

Abstract
Aims
Plasma xanthine oxidoreductase (XOR) activity during the acute phase of acute heart failure (AHF) requires further elucidation.
Methods and results
One hundred eighteen AHF patients and 231 control patients who attended a cardiovascular outpatient clinic were prospectively analysed. Blood samples were collected within 15 min of admission from AHF patients (AHF group) and control patients who visited a daily cardiovascular outpatient clinic (control group). Plasma XOR activity was compared between the two groups, and factors independently associated with extremely elevated XOR activity were identified using a multivariate logistic regression model. Plasma XOR activity in the AHF group (median, 104.0 pmol/h/mL; range, 25.9–423.5 pmol/h/mL) was significantly higher than that in the control group (median, 45.2 pmol/h/mL; range, 19.3–98.8 pmol/h/mL). The multivariate logistic regression model showed that serum uric acid (per 1.0 mg/dL increase, odds ratio: 1.280; 95% confidence interval: 1.066–1.536; P = 0.008) and lactate levels (per 1.0 mmol/L increase, odds ratio: 1.239; 95% confidence interval: 1.040–1.475; P = 0.016) were independently associated with high plasma XOR activity (>300 pg/h/mL) during the acute phase of AHF.
Conclusions
Plasma XOR activity was extremely high in patients with severely decompensated AHF. This would be associated with a high lactate value and would eventually lead to hyperuricaemia in patients with AHF.
Keywords: Acute decompensated heart failure, Reactive oxygen species, Uric acid, XOR inhibitor
Introduction
Elevated serum uric acid (UA) has been established as an important biomarker associated with high mortality and adverse outcomes in patients with chronic and acute heart failure (AHF).1, 2, 3 We previously reported that serum UA levels upon admission of patients with severely decompensated AHF requiring intensive care were an independent predictor of the patients' midterm prognosis, including all‐cause death and heart failure (HF) events.3 However, the mechanism underlying the association between elevated serum UA levels and poor prognosis remained unclear.4
Serum UA is the final product of purine metabolism. Xanthine oxidase (XO) and xanthine dehydrogenase (XDH), two interconvertible forms of xanthine oxidoreductase (XOR),5, 6 are the most important enzymes in this metabolic system. While XO and XDH both catalyse UA production, their electron acceptors differ, with XO requiring NAD+ as its electron acceptor and XDH requiring reduced molecular oxygen.5 Reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) and superoxide anion (O2 −), are generated through the production of UA from xanthine, a reaction catalysed by XO.5 It is the ROS by‐products that lead to cell damage. Although the ratio of XDH to XO activity in the blood had not been sufficiently studied,7 nowadays, the major hypothesis is that XDH is quickly converted to XO after being transferred to the bloodstream.8 Therefore, measurement of plasma XOR activity may reflect the amount of XO in the blood. From this point of view, an excessive increase in XOR activity would not only lead to elevated serum UA levels but also induce increased oxidative stress. Thus, the production of ROS via the activation of XOR may be one mechanism that leads to an adverse outcome in hyperuricaemic AHF. We therefore hypothesized that increased XOR activity would occur during the acute phase of AHF. Recently, a strategy for measuring XOR levels was established, and several reports have been published regarding XOR levels observed in patients with chronic HF and cardiac disease and in normal volunteers.9, 10, 11 In this study, we evaluated plasma XOR activity immediately after admission in patients with severely decompensated AHF patients requiring intensive care.
Methods
Subjects
A total of 118 consecutive AHF patients who were admitted to the intensive care unit of Nippon Medical School Chiba Hokusoh Hospital between December 2016 and March 2018 were prospectively enrolled in this study. AHF was defined as either new‐onset HF or the decompensation of chronic HF with symptoms sufficient to warrant hospitalization.12 Based on the European Society of Cardiology guidelines for the diagnosis of AHF, an abnormal electrocardiogram or the presence of pulmonary oedema on chest X‐ray and a B‐type natriuretic peptide (BNP) level of ≥100 pg/mL are required to diagnose AHF.13 The treating physician in the emergency department diagnosed AHF based on these criteria within 30 min of admission.
All of the patients had a New York Heart Association (NYHA) functional class of either III or IV. Patients who met any of the following criteria were admitted to the intensive care unit: (i) require high‐flow oxygen inhalation (including mechanical support) to treat orthopnea; (ii) require inotrope or mechanical support due to low blood pressure; and (iii) require various types of diuretics to improve generalized or pulmonary oedema. All patients in the present study received either diuretics or vasodilators after admission for the treatment of AHF.
In addition, a total of 231 patients who attended the cardiovascular outpatient clinic of Nippon Medical School Chiba Hokusoh Hospital, Hasegawa Hospital, and Toho Kamagaya Hospital were enrolled as a control group. The period of enrolment was the same for the control and AHF groups. Patients in the control group included those with cardiovascular disease (i.e. prior myocardial infarction, compensated HF, arrhythmia, hypertensive heart disease, and cardiomyopathy) and without pre‐existing cardiovascular conditions (i.e. hypertension, dyslipidaemia, or diabetes mellitus).
