

Abstract
Aims
Left ventricular diastolic dysfunction (LVDD) is common in obese subjects, and a relationship between epicardial adipose tissue (EAT), increased adipocytokines, and cardiovascular diseases has been reported. This study sought to examine as to whether the adipo‐fibrokine activin A is a link between increased EAT, the metabolic syndrome (MetS), and LVDD in severely obese subjects.
Methods and results
In 236 obese subjects (ø body mass index 39.8 ± 7.9 kg/m2) with a variable degree of the MetS and in 60 healthy non‐obese controls (ø body mass index 24.8 ± 3.4 kg/m2), serum activin A levels were measured and correlated with parameters of the MetS, epicardial fat thickness (EFT), and echocardiographic parameters of LVDD. Activin A levels were higher in obese than in non‐obese subjects (362 ± 124 vs. 301 ± 94 pg/mL, P = 0.0004), increased with the number of MetS components (from 285 ± 82 with no MetS component, 323 ± 94 with one or two MetS components, to 403 ± 131 pg/mL with ≥3 MetS components, P < 0.0001) and correlated with EFT (r = 0.41, P < 0.001). Furthermore, activin A levels were related to several parameters of LVDD [e.g. left atrial size (382 ± 117 vs. 352 125 pg/mL, P = 0.024), E/e′ (394 ± 108 vs. 356 ± 127 pg/mL, P = 0.005)]. LVDD was highest in MetS obese subjects with high EFT (44.3%) compared with MetS obese subjects with low EFT (27.0%), non‐MetS obese subjects with high EFT (24.2%), and non‐MetS obese subjects with low EFT (10.6%, P < 0.0001).
Conclusions
In severe obesity, activin A was significantly related to EFT, MetS, and LVDD, implicating MetS‐related alterations in the secretory profile of EAT in the pathogenesis of obesity‐related heart disease.
Keywords: Activin A, Epicardial fat, Obesity, Metabolic syndrome, Diastolic dysfunction
Introduction
Obesity and the metabolic syndrome (MetS) are rapidly increasing disorders in western countries and are associated with a high risk of cardiovascular morbidity and mortality and of developing heart failure.1, 2 Understanding the various underlying pathophysiological mechanism that could lead to heart failure in the obese state is critical. Obesity leads to systemic inflammation and alteration of the immune system network with release of specific immune cell‐derived cytokines in terms of cardiovascular dysfunction.3 Adipose tissue is divided into two groups (white and brown) according to morphology, physiology, and embryological origin. White adipose tissue is further divided into visceral and subcutaneous tissue. Visceral adipose tissue is surrounding the internal organs and is, in contrast to subcutaneous fat, which serves as energy resource, metabolically active with release of different cytokines and plays an important role in the development of cardiovascular diseases. The epicardial fat depot is part of the visceral adipose tissue; it is not separated by fascias from the myocardium with a close and strong interaction between both.4 Excessive epicardial fat is common in obesity and has a strong relationship with visceral adiposity.5 In fact, epicardial fat is a source of adipokines and inflammatory cytokines, and there seems to be interactions through paracrine and vasocrine mechanisms between epicardial fat and the myocardium, particularly in subjects with the MetS.4, 6, 7
In heart failure patients, increasing expression and blood levels of the TGF‐β cytokine member activin A could be demonstrated.8 Activins are expressed in various tissues where they have been shown to regulate multiple biological processes including regulation of apoptosis, proliferation, and differentiation of different cell types.9 Activin A is a growth factor with various activities and has been found in a variety of cell types at all stages of cell and human growth.
