

Abstract
Direct functionalization of C–H bond is rapidly becoming an indispensible tool in chemical synthesis. However, due to the ubiquity of C–H bonds, achieving site-selective functionalization remains an arduous task, especially on advanced synthetic intermediates or natural products. In contrast, Nature has evolved a multitude of enzymes capable of performing this task with extraordinary selectivity, and the use of these enzymes in organic synthesis may provide a viable solution to contemporary challenges in site-selective functionalization of complex molecules. This review covers recent applications of enzymatic C–H functionalization strategies in natural product synthesis, both in the context of key building block preparation and late-stage functionalization of advanced synthetic intermediates.
Graphical Abstract
Introduction
Direct functionalization of C–H bond constitutes a highly powerful strategy for the synthesis of organic compounds [1]. Under this paradigm, an inert C–H bond is viewed as a functional handle for the construction of a new C–C or C–X (X = heteroatom) bond in one step. This approach stands in stark contrast to traditional multistep sequences, which entail independent preparation of prefunctionalized substrates and their use in subsequent C–C or C–X (X = heteroatom) bond formation step. Thus, judicious application of C–H functionalization in complex molecule synthesis offers numerous strategic benefits that ultimately will allow chemical synthesis to be performed with greater efficiency [2,3].
The field of natural product synthesis is often regarded as the ultimate proving ground for new synthetic methods. Unsurprisingly, the number of total syntheses featuring C–H functionalization as the key step has risen dramatically in the past decade. Despite these successes, achieving chemo and regioselective C–H bond functionalization remains a formidable challenge, especially in the context of complex synthetic intermediates which contain numerous C–H bonds with similar bond energies. Successful applications of C–H bond functionalization in natural product synthesis typically rely on the use of intramolecular reactions [4,5], preinstalled directing groups [6] or innate reactivity differences within the molecular framework of interest [7]. Meanwhile, case studies that demonstrate catalyst-controlled selectivity remain rare [8]. In contrast, natural product biosynthesis pathways are replete with tailoring enzymes capable of performing different types of C–H bond functionalization—including hydroxylation [9,10], halogenation [11,12], alkylation [13], and desaturation with exquisite selectivity profiles unmatched by conventional small-molecule catalysts.
Aided by advances in microbial genetics and enzyme engineering, practitioners of organic chemistry and biocatalysis have recently begun to explore the synthetic utility of these catalysts for the synthesis of medicinally relevant molecules and natural products [14]. The application of this strategy can manifest in either early-stage building block synthesis or late-stage functionalization. The former refers to the use of enzymatic functionalization in the upstream portion of a synthetic route to produce a key intermediate, which undergoes subsequent chemical transformations en route to the molecular target. In this case, as the enzyme at hand needs to deliver ample quantities of a particular product for downstream manipulation, high catalytic efficiency/turnover number is paramount. In late-stage functionalization, an advanced intermediate is chemically synthesized and submitted to an enzymatic C–H functionalization step to afford the target natural product. Thus, promiscuous enzymes that can accept different advanced intermediates are typically preferred even if their catalytic efficiency is not particularly high. Herein, we highlight recent applications of biocatalytic C–H functionalization in natural product total synthesis, making clear distinction between examples of building block synthesis and late-stage functionalization for pedagogical purposes.
Building Block Synthesis
As outlined above, building block synthesis requires highly efficient enzymes that utilize inexpensive cosubstrates and/or cofactors so that the desired products can be obtained in the most practical and economical manner. Given these criteria, members of the iron and a-ketoglutarate-dependent dioxygenase (Fe/αKG) superfamily are excellent candidates, as they do not require dedicated reductase partners or expensive cofactors. Since 2016 our laboratory has conducted a series of proof-of-concept studies to examine the biocatalytic potential of these enzymes in the preparation of key building blocks, particularly in the context of nonribosomal peptide and alkaloid syntheses.
Manzacidin C and C avinafungin B
Isolated from the Okinawan sponge Hymeniacidon sp. in 1991, manzacidin C (1) is a bromopyrrole alkaloid that contains a unique tetrahydropyrimidine motif [15]. After the first reported total synthesis in 2000 [16–21], numerous synthetic approaches to 1 have been developed. In 2018, our laboratory reported a formal synthesis of 1 featuring a remoteC–H hydroxylation catalyzed by an Fe/αKG leucine 5-hydroxylase GriE [22] from the griselimycin biosynthesis [23]. By exploiting the substrate promiscuity of GriE, azidoleucine (4, prepared via photocatalytic azidation of L-leucine) could be selectively hydroxylated at the δ position in >95% conversion on 130 mg scale. A one-pot hydrogenation, Boc protection, and intramolecular cyclization provided lactone 6, which is an advanced intermediate (two steps away from 1) from a previous synthesis [16]. This route represents one of the shortest approaches to 1 and illustrates the simplifying power of biocatalytic C–H hydroxylation in complex molecule synthesis. We also found that GriE could catalyze iterative C5 oxidation of L-leucine at high enzyme concentration to give the corresponding imine (7), which was reduced with NH3•BH3 in one-pot to yield (2S,4R)-4-methylproline. Utilizing this method, protected (2S,4R)-4-methylproline 8 was prepared from L-leucine in 57% overall yield on 100 mg scale. With 8 in hand, the first synthesis of cavinafugin B (9), an antiviral aldehyde lipopeptide isolated from C. cavincola [24], was completed in 10 steps and37%overallyieldusingFmoc-basedsolid-phase peptide synthesis (SPPS) [25].