Xanthine oxidoreductase measurement and comparisons
Blood samples were collected from AHF patients within 15 min of admission. For the control group, blood samples were collected during their daily outpatient clinic appointment. The blood samples were centrifuged within 5 min at 4°C and were immediately frozen at −80°C until analysed. A plasma XOR activity assay was performed using a stable isotope‐labelled substrate and liquid chromatography triple quadrupole mass spectrometry (LC‐TQMS; Sanwa Kagaku Kenkyusho Co., Ltd, Japan).
To remove small molecules, including hypoxanthine, xanthine, and UA, 100 μL of each plasma sample was purified using a Sephadex G25 column. The eluate was then mixed with 16 μmol/L [13C2, 15N2]‐xanthine as the substrate and 16 μmol/L NAD+ and 1 μmol/L [13C2, 15N2]‐UA as the internal standard in 250 μL Tris buffer (pH 8.5). Each of the mixtures was incubated at 37°C for 90 min, mixed with 500 μL methanol, and centrifuged at 2000× g for 15 min at 4°C. The supernatants transferred to new tubes were evaporated, reconstituted with 150 μL distilled water, and filtered through an ultrafiltration membrane before undergoing LC/TQMS analysis using the Nano Space SI‐2 LC system (Shiseido, Ltd, Tokyo, Japan) and a TSQ‐Quantum TQM spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with an external systems interface. The amount of [13C2, 15N2]‐UA produced was quantified using the calibration curve, with the XOR activity expressed as [13C2, 15N2]‐UA in pmol/h/mL plasma. The lower limit of detection for XOR activity was 6.67 pmol/h/mL, and the upper limit of detection was 6.670 pmol/h/mL. The inter‐detection assay coefficients of variation of pooled human plasma activity were 6.5% and 9.1%, respectively.14 XOR activity was reported with no addition of NAD+; therefore, it was impossible to measure the actual XO activity. The standard reporting for XO is U/mL of plasma (1 U = 1 μmol of UA formed/min) and is pmol/h/mL of plasma for XOR (600 pmol/h/mL plasma, which equals 10 μU/mL plasma).
Xanthine oxidoreductase activity was compared between AHF patients (AHF group, n = 118) and outpatients (control group, n = 231). We also compared patients' characteristics (gender, age), vital signs (systolic blood pressure, heart rate), risk factors for atherosclerosis and co‐morbidities [diabetes mellitus, hypertension, dyslipidaemia, hyperuricaemia, and chronic kidney disease (CKD)], laboratory data [sodium, potassium, blood urea nitrogen, creatinine, total bilirubin, UA, haemoglobin, BNP, and C‐reactive protein (CRP)], and medications (XOR inhibitor). The factors significantly associated with increased XOR activity were determined by multivariate logistic regression analysis.
In addition, arterial blood gas data (i.e. pH, PO2, PCO2, HCO3 −, and lactate) were measured in AHF patients using an ABL800 FLEX© blood gas analyzer (ABL800; Radiometer Medical ApS, Copenhagen, Denmark). Lactate levels were evaluated in the emergency room using the amperometric measurement method. The left ventricular ejection fraction (LVEF) was evaluated in AHF patients in the emergency room upon admission and calculated using the Teichholz method or modified Simpson's method (Vivid I; GE Yokogawa Medical, Tokyo, Japan).
Statistical analyses
All of the data were statistically analysed using the SPSS 22.0 J software program (SPSS Japan Institute, Tokyo, Japan). All numerical data were expressed as the median and the 25–75% interquartile range, depending on normality. Normality was assessed using the Shapiro–Wilk W‐test. The Mann–Whitney U test was used to compare the two groups (AHF vs. control). Comparisons of all proportions were performed using a chi‐squared test. P‐values <0.05 were considered to indicate statistical significance.
All clinically relevant factors affecting increased XOR activity, including serum UA levels (per 1.0 mg/dL increase), pre‐administrated XOR inhibitor, LVEF upon admission (per 10% increase), serum creatinine levels (per 1.0 mg/dL increase), serum BNP levels (per 10 pg/mL increase), and lactate levels (per 1.0 mmol/L increase), were selected for inclusion in the multivariate logistic regression model. The multivariate logistic regression analysis was performed using backward stepwise selection.
Ethical considerations
The research ethics committee of the Chiba Hokusoh Hospital, Nippon Medical School approved the study protocol. Written informed consent was obtained from all of the participants before commencing the study.
Results
Patient characteristics
The AHF patient cohort consisted of 76 (64.4%) male patients and 42 (35.6%) (median age, 75 years). A total of 76 (64.4%) patients had new‐onset HF, 56 (47.5%) had ischaemic heart disease, and 62 (52.5%) had non‐ischaemic heart disease, including cardiomyopathy (n = 21), hypertensive heart disease (n = 14), and valvular heart disease (n = 21). Most patients (93.2%) were NYHA class IV. The median LVEF upon admission was 37.0% (Table 1). The systolic blood pressure, heart rate, and incidence of CKD in the AHF group were significantly higher than those in the control group. In the AHF group, the serum sodium and haemoglobin levels were significantly decreased, while the serum creatinine, blood urea nitrogen, CRP, and BNP levels were significantly increased, in comparison with the control group (Table 1). Furthermore, the plasma XOR activity in the AHF group (median, 104.0 pmol/h/mL; range, 25.9–423.5 pmol/h/mL) was significantly higher than that in the control group (median, 45.2 pmol/h/mL; range, 19.3–98.8 pmol/h/mL) (Table 1, Figure 1 ).