Activin A levels have been shown to increase in myocardial infarction,10 predict worse left ventricular (LV) remodelling and death in ST‐elevation myocardial infarction,11 and were described as stimulating cardiac fibroses through activating cardiac fibroblasts,12 whereas inhibition of activin A improves cardiac function in mice with dilated cardiomyopathy.13
Activins are highly expressed in adipose tissue and have a role in several steps of physiologic and pathological development of adipose tissue, supporting the hypothesis that increased activity of activin A may lead to insulin resistance and inflammatory status. Moreover, in type 2 diabetes patients, activin A levels were inversely associated with myocardial glucose metabolism and positively with left ventricular mass (LVM), indicating a detrimental role in early diastolic cardiomyopathy.14 Therefore, the activin A pathway could represent a potential therapeutic target for obesity‐associated, cardiometabolic complications. Impaired diastolic functioning of the left ventricle appears in large percentages of the cardiovascular asymptomatic population with diabetes and severe obesity15, 16 and represents usually an early stage in the progression to heart failure in these subjects.15 Thus, the aim of our study was to examine as to whether activin A is linked between increased epicardial adipose tissue, the MetS, and LV diastolic function in severely obese subjects.
Methods
Study population
Subjects were participants of the ‘Regensburg Weight Loss Study’, a prospective longitudinal study evaluating excessive body fat for its pathogenic potential in terms of cardiometabolic diseases and assessing the effects of a considerable weight reduction on interactions in systems biology.
Obese patients intending to participate in a weight reduction programme were offered enrolment in our research study prior to the start of the programme. Patients were eligible for enrolment if they were 18–65 years old, presented with a body mass index (BMI) ≥ 30 kg/m2 and a constant body weight in the last 3 months, and if they gave written informed consent to participate in the study. Exclusion criteria were weight reduction ≥10% of body weight in the last 6 months, cancer, pregnancy, therapy with steroids or thyroid hormones, known heart disease, known diabetes mellitus type 1 or 2, known inflammatory bowel, rheumatoid, or systemic diseases, known chronic renal failure, known liver diseases, mental disorders, or addiction to drugs or alcohol. The participants were recruited by flyers and advertisements. The study was approved by the local ethics committee. The investigation conforms with the principles outlined in the Declaration of Helsinki.17
Metabolic syndrome
The MetS was defined according to the National Cholesterol Education Program–Adult Treatment Panel III guidelines if at least three of the following criteria were fulfilled: central obesity with a waist circumference ≥ 102 cm in men and ≥88 cm in women, dyslipidaemia with triglyceride levels ≥ 150 mg/dL, HDL cholesterol levels < 40 mg/dL in men and <50 mg/dL in women, blood pressure ≥ 130/85 mmHg or treatment for hypertension, and fasting plasma glucose levels ≥ 110 mg/dL or presence of diabetes mellitus type 2.18
Physical performance
Patients performed the 6 min walk test at study entry. Patients were instructed to walk from end to end in an enclosed corridor, covering as much ground as they could during 6 min, and the distance walked was measured using an electronic metre counter.
Laboratory analysis
Serum samples were collected after a 12 h overnight fast and immediately stored at −70°C. The samples had not been thawed before the present measurements. Fasting glucose, insulin, and blood lipids were determined by standard methods in the certified clinical chemistry laboratory of the University Hospital. Insulin resistance was estimated by homeostasis model assessment index. The N‐terminal pro‐brain natriuretic peptide levels were determined by a standard chemiluminescence immunoassay (Roche Diagnostics, Mannheim, Germany). Activin A levels were determined by enzyme‐linked immunosorbent assay (Quantikine, R&D Systems Europe, Wiesbaden‐Nordenstadt, Germany) and were performed at the Medical Clinic III, Dresden University of Technology, Germany, by investigators that were not aware of patients' characteristics and outcomes. Matrix metalloproteinase 9 (MMP‐9) levels were determined by enzyme‐linked immunosorbent assay (Quantikine, R&D Systems Europe, Abingdon, UK); likewise, adiponectin levels were determined by enzyme‐linked immunoassay (BioVendor GmbH, Heidelberg, Germany).