Tambromycin
Isolated from several Streptomyces strains, tambromycin (10) is a nonribosomal peptide natural product with antiproliferative activity against cancerous B- and -cell lines [26]. Structurally, tambromycin contains a trisubstituted indole fragment, a methyloxazoline moiety and an unusual pyrrolidine-containing amino acid named tambroline. Our laboratory recently developed a chemoenzymatic synthesis of 10 by enlisting a biocatalytic C–H functionalization approach to construct the tambroline monomer [27]. Utilizing L-lysine (11) as the starting material, regio- and stereoselective C3 hydroxylation employing an Fe/αKG lysine hydroxylase KDO1 [28] produced 3-hydroxylysine (12) on multigram scale. Subsequent three-step transformation of this intermediate generated the corresponding sulfamidate 13, which was heated at 70 ºC in DMA for 24 h to form protected tambroline (14). In parallel, a chemocatalytic C–H borylation [29] was devised to prepare the trisubstituted indole fragment. With the combination of chemocatalytic and enzymatic C–H functionalization, the total synthesis of tambromycin was completed in 10 steps (longest linear sequence) with 2.4% overall yield.
Late-Stage Functionalization
To allow flexibility in synthesis design, enzymes utilized in late-stage functionalization should ideally be able to accept a range of structurally related advanced intermediates as substrates. Furthermore, substrate promiscuity is a desirable trait if a late-stage enzymatic functionalization is to be employed in subsequent analogue development for medicinal chemistry exploration. Exemplified by the versatile P450BM3 [30], members of the cytochrome P450 superfamily have garnered significant attention from the synthetic community due to their substrate and catalytic promiscuity and thus are generally viewed as well suited for applications in late-stage functionalization. In the past few years, several P450s have been utilized in late-stage biocatalytic oxidation en route to complex natural products.
Nigelladine A
The first enantioselective total synthesis of nigelladine A (15), a highly conjugated norditerpene alkaloid isolated from the Nigella glandulifera plant [31], was achieved chemoenzymatically by the Arnold and Stoltz groups through a P450-catalyzed allylic oxidation of a late-stage imine intermediate [32]. The enantiopure enone 17 was synthesized from racemic 16 in 3 steps through the use of Tsuji–Trost asymmetric allylic alkylation [33], Tsuji–Wacker oxidation [34], and Robinson annulation. Subsequent bromination, Suzuki coupling, and condensation completed the construction of the tricyclic skeleton in 55% yield. Disappointingly, various traditional allylic oxidation conditions gave a mixture of inseparable regioisomers or overoxidized byproducts. The selectivity issue was addressed by the use of P450BM3 variant 8C7, which was previously evolved for regioselective deprotection of methoxymethyl-protected glycosides [35]. Employing 8C7, the desired hydroxylation product could be obtained with 2.8:1 regioselectivity on 160 mg scale. Subsequent DMP oxidation gave nigelladine in 43% yield over 2 steps based on recovered 20.
The juvenimicins
Tylosin and juvenimicins are a family of potent antibiotics featuring a 16-membered macrolide tylactone (21) [36]. In 2017, Sherman and coworkers reported chemoenzymatic total syntheses of tylactone and the juvenimicins by late-stage polyketide assembly, tailoring, and C–H functionalizations [37]. The two enantiopure fragments 22 and 23, prepared via Evans’ and Myers’ chiral auxiliary methodologies respectively [38,39], underwent Horner–Wadsworth–Emmons olefination, thioesterification, and desilylation to afford the key hexaketide intermediate 24 in 32% yield over 3 steps. Subsequent one-pot in vitro reaction catalyzed by the P450s JuvEIV and JuvEV introduced the final 4 carbon atoms and forged the desired macrocycle, affording tylactone, the corresponding aglycone of the juvenimicins in 69% yield. Further feeding of tylactone into DHS316, a mutated S. venezuelae strain [40,41], produced M- 4365 G1 (25) in more than 60% yield on 100 mg scale. With a large amount of 25 in hand, a divergent enzymatic synthesis of other juvenimicin family members was achieved via late-stage biotransformations with P450 oxygenases TylI, JuvD, and MycCI.