Table 1.
Patient characteristics
| Overall | Control group | AHF group | P value | |||
|---|---|---|---|---|---|---|
| (n = 349) | (n = 231) | (n = 118) | ||||
| General status and vital signs | ||||||
| Gender (male, %) | 243 (69.4%) | 167 (72.3) | 76 (64.4%) | 0.141 | ||
| Age (years) | 74 (66–80) | 73 (66–79) | 75 (65–82) | 0.257 | ||
| Systolic blood pressure (mmHg) | 130 (117–148) | 127 (116–138) | 156 (119–186) | <0.001 | ||
| Heart rate (bpm) | 80 (69–98) | 75 (67–83) | 108 (91–120) | <0.001 | ||
| Co‐morbidities | ||||||
| Hypertension (yes, %) | 255 (73.1%) | 163 (70.6%) | 92 (78.0%) | 0.161 | ||
| Dyslipidaemia (yes, %) | 221 (63.1%) | 157 (68.0%) | 64 (54.2%) | 0.014 | ||
| Diabetes mellitus (yes, %) | 136 (39.0%) | 86 (37.2%) | 50 (42.4%) | 0.356 | ||
| Hyperuricaemia (yes, %) | 129 (37.0%) | 82 (35.5%) | 47 (39.8%) | 0.482 | ||
| CKD (yes, %) | 116 (33.2%) | 65 (28.1%) | 51 (43.2%) | 0.006 | ||
| Feature of AHF | ||||||
| New‐onset HF (yes, %) | — | 76 (64.4%) | — | |||
| NYHA IV (yes, %) | — | 112 (75.2%) | — | |||
| LVEF (%) | — | 37 (25–45) | — | |||
| Aetiology of AHF | ||||||
| Ischaemic HF (yes, %) | — | 56 (47.5%) | — | |||
| Valvular HF (yes, %) | — | 20 (16.9%) | — | |||
| Hypertensive HF (yes, %) | — | 14 (11.9%) | — | |||
| Cardiomyopathy (yes, %) | — | 21 (17.8%) | — | |||
| Laboratory data | ||||||
| Sodium (mEq/L) | 141 (139–143) | 142 (140–143) | 139 (136–143) | <0.001 | ||
| Potassium (mEq/L) | 4.3 (4.0–4.7) | 4.3 (4.0–4.7) | 4.4 (3.9–4.8) | 0.757 | ||
| BUN (mg/dL) | 18.0 (14.5–25.4) | 16.8 (13.9–20.0) | 25.2 (18.4–41.4) | <0.001 | ||
| Creatinine (mg/dL) | 0.97 (0.77–1.26) | 0.93 (0.76–1.11) | 1.22 (0.83–2.15) | <0.001 | ||
| Total bilirubin (mg/dL) | 0.7 (0.5–0.9) | 0.7 (0.5–0.9) | 0.6 (0.4–1.0) | 0.109 | ||
| Uric acid (mg/dL) | 5.8 (4.8–7.0) | 5.6 (4.7–6.5) | 6.7 (5.4–8.3) | <0.001 | ||
| Haemoglobin (mg/dL) | 13.3 (11.9–14.7) | 13.6 (12.4–14.9) | 12.2 (10.0–14.3) | <0.001 | ||
| CRP (mg/dL) | 0.20 (0.07–0.84) | 0.10 (0.05–0.24) | 1.05 (0.28–4.12) | <0.001 | ||
| BNP (pg/mL) | 147 (32–663) | 48 (17–122) | 989 (472–1516) | <0.001 | ||
| XOR activity (pmol/h/mL) | 57.2 (20.7–141.0) | 45.2 (19.3–98.8) | 104.0 (25.9–423.5) | <0.001 | ||
| Arterial blood gas | ||||||
| pH | — | 7.36 (7.26–7.44) | — | |||
| PO2 (mmHg) | — | 106 (79–161) | — | |||
| PCO2 (mmHg) | — | 37 (31–51) | — | |||
| HCO3 (mEq/L) | — | 21.7 (18.6–25.1) | — | |||
| BE (mmol/L) | — | −3.1 (−6.2–0.2) | — | |||
| Lactate (mmol/L) | — | 1.95 (1.40–3.48) | — | |||
| Medication | ||||||
| XOR inhibitors (yes, %) | 99 (28.4%) | 60 (26.0%) | 39 (33.1%) | 0.170 | ||
| Febuxostat (yes, %) | 91 (26.1%) | 53 (22.9%) | 38 (32.2%) | |||
| Allopurinol (yes, %) | 5 (1.4%) | 4 (1.7%) | 1 (0.8%) | |||
| Topiroxostat (yes, %) | 2 (0.6%) | 2 (0.9%) | 0 (0.0%) | |||
AHF, acute heart failure; BUN, blood urea nitrogen; CKD, chronic kidney disease; CRP, C‐reactive protein; HF, heart failure; LVEF, left ventricular ejection fraction measured by echocardiography; NYHA, New York Heart Association; XOR, xanthine oxidoreductase.