Echocardiography
Echocardiography was performed using a standard ultrasound system (Philips iE33 Philips Medical Systems, Hamburg, Germany). The LV ejection fraction was measured based on the modified biplane Simpson's method. The following parameters were measured according to previous American Society of Echocardiography guidelines19: parasternal long axis diameter and apical four‐chamber area. Measurements were done just before mitral valve opening. LVM was calculated by the Devereux formula indexed to the body surface area. Conventional transmitral flow was measured with pulsed wave Doppler. Early (E) and late atrial (A) transmitral peak flow velocities, their ratio (E/A), and the deceleration time (DT) of the early transmitral flow velocity were measured, and three consecutive beats were averaged. Pulsed wave tissue Doppler imaging was performed at the junction of the lateral mitral annulus, and three consecutive beats were averaged. Early diastolic velocities (e′ lateral) were recorded. Ratios of E/e′ lateral and e′/a′ were calculated. All measurements were recorded by two expert echocardiographers. We determined the agreement between E/e′ measurements of the two expert cardiologists by the use of the concordance correlation coefficient (CCC). The CCC combines a measure for precision and accuracy to evaluate reproducibility and inter‐rater reliability.20 The CCC for 20 duplicate E/e′ measurements of the two expert sonographers was 0.976 ± 0.014 (P < 0.0001).
Left ventricular diastolic dysfunction
Diastolic dysfunction was defined as fulfilment of the following criteria: (i) a preserved systolic LV function (ejection fraction > 50%) and (ii) the presence of at least two of the following criteria consistent with abnormal LV relaxation, filling, diastolic distensibility, diastolic stiffness, or increased natriuretic peptides (N‐terminal pro‐brain natriuretic peptide > 220 pg/mL): E/e′ lat. > 8, E/A ≤ 0.8 together with a DT > 200 ms, a difference between the duration of reversed pulmonary vein atrial systole flow (Ard) and duration of mitral A wave flow (Ad): Ard – Ad > 30 ms, LV mass > 149 g/m2 (in men), >122 g/m2 (in women) together with (iii) an enlarged left atrial (LA) size [LA area (4 chamber planimetry)] > 20 cm2 or LA diameter (parasternal long axis) > 48 mm, and an e′ < 10 cm/s. In contrast, subjects with a normal lateral e′ ≥ 10 cm/s together with a normal LA size were classified as having normal LV function according to the American Society of Echocardiography 2009 and European Society of Cardiology 2007 consensus criteria.21, 22
Epicardial adipose tissue
Epicardial fat thickness (EFT) was identified as the low echo space between the outer wall of the myocardium and the visceral layer of the pericardium in the parasternal long axis view. It was measured during end‐systole on the free wall of the right ventricle in a perpendicular line to the aortic annulus, used as an anatomic landmark. Pericardial fat, which does not appear hyperechoic and usually is not deformed substantially with cardiac cycles, was not included in the measurement of epicardial fat.
Statistical analysis
Statistical analyses were performed with SPSS version 18.0 software (SPSS Inc., Chicago, IL, USA). Descriptive statistics are presented as mean ± standard deviation for continuous data and as number and percentages for categorical data. Student's t‐test was used for comparison of different biometric, laboratory, and MetS as well as echocardiographic parameters for normal distributed and independent data. Multiple linear regression analysis was performed to account for covariates such as age, sex, and BMI. Skewed data were evaluated by the non‐parametric Wilcoxon (Mann–Whitney) test. Correlation analyses were assessed by Pearson's correlation coefficient. Any P‐value < 0.05 was considered to be statistically significant.
Results
Baseline characteristics
Baseline characteristics of the study population stratified by obesity are displayed in Table 1. As expected in obese subjects (ø BMI 39.8 ± 7.9 kg/m2), all components of the MetS were significantly different with higher systolic and diastolic blood pressure levels, higher blood glucose levels, higher homeostasis model assessment index levels, higher triglyceride levels, and lower HDL cholesterol levels. Similarly, activin A levels were significantly higher in obese than in non‐obese subjects (362 ± 124 vs. 301 ± 94 pg/ml, P < 0.001).
Table 1.
