

Abstract
A broad range of proteins employ nucleotide flipping to recognize specific sites in nucleic acids, including DNA glycosylases, which remove modified nucleobases to initiate base excision repair. Deamination, a pervasive mode of damage, typically generates lesions that are recognized by glycosylases as being foreign to DNA. However, deamination of 5-methylcytosine (mC) generates thymine, a canonical DNA base, presenting a challenge for damage recognition. Nevertheless, repair of mC deamination is important because the resulting G·T mispairs cause C→T transition mutations, and mC is abundant in all three domains of life. Countering this threat are three types of glycosylases that excise thymine from G·T mispairs, including thymine DNA glycosylase (TDG). These enzymes must minimize excision of thymine that is not generated by mC deamination, in A·T pairs and in polymerase-generated G·T mispairs. TDG preferentially removes thymine from DNA contexts in which cytosine methylation is prevalent, including CG and one non-CG site. This remarkable context specificity could be attained through modulation of nucleotide flipping, a reversible step that precedes base excision. We tested this idea using fluorine NMR and DNA containing 2′-fluoro-substituted nucleotides. We find that dT nucleotide flipping depends on DNA context and is efficient only in contexts known to feature cytosine methylation. We also show that a conserved Ala residue limits thymine excision by hindering nucleotide flipping. A linear free energy correlation reveals that TDG attains context specificity for thymine excision through modulation of nucleotide flipping. Our results provide a framework for characterizing nucleotide flipping in nucleic acids using 19F NMR.
Graphical Abstract
INTRODUCTION
Nucleotide flipping is employed by a broad range of proteins to bind specific sites of nucleic acids. Also known as base flipping, this reversible conformational change involves the rotation of one or more nucleotides, by up to 180 degrees, out of the helical stack and into a protein cavity or enzyme active site.1–2 Nucleotide flipping is used by many different types of enzymes that act on DNA or RNA, including restriction endonucleases3 and DNA-modifying enzymes such as cytosine methyltransferases, which convert cytosine to 5-methylcytosine (mC).4 In addition to such “writers” of DNA modifications, nucleotide flipping is employed by many proteins and enzymes that “read” or “erase” DNA modifications.5 Many DNA repair proteins employ nucleotide flipping, including DNA glycosylases and other factors in base excision repair (BER),6–7 the Rad4/XPC protein in nucleotide excision repair,8 and enzymes that perform direct DNA repair such as photolyases and O6-alkylguanine DNA alkyltransferases.9–11
DNA glycosylases use nucleotide flipping to find and remove modified bases from DNA, thereby initiating BER. Deamination is a pervasive type of damage leading to lesions that are processed by DNA glycosylases. While deamination of guanine, adenine, or cytosine generates a lesion that is clearly foreign to DNA, deamination of 5-methylcytosine (mC) produces thymine, one of the four canonical DNA bases. This spontaneous event is mutagenic, because it converts a normal G·mC base pair to a G·T mismatch, a lesion that can generate C→T transitions upon processing by a DNA polymerase.12–13 Protecting against the threat posed by mC deamination are three types of DNA glycosylases that excise thymine from G·T mispairs, one of which is represented by human thymine DNA glycosylase (TDG),14–15 the focus of this work.
Among DNA glycosylases, the G·T mismatch enzymes face a particular challenge in selectively removing the rare thymine bases that arise through mC deamination while not acting on the vast background of “normal” thymine. This is not simply a matter of distinguishing between A·T pairs and G·T mispairs, which is itself a non-trivial and poorly understood feat. Rather, G·T mismatch glycosylases must also avoid acting on G·T mispairs that are generated erroneously by DNA polymerases, because faithful repair of polymerase errors must be directed at the misincorporated nucleotide, which can be dG or dT. Indeed, processing of polymerase-generated G·T mispairs by a mismatch glycosylase could lead to A→G transition mutations, if dG (rather than dT) was incorporated by the polymerase. The question of how G·T mismatch glycosylases attain the specificity to excise thymine arising from mC deamination remains a fundamental problem. It is a question of broad significance given that cytosine methylation is the most abundant DNA modification in the three domains of life, serving as an epigenetic mark in animals and plants and functioning in restriction modification systems of archaea and bacteria.16
In mammals, cytosine methylation occurs predominantly at palindromic CG (or CpG) dinucleotides, producing mCG,17 and mammalian TDG efficiently removes thymine from DNA contexts that are consistent with deamination at mCG sites.18–21 Cytosine methylation also occurs at some non-CG sites, giving mCH or mCHH (where H = G, A, T, C).22 Non-CG methylation, including mCAC and mCAG, is found in embryonic stem cells, induced pluripotent stem cells, oocytes, and neurons of the adult brain.23–25 However, the ability of TDG to protect against mC deamination in most mCHH contexts remains unknown. As such, we used single turnover kinetics experiments to define the activity of TDG for excising thymine from DNA that represents the result of mC deamination in 16 different mCHH contexts. We also investigated a potential role in context specificity for a conserved Gln residue, which forms contacts with bases located on the 3′ side of the flipped nucleotide (dU) in crystal structures of TDG-DNA complexes.26–27
We also sought to determine the mechanism by which TDG attains its remarkable context specificity for thymine excision. Prior studies and results here obtained from single turnover kinetics experiments indicate that specificity could be attained at two steps of the TDG reaction, nucleotide flipping or the subsequent chemical step (base excision). However, efforts to discriminate between these possibilities has been impeded in part by the lack of a robust method to directly monitor dT nucleotide flipping for TDG. Here, we describe an approach to characterize nucleotide flipping using fluorine (19F) NMR and DNA containing a single 2′-fluoro-substituted deoxynucleotide. The 19F NMR spectra provide the relative population of nucleotide in the stacked and flipped states, defining the equilibria and informing on the exchange rate for this reversible conformational change. Using 19F NMR, we characterized nucleotide flipping and defined its role in context-dependent thymine excision by TDG. 19F NMR was also used to directly test the possibility that a conserved Ala residue suppresses thymine excision by hindering the flipping of dT nucleotides into the TDG active site, as suggested by previous structural and biochemical studies.28 Our results provide a general framework for using 19F NMR to characterize nucleotide flipping in free or protein-bound nucleic acids.
RESULTS
Experimental Considerations.
The studies presented here were performed using a construct of human TDG that includes residues 82–308 (TDG82−308), which exhibits substrate binding affinity and glycosylase activity that is equivalent to full length TDG (410 residues) for G·T mispairs and other substrates.26–27 The greater thermal stability and smaller size of TDG82−308 (26 kDa) relative to TDG (46 kDa) was beneficial for collecting single turnover experiments at physiological temperature (37 °C) and enhancing the signal in 19F NMR spectra. TDG activity was defined using single turnover kinetics experiments performed under saturating TDG conditions. The resulting rate constants reflect the maximal rate of product formation (kobs ≈ kmax) and report on steps that include the chemical step and any preceding conformational changes that occur after the initial association of enzyme and substrate, such as nucleotide flipping (Figure 1).29 This avoids complications arising from characterization of TDG activity using multiple-turnover kinetics experiments, which give steady-state rate constants that are dominated by steps after chemistry, due to slow release of the abasic DNA product and potent product inhibition.30–31
Figure 1.
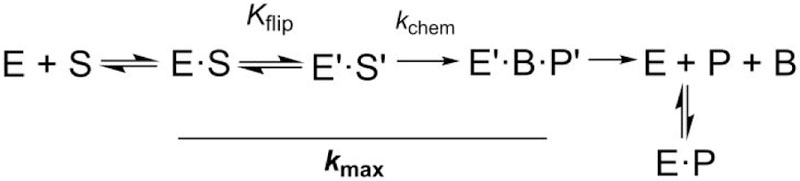
Minimal kinetic mechanism for the TDG reaction. Association of enzyme (E) and DNA substrate (S) gives an initial collision complex (E·S), then nucleotide flipping (Kflip) and potentially other conformational changes give the reactive enzyme-substrate complex (E′·S′). In the chemical step (kchem), cleavage of the N-glycosyl bond and addition of the nucleophile (water) generate the ternary product complex (E′·B·P′, where B is the excised base and P is abasic DNA). Dissociation of E′·B·P′ likely involves rapid release of B and slow release of abasic DNA. The solid line denotes reaction steps that contribute to kmax, the rate constant obtained from the single turnover kinetics experiments.
Effect of 3′-neighboring bases on thymine excision.
We sought to determine the capacity of TDG to protect against mC deamination when cytosine methylation occurs at CG and non-CG sites. There are 16 potential contexts for cytosine methylation when the two 3′-neighboring bases (+1, +2) can vary, and mC deamination would give 16 contexts for a G·T mispair. The nomenclature used here for DNA containing a G·T mispair in different contexts is “G·Txy”, where x and y are the 3′ bases at the +1 and +2 sites, respectively, relative to the mismatched T (Figure 2). For DNA in which only the base at +1 is varied, the nomenclature is G·Tx (or G·Ux).
Figure 2.
