

Abstract
Aims
Cardiac myosin light chain kinase (cMLCK) phosphorylates ventricular myosin regulatory light chain 2 (MLC2v) and regulates sarcomere and cardiomyocyte organization. However, few data exist regarding the relationship between cMLCK mutations and MLC2v phosphorylation, particularly in terms of developing familial dilated cardiomyopathy (DCM) in whom cMLCK gene mutations were identified. The purpose of the present study was to investigate functional consequences of cMLCK mutations in DCM patients.
Methods and results
The diagnosis of DCM was based on the patients' history and on echocardiography. We screened cMLCK gene mutations in DCM probands with high resolution melting analysis. Known DCM‐causing genes mutations were excluded by exome sequencing of family members. MLC2v phosphorylation was analysed by Phos‐tag sodium dodecyl sulfate–polyacrylamide gel electrophoresis assays. We also performed ADP‐Glo assays for determining the total amount of adenosine triphosphate used in the kinase reaction. Unrelated DCM probands (109 males and 40 females) were enrolled in this study, of which 16 were familial and 133 sporadic. By mutation screening, a truncation variant of c1915‐1 g>t (p.Pro639Valfs*15) was identified, which was not detected in 400 chromosomes of 200 healthy volunteers; it is listed in the Human Genetic Variation Database with an allele frequency < 0.001. In the proband, the presence of mutations in known DCM‐causing genes was excluded with exome analysis. Familial analysis identified a 19‐year‐old male carrier who manifested slight left ventricular dilation with preserved systolic function. Phosphorylation assays analysed by Phos‐tag SDS‐PAGE revealed that the identified p.Pro639Valfs*15 mutation results in a complete lack of kinase activity, although it did not affect wild‐type cMLCK activity. ADP‐Glo assays confirmed that the mutant cMLCK had no kinase activity, whereas wild‐type cMLCK had a Km value of 5.93 ± 1.47 μM and a V max of 1.28 ± 0.03 mol/min/mol kinase.
Conclusions
These results demonstrate that a truncation mutation in the cMLCK gene p.Pro639Valfs*15 can be associated with significant impairment of MLC2v phosphorylation and possibly with development of DCM, although a larger study of DCM patients is required to determine the prevalence of this mutation and further strengthen its association with disease development.
Keywords: Dilated cardiomyopathy, Genetics, MYLK3
Introduction
Dilated cardiomyopathy (DCM) includes a large group of heterogeneous myocardial disorders characterized by ventricular dilation and depressed myocardial contractility.1, 2 Although DCM is a leading cause of refractory heart failure, no fundamental therapy exists due to extremely heterogeneous genetic aetiologies. Indeed, over 40 genes have been reported as a cause of DCM, resulting in the identification of disease‐causing mutations in 20% of DCM probands.3 Nonetheless, continued efforts to identify novel disease‐causing genes are important because they provide valuable information on novel disease pathways and functional relevance that may contribute to novel therapeutic strategies.4
Recently, cardiac myosin light chain kinase (cMLCK), which phosphorylates ventricular myosin regulatory light chain 2 (MLC2v) and regulates sarcomere and cardiomyocyte organization,5, 6 was cloned from the myocardia of patients with heart failure.5 Interestingly, in animal models, knockout mice show reduced MLC2v phosphorylation and DCM‐like phenotypes such as reduced left ventricular (LV) systolic function and LV dilation.7, 8, 9, 10 In humans, reduced MLC2v phosphorylation was observed in LV biopsy samples from end‐stage heart failure including DCM.11, 12, 13 However, few data exist regarding the information for the relationship between cMLCK mutations and MLC2v phosphorylation, despite the fact that cMLCK gene (MYLK3) mutations have been identified in DCM pedigrees.14 Therefore, we screened for mutations in the cMLCK gene and investigated the functional consequences of cMLCK mutations in DCM patients.
Methods
Study populations and clinical evaluations
This investigation conforms with the principles outlined in the Declaration of Helsinki.15 The study was approved by the Bioethical Committee on Medical Research, Kanazawa University. Informed consent was obtained from all subjects or their guardians.
Standard M‐mode and two‐dimensional echocardiographic studies were performed to identify and quantify morphological features of the LV. The LV ejection fraction was calculated using Teicholtz's method.16 The diagnosis of DCM was based on the patients' history and on echocardiographic demonstration of a dilated and thinned LV with decreased systolic dysfunction in the absence of coronary artery disease.1
Mutation screening and confirmation
Genomic DNA was isolated from peripheral white blood cells of all subjects using the DNA extractor 341 Nucleic Acid Purification System (GENEPURE, PE Biosystems) or a Wizard Genomic DNA Purification Kit (Promega). Exon coding sequences and exon‐intron boundaries for MYLK3 were amplified by PCR. The primers are shown in Table 1. High resolution melting analysis (LightScanner System, Idaho Technology) was used for screening mutations. We performed direct sequencing using an ABI PRISM 310 Genetic Analyzer (Applied Biosystems) when irregular profiles were detected. The presence or absence of mutations was confirmed by digestion of PCR products with restriction enzyme Bccl. This method was also used to determine the genotype of DNA from family members of the probands and 400 chromosomes of 200 healthy individuals without cardiovascular disease.
Table 1.