Baseline characteristics of the obese study population in comparison with the healthy non‐obese control group
| BMI range | Non‐obese group | Obese group | P‐value |
|---|---|---|---|
| 19.0–29.9 | 30.0–76.0 | ||
| N | 60 | 236 | |
| Age (years) | 46.6 ± 12.9 | 48.3 ± 12.5 | 0.341 |
| Male [n (%)] | 20 (33.3) | 95 (40.3) | 0.320 |
| BMI (kg/m2) | 24.8 ± 3.4 | 39.8 ± 7.9 | <0.001 |
| Waist (cm) | 85 ± 10 | 118 ± 19 | <0.001 |
| WHR | 0.82 ± 0.07 | 0.91 ± 0.10 | <0.001 |
| HR (beats/min) | 68 ± 14 | 73 ± 13 | 0.011 |
| SBP (mmHg) | 128 ± 16 | 139 ± 18 | <0.001 |
| DBP (mmHg) | 80 ± 11 | 88 ± 12 | <0.001 |
| Fasting glucose (mg/dL) | 85 ± 8 | 100 ± 30 | <0.001 |
| Fasting insulin (U/mL) | 8 ± 5 | 23 ± 19 | <0.001 |
| HOMA‐IR | 1.7 ± 1.1 | 6.5 ± 8.1 | <0.001 |
| Triglycerides (mg/dL) | 105 ± 66 | 144 ± 81 | <0.001 |
| HDL (mg/dL) | 85 ± 19 | 48 ± 14 | <0.001 |
| NT‐proBNP (ng/L) | 58 ± 51 | 87 ± 233 | 0.352 |
| Activin A level (pg/mL) | 301 ± 94 | 362 ± 124 | <0.001 |
| Adiponectin (μg/mL) | 11.4 ± 7.5 | 8.9 ± 3.9 | 0.016 |
| MMP‐9 level (ng/mL) | 503 ± 253 | 614 ± 331 | 0.016 |
BMI, body mass index; DBP, diastolic blood pressure; HDL, high density lipoprotein; HR, heart rate; HOMA‐IR, homeostasis model assessment–insulin resistance; MMP‐9, matrix metallopeptidase 9; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; SBP, systolic blood pressure; WHR, waist‐to‐hip ratio.
Values represent the mean ± standard deviation or numbers (percentages).
Activin A and metabolic syndrome
The association of activin A levels to different MetS parameters is displayed in Supporting Information, Table S1 . Participants with MetS had significantly higher activin A levels. Specifically, obese subjects with increased waist circumference, elevated systolic blood pressure, and impaired glucose metabolism showed higher activin A levels, whereas no differences in activin A levels were observed for elevated triglycerides and lower HDL cholesterol phenotypes.
As no validated reference limits for activin A levels exist, a cut‐off level of 325 pg/mL corresponding to the upper limit value of the 95% confidence interval in healthy non‐obese subjects was specified. In obese subjects, higher activin A levels (>325 pg/mL) were associated with increasing age, higher BMI, waist and hip circumference, systolic blood pressure, glucose and fasting insulin levels, and higher MMP‐9 levels (Table 2).
Table 2.
Comparison of metabolic syndrome parameters, serum markers, and physical performance by dichotomized activin A serum levels
| Activin A level ≤ 325 pg/mLa | Activin A level > 325 pg/mLa | P‐value | |
|---|---|---|---|
| N (%) | 97 (41.1) | 139 (58.9) | |
| Age (years) | 42.6 ± 11.2 | 52.3 ± 11.8 | <0.001 |
| Male [n (%)] | 36 (37.1) | 59 (42.4) | 0.413 |
| BMI (kg/m2) | 37.6 ± 5.7 | 41.3 ± 8.8 | <0.001 |
| Hip circumference (cm) | 126 ± 14 | 133 ± 19 | 0.001 |
| Waist circumference (cm) | 112 ± 17 | 122 ± 20 | 0.001 |
| WHR | 0.90 ± 0.11 | 0.93 ± 0.09 | 0.026 |
| SBP (mmHg) | 136 ± 16 | 142 ± 18 | 0.007 |
| DBP (mmHg) | 86 ± 11 | 89 ± 12 | 0.072 |
| HR (beats/min) | 73 ± 13 | 72 ± 13 | 0.855 |
| Fasting glucose (mg/dL) | 94 ± 31 | 104 ± 29 | 0.017 |
| Fasting insulin (U/mL) | 18 ± 12 | 26 ± 22 | <0.001 |
| HOMA‐IR | 4.3 ± 3.8 | 7.3 ± 9.0 | 0.001 |
| Triglycerides (mg/dL) | 133 ± 68 | 152 ± 88 | 0.057 |
| HDL (mg/dL) | 49 ± 15 | 47 ± 13 | 0.391 |
| NT‐proBNP (ng/L) | 58 ± 61 | 105 ± 292 | 0.192 |
| Adiponectin (μg/mL) | 8.7 ± 3.2 | 9.1 ± 4.4 | 0.425 |
| MMP‐9 (ng/mL) | 562 ± 261 | 677 ± 348 | 0.005 |
Abbreviations as in Table 1. Values represent the mean ± standard deviation or numbers (percentages).