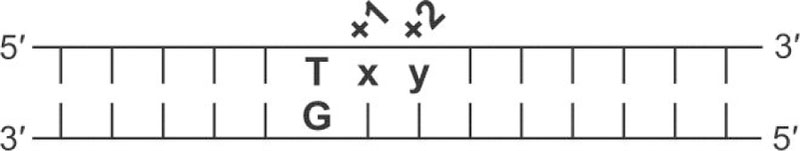
DNA nomenclature. DNA that contains a G·T mispair is referred to as “G·Txy”, where x and y represent bases at the +1 and +2 sites, respectively, relative to the mismatched T. For example, “G·TGT” has G at the +1 and T at the +2 site. DNA for which only the +1 base varies is denoted as G·Tx (or G·Ux). The full DNA sequence is given in Materials and Methods.
Performing single turnover kinetics experiments with 16 G·Txy substrates, we find that thymine excision by TDG is highest for substrates with G at the +1 site (Figure 3, Supporting Information Table S1, Figure S1). Moreover, the dependence of activity on the +1 base gives an overall trend that is independent of the base at the +2 site, where G·TGy > G·TAy > G·TCy > G·TTy, for any given base (y) at +2. This trend is in agreement with previous studies which examined four G·Tx substrates in which only the base at the +1 site was varied.18,21, 32 We find a vast 300-fold difference in activity between substrates that are the most and least efficient (G·TGG, G·TTC), revealing a striking degree of context specificity for a DNA glycosylase. Our results underscore the high specificity of TDG for excising thymine from DNA contexts for which cytosine methylation is most prevalent, including CG sites, as indicated by robust activity for each of four G·TGy substrates.
Figure 3.
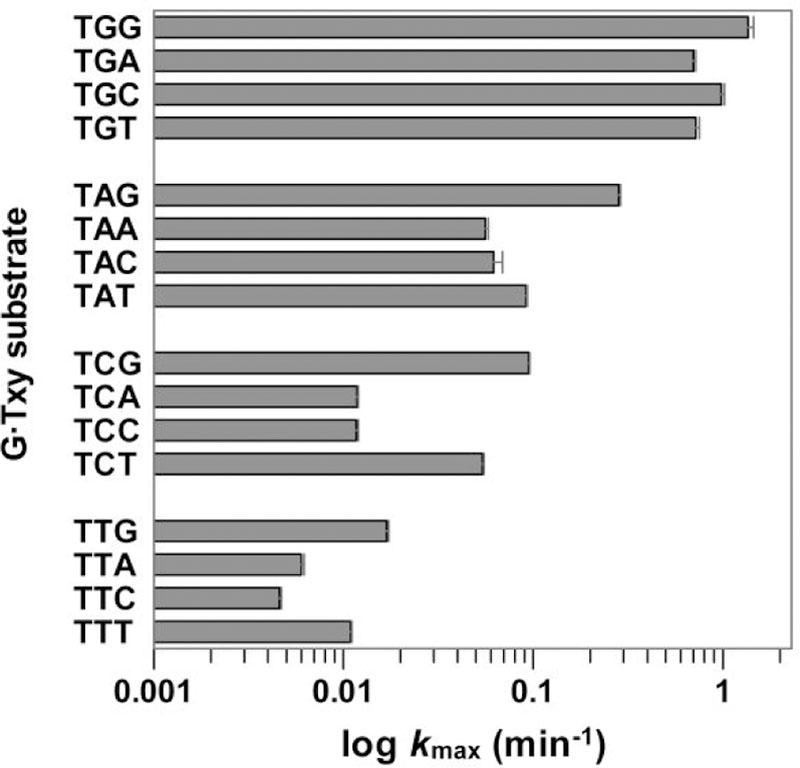
TDG thymine excision activity (log kmax) for G·Txy substrates (abbreviated as Txy) at 37 °C. Substrates are clustered in groups of four, according to the base at +1. Data fitting and kmax values are given in Supporting Information Figure S1, Table S1.
Our results reveal that TDG thymine excision is also modulated by the +2 base, albeit to a lesser extent than for the +1 base (Figure 3). For any given base at +1, the highest thymine excision activity is observed for substrates with G at the +2 site. We note that G·TAG is a remarkably good substrate, much better than the three other G·TAy substrates and nearly as good as the four G·TGy substrates. This is significant because CAG is a prevalent site of non-CG cytosine methylation.23–25 Thymine is the second most preferred base at the +2 site, consistent with structural findings that TDG contacts a thymine base at the +2 site, via the Gln278 side chain, which also contacts guanine at the +1 site (Figure 4).26–27
Figure 4.

Structure of TDG82−308 bound to DNA containing a G·U mispair (PDB ID: 5HF7). Interactions with Gua at the +1 site and Thy at +2 site involve the Q278 side chain and the backbone amide of A277. The dU nucleotide is flipped into the active site and the guanine that it had been paired with (opposing base) is in green. The methyl group of A145 is shown as a sphere, and the dotted line connects this group with C5 of flipped dU (methyl of T is located at C5). No structure has been reported for TDG with dT flipped into the active. Previous findings and results here support a model whereby flipping of dT is partially hindered by a clash between its methyl and that of A145 (see text).
Role of Gln278 in DNA-context specificity.
To investigate the role of Gln278 in the context specificity of TDG, we determined the thymine excision activity of Q278A-TDG using four G·Tx substrates (Figure 5, Supporting Information Table S2, Figure S2). TDG specificity for the 3′ +1 base is diminished by the Q278A mutation, as evidenced by reduced activity for the optimal G·TG substrate and enhanced activity for two of the three other G·Tx substrates. Moreover, the difference in activity between the best and worst substrates is 65-fold for TDG and only 14-fold for Q278A-TDG. Still, the mutation does not alter the qualitative trend in activity imparted by the +1 base. Our finding suggests that TDG specificity for a guanine at the +1 site depends on factors in addition to Gln278, which might include a contact from the backbone nitrogen of Ala277 (Figure 4).26–27 Notably, the glycosylase domain of MBD4 does not contact bases at the +1 or +2 sites, even as thymine excision is modestly dependent on the +1 base (kmax is reduced by <3-fold for G·TA, G·TC, and G·TT relative to G·TG).33 This raises the possibility of inherent differences in the “reactivity” of G·Tx substrates, due perhaps to differential effects of the +1 base on dT flipping.
Figure 5.
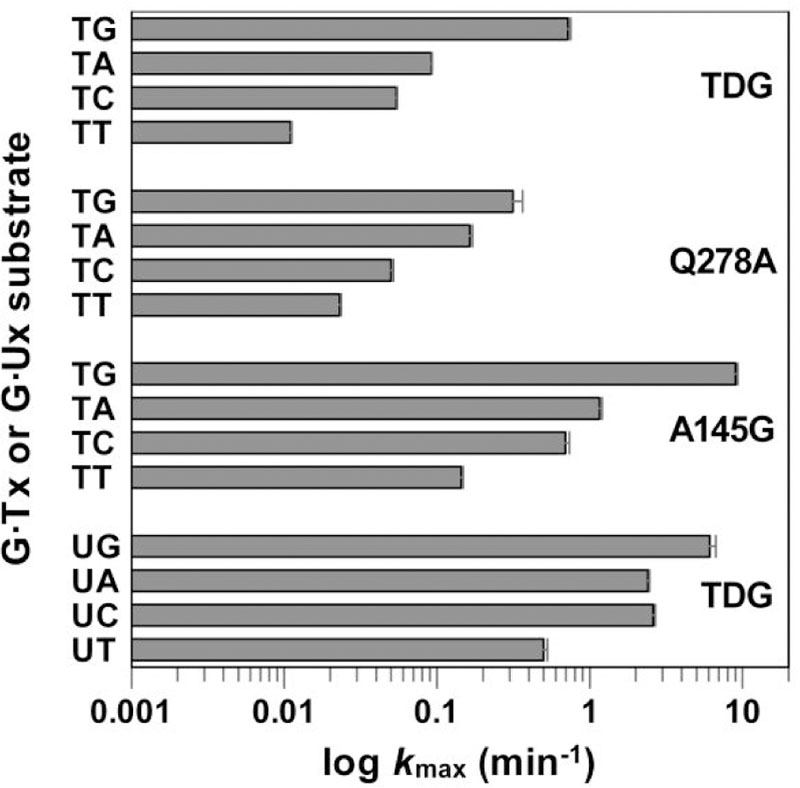
Thymine excision activity (log kmax) of TDG and two variants acting on G·Tx or G·Ux substrates (abbreviated as Tx, Ux) at 37 °C. For all substrates the base at +1 varied and the base at +2 was T (G·TxT, G·UxT). Data fitting and kmax values are given in Supporting Information Figure S2, Table S2.
Does Ala145 impact TDG context specificity?
Previous structural and biochemical studies suggested that Ala145, which is strictly conserved in vertebrate TDG, hinders the flipping of dT nucleotides through a steric clash involving its methyl group with that of dT (Figure 4).28 This idea was supported by findings that the A145G mutation causes a large (13-fold) increase in glycosylase activity for G·T substrates but has no effect on activity for G·U substrates (U lacks the methyl of T).28 Another study showed that the impact of DNA context (3′ +1 base) on uracil excision depends on the size of substituents at C5 of uracil (Figure 4).21 For example, the context effect is much greater for excision of 5-BrU relative to 5-FU (both paired with G). Those prior results are supported by findings here that the DNA context has a much greater impact on excision of T relative to U; activity varies by 65-fold for G·Tx substrates compared to 12-fold for G·Ux (Figure 5, Supporting Information Table S2). These previous findings prompted the question of whether the A145G mutation might reduce the strong dependence of thymine excision on the +1 base (by relieving the steric hindrance to dT flipping). On the contrary, we find that the A145G mutation increases thymine excision activity dramatically but uniformly, by 13-fold for all four G·Tx substrates (Figure 5). Thus, while removing the methyl of T reduces the DNA context specificity of base excision, the same result is not obtained by removing the methyl of Ala145.