Primers used for amplifying MYLK3 exons
| Exon | Sequence (5′‐3′) | Annealing temperature (°C) | |
|---|---|---|---|
| 2 | F | AGCTGGGCGCCTCCTCTTT | 64–67 (touchdown) |
| R | CCTGGCATCAGACTGCACC | ||
| 5 | F | GTGCCGGGAGACCTGGGTTTGA | 64–67 (touchdown) |
| R | CCTGCCCCGTGACTCCTGCTCTAA | ||
| 6 | F | TTCGGGATAGCCAGCAGGGTTTTG | 62 |
| R | AGGCCCCACAGGCAGCTCTACAGA | ||
| 7 | F | GTGGGGCCTGGGGCAAGAAGGT | 64 |
| R | GCCGCCCCGCCTCTGATACTCA | ||
| 8 | F | CTACCCGGGCTGGCCTCTTTTG | 67 |
| R | GTGGCCTGTCCCAGACTCCAGAAC | ||
| 9 | F | CCAGCCCCTTCATGTATTTCCATT | 67 |
| R | AACAAGGCCTGTCTGCAAGTCAAA | ||
| 10 | F | AGCCCCAGCACCACATCTCC | 70 |
| R | CCTTCCCTGGCTCCTTTCCTTC | ||
| 11 | F | CCGCTTCCTCCCTTTAATGAACA | 69 |
| R | TGCCCAGCTCCCAGACCTG | ||
To identify an aberrant transcript that resulted from the splice acceptor site mutation, total RNA was isolated from lymphocytes using a BiOstic® Blood Total RNA Isolation Kit (MO BIO Laboratories). RNA was subjected to reverse transcription, and the DNA products were amplified by PCR. The primers were forward: 5′‐ggaccgggaggacgtgaagaac‐3′; reverse: 5′‐aagtccttggcctcctccgagag‐3′.
Exome sequencing
Whole exome sequencing of the proband and a clinically unaffected sibling were performed. Genomic DNA was fragmented using a DNA shearing system (Covaris Inc.). Genomic DNA was captured using the SureSelectXT Human All Exon V4 (51 Mb) kit (Agilent Technologies) and sequenced on a HiSeq 2000 (Illumina) with 100 bp paired‐end reads. The reads were mapped to the hg19 human reference using BWA‐0.6.1.17 Optical or PCR duplicates were removed using Picard.18 Single nucleotide variants (SNVs) and insertions and/or deletions were called using the Genome Analysis Toolkit (GATK) v. 1.6–13.19, 20 Variants were annotated using ANNOVAR.21
Non‐synonymous SNVs, splice site mutations, and insertions and/or deletions were retained and filtered for a minor allele frequency < 0.001 in the dbSNP135, 1000 Genomes database, the NHLBI Exome Sequencing Project (ESP6500), and the Human Genetic Variation Database22 for discovery of rare de novo mutations. Variants potentially affecting protein function, including non‐synonymous variants, frameshifts in the coding sequence, or variants potentially affecting splicing were analysed. Variants were filtered against the quality of exome sequencing, rarity, functional significance predicted high impact by SnpEff23, and segregation. Further filtering using Ingenuity Pathway Analysis software (Ingenuity Systems) was performed to examine the association with cardiac function and/or structure.
Purification of recombinant cardiac myosin light chain kinase proteins from HEK293T cells
Human MYLK3 cDNA was cloned using pENTR/D‐TOPO Cloning Kits (Invitrogen). Mutant constructs of p.Pro639Valfs*15 were subsequently introduced by primer‐derived mutagenesis. The primers for mutant cMLCK were forward: 5′‐gtacaagcctcgagagaagctgaaggtgaac‐3′; reverse: 5′‐cttgaggtccaggtgcaggatgtagtgctggt‐3′. Wild‐type and mutant plasmids were recombined into N terminal FLAG destination vectors (pEF‐DEST51/nFLAG plasmid) using GATEWAY LR recombinase (Invitrogen). HEK293T cells transfected with wild‐type cMLCK or mutant cMLCK vectors were lysed in lysis buffer (10 mM Tris–HCl, pH 7.2, 0.15 M NaCl, 1% NP40, 1 mM EDTA, and protease inhibitor cocktail) and immunoprecipitated with anti‐FLAG M2 agarose (Sigma‐Aldrich) at 4°C for 30 min. The beads were washed three times with washing buffer (10 mM Tris–HCl, pH 7.2, 0.3 M NaCl, 1% NP40, and 1 mM EDTA) and eluted with elution buffer (10 mM Tris–HCl, pH 7.2, 0.15 M NaCl, 1% NP40, 1 mM EDTA, and 0.05 mg/mL FLAG peptide) at 4°C for 30 min. After centrifugation, the supernatants were used as recombinant FLAG‐tagged proteins.
Purification of recombinant regulatory light chain proteins from Escherichia coli
To prepare substrate proteins, cDNA fragments encoding human MLC2v (gene accession no. NM_000432) was PCR amplified using cDNA from heart. The primers for MLC2v were as follows: the forward, 5′‐caccatggcacctaagaaagcaaagaagagagcc‐3′; the reverse, 5′‐ctagtccttctcttctccgtgggtgatgat‐3′. Amplified cDNA was subcloned into pENTR/D‐TOPO vector and inserted into the pDEST17 vector by Gateway Technology System (Invitrogen) according to the manufacturer's protocol. The pDEST17 constructs possessing each regulatory light chain sequence was transformed into the BL21 chemically competent Escherichia coli. A 5 mL culture using lysogeny broth containing 100 μg/mL of ampicillin was grown overnight shaking at 37°C and used to inoculate 200 mL of lysogeny broth containing ampicillin (in a 1 L flask) until the culture optical density (600 nm) reaches at 0.5. Arabinose was added at final concentration of 0.2% and subsequent culturing for 3 h at 37°C. Cells were harvested by centrifugation at 5000 g at 4°C for 10 min, and resulting cell pellet was resuspended in 10 mL of BugBuster Master Mix (Merck Millipore) containing EDTA‐free protease inhibitor cocktail and rotated at room temperature for 20 min. The inclusion body that contains N‐terminus His‐tagged MLC2v protein was isolated from the supernatant by centrifugation at 16 000 g at 4°C for 10 min.