The activin A cut‐off limit of 325 pg/mL corresponds to the upper limit value of the 95% confidence interval of the healthy non‐obese control group.
Activin A and epicardial fat thickness
Moreover, a gradual increase of EFT could be observed across increasing tertiles of activin A levels (Figure 1A ), and a higher EFT could be observed in subjects with activin A levels > 325 pg/mL (Figure 1B ). This association remained significant after adjusting for age, sex, and BMI (P = 0.015). Echocardiographic evidence of left ventricular diastolic dysfunction (LVDD) was strongly associated with EFT (Figure 1C ), and, in turn, subjects with LVDD had higher activin A levels than subjects with normal LV function (Figure 1D ).
Figure 1.
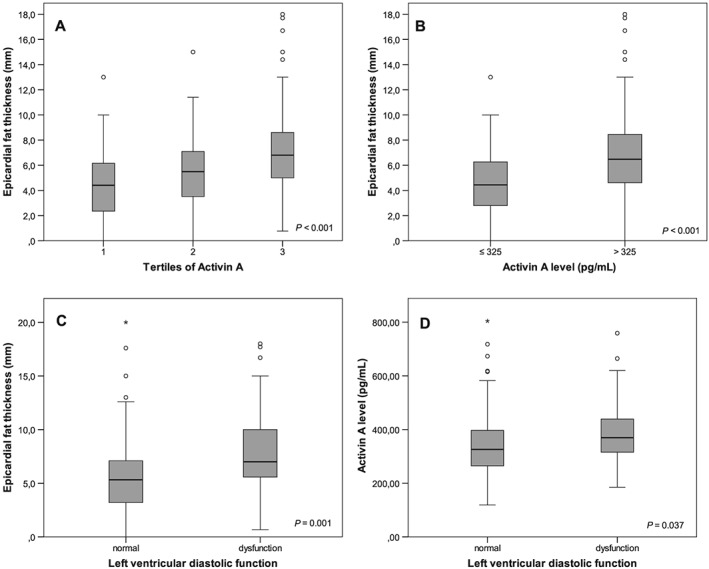
(A) Association of tertiles of activin A serum levels with epicardial fat thickness (EFT). (B) Association of dichotomized activin A levels with EFT. (C) Association of left ventricular diastolic function with EFT. (D) Association of left ventricular diastolic function with activin A levels.
Furthermore, activin A levels increased significantly across the spectrum of obesity (Figure 2 ): specifically, non‐obese control subjects followed by obese subjects with low EFT showed the lowest activin A levels, and MetS obese subjects with high EFT (>7.4 mm thickness, corresponding to the 95% confidence interval in the total obese group) showed the highest activin A levels. The same relationship could be observed with respect to the frequency of LVDD (Figure 2 ).
Figure 2.

Association of different obesity groups with low and high epicardial fat thickness (EFT) with activin A serum levels and left ventricular diastolic dysfunction (LVDD). MetS, metabolic syndrome; OB, obese; low EFT, EFT ≤ 7.4 mm; high EFT, EFT > 7.4 mm (the cut‐off corresponds to the 95% confidence interval of all obese study participants in the Regensburg Weight loss study).