Limitations in the results from single turnover kinetics.
Rate constants (kmax) obtained from single turnover experiments can be influenced by multiple steps of the TDG reaction, including nucleotide flipping, the chemical step(s), and any post-flipping conformational changes that may lead to a productive E·S complex (Figure 1). An experimental approach to directly monitor the fraction of nucleotide in the flipped versus stacked conformations could potentially reveal which step of the TDG reaction confers its stringent context specificity for thymine excision. While fluorescent base analogs such as 2-aminopurine are used widely to monitor nucleotide flipping,34 these probes are typically indirect since they are usually positioned adjacent to the nucleotide that is flipped. Moreover, the data can be complicated to interpret and often do not readily give the relative population of the nucleotide in the stacked and flipped conformations (Kflip) for protein-bound DNA. To address this problem, we developed an approach to characterize nucleotide flipping using fluorine NMR.
Characterizing nucleotide flipping by fluorine NMR.
19F NMR is a powerful approach for studying structure and dynamics in biological systems.35 Its power stems from the absence of fluorine in most biomolecules, the 100% natural abundance of 19F, and the hypersensitivity of 19F to its local environment, where the range of chemical shifts is up to 100-fold greater than that observed for corresponding 1H nuclei. Our approach for monitoring nucleotide flipping involves simple 1D NMR experiments, using DNA in which 19F replaces 1H at the 2′ position of a nucleotide. 2′-F-substituted nucleotides are synthesized in two forms, 2′-F-ribo (α) and 2′-F-arabino (β) (Chart 1). The sugar pucker of 2′-F-α nucleotides is compatible with A-form DNA or RNA, while that of 2′-F-β nucleotides is compatible with B-form DNA.26, 36 Our studies employ the 2′-F-β forms exclusively, including 2′-F-β-dT and 2′-F-β-dU, hereafter referred to as dT2′F and dU2′F.
Chart 1.
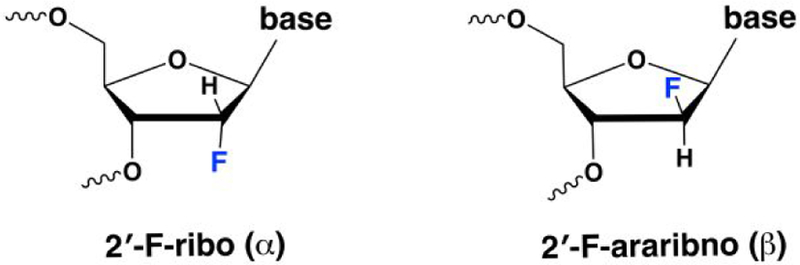
Two types of 2′-F substitutions in deoxynucleotides. Studies here used the 2′-F-arabino (2′-F-β) forms of dT or dU, referred to as dT2′F and dU2′F.
Importantly, 2′-F-β-substituted deoxynucleotides have been used widely in structural and biochemical studies of DNA glycosylases, because the 2′-F substitution precludes hydrolysis of the N-glycosyl bond due to transition-state destabilization.37–40 This enables formation of a stable enzyme-substrate complex, where the 2′-F nucleotide flips into the active site in the absence of bond cleavage, as demonstrated for TDG and many other DNA glycosylases.26–27, 41–43 However, to our knowledge, 2′-F-substituted nucleic acids have not been used for 19F NMR studies of nucleotide flipping. We reasoned that the chemical environment of the fluorine reporter would likely differ substantially for the stacked and flipped conformations of dT2′F (or dU2′F), leading to distinct 19F chemical shifts. The studies below confirm this idea and show that dT2′F and dU2′F are excellent probes of nucleotide flipping using 19F NMR.
We illustrate the 19F NMR method for studying nucleotide flipping through studies of DNA containing dT2′F and dU2′F (Figure 6). We consider initially the spectrum for free DNA containing dT2′F in an A·T base pair, which exhibits a single peak. This is consistent with the expectation that, although A·T pairs exhibit rapid opening and closing, the nucleotides are predominantly stacked in the DNA duplex, as shown by NMR imino exchange studies.36, 44–45 The spectrum for TDG-bound A·T DNA exhibits a single peak at the same chemical shift but with increased linewidth relative to the peak for free DNA. While TDG does not efficiently remove T from A·T pairs it has relatively high affinity for nonspecific DNA, binding with a dissociation constant (Kd <0.3 μM) that is at least 1000-fold below the TDG concentration used in the NMR sample.40, 46 As such, the NMR results are most reasonably explained by nonspecific binding of TDG to the A·T DNA, increasing its overall rotational correlation time and increasing the 19F resonance linewidth for dT2′F while not substantially altering its chemical environment. The peak broadening could also reflect binding of TDG to multiple sites on the DNA, which may cause minor changes to the environment of dT2′F. The absence of a substantial chemical shift difference for dT2′F in free versus TDG-bound A·T DNA suggests that TDG does not stably flip dT from A·T pairs. While novel, this finding is not unexpected given that TDG does not efficiently excise thymine from A·T pairs.
Figure 6.
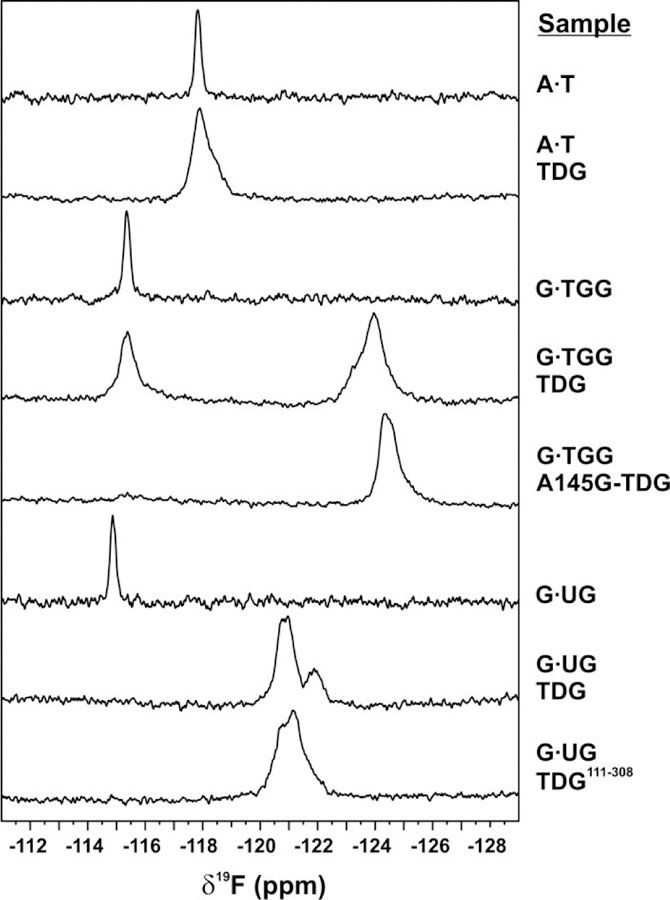
19F NMR spectra for samples of DNA containing dT2′F or dU2′F, in the base pairs as indicated, with or without TDG, collected at 18 °C. Labels (right) indicate the type of DNA and enzyme (if present) in the NMR sample. One sample contained TDG111−308 while others contained the TDG82−308 construct (TDG, A145G-TDG).
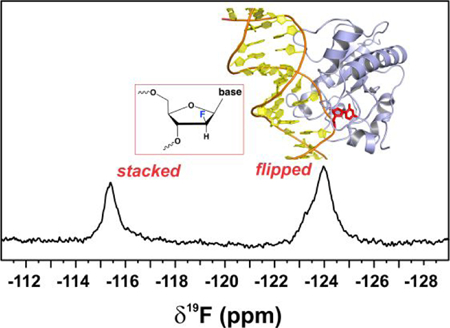
We next consider NMR spectra for free and TDG-bound DNA that contains dT2′F in a G·T mispair (G·TGG; Figure 6). The spectrum for dT2′F in free DNA features a single peak, consistent with the expectation that nucleotides in a G·T mispair, although kinetically and thermodynamically destabilized relative to Watson-Crick pairs, are still predominantly stacked in the DNA duplex (K = 7 × 10−4 for G·T opening), as shown by NMR imino exchange studies.47 The spectrum for TDG-bound G·T DNA exhibits two broad peaks with a large difference in chemical shift (Δδ19F of 8.6 ppm), indicating that the dT2′F nucleotide partitions between two distinct chemical environments. The downfield peak is attributed to the stacked conformation of dT2′F for TDG-bound DNA because it has the same δ19F as the peak for free DNA; the upfield peak (δ19F −124 ppm) is attributed to dT2′F flipped into the TDG active site. Notably, assignment of these peaks to the stacked and flipped states of dT2′F for TDG-bound DNA are supported by the other NMR results described below. The possibility that the NMR sample contains a substantial amount of free DNA is excluded by the presence of TDG at a concentration that is at least 104-fold above the Kd for its binding to G·T DNA.26, 40 Thus, the 19F NMR spectra reveal that a substantial fraction of the mismatched dT nucleotide remains stacked for TDG-bound DNA, even for a G·T mispair in the context that gives maximal thymine excision activity (G·TGG). Similar findings are provided below for G·T mispairs in other DNA contexts.