The resulting inclusion body was washed with two‐fold diluted BugBuster Master Mix one time and 10‐fold diluted BugBuster Master Mix twice. The inclusion body pellet was solubilized in immobilized metal ion affinity chromatography binding buffer [10 mM HEPES (pH 7.4), 0.5 M NaCl, 1 mM MgCl2, and 6 M urea] by repeatedly passing the inclusion body through an 18 g syringe and incubated at 4°C for 1 h. After centrifugation at 20 000 g at 4°C for 20 min, any insoluble material was centrifuged out. The resulting resuspended protein solution was loaded onto a column of TALON Metal Affinity Resin (Clontech) equilibrated with immobilized metal ion affinity chromatography binding buffer at 4°C. The bound His‐tagged MLC2v protein was eluted with elution buffer [50 mM sodium phosphate (pH 8.0), 0.3 M NaCl, 0.1% CHAPS, and 0.15 M imidazole], refolded, and concentrated by centrifugation at 5000 g at 4°C using centrifugal filter (Amicon Ultra‐15, Millipore) and stored at −80°C until use.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
The stacking gel was composed of 12% (wt/vol) acrylamide, 0.1% (wt/vol) sodium dodecyl sulfate (SDS), 125 mM Tris–HCl (pH 6.8), 0.1% (wt/vol) ammonium persulfate, and 0.5% (vol/vol) N,N,N′,N′‐tetramethylethylenediamine. The resolving gel was composed of 12% (wt/vol) acrylamide, 0.1% (wt/vol) SDS, 375 mM Tris–HCl (pH 8.8), 0.05% (wt/vol) ammonium persulfate, and 0.25% (wt/vol) N,N,N′,N′‐tetramethylethylenediamine. Electrophoresis was performed in 0.1% (wt/vol) SDS, 25 mM tris, and 192 mM glycine at 180 V per gel for 50 min. Proteins were transferred to poly(vinylidene difluoride) membranes (0.2 μm; Bio‐Rad) at 15 V for 30 min in transfer buffer. Membranes were incubated with 3% (wt/vol) non‐fat dry milk in Tris‐buffered saline containing Tween [TTBS; 50 mM Tris–HCl (pH 7.8), 150 mM NaCl, and 0.1% (vol/vol) Tween 20] for 1–2 h and then in anti‐FLAG M2‐HRP antibody (1:4,000; Sigma‐Aldrich, A8592) in TTBS for 2 h at room temperature. After washing with TTBS (three times for 5 min each), membranes were incubated with the ECL Western Blotting Detection Reagent (GE Healthcare). The emitted light was detected and quantified with a chemiluminescence imaging analyzer (LAS4000, Fujifilm), and images were analysed with Adobe Photoshop Elements 13.
In vitro kinase activity
Kinase activities were assayed in 20 mM HEPES (pH 7.5), 1 mM CaCl2, 5 mM MgCl2, 2 mM dithiothreitol, 150 μM ATP, 0.01% Tween 20, and 150 nM calmodulin with 2.5 or 5 nM purified FLAG‐tagged MLCKs and indicated concentration of purified nHis‐MLC2v in 40 μL total volume. Reaction mixtures were pre‐incubated for 5 min, and the kinase reactions were started by the addition of ATP and incubated for 1 or 3 h at 25°C. Reactions were terminated, and kinase activities were measured by Phos‐tag sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) or ADP‐Glo assay. For measurement of MLC2v Km values of wild‐type and mutant cMLCK, 5 nM kinase were assayed with 0.625–20 μM nHis‐MLC2v. Km values were calculated by non‐linear fit to the Michaelis–Menten equation by using Igor Pro software.
Phos‐tag sodium dodecyl sulfate–polyacrylamide gel electrophoresis
For analysis of MLC2v phosphorylation, samples of kinase reactions were subjected to phosphate affinity SDS‐PAGE using an acrylamide‐pendant phosphate‐binding tag (Phos‐tag SDS‐PAGE24). The stacking gel was composed of 12% (wt/vol) acrylamide, 0.1% (wt/vol) SDS, 125 mM Tris–HCl (pH 6.8), 0.1% (wt/vol) ammonium persulfate, and 0.5% (vol/vol) N,N,N′,N′‐tetramethylethylenediamine. The resolving gel was composed of 12% (wt/vol) acrylamide, 30 μM Phos‐tag acrylamide (Wako), 60 μM MnCl2, 0.1% (wt/vol) SDS, 375 mM Tris–HCl (pH 8.8), 0.05% (wt/vol) ammonium persulfate, and 0.25% (wt/vol) N,N,N′,N′‐tetramethylethylenediamine. Electrophoresis was performed in 0.1% (wt/vol) SDS, 25 mM Tris, and 192 mM glycine at 150 V per gel for 80 min. After electrophoresis, the gel was soaked in EDTA (+) transfer buffer [10 mM EDTA, 50 mM Tris, 380 mM glycine, 0.000375% (wt/vol) SDS, and 20% (wt/vol) EtOH] for 10 min and then in transfer buffer for 10 min. Proteins were transferred to poly(vinylidene difluoride) membranes (0.2 μm; Bio‐Rad) at 15 V for 30 min in transfer buffer. Membranes were then blocked with non‐fat dry milk, incubated with primary antibody (anti‐MLC2v 1:4000, Abcam ab92721) followed by secondary antibody (HRP‐coupled goat anti‐rabbit 1:8000; Cappel, #55696) and detection as with SDS‐PAGE. Phosphorylated and non‐phosphorylated MLC2v were quantified by densitometry using ImageJ software.25
ADP‐Glo kinase assay
Kinase activity was measured by quantifying the amount of adenosine diphosphate (ADP) produced during a kinase reaction using the ADP‐Glo Kinase Assay (Promega) according to the manufacturer's protocol. In brief, the kinase reactions were stopped by adding 40 μL of ADP‐Glo reagent. After incubation at room temperature for 40 min, 80 μL of kinase detection reagent was added per well and incubated for 30 min. Luminescence was measured using the chemiluminescence imaging analyzer TriStar2 LB942 (Berthold) with an integration time of 0.3 s/well. The luminescence intensity was converted to ADP amount using calibration curve. The amount of phosphates used for MLC2v phosphorylation was calculated as follows: total ADP produced during kinase reaction was measured as earlier; background ADP including cMLCK autophosphorylation was measured by the reaction without MLC2v; and the amount of ADP used for MLC2v phosphorylation was assessed by subtracting background ADP from total ADP.