Activin A and left ventricular diastolic dysfunction
In Table 3, different echocardiographic measurements of LA and LV geometry as well as parameters of LV systolic and diastolic function are displayed by dichotomized activin A levels. A significant association of elevated activin A levels with LA size, LV end‐diastolic diameter, interventricular and posterior wall thickness, and LVM as well as E/A, E/e′, and e′/a′ could be assessed. Moreover, obese participants with higher activin A levels > 325 pg/mL covered a significant shorter walking distance during the 6 min walk test (Table 3).
Table 3.
Comparison of echocardiographic parameters in obese subjects by dichotomized activin A serum levels
| Activin A level ≤ 325 pg/mLa | Activin A level > 325 pg/mLa | P‐value | |
|---|---|---|---|
| N (%) | 97 (41.1) | 139 (58.9) | |
| LAA (cm2) | 18.3 ± 3.7 | 20.3 ± 5.0 | 0.003 |
| LVEDD (mm) | 50.8 ± 5.3 | 52.3 ± 5.4 | 0.042 |
| LVESD (mm) | 32.0 ± 4.4 | 32.6 ± 5.3 | 0.337 |
| IVS (mm) | 10.6 ± 1.7 | 11.2 ± 2.0 | 0.017 |
| PW (mm) | 10.1 ± 1.5 | 10.6 ± 1.6 | 0.027 |
| LVM (g) | 114 ± 26 | 124 ± 30 | 0.018 |
| EF (%) | 65 ± 7 | 64 ± 7 | 0.217 |
| DT (ms) | 190 ± 44 | 215 ± 68 | 0.001 |
| E/A | 1.2 ± 0.4 | 1.1 ± 0.4 | 0.035 |
| e′ (cm/s) | 13.0 ± 3.9 | 11.8 ± 4.9 | 0.058 |
| E/e′ | 6.6 ± 2.2 | 7.4 ± 2.5 | 0.028 |
| e′/a′ | 1.5 ± 0.7 | 1.2 ± 0.5 | 0.001 |
| 6MWT (m) | 580 ± 68 | 528 ± 97 | <0.001 |
A, late diastolic mitral inflow; e′, early diastolic mitral annular tissue velocity; a′, late (atrial contraction) diastolic mitral annular tissue velocity; DT, deceleration time; E, early diastolic mitral inflow; EF, ejection fraction; IVS, interventricular septum thickness; LAA, left atrial area; LVEDD, left ventricular end‐diastolic diameter; LVESD, left ventricular end‐systolic diameter; LVM, left ventricular mass; PW, posterior wall; 6MWT, 6 min walking test.
Values represent the mean ± standard deviation.
The activin A cut‐off limit corresponds to the upper limit value of the 95% confidence interval of the healthy non‐obese control group.
Additionally, impaired values of e′, E/e′, E/A + DT, and LA size were associated with significantly elevated activin A levels (Table 4).
Table 4.
Activin A serum levels compared in normal and pathologic diastolic function parameters
| Activin A level (pg/mL) when diastolic function parameter is normal according to prespecified criteria | Activin A level (pg/mL) when diastolic function parameter is pathologic according to prespecified criteria | P‐value | |
|---|---|---|---|
| LVDD | 355 ± 125 | 389 ± 117 | 0.037 |
| Low e′ | 353 ± 125 | 392 ± 117 | 0.010 |
| High E/e′ | 356 ± 127 | 394 ± 108 | 0.005 |
| Impaired E/A + DT | 357 ± 123 | 4265 ± 112 | 0.007 |
| Low e′/a′ | 355 ± 125 | 397 ± 113 | 0.009 |
| Increased LA size | 352 ± 125 | 382 ± 117 | 0.024 |
| Increased LVM | 352 ± 116 | 381 ± 125 | 0.060 |
Abbreviations as in Table 3; LA, left atrial; LVDD, left ventricular diastolic dysfunction; LVDD as defined in the methods section according to the American Society of Echocardiography 2009 and European Society of Cardiology 2007 consensus criteria19, 20; e′ was pathologic < 10 cm/s; E/e′ was pathologic ≥ 8; E/A ≤ 0.8 + DT > 200 was pathologic; e′/a′ was pathologic ≤ 0.8; LA size (= left atrial size) was pathologic with LAA (4 chamber planimetry) > 20 cm2 or LA diameter (parasternal long axis) > 48 mm; LVM was pathologic > 149 g/m2 in men and >122 g/m2 in women.