The spectrum for G·T DNA bound to A145G-TDG features only one significant peak, which resonates slightly upfield from the peak for flipped dT2′F for the same DNA bound to wild-type TDG. This observation and additional findings below support the idea that Ala145 hinders flipping of dT into the TDG active site (Figure 4). Notably, peaks for both the stacked and flipped states of dT2′F are observed in spectra for other G·T DNA bound to A145G-TDG, as shown below (Figure 6).
The 19F NMR spectrum for DNA containing dU2′F in a G·U pair features a single peak (Figure 6), consistent again with the expectation that the nucleotides are predominantly stacked in the duplex, based on NMR imino exchange studies for G·T mispairs (see above).47 The spectrum for TDG-bound G·U DNA exhibits no peak at the expected δ19F for stacked dU2′F but has two broad and overlapping peaks located about 6 ppm upfield of the peak for TDG-free DNA. Together, the spectra indicate that for TDG-bound G·U DNA, the vast majority of dU is flipped, with no evidence for a substantial population of stacked dU. Observation of two overlapping peaks suggests multiple conformations for the flipped state of dU2′F, which is notable because a single conformation for flipped dU2′F is observed in a high-resolution (1.54 Å) crystal structure of the identical complex (TDG82−308, G·U DNA; Figure 4).26 These observations suggest that one of the conformations indicated by 19F NMR predominates in the crystal state. We also collected 19F NMR data for G·U DNA bound to a smaller TDG construct (TDG111−308) that has 30 fewer N-terminal residues and weaker substrate binding relative to the larger TDG82−308 construct.26 The spectrum exhibits a single though broad peak for dU2′F, suggesting multiple conformations for the flipped state and faster conformational exchange relative to TDG82–308.
Equilibria and dynamics of nucleotide flipping.
The 19F NMR spectra provide the equilibrium constant for reversible nucleotide flipping (Kflip), from the ratio of integrals for peaks corresponding to the flipped (IF) and stacked (IS) states (Kflip = IF/IS). For example, the spectrum for G·TGG DNA bound to TDG (Figure 6) yields Kflip of 2.1, while a Kflip of 30 is obtained for the same DNA bound to A145G-TDG. An upper or lower limit for Kflip can be assigned in the absence of a significant peak for the flipped or stacked state, respectively.
The 19F NMR spectra also inform the dynamics of nucleotide flipping. Observation of well resolved peaks for the stacked and flipped conformations of dT2′F indicates that the conformational exchange rate (kex) for these two states is slow relative to the difference in resonance frequency (Δv) for the two peaks (that is, slow on the NMR timescale).48 As such, the NMR spectra yield an upper limit for kex (eq. 1).
| (1) |
Considering the spectrum for G·TGG DNA bound to TDG (Figure 6), the difference in resonance frequency for peaks representing the stacked and flipped conformations of dT2′F (Δv = 4043 Hz; Δδ19F = 8.6 ppm) gives kex <9000 s−1. Similarly, NMR spectra for G·U DNA, free and TDG-bound, suggest that kex for dU flipping is well below 6800 s−1 (assuming δ19F for the stacked conformation of dU is equivalent for free and TDG-bound G·U DNA). As noted above, NMR spectra for G·U DNA bound to TDG82−308 reveal two overlapping peaks, indicating multiple conformations for flipped dU2′F in slow exchange; the difference in resonance frequency (Δv ~431 Hz) indicates kex < 960 s−1. Observation of a broader peak for G·U DNA bound to TDG111−308, with a chemical shift at about the weighted average of the two peaks for TDG82−308, suggests flipped dU samples multiple conformations in fast exchange. Notably, structures of the two TDG constructs (TDG82−308, TDG111−308) bound to G·U DNA reveal differences in the conformation of a catalytic loop (residues 192–204),26 among other moieties, which might account for differences in the dynamics of flipped dU indicated here by 19F NMR.
Dependence of nucleotide flipping on DNA context.
Using this 19F NMR approach we sought to determine the dependence of dT flipping (from a G·T mispair) on the 3′-neighboring bases, examining the four potential bases at +1 and some at the +2 site, for TDG and A145G-TDG (Figure 7), giving a representative look at the 16 G·Txy substrates examined in the activity assays. As noted above, the downfield peaks (near −115.5 ppm) report on the stacked conformation of dT2′F and the upfield peaks (near –124 ppm) report on the flipped state. Results for TDG indicate that a large fraction of dT2′F is flipped for G·TGG and G·TGT while no substantial flipping is observed for G·TAT or G·TTT DNA. Notably, a much higher fraction of dT2′F is flipped for G·TAG relative to G·TAT, demonstrating that the +2 base can impact flipping substantially. Flipping for G·TAG is relevant because CAG is a prominent site of non-CG methylation. Notably, the dependence of nucleotide flipping on DNA context exhibits a trend (G·TGG > G·TGT > G·TAG > G·TAT > G·TTT) that is identical to that observed for the dependence of thymine excision activity (kmax) on DNA context (Figure 3).
Figure 7.
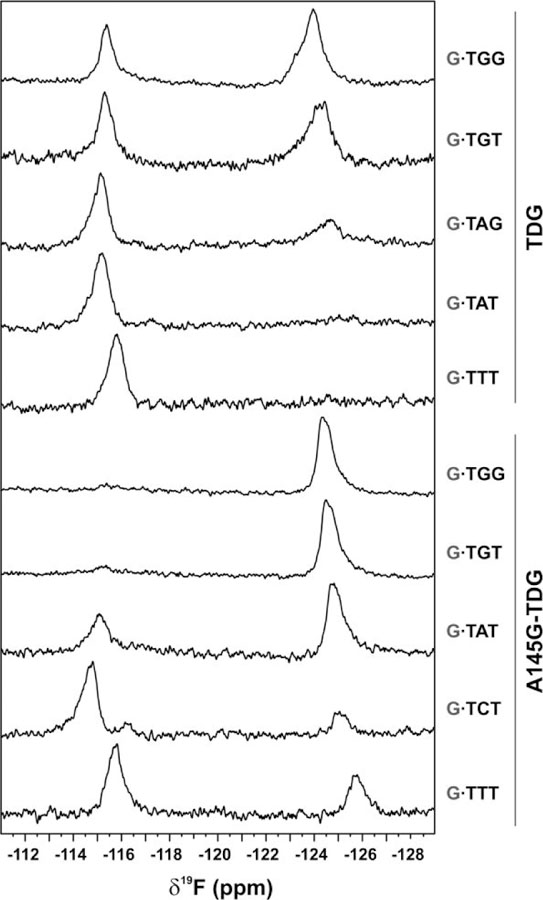
19F NMR spectra for 2′-F-dT in G·Txy DNA, bound to either TDG (top half) or to A145G-TDG (bottom half), collected at 18°C. Downfield peaks (near −115 ppm) reflect the stacked (non-flipped) conformation of 2′-F-dT, while the upfield peaks (near −125 ppm) report on the flipped conformation.
Remarkably, for any given G·Txy DNA, the fraction of dT2′F flipped is much greater for A145G-TDG relative to TDG (Figure 7). Indeed, the vast majority of dT2′F is flipped for G·TGG or G·TGT bound to A145G-TDG, and the fraction dT2′F flipped is relatively high for DNAs contexts that exhibit little or no flipping for wild-type TDG, including G·TAT and G·TTT. This provides the first direct evidence that Ala145 hinders dT flipping, an idea that had been suggested by previous studies.28 As observed for wild-type TDG, we find a strong qualitative correlation between the fraction of dT flipped and thymine excision activity for A145G-TDG.
Linear dependence of glycosylase activity (kmax) on Kflip.
The similar DNA-context dependence observed for dT flipping and thymine excision suggested that a quantitative analysis could be informative. Indeed, we find a linear correlation between log kmax and log Kflip for TDG and for A145G-TDG (Figure 8, Supporting Information Table S3, Figure S3). We note that kmax values used in this analysis were obtained from enzyme activity assays performed at the same temperature as the NMR experiments (18 °C).
Figure 8.

Dependence of thymine excision activity (log kmax) on nucleotide flipping (log Kflip) for TDG or A145G and G·Txy substrates, at 18 °C. Linear fitting gives a slope of m = 1.03 ± 0.05 (r = 0.996) for TDG (●, black line). Data for A145G-TDG (■) were fitted with or without G·TTT (★). Fitting that excludes G·TTT (solid red line) gives a slope of m = 0.97 ± 0.11 (r = 0.988); fitting that includes G·TTT (dotted red line) gives a slope of m = 1.20 ± 0.23 (r = 0.949). Data fitting and kmax and Kflip values are in Supporting Information Table S3, Figure S3.