Figure 1.
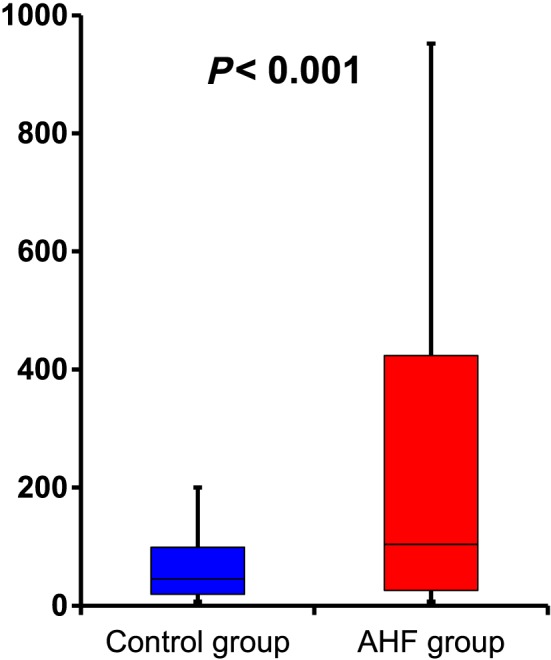
Plasma xanthine oxidoreductase activity in each group. The plasma xanthine oxidoreductase activity in the acute heart failure (AHF) group [104.0 (25.9–423.5) pmol/h/mL] was significantly higher than that in the control group [45.2 (19.3–98.8) pmol/h/mL; P < 0.001].
Plasma xanthine oxidoreductase activity in acute heart failure and control patients
The distribution of XOR activity in the control group and in AHF patients is illustrated in Figure 2 . For the control group (n = 231), plasma XOR activity was <25 pmol/h/mL in 71 (30.7%) patients and >300 pmol/h/mL in seven (3.0%). For the AHF group (n = 118), plasma XOR activity was <25 pmol/h/mL in 29 (24.6%) patients and >300 pmol/h/mL in 34 (28.8%). Multivariate logistic regression analysis revealed that serum UA levels (per 1.0 mg/dL increase, odds ratio: 1.280; 95% confidence interval: 1.066–1.536; P = 0.008) and lactate (per 1.0 mmol/L increase, odds ratio: 1.239; 95% confidence interval: 1.040–1.475; P = 0.016) were independently associated with high plasma XOR activity during the acute phase of AHF (Table 2).
Figure 2.
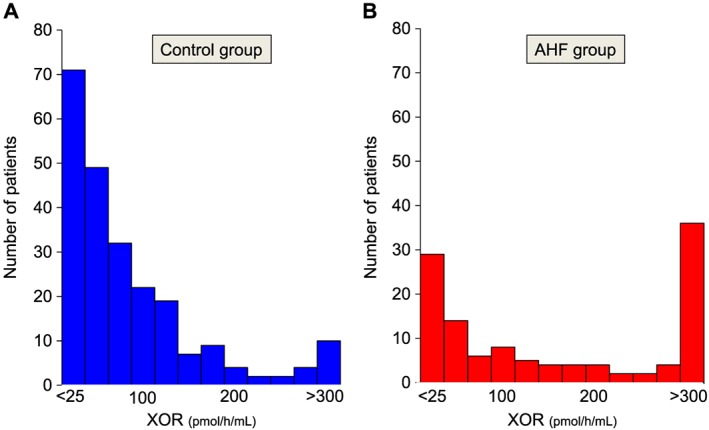
Distribution of plasma xanthine oxidoreductase (XOR) activity in each group. (A) In the control group, the plasma XOR activity was <25 pmol/h/mL in 71 (30.7%) patients and >300 pmol/h/mL in seven (3.0%). (B) In the acute heart failure (AHF) group, the plasma XOR activity was <25 pmol/h/mL in 29 (24.6%) patients and >300 pmol/h/mL in 34 (28.8%).
Table 2.
The multivariate logistic model of the associations xanthine oxidoreductase >300 pmol/h/min
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| OR | 95% CI | P value | OR | 95% CI | P value | |
| Influence factor | ||||||
| Uric acid (per 1.0 mg/dL increase) | 1.339 | 1.118–1.605 | 0.002 | 1.280 | 1.066–1.536 | 0.008 |
| Pre‐administration of XO‐I (yes) | 0.401 | 0.157–1.025 | 0.056 | |||
| LVEF (per 10% increase) | 0.695 | 0.519–0.931 | 0.015 | |||
| Creatinine (per 1.0 mg/dL increase) | 0.991 | 0.829–1.186 | 0.925 | |||
| Lactate (per 1.0 mmol/L increase) | 1.305 | 1.103–1.543 | 0.002 | 1.239 | 1.040–1.475 | 0.016 |
| BNP (per 10 pg/mL increase) | 0.999 | 0.994–1.004 | 0.580 | |||
CI, confidence interval; LVEF, left ventricular ejection fraction measured by echocardiography; OR, odds ratio; XO‐I, xanthine oxidase inhibitor.