Values represent the mean ± standard deviation.
Discussion
In our obese study population (ø BMI 39.8 ± 7.9 kg/m2), activin A levels were positively associated with most parameters of the MetS, increased with the number of MetS components, and were positively correlated with EFT. Moreover, elevated activin A levels were significantly associated with different parameters of LA and ventricular geometry as well as parameters of diastolic dysfunction. As a gradual increase of elevated activin A levels across increasing EFT could be observed independently from body mass, we conclude that activin A released from epicardial adipose tissue cells may be involved in the cardiometabolic dysfunctioning, given the findings from experimental animal models, the fact that activin A is abundantly secreted by human epicardial adipose tissue of diabetic and obese patients,23 and the consistent results of human epicardial adipose tissue secretome analysis.7 To the best of our knowledge, this is the first clinical study correlating activin A levels with cardiometabolic disturbances that may ultimately lead to heart failure.
Epicardial adipose tissue is a source of several inflammatory mediators and shares the same microcirculation with myocardial cells.24 Thus, mediators expressed by epicardial adipose tissue may directly influence myocardial function. Activin A is expressed in many immune cells, such as monocytes, macrophages, dentritic cells, T and B lymphocytes, and mast cells, and its expression increases by the activation of various immune stimuli.9
Preceding experimental investigations have suggested that activin A was associated with the occurrence and severity of heart failure8, 25; however, the underlying mechanisms are still unclear. Previous experimental studies on the relationship between heart failure and activin A focused on promoting myocardial hypertrophy and fibrosis26, 27 and investigated the role of the activin A–follistatin system in myocardial cell apoptosis,28, 29 whereby follistatin directly binds to activin A, inhibiting the physiological action of activin A.30 From the latter studies, it has been suggested that an imbalance of the activin A–follistatin system in animal models of heart failure causes up‐regulation of activin A and down‐regulation of follistatin, proposing that the activin A–follistatin system is involved in endoplasmic reticulum stress‐mediated myocardial cell apoptosis in heart failure.
However, most available evidence came from experimental studies suggesting that activin A is an important factor of inducing myocardial fibrosis.12, 31 Specifically, it has been shown that overexpression of activin A leads to ventricular remodelling in rat models of heart failure.12 Moreover, activin A promoted gene expression of atrial natriuretic peptides and MMP‐9, which are known to be factors playing an important role in myocardial remodelling and interstitial fibrosis,8 are associated with severity of chronic heart failure, and have been identified to predict heart failure events.32 Recent research has also demonstrated that the secretome from human epicardial (but not subcutaneous) adipose tissue can induce myocardial fibrosis and that the adipo‐fibrokine activin A constitutes an important mediator of this pro‐fibrotic effect, which can be neutralized by an anti‐activin A antibody.7 Interestingly, further findings have suggested that the angiotensin‐converting enzyme inhibitor Ramipril benefited LV remodelling by reducing fibrosis and collagen accumulation in the left ventricle of rats after myocardial infarction, an observation which was also associated with down‐regulation of activin A expression.33 Besides these effects, activin A has been described as an additional factor contributing to diabetic cardiomyopathy.14, 23 Activin A released from epicardial adipose tissue of obese type 2 diabetic patients provoked impairment of myocardial contractility by reduction of sarcomer shortening, cytosolic calcium flow and expression of sarco/endoplasmic reticulum Ca2+ ATPase and decreased insulin‐mediated glucose uptake in atrial rat cardiomyocytes.23
Our results are consistent with these earlier findings implicating a relationship between obesity (particularly when there is evidence of insulin resistance or the MetS is present), epicardial tissue, activin A, MMP‐9 levels, and signs of LVDD. Taken together, observations from the literature together with our present clinical data provide compelling evidence that activin A is an important mediator of the pro‐fibrotic effect of epicardial adipose tissue on human myocardium and LV structure and function.