For TDG, the linear free energy (LFE) correlation has a slope of 1.03 (r = 0.996), indicating that regulation of thymine excision activity by DNA context (3′ bases) is derived through modulation of nucleotide flipping. The results for A145G-TDG reveal a similar LFE correlation, and further analysis suggests that for at least one DNA context, regulation of thymine excision by 3′ bases involves perturbation of a post-flipping step, in addition to modulation of nucleotide flipping. Thus, a fitting that includes all data for A145G-TDG gives a slope of m = 1.20 (r = 0.949; dotted red line) and appears to be skewed by G·TTT (red star). By contrast, fitting that excludes G·TTT gives an improved correlation, with a slope of m = 0.97 (r = 0.988; solid red line). Notably, kmax for G·TTT is ~6-fold lower than predicted by the latter LFE correlation, suggesting that for substrates with thymine at the +1 site (G·TT), thymine excision could be suppressed by a mechanism that acts not only on dT flipping but also on a post-flipping step, which could include a conformational change needed to give a productive E·S complex or the chemical step(s). Notably, fitting for wild-type TDG excluded G·TTT due to the absence of a peak for flipped dT in the 19F NMR spectrum (Figure 7).
The possibility that a post-flipping step may be impaired for substrates with T at the +1 site was also investigated using G·U DNA. We collected 19F NMR spectra for TDG-bound DNA that contained dU2′F in one of four G·Ux pairs (Figure 9, Supporting Information Table S4, Figure S4). Remarkably, the NMR spectra indicate that dU2′F is predominantly flipped into the TDG active site, with little evidence for the stacked state, regardless of the +1 base. While the NMR spectra indicate that the fraction of dU flipped is similar for all G·Ux DNA, uracil excision is dramatically reduced for G·UT relative to the three other G·Ux substrates. Indeed, compared to G·UG, kmax is reduced by 20-fold for G·UT but by less than 3-fold for G·UA and G·UC. Together, our findings suggest that impairment of T (or U) excision by a thymine at the 3′ +1 site involves effects on both nucleotide flipping and on a post-flipping step.
Figure 9.
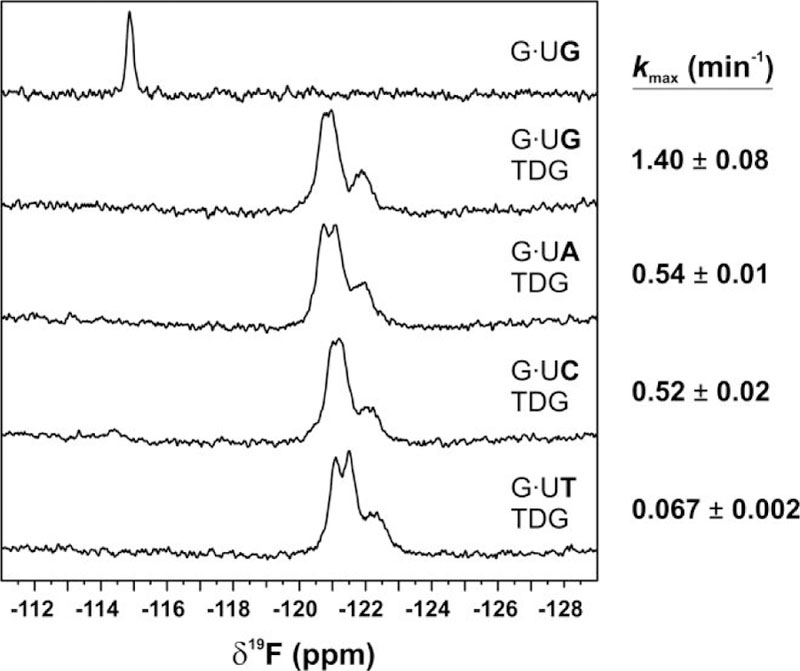
Analysis of dU nucleotide flipping and uracil excision at 18 °C. 19F NMR spectra for dU2′F in G·Ux DNA, free and TDG-bound. Uracil excision activity (kmax) for G·Ux substrates (at 18 °C) is shown to the right of corresponding NMR spectra.
Temperature dependence of nucleotide flipping.
Previous studies and results here reveal that the temperature dependence of TDG activity is far more pronounced for G·T relative to G·U substrates,49 particularly for mispairs in a non-CG context. Indeed, the fold increase in G·T activity at 37 °C relative to 18 °C (kmax37C/kmax18C) is 11, 13, 34, and 44 for G·TG, G·TA, G·TC, and G·TT substrates, respectively (Supporting Information Table S4). The corresponding changes are much smaller for G·U substrates, with kmax37C/kmax18C ratios of 4 to 7 for G·Ux substrates. Notably, the 19F NMR results obtained at 18 °C indicate that for any TDG-bound G·Ux DNA, the dU2′F nucleotide is predominantly flipped, with no substantial peak observed for the stacked state (Figure 9). By contrast, for TDG-bound to the optimal G·TGG DNA, a substantial fraction (~32%) of dT2′F remains stacked (Figure 6). These observations prompted the question of whether the equilibria for dT flipping (Kflip) increases with temperature for TDG-bound G·T DNA, which we investigated using 19F NMR. Indeed, the spectra reveal that the fraction of dT2′F nucleotide in the flipped state increases steadily with temperature, with dT2′F predominantly stacked at 5 °C and largely flipped at 34 °C (Figure 10). The robust temperature-dependent increase in the fraction of dT2′F in the flipped conformation provides a reasonable explanation for the more pronounced temperature dependence of TDG glycosylase activity (kmax) for G·T relative to G·U substrates. In other words, the contribution of nucleotide flipping to the temperature dependence of kmax is greater for G·T relative to G·U substrates.
Figure 10.
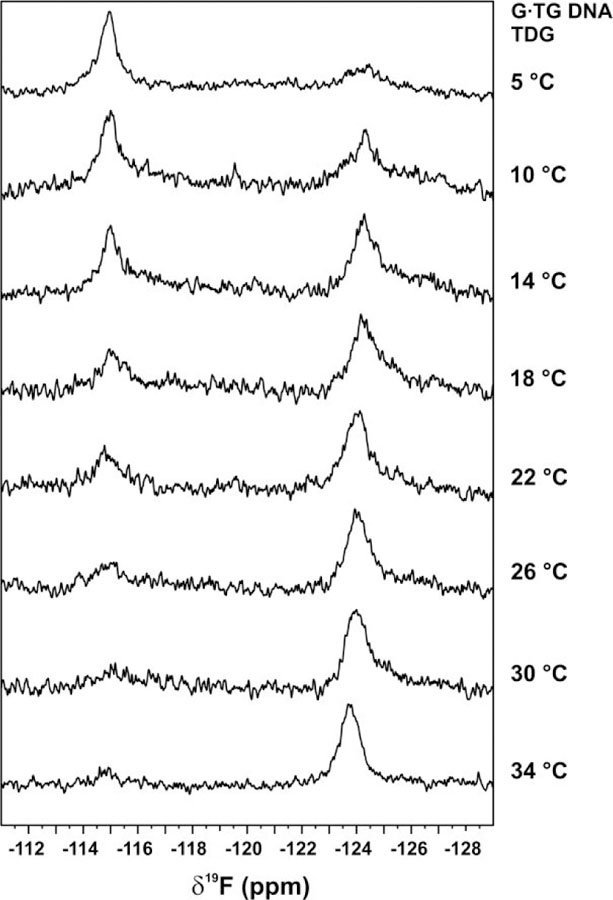
19F NMR spectra for dT2′F in G·TGC DNA (0.15 mM) bound to TDG (0.3 mM) collected with varying temperature. The spectra exhibit two peaks, reporting on the stacked state (downfield) and the flipped state (upfield) for dT2′F.
DISCUSSION
Protection against mC deamination by TDG.
We investigated the capacity of human TDG to protect against mC deamination for 16 possible mCHH contexts, using a series of 16 G·Txy substrates. Our results indicate that TDG thymine excision activity is greatest for G·T mispairs located in a context for which cytosine methylation is most prevalent, including all four CG(H) contexts and CAG, a prominent site for non-CG methylation in embryonic stem cells, induced pluripotent stem cells, and oocytes,23–25 among other possible cell types. By contrast, TDG activity is reduced by 11- to 22-fold for G·TAC relative to the optimal G·TG(H) substrates, which is notable because CAC is a prominent site of non-CG methylation, particularly in the adult mammalian brain.23–25 Our finding raises the question of whether TDG offers substantial protection against mC deamination for mCAC sites. Our results will inform the ability of TDG to protect against mC deamination at other non-CG methylation sites that may be identified in future studies.