Vancomycin
The glycopeptide vancomycin is used as a drug of last resort to treat serious bacterial infections [42]. Structurally, it is a rigid heptapeptide with three macrocyclic rings, a biaryl linkage and two aryl ether crosslinks. Because of the structural complexity and outstanding clinical value, vancomycin has attracted much attention of synthetic chemists [43], culminating in three total syntheses in the late 1990s [44–50]. In 2018, Seyedsayamdost and co-worker reported a chemoenzymatic synthetic approach towards vancomycin aglycone variants [51]. A 7mer substrate 30 was prepared by SPPS, followed by thioesterification with coenzyme A and pantetheinylation with an X-domain peptidyl carrier protein (X-PCP) to give precursor 31. Treatment of 31 with three P450s, OxyA, OxyB, and OxyC installed the three synthetically challenging aromatic crosslinks via a sequence consisting of: (i) C-O-D aryl ether bond formation by OxyB, (ii) D-O-E aryl ether bond formation by OxyA, and (iii) -B biaryl linkage formation by OxyC. In addition, the synthesis of a thioamide-containing analogue via the same method suggested the potential of this biocatalytic cascade in the creation of vancomycin analogue libraries.
Future Directions and Outlook
The case studies outlined above suggest that the use of enzymatic C–H functionalization in natural product synthesis has mainly revolved around hydroxylation chemistry. For the field to continue to flourish, it is crucial that we begin tapping into a wider range of biocatalytic transformations. In the last decade, significant progress has been made in the discovery, characterization, and engineering of various enzyme families that catalyze other types of C–H functionalization, including halogenation [10,11] and alkylation [12]. Despite early work by Kirschning on the ansamitocins [52] and recent success of alkene/arene halofunctionalization by Moore [53], the use of enzymatic C–H halogenation in chemoenzymatic total synthesis has remained underexplored. Cursory examination of synthetic strategies to access certain natural product motifs, however, quickly reveals the decided advantage of applying these transformations in chemoenzymatic synthesis. For example, many prenylated indole and monoterpene indole alkaloids contain distinct halogenation patterns that are nontrivial to introduce via traditional chemical methods. As an alternative, one can envision an alternative strategy involving late-stage enzymatic halogenation on advanced synthetic intermediates, which in turn can be accessed using established chemical methods. This strategy can be implemented in the synthesis of spiromalbramide (33), a dichlorinated spirooxindole alkaloid from M. graminicola [54], by first targeting the construction [55] of the nonhalogenated precursor (premalbrancheamide, 37). Subjecting 37 to enzymatic halogenation with MalA [56] would furnish the corresponding dichlorinated product, which can in turn be oxidized using established procedures to generate the spirooxindole motif [57]. A similar strategy can also be conceived to access welwitindolinone A (39) via enzymatic chlorination of synthetic [58] 12-epi-fischerindole U (42) with the halogenase WelO5/AmbO5 [59], followed by spirooxindole ring formation. In the same vein, enzymatic C–H alkylation can be used to construct otherwise challenging C–C bonds in natural product synthesis. While radical M enzymes have gained notoriety due to the general sensitivity of the Fe C– cluster, we believe that in certain cases, their use in chemoenzymatic synthesis can be strategically enabling. One potential application can be found in the topological problem presented by the streptide family of natural product [60], which contains a highly unique Lys-Trp crosslink. Here, one can conceive a chemoenzymatic approach towards streptide (44) involving solid-phase synthesis or a combination of solid-phase synthesis and native chemical ligation to assemble the linear precursor peptide, which can subsequently be cyclized through the use of StrB [61], the native radical SAM enzyme within the streptide biosynthesis pathway.
The examples presented in this review serve to highlight the power of enzymatic C–H functionalization in solving challenging problems in natural product synthesis. These developments notwithstanding, we believe that we are nowhere close to realizing the full potential of this platform. Advances in sequencing technology have provided an abundance of genomic data that is now at our disposal. New techniques in DNA synthesis and protein and metabolic engineering [63,64] also hold promise in accelerating the discovery of new, synthetically useful C–H functionalization biocatalysts. These developments will facilitate more widespread incorporation of enzymatic C–H functionalization in natural product synthesis and will enable the invention of creative biocatalytic retrosynthetic disconnections that will bring us closer to achieving “ideality” in synthesis [65].
Figure 1.