Data analysis
Statistical analyses were performed using SPSS statistics software version 22 (IBM). Data were analysed using one‐way analysis of variance and Bonferroni post hoc testing. A P value below 0.01 was considered statistically significant.
Results
Genetics
One hundred and forty‐nine unrelated DCM probands (109 male and 40 female) were enrolled in this study. There were 16 familial and 133 sporadic DCM cases. Among these patients, we identified one SNV at the splice acceptor site of exon 9 (c.1915‐1G>A; p.Pro639Valfs*15 in DCM‐155) (Table 2 and Figure 1 ). This variant was not detected in 400 chromosomes of 200 healthy volunteers and is listed in the Human Genetic Variation Database15 with an allele frequency < 0.001. Sequence analysis of the reverse transcription PCR product derived from mRNA demonstrated that p.Pro639Valfs*15 caused exon skipping and created a premature terminal codon (Figure 1 B and C) that is predicted to result in protein truncation. Pro639 locates to the kinase active site of the serine/threonine kinase domain (Figure 2 ).
Table 2.
Clinical characteristics of pedigrees of DCM‐155
| Member | Genotype | Age | Gender | Echocardiographic data | ECG findings | |||
|---|---|---|---|---|---|---|---|---|
| LVDd/Ds | LVEF | IVS/PW | LAD | |||||
| (mm) | (%) | (mm) | (mm) | |||||
| II‐1 | Hetero | 49 | M | 70/55 | 42 | 8/9 | 42 | IVCD, mild ST‐T change |
| II‐3 | WT | 47 | M | 50/33 | 63 | 10/10 | 38 | Early repolarization |
| III‐1 | WT | 24 | M | 51/35 | 59 | 10/10 | 31 | WNL |
| III‐2 | WT | 22 | M | 51/36 | 56 | 10/10 | 31 | WNL |
| III‐3 | WT | 21 | F | 50/34 | 60 | 9/9 | 33 | PAC |
| III‐4 | Hetero | 19 | M | 55/36 | 63 | 8/8 | 31 | CR, IVCD, mild ST‐T change |
CR, clockwise rotation; DCM, dilated cardiomyopathy; Ds, end‐systolic dimension; ECG, electrocardiogram; IVCD, intraventricular conduction delay; IVS, intraventricular septum; PW, posterior wall; LAD, left atrial diameter; LVDd, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; PAC, premature atrial contraction; WNL, within normal limits; WT, wild type.
Figure 1.
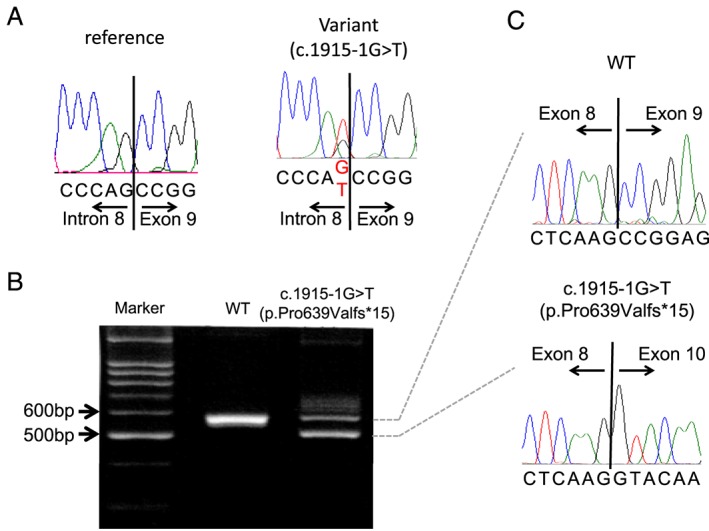
MYLK3 variant identified in a patient with dilated cardiomyopathy (DCM). (A) Sequence trace of the MYLK3 variant p.Pro639Valfs*15 (DCM‐155). (B and C) Reverse transcription PCR and sequence analysis of the reverse transcription PCR product derived from mRNA of the DCM‐155 proband and a control showing out‐frame skipping of exon 9 (71 bp) caused by the c.1915‐1g>t mutation. WT, wild type.
Figure 2.