Study limitations
Our study has several limitations. First, the way of recruitment of study participants may be lead to selection bias. Second, a large variability of echocardiographic parameters, especially in consequence of limited testing conditions in extremely obese subjects, could impact our results. However, concordance for 20 duplicate measurements of two expert echocardiographers was very high for tissue Doppler measurements. Third, we can present only associations between activin A, EFT, and diastolic function parameters and not give evidence of a causal link.
Conclusions
In conclusion, our data confirm the pre‐existing investigations that epicardial adipose tissue in severe obesity leads to enhanced release of the inflammatory marker activin A, especially in patients with insulin resistant MetS. In these patients, activin A may be involved in the pathogenesis of myocardial fibrosis leading to impaired LV diastolic function, which represents an early sign of obesity‐related heart disease.
Conflict of interest
None declared.
Funding
None.
Supporting information
Table S1. Activin A levels compared in normal and pathologic metabolic syndrome (MetS) parameters in the obese.
Acknowledgements
We appreciate the invaluable contribution of all study participants. We gratefully acknowledge the excellent technical assistance of Ingrid Lugauer and Ute Hubauer. Permission to acknowledge was obtained from the mentioned persons.
Zeller J., Krüger C., Lamounier‐Zepter V., Sag S., Strack C., Mohr M., Loew T., Schmitz G., Maier L., Fischer M., and Baessler A. (2019) The adipo‐fibrokine activin A is associated with metabolic abnormalities and left ventricular diastolic dysfunction in obese patients, ESC Heart Failure, 6, 362–370. 10.1002/ehf2.12409.
The work was performed at the Clinic of Internal Medicine II at University Hospital of Regensburg, Germany.
References
- 1. The GBD 2015 Obesity Collaborators . Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med 2017; 377: 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Caleyachetty R, Thomas GN, Toulis KA, Mohammed N, Gokhale KM, Balachandran K, Nirantharakumar K. Metabolically healthy obese and incident cardiovascular disease events among 3.5 million men and women. J Am Coll Cardiol 2017; 70: 1429–1437. [DOI] [PubMed] [Google Scholar]
- 3. Halade GV, Kain V. Obesity and cardiometabolic defects in heart failure pathology. Compr Physiol 2017; 7: 1463–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sacks HS, Fain JN. Human epicardial adipose tissue: a review. Am Heart J 2007; 153: 907–917. [DOI] [PubMed] [Google Scholar]
- 5. Iacobellis G, Ribaudo MC, Assael F, Vecci E, Tiberti C, Zappaterreno A, Di Mario U, Leonetti F. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: a new indicator of cardiovascular risk. J Clin Endocrinol Metab 2003; 88: 5163–5168. [DOI] [PubMed] [Google Scholar]
- 6. Ouwens DM, Sell H, Greulich S, Eckel J. The role of epicardial and perivascular adipose tissue in the pathophysiology of cardiovascular disease. J Cell Mol Med 2010; 14: 2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, Amour J, Leprince P, Dutour A, Clément K, Hatem SN. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo‐fibrokines. Eur Heart J 2015; 36: 795–805a. [DOI] [PubMed] [Google Scholar]
- 8. Yndestad A, Ueland T, Øie E, Florholmen G, Halvorsen B, Attramadal H, Simonsen S, Frøland SS, Gullestad L, Christensen G, Damås JK, Aukrust P. Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation 2004; 109: 1379–1385. [DOI] [PubMed] [Google Scholar]
- 9. Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. J Endocrinol 2009; 202: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miyoshi T, Hirohata S, Uesugi T, Hirota M, Ohnishi H, Nogami K, Hatanaka K, Ogawa H, Usui S, Kusachi S. Relationship between activin A level and infarct size in patients with acute myocardial infarction undergoing successful primary coronary intervention. Clin Chim Acta 2009; 401: 3–7. [DOI] [PubMed] [Google Scholar]
- 11. Lin JF, Hsu SY, Teng MS, Wu S, Hsieh CA, Jang SJ, Liu CJ, