]
- [9].Nemeth-Cahalan KL, Hall JE, pH and calcium regulate the water permeability of aquaporin 0, J Biol Chem, 275 (2000) 6777–6782. [DOI] [PubMed] [Google Scholar]
- [10].Nemeth-Cahalan KL, Kalman K, Hall JE, Molecular basis of pH and Ca2+ regulation of aquaporin water permeability, J Gen Physiol, 123 (2004) 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Reichow SL, Clemens DM, Freites JA, Nemeth-Cahalan KL, Heyden M, Tobias DJ, Hall JE, Gonen T, Allosteric mechanism of water-channel gating by Ca(2+)-calmodulin, Nat Struct Mol Biol, 20 (2013) 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Reichow SL, Gonen T, Noncanonical binding of calmodulin to aquaporin-0: implications for channel regulation, Structure, 16 (2008) 1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fields JB, Nemeth-Cahalan KL, Freites JA, Vorontsova I, Hall JE, Tobias DJ, Calmodulin Gates Aquaporin 0 Permeability through a Positively Charged Cytoplasmic Loop, J Biol Chem, 292 (2017) 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Németh-Cahalan KL, Clemens DM, Hall JE, Regulation of AQP0 water permeability is enhanced by cooperativity, J Gen Physiol, 141 (2013) 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wambo TO, Rodriguez RA, Chen LY, Computing osmotic permeabilities of aquaporins AQP4, AQP5, and GlpF from near-equilibrium simulations, Biochim Biophys Acta Biomembr, 1859 (2017) 1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sui H, Han BG, Lee JK, Walian P, Jap BK, Structural basis of water-specific transport through the AQP1 water channel, Nature, 414 (2001) 872–878. [DOI] [PubMed] [Google Scholar]
- [17].Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, Forte A, Gagliardo J, Gill G, Greskamp B, Ho CR, Ierardi DJ, Iserovich L, Kuskin JS, Larson RH, Layman T, Lee L, Lerer AK, Li C, Killebrew D, Mackenzie KM, Mok SY, Moraes MA, Mueller R, Nociolo LJ, Peticolas JL, Quan T, Ramot D, Salmon JK, Scarpazza DP, Schafer UB, Siddique N, Snyder CW, Spengler J, Tang PTP, Theobald M, Toma H, Towles B, Vitale B, Wang SC, Young C, Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer, in: SC ‘14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, 2014, pp. 41–53. [Google Scholar]
- [18].Phillips JC, Braun B, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K, Scalable molecular dynamics with NAMD, Journal of Computational Chemistry, 26 (2005) 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, MacKerell AD Jr., CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat Methods, 14 (2017) 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., Pastor RW, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, The journal of physical chemistry. B, 114 (2010) 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, J.Chem.Phys, 79 (1983) 926–935. [Google Scholar]
- [22].Tuckerman M, Berne BJ, Martyna GJ, Reversible multiple time scale molecular dynamics, J.Chem.Phys, 97 (1992) 1990–2001. [Google Scholar]
- [23].Martyna GJ, Klein ML, Tuckerman M, Nosé-Hoover chains: The canonical ensemble via continuous dynamics, J.Chem.Phys, 97 (1992) 2635–2643. [Google Scholar]
- [24].Lippert RA, Predescu C, Ierardi DJ, Mackenzie KM, Eastwood MP, Dror RO, Shaw DE, Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure, The Journal of Chemical Physics, 139 (2013) 164106. [DOI] [PubMed] [Google Scholar]
- [25].Humphrey W, Dalke A, Schulten K, VMD: visual molecular dynamics, J Mol Graph, 14 (1996) 33–38, 27-38. [DOI] [PubMed] [Google Scholar]
- [26].Team RC, R: A language and environment for statistical computing R Foundation for Statistical Computing, in, R Foundation for Statistical Computing, Vienna, Austria, 2016. [Google Scholar]
- [27].Butts CT, Social network analysis with sna, J. Stat. Soft, 24 (2008) 1–51. [Google Scholar]
- [28].Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, Caves LS, Bio3d: an R package for the comparative analysis of protein structures, Bioinformatics, 22 (2006) 2695–2696. [DOI] [PubMed] [Google Scholar]
- [29].Hill WG, Southern NM, MacIver B, Potter E, Apodaca G, Smith CP, Zeidel ML, Isolation and characterization of the Xenopus oocyte plasma membrane: a new method for studying activity of water and solute transporters, American Journal of Physiology-Renal Physiology, 289 (2005) F217–F224. [DOI] [PubMed] [Google Scholar]
- [30].Horner A, Zocher F, Preiner J, Ollinger N, Siligan C, Akimov SA, Pohl P, The mobility of single-file water molecules is governed by the number of H-bonds they may form with channel-lining residues, Sci Adv, 1 (2015) e1400083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].David CC, Jacobs DJ, Principal component analysis: a method for determining the essential dynamics of proteins, Methods Mol Biol, 1084 (2014) 193–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Amadei A, Ceruso MA, Di Nola A, On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins’ molecular dynamics simulations, Proteins, 36 (1999) 419–424. [PubMed] [Google Scholar]
- [33].Zhu F, Tajkhorshid E, Schulten K, Collective diffusion model for water permeation through microscopic channels, Phys Rev Lett, 93 (2004) 224501. [DOI] [PubMed] [Google Scholar]
- [34].Saboe PO, Rapisarda C, Kaptan S, Hsiao YS, Summers SR, De Zorzi R, Dukovski D, Yu J, de Groot BL, Kumar M, Walz T, Role of Pore-Lining Residues in Defining the Rate of Water Conduction by Aquaporin-0, Biophys J, 112 (2017) 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Qiu H, Ma S, Shen R, Guo W, Dynamic and energetic mechanisms for the distinct permeation rate in AQP1 and AQP0, Biochim Biophys Acta, 1798 (2010) 318–326. [DOI] [PubMed] [Google Scholar]
- [36].Jensen MO, Dror RO, Xu H, Borhani DW, Arkin IT, Eastwood MP, Shaw DE, Dynamic control of slow water transport by aquaporin 0: implications for hydration and junction stability in the eye lens, Proc Natl Acad Sci U S A, 105 (2008) 14430–14435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hashido M, Kidera A, Ikeguchi M, Water transport in aquaporins: osmotic permeability matrix analysis of molecular dynamics simulations, Biophys J, 93 (2007) 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.