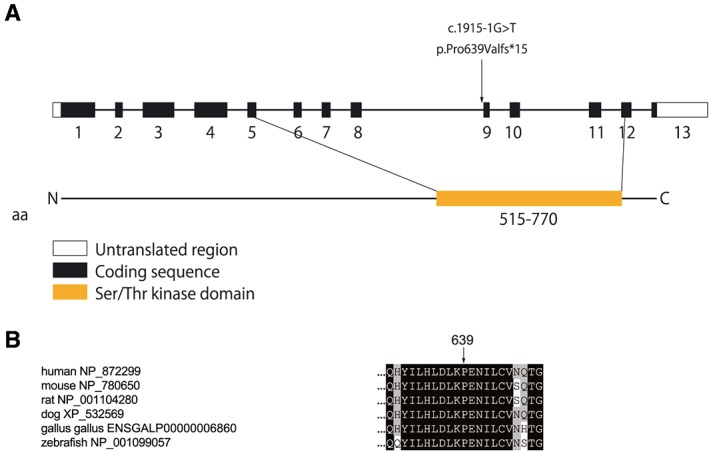
MYLK3 variant and scheme of cardiac myosin light chain kinase structure. (A) Schematic of the genomic structure of human MYLK3 and functional domains of myosin light chain kinase. p.Pro639Valfs*15 mutations locate in the serine/threonine kinase domain. (B) Protein sequence alignment of cardiac myosin light chain kinase in vertebrates. Residue 639 is highly conserved across species. DCM, dilated cardiomyopathy.
The family analysis of patient DCM‐155 revealed a 19‐year‐old male carrier (DCM‐155; III‐4) with borderline dilation and preserved ejection fraction of the LV on echocardiography and ST‐T abnormalities on electrocardiography (Figure 3 and Table 2), whereas all non‐carriers showed normal echocardiographic and electrocardiographic findings. The mother of the proband (DCM‐155; I‐2) had died from gastric cancer at the age of 35.
Figure 3.
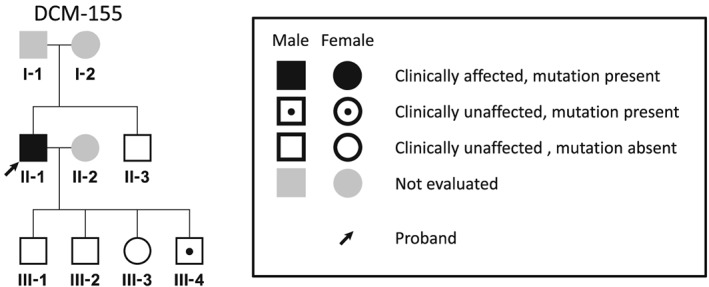
Pedigree of the proband identified with MYLK3 mutations. Familial analysis was performed in the pedigree of DCM‐155. A 19‐year‐old male carrier (III‐4) showed dilation of the left ventricle on echocardiography and ST‐T abnormalities on electrocardiography, although these findings did not meet clinical criteria for DCM. All non‐carriers had the normal ventricular size on echocardiographic and also had normal electrocardiographic findings. DCM, dilated cardiomyopathy.
To exclude the possibility of the presence of mutations in known DCM‐causing genes in the DCM‐155 pedigree, we performed whole exome sequencing in the proband (II‐1) and his clinically unaffected sibling (II‐3) (Table 3). This revealed 80 variants from 74 genes that were not shared between these two subjects. However, none of the 74 genes has been reported as a DCM disease‐causing gene or implicated in other forms of cardiomyopathies. Ingenuity Pathway Analysis software (Ingenuity Systems) showed that only 2 of the 74 genes (STAB1 and AKAP9) were associated with cardiac function and/or structure. STAB1 encodes stabilin 1, a multifunctional scavenger receptor selectively expressed in tissue macrophages and sinusoidal endothelial cells, and is reported to be a candidate gene for bipolar disorder.26 AKAP9 mutations were reported as a cause of long QT syndrome.27 Thus, the presence of mutations in known DCM‐causing genes was excluded in the pedigree of DCM‐155, suggesting that MYLK3 p.Pro639Valfs*15 could be a disease‐causing mutation.
Table 3.
Whole exome sequencing analyses in the pedigree of DCM‐155
| Proband (DCM‐155; II‐1) | Sibling (DCM‐155; II‐3) | |
|---|---|---|
| All variants | 85 682 | 85 982 |
| Quality passed | 52 681 | 53 126 |
| Non‐synonymous mutation, not in dbSNV151, and <0.5% in 1000 genome | 1091 | 1107 |
| Deleterious variants (predicted high impact by SnpEff) | 169 | 178 |
| Segregation | 80 variants (in 74 genes) | |
| ABCA2a, ACAD9, AKAP9, AKD1a, ATAD3B, ATG4B, C1R, CAMKK2, CAPN8, CCDC17, CD151, CDH23, CHL1a, CLTC, CLTCL1, CNKSR1, D2HGDHa, DCHS2a, DNAH17, DNM1L, DTL, EPSTI1, EWSR1, EYS, FAM81B, FAP, HLA‐DQA1, HLA‐DQB1, HLA‐DRB1, HPS3, HYOU1, ITPR1, KALRN, KIAA0430, KIR3DX1, LAPTM4B, LCMT2, LDLRAP1, LILRB5, LRP5L, LTF, MICAL1, MON2, MYLK3, MYO7B, NADK, NEDD4, NFIA, NHLRC3, NLRP8, NOTCH2, NPHP1, OLFM1, OXCT1, PDC, PDE4DIP, PNPLA7, PTGES, RGS11, RNF212, SAT1, SCARF2, SELPLG, SERINC4, SLC25A25, SNF8, SPEF2, STAB1a, TUBGCP6, UNC50, VLDLR, WDR90, ZDHHC11, ZNF527 | ||
| On IPA gene list | Three variants in two genes (STAB1a and AKAP9) | |
DCM, dilated cardiomyopathy; IPA, Ingenuity Pathway Analysis.