Discussion
Xanthine oxidoreductase activity and oxidative stress in acute heart failure patients
Plasma XOR activity is a novel biomarker of metabolic disorders, developed in 2016 by the Sanwa Kagaku Kenkyusho group.15, 16 Their technique enables the stable measurement of tiny amounts of human XOR activity in vivo.14 However, the value of human XOR activity has not been previously evaluated in patients with severely decompensated AHF.
Xanthine oxidoreductase is expressed primarily in the liver and intestine, as well as in other major organs, including adipose tissue, vascular tissue, and the kidneys. XOR is sometimes used to refer to both XO and XDH, where XO, in particular, produces ROS that might induce organ dysfunction. XOR activity is enhanced by various stimuli, including inflammatory cytokines (i.e. interferon‐γ, interleukin‐1, and interleukin‐6), hypoxic stimulation, virus infection, ischaemia, and transplantation.8, 17 A previous study showed that a certain level of XOR was expressed on the vascular endothelium, wherein XO binds to glycosaminoglycan residues as a consequence of tissue damage such as ischaemia and inflammation.18
It is believed that the enhancement of XOR activity produces ROS and ultimately induces oxidative stress. These mechanisms adversely affect various types of disease conditions.
In the present study, the XOR activity of AHF patients was extremely high compared with cardiovascular outpatients. As oxidative stress was associated with enhanced XOR activity, this pathway may be one of the mechanisms by which oxidative stress is enhanced in AHF patients. Several other reports have suggested that oxidative stress is up‐regulated by other mechanisms in AHF patients.19, 20
Xanthine oxidoreductase values reported in previous studies
Even though plasma XOR activity is a new surrogate biomarker that reflects oxidative stress, some interesting articles on human plasma XOR activity have recently been published.9, 10, 11, 21, 22 The first report on plasma XOR activity in humans by Otaki et al. evaluated plasma XOR activity in 440 patients with chronic HF whose status was mainly NYHA class II.9 The median XOR activity was 73.2 pmol/h/mL, and chronic HF patients with high XOR activity tended to have a worse grade of NYHA and to experience adverse cardiovascular events.9 Washio et al. investigated plasma XOR activity in 29 healthy young volunteers and found the natural logarithmic XOR activity value to be 3.4 ± 0.8 pmol/h/mL.11 According to their analysis, XOR activity positively correlated with body mass index, serum UA levels, and high sensitive CRP, which is considered an important inflammatory biomarker. Terawaki et al. reported that the mean XOR activity in 13 patients with CKD was 23.1 ± 15.9 pmol/h/mL.22 Furthermore, Nakatani et al. reported that the mean XOR activity of 163 patients with end‐stage renal disease on haemodialysis was 21.4 ± 13.5 pmol/h/mL and demonstrated that plasma XOR activity was associated with diabetes mellitus, leading to the conclusion that it may be necessary to reduce the ROS induced by enhanced XOR activity in patients with diabetes mellitus who required haemodialysis.21 Finally, Fujimura et al. investigated the relationship between XOR activity and clinical features observed in patients with cardiac diseases. Among the 207 patients in a subgroup of their cohort who had not taken XOR inhibitors, the median XOR activity was approximately 36.1 pmol/h/mL, indicating higher liver enzyme and HbA1c levels in the group with high XOR activity in this population.10
In comparison with previous reports, the median value of XOR activity observed in the present study was extremely high at 104 pmol/h/mL. This suggests that patients with severely decompensated AHF suffered from excessive oxidative stress, which could have been caused by enhanced XOR activity during the acute phase.
Factors associated with high xanthine oxidoreductase activity in acute heart failure patients
In the present study, the multivariate logistic regression model showed that serum UA and lactate levels upon admission were independent predictors of high plasma XOR activity (≥300 pmol/h/mL) (Table 2). It is reasonable that patients with extremely high XOR activity were included among patients who also had high serum UA levels. Theoretically, XOR activity should be positively correlated with the serum UA levels because the production of UA requires activation of XOR. While the results of some recent studies support this hypothesis,11, 21, 23 other studies report conflicting results.9, 10, 22 Nevertheless, although XOR activity and serum UA levels were significantly correlated in the present study, the correlation coefficient was not definitive [r = 0.335 (Spearman), data not shown]. There are several possible explanations for these inconsistencies. First, patients with elevated serum UA levels included those with not only excessive production but also decreased excretion of UA. According to the current guidelines, hyperuricaemia caused by excretory failure accounts for approximately 60% of all cases of hyperuricaemia.24 Although XOR activity should theoretically increase in patients in whom UA is overproduced, patients with decreased excretion due to conditions such as advanced renal failure or cardiac dysfunction may not necessarily show elevated XOR activity. Indeed, we previously reported that the majority of AHF patients also had acute kidney injury and/or worsening renal function.25 Second, some of the previous studies excluded patients who had taken XOR inhibitors. In this present study, 18 (62.1%) of the 29 patients in the AHF group who had extremely low XOR activity (≤25 pmol/h/mL) also had a history of XOR inhibitor intake before admission.