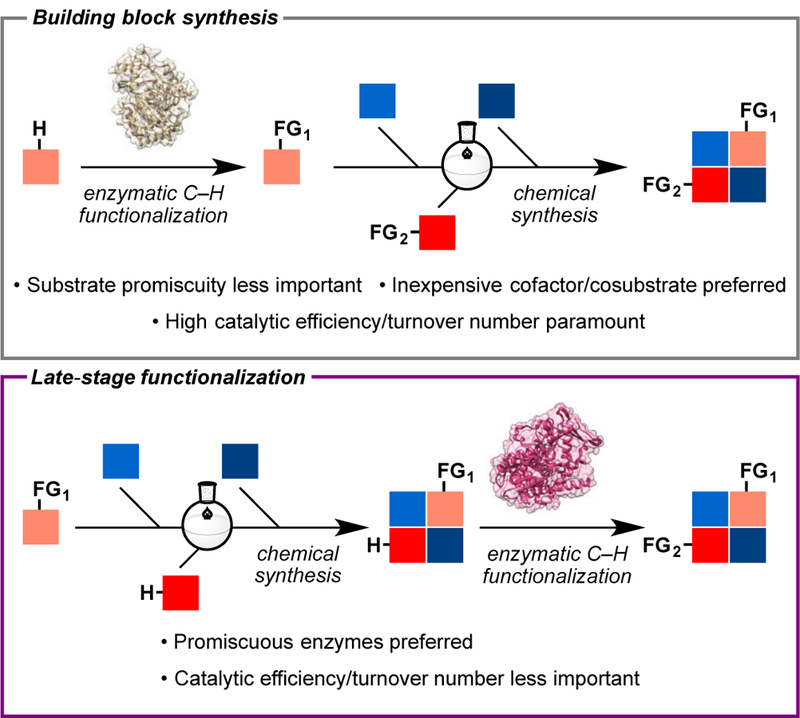
Schematic illustration of biocatalytic C–H functionalization for building block synthesis and late-stage modification in multi-step synthesis.
Figure 2.
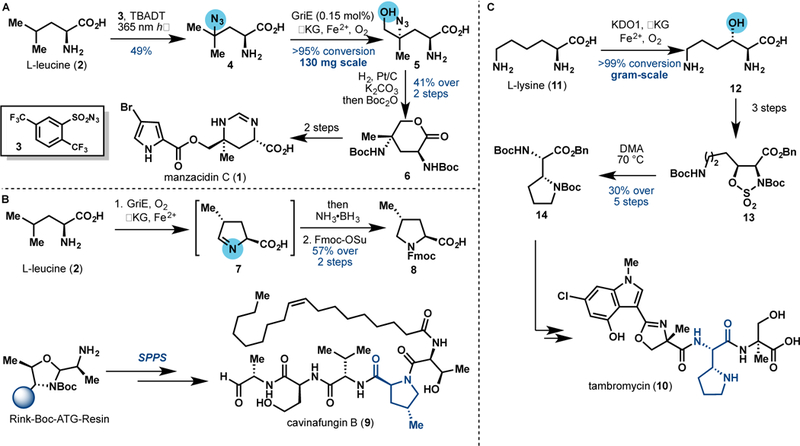
A. Formal synthesis of manzacidin C featuring remote hydroxylation of 4 with the Fe/αKG GriE. B. Chemoenzymatic cascade with GriE for the preparation of protected (2S,4R)-4-methylproline (8) in the synthesis of cavinafungin B. C. Selective C3 hydroxylation of L-lysine with the Fe/αKG KDO1 for the preparation of protected tambroline monomer (14) in the total synthesis of tambromycin.
Figure 3.

A. Application of late-stage biocatalytic oxidation with an engineered P450BM3 8C7 in the total synthesis of nigelladine A. B. Late-stage oxidative diversification of M4365 G1 with the P450s TylI, MycCI, and JuvD. C. Oxidative phenol couplings catalyzed by the P450s OxyA, OxyB, and OxyC in the chemoenzymatic synthesis of vancomycin aglycone.
Figure 4.
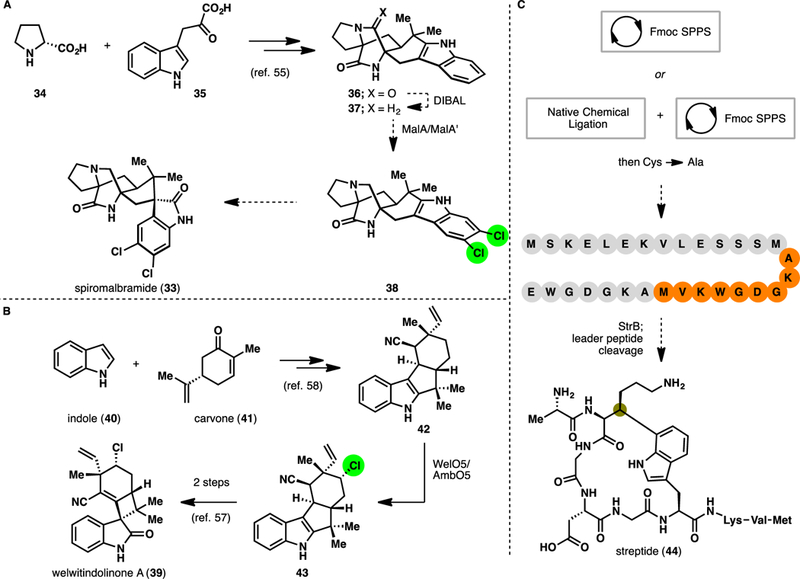
A. Proposed use of late-stage enzymatic chlorination for the chemoenzymatic synthesis of spiromalbramide. B. Proposed use of late-stage enzymatic chlorination for the chemoenzymatic synthesis of welwitindolinone A. C. Proposed application of StrB in late-stage Lys-Trp crosslinking for the chemoenzymatic synthesis of streptide.
Acknowledgments
The authors acknowledge the support of the National Institute of Health (1R35GM128895) and The Scripps Research Institute. The content is solely the responsibility of the authors and does not represent the official views of any of the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yamaguchi J, Yamaguchi AD, Itami K: C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew Chem Int Ed 2012, 51:8960–9009. [DOI] [PubMed] [Google Scholar]
- 2.Gutekunst WR, Baran PS: C–H functionalization logic in total synthesis. Chem Soc Rev 2011, 40:1976–1991. [DOI] [PubMed] [Google Scholar]
- 3.McMurray L, O’Hara F, Gaunt MJ: Recent developments in natural product synthesis using metal-catalyzed C–H bond functionalization. Chem Soc Rev 2011, 40:1885–1898. [DOI] [PubMed] [Google Scholar]
- 4.Chen K, Baran PS: Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 2009, 459:824–828. [DOI] [PubMed] [Google Scholar]
- 5.Hinman A, Du Bois J: A stereoselective synthesis of (–)-tetrodotoxin. J Am Chem Soc 2003, 125:11510–11511. [DOI] [PubMed] [Google Scholar]
- 6.Feng Y, Chen G: Total synthesis of celogentin by stereoselective C–H activation. Angew Chem Int Ed 2010, 49:958–961. [DOI] [PubMed] [Google Scholar]
- 7.Kawamura S, Chu H, Felding J, Baran PS: Nineteen-step total synthesis of (+)-phorbol. Nature 2016, 532:90C–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamaguchi AD, Chepiga KM, Yamaguchi J, Itami K, Davies HML: Concise syntheses of dictyodendrins A and F by a sequential C–H functionalization strategy. J Am Chem Soc 2015, 137:644–647. [DOI] [PubMed] [Google Scholar]
- 9.Podust LM, Sherman DH: Diversity of P450 enzymes in the biosynthesis of natural products. Nat Prod Rep 2012, 29:1251–1256.• This review discusses a range of intriguing transformations catalyzed by P450 enzymes in natural product biosynthesis.
- 10.Gao S-S, Naowarojna N, Cheng R, Liu X, Liu P: Recent examples of α-ketoglutarate-dependent mononuclear non-haem iron enzymes in natural product biosynthesis. Nat Prod Rep 2018, 35:792–837.• This article provides an excellent overview of the wealth of biocatalytic transformations catalyzed by Fe/ αKG dioxygenases in natural product biosynthesis.
- 11.Latham J, Brandenburger E, Shepherd SA, Menon BRK, Micklefield J: Development of halogenase enzymes for use in synthesis. Chem Rev 2018, 118:232–269.• This review provides a comprehensive look at Nature’s inventory of halogenation biocatalysts, with special emphasis on their use in the context of organic synthesis.
- 12.Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, Moore BS: Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chem Rev 2017,117:5619–5674.•A magnificent review that covers Nature’s enzymatic strategies for halogenation and dehalogenation, focusing on the mechanistic features of the transformations.
- 13.Yokoyama K, Lilla EA: C–C bond forming radical SAM enzymes involved in the construction of carbon skeletons of cofactors and natural products. Nat Prod Rep 2018, 35:660–694.• This review provides an excellent summary of the different types of C–C bond forming reactions catalyzed by radical SAM enzymes in the biosynthesis of natural cofactors and natural products.
- 14.King-Smith E, Zwick CR III, Renata H: Applications of oxygenases in the chemoenzymatic total synthesis of complex natural products. Biochemistry 2018, 57:403–412. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi J, Kanda F, Ishibashi, Shigemori H: Manzacidins -A-C, novel tetrahydropyrimidine alkaloids from the Okinawan marine sponge Hymeniacidon sp. J Org Chem 1991, 56:4574–4576. [Google Scholar]
- 16.Namba K, Shinada T, Teramoto T, Ohfune Y: Total synthesis and absolute structure of manzacidin A and C. J Am Chem Soc 2000, 122:10708–10709. [Google Scholar]
- 17.Hashimoto T, Maruoka K: Syntheses of manzacidins: a stage for the demonstration of synthetic methodologies. Org Biomol Chem 2008, 6:829–835. [DOI] [PubMed] [Google Scholar]
- 18.Tran K, Lombardi PJ, Leighton JL: An efficient asymmetric synthesis of manzacidin C. Org Lett 2008, 10:3165–3167. [DOI] [PubMed] [Google Scholar]
- 19.Ichikawa Y, Okumura K, Matsuda Y, Hasegawa T, Nakamura M, Fujimoto A, Masuda T, Nakano K, Kotsuki H: Synthesis of manzacidin A and C: efficient construction of quaternary carbon stereocenters bearing nitrogen substituents. Org Biomol Chem 2012, 10:614–622. [DOI] [PubMed] [Google Scholar]
- 20.Bretzke S, Scheeff S, Vollmeyer F, Eberhagen F, Rominger F, Menche D: Modular synthesis of the pyrimidine core of the manzacidins by divergent Tsuji-Trost coupling. Beilstein J Org Chem 2016, 12:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong TMT, Soeta T, Suga T, Kawamoto K, Hayashi Y, Ukaji Y: Formal total synthesis of manzacidin C based on asymmetric 1,3-dipolar cycloaddition of azomethine imines. J Org Chem 2017, 82:1969–1976. [DOI] [PubMed] [Google Scholar]
- 22.Zwick CR III, Renata H: Remote C−H hydroxylation by an α-ketoglutarate dependent dioxygenase enables efficient chemoenzymatic synthesis of manzacidin C and proline analogs. J Am Chem Soc 2018, 140:1165–1169.•• Here, the synthetic utility of an amino acid hydroxylase is showcased in the gramscale synthesis of 5-hydroxyleucine and in the hydroxylation of a non-native substrate, 4-azidoleucine, en route to a formal synthesis of manzacidin C.
- 23.Lukat P, Katsuyama Y, Wenzel S, Binz T, König C, Blankenfeldt W, Brönstrup M, Müller R: Biosynthesis of methyl-proline containing griselimycins, natural products with anti-tuberculosis activity. Chem. Sci 2017, 8:7521–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ortíz-López FJ, Monteiro MC, González-Menéndez V, Tormo JR, Genilloud O, Bills GF, Vicente F, Zhang C, Roemer T, Singh SB, Reyes F: Cyclic colisporifungin and linear cavinafungins, antifungal lipopeptides isolated from Colispora cavincola. J Nat Prod 2015, 78:468–475. [DOI] [PubMed] [Google Scholar]
- 25.Zwick CR III, Renata H: A one-pot chemoenzymatic synthesis of (2S,4R)-4methylproline enables the first total synthesis of antiviral lipopeptide cavinafungin B. ChemRxiv 2018, doi: 10.26434/chemrxiv.6405761.v1. [DOI]
- 26.Goering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang Y, Ju K-S, Thomson RJ, Metcalf WM, Kelleher NL:Metabologenomics: correlation of microbial gene clusters with metabolites drives discovery of a nonribosomal peptide with an unusual amino acid monomer. ACS Cent Sci 2016, 2:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, King-Smith E, Renata H: Total synthesis of tambromycin by combining chemocatalytic and biocatalytic C−H functionalization. Angew Chem Int Ed 2018, 57:5037–5041.•• This example illustrates the synthetic utility and practicality of amino acid hydroxylation with Fe/αKG dioxygenase as 4 grams of L-lysine can be converted to the hydroxylated counterpart in single pass.
- 28.Baud D, Saaidi P-L, Monfleur A, Harari M, Cuccaro J, Fossey A, Besnard M, Debard A, Mariage A, Pellouin V, Petit J-L, Salanoubat M, Weissenbach J, de Berardinis V, Zaparucha A: Synthesis of mono‐ and dihydroxylated amino acids with new α‐ ketoglutarate‐ dependent dioxygenases: biocatalytic oxidation of C-H bonds. ChemCatChem 2014, 6:3012–3017. [Google Scholar]
- 29.Feng Y, Holte D, Zoller J, Umemiya S, Simke LR, Baran PS: Totalsynthesis of erruculogen and fumitremorgin enabled by ligand-controlled C−H borylation. J Am Chem Soc 2015, 137:10160–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitehouse CJC, Bell SG, Wong L-L: P450BM3 (CYP102A1): connecting the dots. Chem Soc Rev 2012, 41:1218–1260. [DOI] [PubMed] [Google Scholar]
- 31.Chen QB, Xin XL, Yang Y, Lee SS, Aisa HA: Highly conjugated norditerpenoid and pyrroloquinoline alkaloids with potent PTP1B inhibitory activity from Nigella glandulifera. J Nat Prod 2014, 77:807–812. [DOI] [PubMed] [Google Scholar]
- 32.Loskot SA, Romney DK, Arnold FH, Stoltz BM: Enantioselective total synthesis of nigelladine A via late-stage C−H oxidation enabled by an engineered P450 enzyme. J Am Chem Soc 2017, 139:10196–10199.•• The authors identify a P450BM3 variant that provides superior chemo- and regioselectivity over small-molecule catalysts for late-stage oxidation of a synthetic intermediate. Furthermore, preparative scale synthesis could be achieved by enlisting the use of NADPH recycling system.
- 33.Hong AY, Stoltz BM: The construction of all-carbon quaternary stereocenters by use of d-catalyzed asymmetric allylic alkylation reactions in total synthesis. Eur J Org Chem 2013, 14:2745–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong JJ, Browne WR, Feringa BL: Palladium-catalyzed anti-Markovnikov oxidation of terminal alkenes. Angew Chem Int Ed 2015, 54:734–744. [DOI] [PubMed] [Google Scholar]
- 35.Lewis JC, Manotvani Sm, Fu Y, Snow CD, Komor RS, Wong C-H, Arnold FH: Combinatorial alanine substitution enables rapid optimization of cytochrome P450BM3 for selective hydroxylation of large substrates. ChemBioChem 2010, 11:2502–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kishi T, Harada S, Yamana H, Miyake A: Studies on juvenimicin, a new antibiotic. II. Isolation, chemical characterization and structures. J. Antibiot 1976, 29:1171–1181. [DOI] [PubMed] [Google Scholar]
- 37.Lowell AN, DeMars MD II, Slocum ST, Yu F, Anand K, Chemler JA, Korakavi N, Priessnitz JK, Park SR, Koch AA, Schultz PJ, Sherman DH: Chemoenzymatic total synthesis and structural diversification of tylactone-based macrolide antibiotics through late-stage polyketide assembly, tailoring, and C−H functionalization. J Am Chem Soc 2017, 139:7913–7920.•• This work illustrates the synthetic utility of P450-catalyzedC–H hydroxylation in the late-stage diversification of a macrolide scaffold.
- 38.Shirokawa S, Shinoyama M, Ooi I, Hosokawa S, Nakazaki A, Kobayashi S: Total synthesis of kharefungin using highly stereoselective vinylogous Mukaiyama aldo reaction. Org Lett 2007, 9:849–852. [DOI] [PubMed] [Google Scholar]
- 39.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL: Pseudoephedrine as a practical chiral auxiliary for the synthesis of highly enantiomerically enriched carboxylic acids, alcohols, aldehydes, and ketones. J Am Chem Soc 1997, 119:6496–6511. [Google Scholar]
- 40.Borisova SA, Zhao L, Sherman DH, Liu H-W: Biosynthesis of desosamine: construction of a new macrolide carrying a genetically designed sugar moiety. Org Lett 1999, 1:133–136. [DOI] [PubMed] [Google Scholar]
- 41.DeMars MD II, Sheng F, Park SR, Lowell AN, Podust LM, Montgomery J, Sherman DH: Biochemical and structural characterization of MycCI, a versatile P450 biocatalyst from the mycinamicin biosynthetic pathway. ACS Chem Biol 2016, 11:2642–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butler MS, Hansford KA, Blaskovich MA, Halai R, Cooper MA: Glycopeptide antibiotics: back to the future. J Antibiot 2014, 67:631–644. [DOI] [PubMed] [Google Scholar]
- 43.Okano A, Isley NA, Boger DL: Total syntheses of vancomycin-related glycopeptide antibiotics and key analogues. Chem Rev 2017, 117:11952–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL: Total syntheses of vancomycin and eremomycin aglycons. Angew Chem Int Ed 1998, 37:2700–2704. [DOI] [PubMed] [Google Scholar]
- 45.Evans DA, Dinsmore CJ, Watson PS, Wood MR, Richardson TI, Trotter BW, Katz JL: Nonconventional stereochemical issues in the design of the synthesis of the vancomycin antibiotics: Challenges imposed by axial and nonplanar chiral elements in the heptapeptide aglycons. Angew Chem Int Ed 1998, 37:2704–2708. [DOI] [PubMed] [Google Scholar]
- 46.Nicolaou KC, Natarajan S, Li H, Jain NF, Hughes R, Solomon ME, Ramanjulu JM, Boddy CNC, Takayanagi M: Total synthesis of vancomycin aglycon−Part 1: Synthesis of amino acids 4−7 and construction of the AB-COD ring skeleton. Angew Chem Int Ed 1998, 37:2708–2714. [DOI] [PubMed] [Google Scholar]
- 47.Nicolaou KC, Jain NF, Natarajan S, Hughes R, Solomon ME, Li H, amanjulu JM, Takayanagi M, Koumbis AE, Bando T: Total synthesis of vancomycin aglycon−Part 2: Synthesis of amino acids 1−3 and construction of the AB-COD-DOE ring skeleton. Angew Chem Int Ed 1998, 37:2714–2716. [DOI] [PubMed] [Google Scholar]
- 48.Nicolaou KC, Takayanagi M, Jain NF, Natarajan S, Koumbis AE, Bando T, Ramajulu JM: Total synthesis of vancomycin aglycon−Part 3: final stages. Angew Chem Int Ed 1998, 37:2717–2719. [DOI] [PubMed] [Google Scholar]
- 49.Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL: Diastereoselective total synthesis of the vancomycin aglycon with ordered atropisomer equilibrations. J Am Chem Soc 1999, 121:3226–3227. [Google Scholar]
- 50.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q: Total synthesis of the vancomycin aglycon. J Am Chem Soc 1999, 121:10004–10011. [Google Scholar]
- 51.Forneris CC, Seyedsayamdost MR: In vitro reconstitution of OxyC activity enables total chemoenzymatic syntheses of vancomycin aglycone variants. Angew Chem Int Ed 2018, 57:8048–8052.•• Through in vitro experiments, the authors were able to demonstrate the catalytic activity of one of the P450s in the vancomycin biosynthesis for the first time, which allowed them to subsequently develop a biocatalytic cascade to rapidly generate the core scaffold of vancomycin.