The DNA-context specificity of TDG could reflect a biological imperative to maximize its capacity to excise “damaged” thymine bases that arise through mC deamination while minimizing activity on undamaged thymine, in A·T base pairs or G·T mispairs generated by a DNA polymerase. Faithful repair of polymerase-generated G·T mispairs must be directed at the newly incorporated nucleotides (nascent strand), as observed for mismatch repair (MMR). TDG processing of polymerase-generated G·T mispairs could generate an A→G transition mutation if it excises thymine from a G·T mispair for which dG was mistakenly incorporated. TDG is degraded in S phase,50–52 when G·T mispairs generated during bulk DNA replication are expected to arise. Nevertheless, if degradation is incomplete, the context specificity of thymine excision could minimize the activity of any residual TDG on thymine bases that are not generated by mC deamination, including polymerase-generated G·T mispairs. This specificity could also minimize TDG activity on G·T mispairs generated by polymerases functioning outside of DNA replication, in translesion synthesis, DNA repair, or DNA recombination.
The context specificity of TDG might also serve to minimize aberrant thymine excision from the vast background of A·T pairs. Although TDG activity is very weak for A·T pairs at room temperature (kmax = ~1 × 10−5 min−1),21 the activity is likely to be substantially higher at 37 °C. While TDG activity for A·T pairs is too weak to define the effect of DNA context (e.g., 3′ +1 base), the activity is substantial for A·U pairs and, more generally, on A·5xU pairs (x is H, F, Cl, Br).21 TDG activity for A·5xU pairs depends strongly on sequence context (3′ +1 base), giving a trend (A·5xUG > A·5xUA > A·5xUC > A·5xUT) that is identical to that observed here for G·U and G·T mispairs. Thus, the strong context specificity observed for G·T mispairs is also likely to occur for residual activity on the vast background of A·T pairs. This could serve to limit any residual A·T activity to those in an A·TG context.
The robust specificity of TDG for excising thymine from DNA contexts that are prevalent for cytosine methylation, shown here and in previous studies,18–21 indicates that TDG has evolved with the capacity to initiate base excision repair of deaminated mC. However, evidence for this activity in vivo is somewhat limited, due perhaps to experimental challenges posed by the fact that TDG deficiency causes embryonic lethality (in mice),53–54 and that another mammalian DNA glycosylase, methyl binding domain IV (MBD4), also excises thymine from G·T mispairs.55–56 Nevertheless, one study found that depletion of TDG in mammalian cell (MEF) extracts abrogates detectable excision of T from a G·T mispair in DNA, suggesting TDG is the predominant glycosylase acting on deaminated mC (in MEF extracts).53 In another study, a rectal cancer patient with MMR deficiency exhibited elevated C→T mutations at CpG sites (methylated) in tumor suppressor genes, which was attributed to reduced TDG protein levels in the tumor cells (due to a D284Y mutation).57 Two studies found that while depletion of MBD4 causes an increase in C→T transitions at CpG sites, the results also suggested that a factor in addition to MBD4 contributes to repair of deaminated mC.58–59 The other factor is likely to be TDG, since it is the only other mammalian enzyme that has specificity for excising deaminated mC. While these findings collectively suggest that TDG repairs deaminated mC in vivo, additional studies are clearly warranted.
Mechanism of context specificity elucidated by 19F NMR.
Among the 16 G·Txy substrates examined here, we find a vast 300-fold difference in activity (kmax) between the best and worst (G·TGG and G·TTC). This reveals a strikingly high level of DNA-context specificity for a DNA glycosylase. Addressing the fundamental question of how a DNA glycosylase attains such remarkable context specificity necessitated a new approach to directly monitor nucleotide flipping. We developed such an approach using 19F NMR, a powerful probe of conformational change in biological systems. Our method involves simple 1D NMR experiments and samples that feature a single fluorine atom, using DNA containing a 2′-F-β-substituted nucleotide. These nucleotide analogs are fully compatible with B form DNA and resistant to N-glycosyl bond cleavage by DNA glycosylases. Our 19F NMR results show that 2′-F-dT and -dU are outstanding probes of TDG-mediated nucleotide flipping. The NMR spectra reveal well resolved peaks corresponding to the stacked and flipped conformations of the nucleotide, providing the relative population in each state. As such, the results provide the equilibrium constant for nucleotide flipping and an upper limit for the exchange rate (kex).
Using this 19F NMR approach we show that dT flipping from a G·T mispair is strongly dependent on 3′-neighboring bases (+1, +2), for TDG and A145G-TDG. Our results provide the first direct evidence that TDG attains its stringent DNA context specificity for thymine excision by modulating the equilibria for dT flipping (Kflip). More broadly, our findings constitute the first direct evidence, to our knowledge, that a glycosylase attains specificity for acting on a particular DNA context through modulation of nucleotide flipping (Kflip).
We observe a linear free energy (LFE) correlation for the dependence of thymine excision activity (log kmax) on dT nucleotide flipping (log Kflip) by TDG. The slope of unity indicates that the regulation of thymine excision by DNA context (3′ +1, +2 base) is attained through modulation of nucleotide flipping. The results also indicate that for G·T (or G·U) substrates with thymine at the +1 site (e.g., G·TTy, G·UTy), base excision activity (kmax) is reduced by a post-flipping mechanism that acts in addition to effects on nucleotide flipping. This could involve a mechanism that perturbs the formation of a productive enzyme-substrate complex and/or an effect on the chemical step(s) of the reaction.
Ala145 hinders dT flipping.
The results of our 19F NMR studies provide the first direct evidence that Ala145 hinders flipping of dT into the TDG active site, an idea that had been suggested by previous biochemical and structural studies.28 Indeed, Kflip is 9- to 14-fold higher for A145G-TDG relative to TDG for a G·T mispair in several DNA contexts (Supporting Information Table S3). Moreover, thymine excision is much (13-fold) faster for A145G-TDG versus TDG, for G·T mispairs in a CG context and in the three non-CG contexts examined here. Notably, flipping appears to be somewhat more productive for A145G-TDG relative to TDG, as indicated by the observation in the LFE correlations that for a given degree of flipping (Kflip), thymine excision activity (kmax) is about twofold higher for A145G-TDG. This suggests that Ala145 adversely impacts a step after nucleotide flipping, perhaps by hindering formation of a productive E·S complex or by perturbing the chemical step. These observations are important given that Ala145 is strictly conserved in vertebrate TDG. Previous studies suggest that Ala145 serves to minimize excision of thymine from A·T pairs,28 even as it greatly reduces TDG activity on G·T mispairs and thus its capacity to protect against mutations caused by mC deamination. As noted previously, the compromised G·T activity of TDG offers a reasonable explanation for the high frequency of C→T transition mutations at CG dinucleotides in cancer and genetic disease.19, 60–62
19F NMR to study nucleotide flipping in other proteins.
We anticipate that the 19F NMR studies presented here will provide a general approach to monitor nucleotide flipping by proteins that use this ubiquitous mechanism to bind specific sites of nucleic acids. Nucleotide flipping is employed by a broad range of proteins that perform a variety of functions and are found in all three domains of life. Two previous studies employed 19F NMR to characterize nucleotide flipping, for a cytosine methyltransferase and uracil DNA glycosylase.63–64 However, for these studies the 19F substitution was in the nucleobase, which provides a useful probe of flipping but can substantially perturb protein interactions and thereby alter the equilibria for flipping. Our approach, using 2′-F-substituted nucleotides in DNA can, in principle, be employed for any canonical DNA or RNA nucleotide, or analogues thereof. In practice, the utility of our approach for a given system will depend on factors including the amenability of the protein to NMR studies (solubility, etc.), and the availability of a phosphoramidite to synthesize oligonucleotides containing the desired 2′-F-substitution. We note that phosphoramidites for the 2′-F-substituted forms of the four canonical deoxynucleotides in DNA are commercially available and synthesis of phosphoramidites for 2′-F-substituted nucleotides has been described for several modified nucleotides, including 5-formyl-dC, 5-carboxyl-dC, and N7-methyl-dG.27, 65–66
MATERIALS AND METHODS
Materials.
TDG82−308 was expressed and purified as described.26 The expression vector for A145G-TDG82−308 was generated via site-directed mutagenesis using the Quickchange II system (Agilent Technologies), as described,67 and the expression vector for Q278A-TDG82−308 was obtained from ATUM (Newark, CA). The variant enzymes were expressed and purified as described for wild-type TDG82−308.26 Purity of the enzymes was >99% as assessed by SDS-PAGE with Coomassie staining. Enzyme concentration was determined by absorbance at 280 nm,40, 68 using an extinction coefficient of ε280 = 17.4 mM−1cm−1 (for TDG82−308 and its variants).69
Oligodeoxynucleotides (ODNs) were obtained from IDT or the Keck Foundation Biotechnology Resource Laboratory of Yale University. ODNs containing the 2′-fluoroarabino analogs of deoxyuridine or deoxythymidine were synthesized at Yale using phosphoramidites obtained from Glen Research or Link Technologies.67 TDG binds productively to DNA containing these analogs, which are fully resistant to N-glycosyl bond cleavage because the single-atom fluorine substitution destabilizes the chemical transition-state of the reaction.37, 40–41, 67 ODNs were purified by reverse phase HPLC using an XBridge OST C18 column (Waters Corp.), mobile phases that contained 0.1 M TEAA pH 7.0 and either 5% (A) or 15% (B) acetonitrile, a flow rate of 3.5 ml/min, and a gradient of 35–60% B over 19 min.70 Purified ODNs were dried in a vacuum concentrator and exchanged into 0.01 M Tris–HCl pH 8, 0.05 M NaCl, 1 mM EDTA; the ODN concentration was determined by absorbance.21 The 28 bp duplex DNA was made by mixing a target strand (5′-ACCAGTCCATCGCTCA XxyACAGAGCTG; X = T, U, dT2′F, or dU2′F, x and y are any of the four DNA bases) and its complement, heating to 80 °C, and slowly cooling to room temperature.
Glycosylase assays.
Glycosylase activity was monitored using single turnover kinetics experiments performed under saturating enzyme conditions ([E] > [S], [E] >> Kd), giving rate constants that are not impacted by enzyme-substrate association or by product release or product inhibition and thereby reflect the maximal rate of product formation (kobs ≈ kmax).29 Reactions were initiated by adding enzyme to DNA substrate (0.5 µM) in HEN.1 buffer (0.02 M HEPES pH 7.5, 0.1 M NaCl, 0.2 mM EDTA), at 37 °C (unless noted otherwise). Aliquots were removed and added to 50% (v:v) quench solution (0.3 M NaOH, 0.03 M EDTA) to immediately halt the reaction, and samples were heated for 5 min at 85 °C to quantitatively cleave the DNA backbone at abasic sites. The resulting DNA fragments were resolved by HPLC and peak integrals were used to determine fraction product.21, 29 Rate constants were determined from fitting progress curves (fraction product versus time) to eq. 2 using non-linear regression:
| (2) |
where A is the amplitude, kobs is the rate constant, and t is reaction time. The presence of saturating enzyme ([E] >> Kd) in the single turnover experiments was confirmed by observation that the rate constants (kobs) were the same (within experimental error) for multiple enzyme concentrations (typically, 1 μM, 2 μM). Previous studies show that TDG and TDG82−308 bind tightly to G·T DNA substrates (Kd < 0.02 μM).26, 40
19F NMR Spectroscopy.
Fluorine NMR experiments were performed on a Varian 500 MHz spectrometer (470.13 MHz for 19F) equipped with four channels, a Z-axis gradient, and a 5 mm HFCN probe (optimized for 1H, 19F, 13C and 15N). 19F NMR experiments were collected with 8192 complex points, an acquisition time of 0.66 s, a relaxation delay of 1.0 s, a carrier frequency of −119 ppm relative to δ19F for trifluoroacetic acid (TFA), and at 18 °C (unless stated otherwise). 19F NMR spectra were collected with 1000–2000 scans for free DNA and 7,700–25,000 scans for protein-DNA complexes. The data were processed by applying exponential multiplication with 25 Hz line broadening prior to Fourier transformation, using NMRpipe.71 The observed 19F chemical shifts (δ19F) are relative to TFA (external). The samples contained ~0.2 mM DNA and 0.3 mM enzyme (TDG82−308 or A145G-TDG82−308). Enzyme-free samples contained 0.05–0.1 mM DNA. All samples were in a buffer consisting of 15 mM Tris-HCl pH 7.5, 0.1 M NaCl, 10% D2O.
Supplementary Material
ACKNOWLEDGMENT
We thank Kellie Hom for collecting NMR experiments. The studies were supported by the University of Maryland School of Pharmacy NMR Facility, and by a grant from the National Institutes of Health (NIH), R01-GM072711 (to A.C.D.).
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Hong S; Cheng X, DNA Base Flipping: A General Mechanism for Writing, Reading, and Erasing DNA Modifications. Advances in experimental medicine and biology 2016, 945, 321–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stivers JT, Site-specific DNA damage recognition by enzyme-induced base flipping. Prog Nucleic Acid Res Mol Biol 2004, 77, 37–65. [DOI] [PubMed] [Google Scholar]
- 3.Bochtler M; Szczepanowski RH; Tamulaitis G; Grazulis S; Czapinska H; Manakova E; Siksnys V, Nucleotide flips determine the specificity of the Ecl18kI restriction endonuclease. EMBO J 2006, 25 (10), 2219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klimasauskas S; Kumar S; Roberts RJ; Cheng X, HhaI methyltransferase flips its target base out of the DNA helix. Cell 1994, 76 (2), 357–69. [DOI] [PubMed] [Google Scholar]
- 5.Drohat AC; Coey CT, Role of Base Excision “Repair” Enzymes in Erasing Epigenetic Marks from DNA. Chemical reviews 2016, 116 (20), 12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slupphaug G; Mol CD; Kavli B; Arvai AS; Krokan HE; Tainer JA, A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature 1996, 384 (6604), 87–92. [DOI] [PubMed] [Google Scholar]
- 7.Yamagata Y; Kato M; Odawara K; Tokuno Y; Nakashima Y; Matsushima N; Yasumura K; Tomita K; Ihara K; Fujii Y; Nakabeppu Y; Sekiguchi M; Fujii S, Three-dimensional structure of a DNA repair enzyme, 3-methyladenine DNA glycosylase II, from Escherichia coli. Cell 1996, 86 (2), 311–9. [DOI] [PubMed] [Google Scholar]
- 8.Min JH; Pavletich NP, Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449 (7162), 570–5. [DOI] [PubMed] [Google Scholar]
- 9.Daniels DS; Woo TT; Luu KX; Noll DM; Clarke ND; Pegg AE; Tainer JA, DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nature structural & molecular biology 2004, 11 (8), 714–20. [DOI] [PubMed] [Google Scholar]
- 10.Park HW; Kim ST; Sancar A; Deisenhofer J, Crystal structure of DNA photolyase from Escherichia coli. Science 1995, 268 (5219), 1866–72. [DOI] [PubMed] [Google Scholar]
- 11.Mees A; Klar T; Gnau P; Hennecke U; Eker AP; Carell T; Essen LO, Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 2004, 306 (5702), 1789–93. [DOI] [PubMed] [Google Scholar]
- 12.Klose RJ; Bird AP, Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 2006, 31 (2), 89–97. [DOI] [PubMed] [Google Scholar]
- 13.Deaton AM; Bird A, CpG islands and the regulation of transcription. Genes Dev 2011, 25 (10), 1010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neddermann P; Jiricny J, The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J Biol Chem 1993, 268 (28), 21218–24. [PubMed] [Google Scholar]
- 15.Neddermann P; Gallinari P; Lettieri T; Schmid D; Truong O; Hsuan JJ; Wiebauer K; Jiricny J, Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem 1996, 271 (22), 12767–12774. [DOI] [PubMed] [Google Scholar]
- 16.Nabel CS; Manning SA; Kohli RM, The Curious Chemical Biology of Cytosine: Deamination, Methylation,and Oxidation as Modulators of Genomic Potential. ACS Chem. Biol 2012, 7, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bird A, DNA methylation patterns and epigenetic memory. Genes Dev 2002, 16 (1), 6–21. [DOI] [PubMed] [Google Scholar]
- 18.Sibghat U; Gallinari P; Xu YZ; Goodman MF; Bloom LB; Jiricny J; Day RS 3rd, Base analog and neighboring base effects on substrate specificity of recombinant human G:T mismatch-specific thymine DNA-glycosylase. Biochemistry 1996, 35 (39), 12926–32. [DOI] [PubMed] [Google Scholar]
- 19.Waters TR; Swann PF, Thymine-DNA glycosylase and G to A transition mutations at CpG sites. Mutat. Res 2000, 462 (2–3), 137–147. [DOI] [PubMed] [Google Scholar]
- 20.Abu M; Waters TR, The main role of human thymine-DNA glycosylase is removal of thymine produced by deamination of 5-methylcytosine and not removal of ethenocytosine. J Biol Chem 2003, 278 (10), 8739–8744. [DOI] [PubMed] [Google Scholar]
- 21.Morgan MT; Bennett MT; Drohat AC, Excision of 5-halogenated uracils by human thymine DNA glycosylase: Robust activity for DNA contexts other than CpG. J Biol Chem 2007, 282 (38), 27578–27586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Y; Ecker JR, Non-CG Methylation in the Human Genome. Annu Rev Genomics Hum Genet 2015, 16, 55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lister R; Mukamel EA; Nery JR; Urich M; Puddifoot CA; Johnson ND; Lucero J; Huang Y; Dwork AJ; Schultz MD; Yu M; Tonti-Filippini J; Heyn H; Hu S; Wu JC; Rao A; Esteller M; He C; Haghighi FG; Sejnowski TJ; Behrens MM; Ecker JR, Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341 (6146), 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo JU; Su Y; Shin JH; Shin J; Li H; Xie B; Zhong C; Hu S; Le T; Fan G; Zhu H; Chang Q; Gao Y; Ming GL; Song H, Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nature neuroscience 2014, 17 (2), 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie W; Barr CL; Kim A; Yue F; Lee AY; Eubanks J; Dempster EL; Ren B, Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148 (4), 816–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coey CT; Malik SS; Pidugu LS; Varney KM; Pozharski E; Drohat AC, Structural basis of damage recognition by thymine DNA glycosylase: Key roles for N-terminal residues. Nucleic Acids Res 2016, 44 (21), 10248–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pidugu LS; Flowers JW; Coey CT; Pozharski E; Greenberg MM; Drohat AC, Structural Basis for Excision of 5- Formylcytosine by Thymine DNA Glycosylase. Biochemistry 2016, 55 (45), 6205–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiti A; Noon MS; Mackerell AD Jr.; Pozharski E; Drohat AC, Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc Natl Acad Sci U S A 2012, 109 (21), 8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coey CT; Drohat AC, Kinetic Methods for Studying DNA Glycosylases Functioning in Base Excision Repair. Methods in enzymology 2017, 592, 357–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waters TR; Gallinari P; Jiricny J; Swann PF, Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem 1999, 274 (1), 67–74. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald ME; Drohat AC, Coordinating the Initial Steps of Base Excision Repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J Biol Chem 2008, 283 (47), 32680–32690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waters TR; Swann PF, Kinetics of the action of thymine DNA glycosylase. J Biol Chem 1998, 273 (32), 20007–20014. [DOI] [PubMed] [Google Scholar]
- 33.Manvilla BA; Maiti A; Begley MC; Toth EA; Drohat AC, Crystal Structure of Human Methyl-Binding Domain IV Glycosylase Bound to Abasic DNA. J Mol Biol 2012, 420, 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones AC; Neely RK, 2-Aminopurine as a fluorescent probe of DNA conformation and the DNA-enzyme interface. Q Rev Biophys 2015, 48 (2), 244–79. [DOI] [PubMed] [Google Scholar]
- 35.Kitevski-LeBlanc JL; Prosser RS, Current applications of 19F NMR to studies of protein structure and dynamics. Progress in nuclear magnetic resonance spectroscopy 2012, 62, 1–33. [DOI] [PubMed] [Google Scholar]
- 36.Berger I; Tereshko V; Ikeda H; Marquez V; Egli M, Crystal structures of B-DNA with incorporated 2’-deoxy-2’-fluoro-arabino-furanosyl thymines: implications of conformational preorganization for duplex stability. Nucl. Acids. Res 1998, 26 (10), 2473–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scharer OD; Kawate T; Gallinari P; Jiricny J; Verdine GL, Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc Natl Acad Sci U S A 1997, 94 (10), 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stivers JT; Pankiewicz KW; Watanabe KA, Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry 1999, 38 (3), 952–63. [DOI] [PubMed] [Google Scholar]
- 39.Chepanoske CL; Porello SL; Fujiwara T; Sugiyama H; David SS, Substrate recognition by Escherichia coli MutY using substrate analogs. Nucleic Acids Res 1999, 27 (15), 3197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan MT; Maiti A; Fitzgerald ME; Drohat AC, Stoichiometry and affinity for thymine DNA glycosylase binding to specific and nonspecific DNA. Nucleic Acids Res 2011, 39 (6), 2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrett TE; Scharer OD; Savva R; Brown T; Jiricny J; Verdine GL; Pearl LH, Crystal structure of a thwarted mismatch glycosylase DNA repair complex. EMBO J 1999, 18 (23), 6599–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L; Lu X; Lu J; Liang H; Dai Q; Xu GL; Luo C; Jiang H; He C, Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol 2012, 8 (4), 328–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S; Verdine GL, Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc Natl Acad Sci U S A 2009, 106 (44), 18497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leroy JL; Kochoyan M; Huynh-Dinh T; Gueron M, Characterization of base-pair opening in deoxynucleotide duplexes using catalyzed exchange of the imino proton. J Mol Biol 1988, 200 (2), 223–38. [DOI] [PubMed] [Google Scholar]
- 45.Lemkul JA; Savelyev A; MacKerell AD Jr., Induced Polarization Influences the Fundamental Forces in DNA Base Flipping. J Phys Chem Lett 2014, 5 (12), 2077–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennett MT; Rodgers MT; Hebert AS; Ruslander LE; Eisele L; Drohat AC, Specificity of Human Thymine DNA Glycosylase Depends on N-Glycosidic Bond Stability. J. Am. Chem. Soc 2006, 128 (38), 12510–12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moe JG; Russu IM, Kinetics and energetics of base-pair opening in 5’-d(CGCGAATTCGCG)-3’ and a substituted dodecamer containing G.T mismatches. Biochemistry 1992, 31 (36), 8421–8. [DOI] [PubMed] [Google Scholar]
- 48.Wuthrich K, NMR of Proteins and Nucleic Acids John Wiley & Sons, Inc.: New York, NY, 1986. [Google Scholar]
- 49.Maiti A; Drohat AC, Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and processing of G.T mispairs. DNA Repair 2011, 10 (5), 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardeland U; Kunz C; Focke F; Szadkowski M; Schar P, Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res 2007, 35 (11), 3859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slenn TJ; Morris B; Havens CG; Freeman RM Jr.; Takahashi TS; Walter JC, Thymine DNA Glycosylase is a CRL4Cdt2 Substrate. J Biol Chem 2014, 289 (33), 23043–23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shibata E; Dar A; Dutta A, CRL4Cdt2 E3 Ubiquitin Ligase and PCNA Cooperate to Degrade Thymine DNA Glycosylase in S-phase. J Biol Chem 2014, 289 (33), 23056–23064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cortellino S; Xu J; Sannai M; Moore R; Caretti E; Cigliano A; Le Coz M; Devarajan K; Wessels A; Soprano D; Abramowitz LK; Bartolomei MS; Rambow F; Bassi MR; Bruno T; Fanciulli M; Renner C; Klein-Szanto AJ; Matsumoto Y; Kobi D; Davidson I; Alberti C; Larue L; Bellacosa A, Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146 (1), 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cortazar D; Kunz C; Selfridge J; Lettieri T; Saito Y; Macdougall E; Wirz A; Schuermann D; Jacobs AL; Siegrist F; Steinacher R; Jiricny J; Bird A; Schar P, Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470 (7334), 419–423. [DOI] [PubMed] [Google Scholar]
- 55.Hendrich B; Hardeland U; Ng HH; Jiricny J; Bird A, The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 1999, 401 (6750), 301–304. [DOI] [PubMed] [Google Scholar]
- 56.Bellacosa A; Cicchillitti L; Schepis F; Riccio A; Yeung AT; Matsumoto Y; Golemis EA; Genuardi M; Neri G, MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A 1999, 96 (7), 3969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vasovcak P; Krepelova A; Menigatti M; Puchmajerova A; Skapa P; Augustinakova A; Amann G; Wernstedt A; Jiricny J; Marra G; Wimmer K, Unique mutational profile associated with a loss of TDG expression in the rectal cancer of a patient with a constitutional PMS2 deficiency. DNA Repair (Amst) 2012, 11 (7), 616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Millar CB; Guy J; Sansom OJ; Selfridge J; MacDougall E; Hendrich B; Keightley PD; Bishop SM; Clarke AR; Bird A, Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 2002, 297 (5580), 403–405. [DOI] [PubMed] [Google Scholar]
- 59.Wong E; Yang K; Kuraguchi M; Werling U; Avdievich E; Fan K; Fazzari M; Jin B; Brown AM; Lipkin M; Edelmann W, Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci U S A 2002, 99 (23), 14937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rideout WM 3rd; Coetzee GA; Olumi AF; Jones PA, 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990, 249 (4974), 1288–1290. [DOI] [PubMed] [Google Scholar]
- 61.Cooper DN; Youssoufian H, The CpG dinucleotide and human genetic disease. Hum Genet 1988, 78 (2), 151–155. [DOI] [PubMed] [Google Scholar]
- 62.Pfeifer GP; Besaratinia A, Mutational spectra of human cancer. Hum Genet 2009, 125 (5–6), 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klimasauskas S; Szyperski T; Serva S; Wuthrich K, Dynamic modes of the flipped-out cytosine during HhaI methyltransferase-DNA interactions in solution. EMBO J 1998, 17 (1), 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang YL; McDowell LM; Poliks B; Studelska DR; Cao C; Potter GS; Schaefer J; Song F; Stivers JT, Recognition of an unnatural difluorophenyl nucleotide by uracil DNA glycosylase. Biochemistry 2004, 43 (49), 15429–38. [DOI] [PubMed] [Google Scholar]
- 65.Dai Q; Lu X; Zhang L; He C, Synthesis of DNA oligos containing 2’-deoxy-2’-fluoro-D-arabinofuranosyl-5-carboxylcytosine as hTDG inhibitor. Tetrahedron 2012, 68 (26), 5145–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee S; Bowman BR; Ueno Y; Wang S; Verdine GL, Synthesis and structure of duplex DNA containing the genotoxic nucleobase lesion N7-methylguanine. J. Am. Chem. Soc 2008, 130 (35), 11570–11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maiti A; Morgan MT; Drohat AC, Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase. J Biol Chem 2009, 284 (52), 36680–36688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gill SC; von Hippel PH, Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem 1989, 182 (2), 319–26. [DOI] [PubMed] [Google Scholar]
- 69.Maiti A; Morgan MT; Pozharski E; Drohat AC, Crystal Structure of Human Thymine DNA Glycosylase Bound to DNA Elucidates Sequence-Specific Mismatch Recognition. Proc Natl Acad Sci USA 2008, 105 (26), 8890–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malik SS; Coey CT; Varney KM; Pozharski E; Drohat AC, Thymine DNA glycosylase exhibits negligible affinity for nucleobases that it removes from DNA. Nucleic Acids Res 2015, 43 (19), 9541–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995, 6 (3), 277–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.