Two variants in one gene.
Functional analysis
To assess the functional consequences of possessing this variant, we examined kinase activity in vitro. Purified nHis‐tagged MLC2v proteins from E. coli were phosphorylated by purified nFlag‐tagged wild‐type or p.Pro639Valfs*15 cMLCK in vitro. We used Phos‐tag SDS‐PAGE24 to separate phosphorylated and non‐phosphorylated MLC2v. Importantly, the kinase activity of the mutant p.Pro639Valfs*15‐cMLCK was completely abrogated, although wild‐type MLCK showed kinase activity in a dose‐dependent manner (Figure 4 A and B). Kinase assays with a mixture of wild‐type and mutant cMLCK showed that mutant cMLCK did not inhibit the former (Figure 4 C and D).
Figure 4.
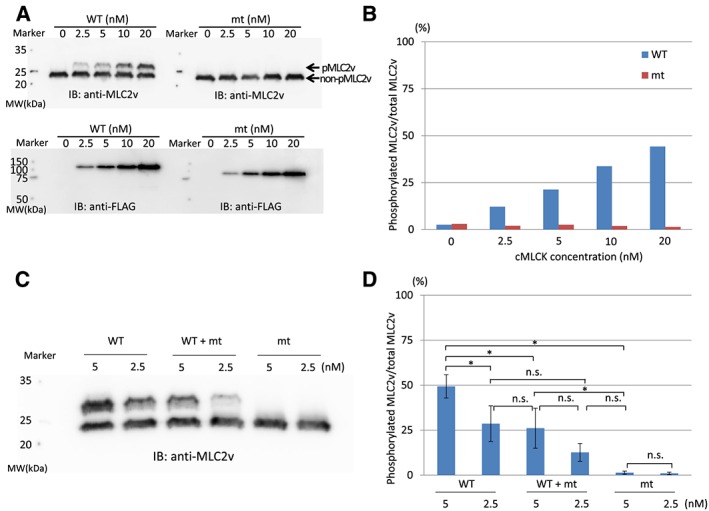
Phosphorylation assays analysed by Phos‐tag sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE). (A) Purified nHis‐tagged ventricular myosin regulatory light chain 2 (MLC2v) proteins were phosphorylated by purified nFlag‐tagged wild‐type (WT) or p.Pro639Valfs*15 cardiac myosin light chain kinase (cMLCK) in vitro at 25°C for 1 h. Phosphorylated MLC2v (pMLC2v) and non‐phosphoryated MLC2v (non‐pMLC2v) were then separated by Phos‐tag SDS‐PAGE followed by immunoblotting for MLC2v. Note that MLC2v is phosphorylated in a dose‐dependent manner only by WT cMLCK. Mutant (mt) cMLCK has completely lost kinase activity. (B) Quantitative analysis also showed that MLC2v is phosphorylated in a dose‐dependent manner only by WT cMLCK. (C) Phos‐tag SDS‐PAGE of WT cMLCK, mixture of WT and mt cMLCK, and mt cMLCK. In vitro kinase assay by cMLCK was performed at 25°C for 3 h; mt cMLCK did not affect WT cMLCK activity. (D) Densitometries of Phos‐tag SDS‐PAGE. WT + mt, the same amount of wild‐type and mutant cMLCKs. *P < 0.01; n = 4 for each point; n.s., not significant.
Calculating MLC2v Km and V max values by ADP‐Glo assay indicated lack of kinase activity of mutant cMLCK, although wild‐type cMLCK had a Km value of 5.93 ± 1.47 and a maximum luminescence of (3.67 ± 0.37) × 104 relative light unit that corresponded to a V max of 1.28 ± 0.03 mol/min/mol kinase (Figure 5 ).
Figure 5.
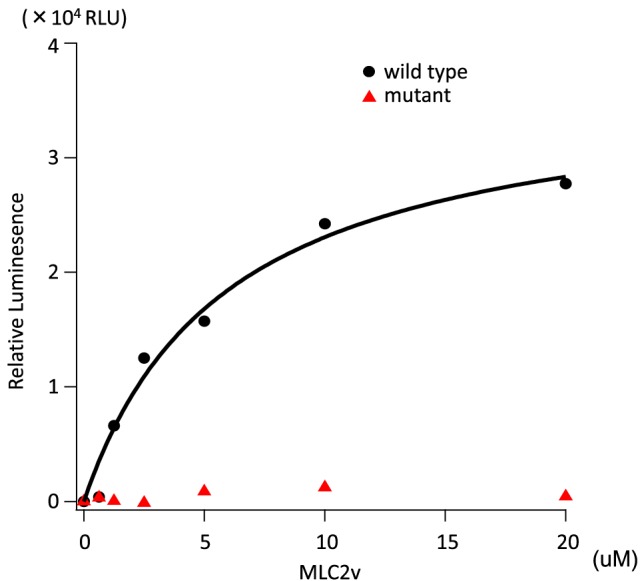
Michaelis–Menten saturation curve measured with the ADP‐Glo assay. MLC2v dose‐dependence of the nFlag‐tagged wild‐type or p.Pro639Valfs*15 cMLCK activities measured by ADP‐Glo assay. The reaction that contained no MLC2v was set as a background. The luminescence readings of the reactions were corrected with background subtraction and finally fit with the Michaelis–Menten equation using Igor Pro software to obtain Km (MLC2v). Wild‐type cMLCK had a Km value of 5.93 ± 1.47 μM and a maximum luminescence of (3.67 ± 0.37) × 104 relative light units that corresponded to a V max of 1.28 ± 0.03 mol/min/mol kinase. Mutant cMLCK had no kinase activity. n = 2 for each point. RLU, relative light unit.
Discussion
In the present study, we identified a p.Pro639Valfs*15 mutation in cMLCK in a DCM family. Assessing cMLCK kinase activity by Phos‐tag SDS‐PAGE and ADP‐Glo assays revealed a complete lack of phosphorylation even at high concentrations of mutant cMLCK. In contrast, kinase activity of wild‐type cMLCK was well preserved. Km value of wild‐type cMLCK was 5.93 ± 1.47 μM, which was almost the same as the previously reported value of 4.3 ± 1.5 μM.6 Additionally, this mutation was observed only in a DCM patient. These results demonstrate that this mutation is closely related to the occurrence of myocardial dysfunction associated with DCM.
Ventricular myosin regulatory light chain 2 functions are regulated by phosphorylation by its specific kinase and some phosphatases.28 Mutations in this gene are reported as a cause of DCM.29 Thus, MLC2v dephosphorylation was associated with heart failure including DCM in humans.11, 12, 13 Mice overexpressing myosin phosphatase exhibited MLC2v dephosphorylation and DCM phenotypes.30 In fact, a conditional MYLK3 knockout mouse showed reduced MLC2v phosphorylation and DCM phenotype like LV dilation, wall thinning, and decreased fractional shortening.8, 9, 10 Ding et al. demonstrated that heterozygous cMLCK knockout mice exhibited decreased systolic function of the LV,7 which also supports our hypothesis that the truncation mutation in MYLK3, p.Pro639Valfs*15, causes DCM in humans. This is reasonable considering that Pro639 locates to the kinase active site of serine/threonine kinase domain6 (Figure 2 ).
Of note, one young carrier in the proband's family also exhibited borderline LV dilation on echocardiography, indicating that the MYLK3 truncation mutation, p.Pro639Valfs*15, may cause pathological LV remodelling even in adolescents. Tobita et al.14 reported five MYLK3 mutation carriers from two pedigrees. Three carriers had read‐through mutation c.2459A>C (p.*820Sext*19); the level of expression of the protein and phosphorylated MLC2v level was greatly decreased. The other two carriers had a truncation variant c.1879_1885del (p.L623 fs*41) with diminished kinase activity. Interestingly, three of the five carriers were diagnosed with DCM in their 40s and 50s. The exact relationship between the onset of cardiac dysfunction and MYLK3 mutation requires further investigation.
Cardiac myosin light chain kinase is a not structural but is a functional protein. Therefore, cMLCK could present a therapeutic modality in patients with DCM caused by MYLK3 mutations. From this point of view, further studies are warranted to determine whether harbouring p.Pro639Valfs*15 is associated with increased risk of heart failure in the general Japanese population, because this variant identified in a DCM family was listed in the reference database of genetic variations in the Japanese population at an allele frequency < 0.001.
There are several limitations to the present study. First, we did not assess the phosphorylation level of MLC2v because of lack of heart tissue samples from biopsy. In addition, determination of the MLC2v phosphorylation level in experimental models is difficult, because it depends on heart load conditions.11, 12, 13 However, under these conditions, it is possible that MLC2v phosphorylation levels could be decreased in the proband, because MCL2v phosphorylation was shown to decrease in a heterozygous cMLCK knockout mouse model.7 Mutant cMLCK did not interfere with the wild‐type activity in vitro, and truncation variants such as this mutation are generally degraded by nonsense‐mediated mRNA decay, suggesting that this mutation can cause DCM with haploinsufficiency. Determination of MLC2v phosphorylation levels in induced pluripotent stem cells derived from mutation carriers may verify these functional data. Second, the prevalence of this cMLCK mutation in clinical DCM is still unclear, although we identified the possible pathogenic mutation of cMLCK in a patient with clinical DCM. Therefore, large‐scale, multicentre studies are needed to clarify the prevalence and spectrum of MYLK3 mutations.
In conclusion, a truncation mutation in the cMLCK gene (p.Pro639Valfs*15) can be associated with clinical DCM. We suggest that cMLCK could be a therapeutic target for DCM, although a larger study of DCM patients to identify the prevalence of this mutation and further strengthen its association with disease through family analysis is necessary.
Conflict of interest
None declared.
Funding
This work was supported by JSPS KAKENHI Grants 24790748 to T.K. and 25860589 to A.H.
Acknowledgements
We thank Tomoya Kaneda for data collection and Takako Obayashi for technical assistance.
Hodatsu A., Fujino N., Uyama Y., Tsukamoto O., Imai‐Okazaki A., Yamazaki S., Seguchi O., Konno T., Hayashi K., Kawashiri M.‐a., Asano Y., Kitakaze M., Takashima S., and Yamagishi M. (2019) Impact of cardiac myosin light chain kinase gene mutation on development of dilated cardiomyopathy, ESC Heart Failure, 6: 406–415. 10.1002/ehf2.12410.