Lactate levels were another factor independently associated with high plasma XOR activity (>300 pmol/h/mL). The lactate level is known to be positively correlated with the severity status of patients in intensive care. Authoritative guidelines, such as the Surviving Sepsis Campaign Guidelines, therefore strongly recommend a proactive approach based on the serum lactate value when managing patients with severe sepsis or septic shock.26 Lactate is mainly produced by anaerobic metabolism and is widely considered to be a marker of peripheral circulation insufficiency, including hypoxaemia. Systemic hypoxia due to pulmonary congestion and peripheral circulatory insufficiency caused by the central shift of the blood flow as a consequence of low cardiac output has been suggested as one of the pathophysiological mechanisms that could lead to the development of AHF. These situations would then result in the production of lactate and in subsequent elevation of serum lactate levels. Thus, the serum lactate level is an indicator of tissue hypoxia in patients with severely decompensated AHF.
There are some reasonable hypotheses as to why a high lactate level might be associated with XOR activity in AHF patients. First, tissue hypoxia directly induces the mobilization of XDH into the blood. Although XDH and XO distribution is still controversial, the primary source of XDH has been reported to be the liver and the intestine.27 XDH is released into the circulation from the liver and the intestine due to tissue ischaemia or hypoxaemia, and it is rapidly and irreversibly converted to XO by proteolysis.27 In fact, it has been previously reported that the XOR level is elevated in situations of tissue hypoxia.28 Second, adenosine triphosphate is produced by anaerobic metabolism in AHF due to tissue hypoxia; however, its levels are insufficient to maintain the diseased state, resulting in the broken down of such adenosine triphosphate. Lactate and hypoxanthine are also produced by this anaerobic pathway, with the latter accelerating this purine metabolism pathway, activating the enzymatic activity of XOR. Serum lactate, an indicator of issue hypoxia, is therefore a factor associated with the increase in XOR activity in AHF patients. Moreover, our results suggest the possibility that lactate directly induces the mobilization of XDH from the liver or the intestine into systemic circulation. Further research will be required to investigate the association between XOR activity and lactate.
The lactate level is not routinely evaluated in patients admitted in the general hospital wards. The present findings may serve useful for elucidating the mechanisms underlying XOR enhancement in patients with severely decompensated AHF requiring intensive care. Previous reports on XOR activity have only included non‐intensive care patients; thus, our study on intensive care patients has greater clinical impact and pioneers the investigation of human XOR activity.
Limitations
This study has several limitations. First, as it was a single‐centre study, some patient‐related biases might have been included. Second, our study cohort included patients administered XOR inhibitors at the time of admission. Sephadex G25 was used to remove small molecules such as xanthine and hypoxanthine, which are competitive inhibitors of stable isotope‐labelled [13C2, 15N2]‐xanthine in XOR activity assay, as well as the interfering drug molecules from plasma samples. However, the study enrolled patients who had been administrated medications that decreased UA, including allopurinol (n = 5), febuxostat (n = 91), and topiroxostat (n = 2). If any of these drugs remained in the samples, the XOR activity may have been underestimated. Furthermore, the time after the administration of XOR inhibitors is an important additional consideration. The percentages of these drugs that remained after exclusion with Sephadex G25 have not been reported. Although the results in patients who were treated with XOR inhibitors were almost the same as those of who did not receive XOR inhibitors (data not shown), further studies are required to investigate this issue. Third, in this paper, we did not consider the influence of other substances that may have affected XOR activity. Vitamins E and C, which are known antioxidants, have been reported to be inversely correlated with XO activity in healthy humans.29 Therefore, including patients who were taking antioxidant supplements could have affected the results of this study. Fourth, the patients enrolled in the control group were heterogeneous, with some having cardiovascular and non‐cardiovascular diseases. This heterogeneous population cohort might not have approximated for a control group. Indeed, there were seven (3.0%) patients with extremely high XOR (≥300 pmol/h/mL) activity in the control group, six of which had cardiovascular diseases, including end‐stage HF, prior myocardial infarction, hypertensive heart disease, myocarditis, and arrhythmia. Although this study's population is too small to be resolved by a statistical approach, we hypothesized that XOR activity was higher in patients with cardiovascular disease than in those with non‐cardiovascular disease. Further study regarding XOR activity in patients seen at the outpatient clinic will be required. Furthermore, because the patients were not completely enrolled consecutively, patient selection may have been biased. Fifth, representative markers of oxidative stress were not evaluated; thus, we were unable to show a direct relationship between XOR activity and oxidative stress in the present study. Finally, we did not present time‐dependent change in XOR activity throughout AHF treatment. This is essential to establish XOR activity as a biomarker influenced by emergency stress.
Conclusions
Plasma XOR activity was extremely high in patients with severely decompensated AHF. This would be associated with a high lactate value and would eventually lead to hyperuricaemia in patients with AHF.
Conflict of interest
None declared.
Funding
None.
Acknowledgements
We are grateful to the staff in the ICU and the medical records office at the Chiba Hokusoh Hospital, Nippon Medical School, for collecting the medical data. We are also grateful to the staff in the Hasegawa Hospital and Toho Kamagaya Hospital (Yuko Suzuki).
Okazaki H., Shirakabe A., Matsushita M., Shibata Y., Sawatani T., Uchiyama S., Tani K., Murase T., Nakamura T., Takayasu T., Asano M., Kobayashi N., Hata N., Asai K., and Shimizu W. (2019) Plasma xanthine oxidoreductase activity in patients with decompensated acute heart failure requiring intensive care, ESC Heart Failure, 6, 336–343. 10.1002/ehf2.12390.
References
- 1. Tamariz L, Harzand A, Palacio A, Verma S, Jones J, Hare J. Uric acid as a predictor of all‐cause mortality in heart failure: a meta‐analysis. Congest Heart Fail 2011; 17: 25–30. [DOI] [PubMed] [Google Scholar]
- 2. Huang H, Huang B, Li Y, Huang Y, Li J, Yao H, Jing X, Chen J, Wang J. Uric acid and risk of heart failure: a systematic review and meta‐analysis. Eur J Heart Fail 2014; 16: 15–24. [DOI] [PubMed] [Google Scholar]
- 3. Okazaki H, Shirakabe A, Kobayashi N, Hata N, Shinada T, Matsushita M, Yamamoto Y, Shibuya J, Shiomura R, Nishigoori S, Asai K, Shimizu W. The prognostic impact of uric acid in patients with severely decompensated acute heart failure. J Cardiol 2016; 68: 384–391. [DOI] [PubMed] [Google Scholar]
- 4. Okazaki H, Shirakabe A, Kobayashi N, Hata N, Shinada T, Matsushita M, Yamamoto Y, Shibata Y, Shibuya J, Shiomura R, Nishigoori S, Asai K, Shimizu W. Are atherosclerotic risk factors associated with a poor prognosis in patients with hyperuricemic acute heart failure? The evaluation of the causal dependence of acute heart failure and hyperuricemia. Heart Vessels 2017; 32: 436–445. [DOI] [PubMed] [Google Scholar]
- 5. Agarwal A, Banerjee A, Banerjee UC. Xanthine oxidoreductase: a journey from purine metabolism to cardiovascular excitation‐contraction coupling. Crit Rev Biotechnol 2011; 31: 264–280. [DOI] [PubMed] [Google Scholar]
- 6. Robert AM, Robert L. Xanthine oxido‐reductase, free radicals and cardiovascular disease. A critical review Pathol Oncol Res 2014; 20: 1–10. [DOI] [PubMed] [Google Scholar]
- 7. Battelli MG, Bolognesi A, Polito L. Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi‐tasking enzyme. Biochim Biophys Acta 2014; 1842: 1502–1517. [DOI] [PubMed] [Google Scholar]
- 8. Cantu‐Medellin N, Kelley EE. Xanthine oxidoreductase‐catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol 2013; 1: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Otaki Y, Watanabe T, Kinoshita D, Yokoyama M, Takahashi T, Toshima T, Sugai T, Murase T, Nakamura T, Nishiyama S, Takahashi H, Arimoto T, Shishido T, Miyamoto T, Kubota I. Association of plasma xanthine oxidoreductase activity with severity and clinical outcome in patients with chronic heart failure. Int J Cardiol 2017; 228: 151–157. [DOI] [PubMed] [Google Scholar]
- 10. Fujimura Y, Yamauchi Y, Murase T, Nakamura T, Fujita SI, Fujisaka T, Ito T, Sohmiya K, Hoshiga M, Ishizaka N. Relationship between plasma xanthine oxidoreductase activity and left ventricular ejection fraction and hypertrophy among cardiac patients. PLoS One 2017; 12: e0182699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Washio KW, Kusunoki Y, Murase T, Nakamura T, Osugi K, Ohigashi M, Sukenaga T, Ochi F, Matsuo T, Katsuno T, Moriwaki Y, Yamamoto T, Namba M, Koyama H. Xanthine oxidoreductase activity is correlated with insulin resistance and subclinical inflammation in young humans. Metabolism: clinical and experimental 2017; 70: 51–56. [DOI] [PubMed] [Google Scholar]
- 12. Gheorghiade M, Zannad F, Sopko G, Klein L, Pina IL, Konstam MA, Massie BM, Roland E, Targum S, Collins SP, Filippatos G, Tavazzi L, International Working Group on Acute Heart Failure S . Acute heart failure syndromes: current state and framework for future research. Circulation 2005; 112: 3958–3968. [DOI] [PubMed] [Google Scholar]
- 13. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, Authors/Task Force M , Document R . 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18: 891–975. [DOI] [PubMed] [Google Scholar]
- 14. Murase T, Nampei M, Oka M, Miyachi A, Nakamura T. A highly sensitive assay of human plasma xanthine oxidoreductase activity using stable isotope‐labeled xanthine and LC/TQMS. J Chromatogr B Analyt Technol Biomed Life Sci 2016; 1039: 51–58. [DOI] [PubMed] [Google Scholar]
- 15. Murase T, Nampei M, Oka M, Ashizawa N, Matsumoto K, Miyachi A, Nakamura T. Xanthine oxidoreductase activity assay in tissues using stable isotope‐labeled substrate and liquid chromatography high‐resolution mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2016; 1008: 189–197. [DOI] [PubMed] [Google Scholar]
- 16. Murase T, Oka M, Nampei M, Miyachi A, Nakamura T. A highly sensitive assay for xanthine oxidoreductase activity using a combination of [(13) C2,(15) N2]xanthine and liquid chromatography/triple quadrupole mass spectrometry. J Labelled Comp Radiopharm 2016; 59: 214–220. [DOI] [PubMed] [Google Scholar]
- 17. Page S, Powell D, Benboubetra M, Stevens CR, Blake DR, Selase F, Wolstenholme AJ, Harrison R. Xanthine oxidoreductase in human mammary epithelial cells: activation in response to inflammatory cytokines. Biochim Biophys Acta 1998; 1381: 191–202. [DOI] [PubMed] [Google Scholar]
- 18. Kelley EE. Dispelling dogma and misconceptions regarding the most pharmacologically targetable source of reactive species in inflammatory disease, xanthine oxidoreductase. Arch Toxicol 2015; 89: 1193–1207. [DOI] [PubMed] [Google Scholar]
- 19. Ayoub KF, Pothineni NVK, Rutland J, Ding Z, Immunity MJL. Inflammation, and oxidative stress in heart failure: emerging molecular targets. Cardiovasc Drugs Ther 2017; 31: 593–608. [DOI] [PubMed] [Google Scholar]
- 20. Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal 2006; 8: 1737–1744. [DOI] [PubMed] [Google Scholar]
- 21. Nakatani A, Nakatani S, Ishimura E, Murase T, Nakamura T, Sakura M, Tateishi Y, Tsuda A, Kurajoh M, Mori K, Emoto M, Inaba M. Xanthine oxidoreductase activity is associated with serum uric acid and glycemic control in hemodialysis patients. Sci Rep 2017; 7: 15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terawaki H, Murase T, Nakajima A, Aoyagi K, Fukushima N, Tani Y, Nakamura T, Kazama JJ. The relationship between xanthine oxidoreductase and xanthine oxidase activities in plasma and kidney dysfunction. J Clin Exp Nephrol 2017; 1: 31. [Google Scholar]
- 23. Furuhashi M, Matsumoto M, Tanaka M, Moniwa N, Murase T, Nakamura T, Ohnishi H, Saitoh S, Shimamoto K, Miura T. Plasma xanthine oxidoreductase activity as a novel biomarker of metabolic disorders in a general population. Circ J 2018; 82: 1892–1899. [DOI] [PubMed] [Google Scholar]
- 24. Teramoto T, Sasaki J, Ueshima H, Egusa G, Kinoshita M, Shimamoto K, Daida H, Biro S, Hirobe K, Funahashi T, Yokote K, Yokode M, Japan Atherosclerosis Society Committee for E , Clinical Management of A . Diagnostic criteria for dyslipidemia. Executive summary of Japan Atherosclerosis Society (JAS) guideline for diagnosis and prevention of atherosclerotic cardiovascular diseases for Japanese. J Atheroscler Thromb 2007; 14: 155–158. [DOI] [PubMed] [Google Scholar]
- 25. Shirakabe A, Hata N, Kobayashi N, Okazaki H, Matsushita M, Shibata Y, Nishigoori S, Uchiyama S, Asai K, Shimizu W. Worsening renal function definition is insufficient for evaluating acute renal failure in acute heart failure. ESC Heart Fail 2018; 5: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, Rochwerg B, Rubenfeld GD, Angus DC, Annane D, Beale RJ, Bellinghan GJ, Bernard GR, Chiche JD, Coopersmith C, De Backer DP, French CJ, Fujishima S, Gerlach H, Hidalgo JL, Hollenberg SM, Jones AE, Karnad DR, Kleinpell RM, Koh Y, Lisboa TC, Machado FR, Marini JJ, Marshall JC, Mazuski JE, McIntyre LA, McLean AS, Mehta S, Moreno RP, Myburgh J, Navalesi P, Nishida O, Osborn TM, Perner A, Plunkett CM, Ranieri M, Schorr CA, Seckel MA, Seymour CW, Shieh L, Shukri KA, Simpson SQ, Singer M, Thompson BT, Townsend SR, Van der Poll T, Vincent JL, Wiersinga WJ, Zimmerman JL, Dellinger RP. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med 2017; 45: 486–552. [DOI] [PubMed] [Google Scholar]
- 27. Day RO, Kamel B, Kannangara DR, Williams KM, Graham GG. Xanthine oxidoreductase and its inhibitors: relevance for gout. Clin Sci (Lond) 2016; 130: 2167–2180. [DOI] [PubMed] [Google Scholar]
- 28. Friedl HP, Smith DJ, Till GO, Thomson PD, Louis DS, Ward PA. Ischemia‐reperfusion in humans. Appearance of xanthine oxidase activity. Am J Pathol 1990; 136: 491–495. [PMC free article] [PubMed] [Google Scholar]
- 29. Shah AA, Khand F, Khand TU. Effect of smoking on serum xanthine oxidase, malondialdehyde, ascorbic acid and alpha‐tocopherol levels in healthy male subjects. Pak J Med Sci 2015; 31: 146–149. [DOI] [PMC free article] [PubMed] [Google Scholar]