- 52.Meyer A, Brünjes M, Taft F, Frenzel T, Sasse F, Kirschning A: Chemoenzymatic approaches toward dechloroansamitocin P-3. Org Lett 2007, 9:1489–1492.• One of the earliest examples of biocatalytic C–H halogenation using whole cell system in the chemoenzymatic synthesis of bioactive natural products.
- 53.Miles ZD, Diethelm S, Pepper HP, Huang DM, George JH, Moore BS: A unifying paradigm for naphthoquinone-based meroterpenoid (bio)synthesis. Nat Chem 2017, 9:1235–1242.• This study illustrates the synthetic utility of vanadium-dependent halogenase in the chemoenzymatic synthesis of meroterpenoid natural products.
- 54.Watts KR, Loveridge ST, Tenney K, Media J, Valeriote FA, Crews P: Utilizing DART mass spectrometry to pinpoint halogenated metabolites from a marine invertebrate-derived fungus. J Org Chem 2011, 76:6201–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frebault FC, Simpkins NS: A cationic cyclization route to prenylated indole alkaloids: synthesis of malbrancheamide B and brevianamide B, and progress towards stephacidin A. Tetrahedron 2010, 66:6585–6596. [Google Scholar]
- 56.Fraley AE, Garcia-Borras M, Tripathi A, Khare D, Mercado-Marin EV, Tran H, Dan Q, Webb GP, Watts KR, Crews P, Sarpong R, Williams RM, Smith JL, Houk KN, Sherman DH: Function and structure of MalA/MalA’, iterative halogenases for late-stage C−H functionalization of indole alkaloids. J Am Chem Soc 2017, 139:12060–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baran PS, Maimone TJ, Richter JM Total synthesis of marine natural products without using protecting groups . Nature 2007, 446:404–408. [DOI] [PubMed] [Google Scholar]
- 58.Richter JM, Ishihara Y, Masuda T, Whitefield BW, Llamas T, Pohjakallio A, Baran PS: Enantiospecific total synthesis of the hapalindoles, fischerindoles, and welwitindolinones via a redox economic approach. J Am Chem Soc 2008, 130:17938–17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hillwig ML, Zhu Q, Ittiamornkul K, Liu X: Discovery of a promiscuous non- heme iron halogenase in ambiguine alkaloid biogenesis: implication for an evolvable enzyme family for late-stage halogenation of aliphatic carbons in small molecules. Angew Chem Int Ed 2016, 55:5780–5784. [DOI] [PubMed] [Google Scholar]
- 60.Schramma KR, Bushin LB, Seyedsayamdost MR: Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat Chem 2015, 7:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schramma KR, Seyedsayamdost MR: Lysine-tryptophan-crosslinked peptides produced by radical SAM enzymes in pathogenic Streptococci. ACS Chem Biol 2017, 12:922–927. [DOI] [PubMed] [Google Scholar]
- 62.Palluk S, Arlow DH, de Rond T, Barthel S, Kang JS, Bector R, Baghdassarian HM, Truong AN, Kim PW, Singh AK, Hillson NJ, Keasling JD: De novo DNA synthesis using polymerase-nucleotide conjugates. Nat Biotechnol 2018, 36:645–650. [DOI] [PubMed] [Google Scholar]
- 63.Plesa C, Sidore AM, Lubock N, Zhang D, Kosuri S: Multiplexed gene synthesis in emulsions for exploring protein functional landscapes. Science 2018, 359:343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lian J, HamediRad M, Hu S, Zhao H: Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat Commun 2017, 8:1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaich T, Baran PS: Aiming for the ideal synthesis. J Org Chem 2010, 75:4657–4673. [DOI] [PubMed] [Google Scholar]