References
- 1. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 1996; 93: 841–842. [DOI] [PubMed] [Google Scholar]
- 2. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62: e147–e239. [DOI] [PubMed] [Google Scholar]
- 3. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013; 10: 531–547. [DOI] [PubMed] [Google Scholar]
- 4. Mestroni L, Taylor MR. Genetics and genetic testing of dilated cardiomyopathy: a new perspective. Discov Med 2013; 15: 43–49. [PMC free article] [PubMed] [Google Scholar]
- 5. Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, Shintani Y, Wakeno M, Minamino T, Kondo H, Furukawa H, Nakamaru K, Naito A, Takahashi T, Ohtsuka T, Kawakami K, Isomura T, Kitamura S, Tomoike H, Mochizuki N, Kitakaze M. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest 2007; 117: 2812–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification of cardiac‐specific myosin light chain kinase. Circ Res 2008; 102: 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding P, Huang J, Battiprolu PK, Hill JA, Kamm KE, Stull JT. Cardiac myosin light chain kinase is necessary for myosin regulatory light chain phosphorylation and cardiac performance in vivo. J Biol Chem 2010; 285: 40819–40829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Warren SA, Briggs LE, Zeng H, Chuang J, Chang EI, Terada R, Li M, Swanson MS, Lecker SH, Willis MS, Spinale FG, Maupin‐Furlowe J, McMullen JR, Moss RL, Kasahara H. Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation 2012; 126: 2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Massengill MT, Ashraf HM, Chowdhury RR, Chrzanowski SM, Kar J, Warren SA, Walter GA, Zeng H, Kang BH, Anderson RH, Moss RL, Kasahara H. Acute heart failure with cardiomyocyte atrophy induced in adult mice by ablation of cardiac myosin light chain kinase. Cardiovasc Res 2016; 111: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Islam YF, Joseph R, Chowdhury RR, Anderson RH, Kasahara H. Heart Failure induced by perinatal ablation of cardiac myosin light chain kinase. Front Physiol 2016; 7: 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+‐sensitivity of the contractile apparatus in end‐stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res 2003; 57: 37–47. [DOI] [PubMed] [Google Scholar]
- 12. van der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PM, Hasenfuss G, Stienen GJ. The effect of myosin light chain 2 dephosphorylation on Ca2+‐sensitivity of force is enhanced in failing human hearts. Cardiovasc Res 2003; 57: 505–514. [DOI] [PubMed] [Google Scholar]
- 13. Scruggs SB, Reisdorph R, Armstrong ML, Warren CM, Reisdorph N, Solaro RJ, Buttrick PM. A novel, in‐solution separation of endogenous cardiac sarcomeric proteins and identification of distinct charged variants of regulatory light chain. Mol Cell Proteomics 2010; 9: 1804–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tobita T, Nomura S, Morita H, Ko T, Fujita T, Toko H, Uto K, Hagiwara N, Aburatani H, Komuro I. Identification of MYLK3 mutations in familial dilated cardiomyopathy. Sci Rep 2017; 7: 17495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rickham PP. Human experimentation: code of ethics of World Medical Association. Declaration of Helsinki. Br Med J 1964; 2: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Teichholz LE, Cohen MV, Sonnenblick EH, Gorlin R. Study of left ventricular geometry and function by B‐scan ultrasonography in patients with and without asynergy. N Engl J Med 1974; 291: 1220–1226. [DOI] [PubMed] [Google Scholar]
- 17. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows–Wheeler transform. Bioinformatics 2010; 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Broad institute . Picard tools. http://broadinstitute.github.io/picard/ (August 10, 2018)
- 19. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011; 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, Tsurusaki Y, Morishita S, Kawaguchi T, Migita O, Nakayama K, Nakashima M, Mitsui J, Narahara M, Hayashi K, Funayama R, Yamaguchi D, Ishiura H, Ko WY, Hata K, Nagashima T, Yamada R, Matsubara Y, Umezawa A, Tsuji S, Matsumoto N, Matsuda F. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet 2016; 61: 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. SnpEff ; Genomic variant annotations and functional effect prediction toolbox. http://snpeff.sourceforge.net/ (August 10, 2018)
- 24. Kinoshita E, Kinoshita‐Kikuta E, Takiyama K, Koike T. Phosphate‐binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 2006; 5: 749–757. [DOI] [PubMed] [Google Scholar]
- 25. Rasband, W.S. , ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997. –2016.
- 26. Witt SH, Juraeva D, Sticht C, Strohmaier J, Meier S, Treutlein J, Dukal H, Frank J, Lang M, Deuschle M, Schulze TG, Degenhardt F, Mattheisen M, Brors B, Cichon S, Nöthen MM, Witt CC, Rietschel M. Investigation of manic and euthymic episodes identifies state‐ and trait‐specific gene expression and STAB1 as a new candidate gene for bipolar disorder. Transl Psychiatry 2014; 4: e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A‐kinase‐anchoring protein causes long‐QT syndrome. Proc Natl Acad Sci U S A 2007; 104: 20990–20995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheikh F, Lyon RC, Chen J. Functions of myosin light chain‐2 (MYL2) in cardiac muscle and disease. Gene 2015; 569: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang W, Liang J, Yuan CC, Kazmierczak K, Zhou Z, Morales A, McBride KL, Fitzgerald‐Butt SM, Hershberger RE, Szczesna‐Cordary D. Novel familial dilated cardiomyopathy mutation in MYL2 affects the structure and function of myosin regulatory light chain. FEBS J 2015; 282: 2379–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizutani H, Okamoto R, Moriki N, Konishi K, Taniguchi M, Fujita S, Dohi K, Onishi K, Suzuki N, Satoh S, Makino N, Itoh T, Hartshorne DJ, Ito M. Overexpression of myosin phosphatase reduces Ca(2+) sensitivity of contraction and impairs cardiac function. Circ J 2010; 74: 120–128. [DOI] [PubMed] [Google Scholar]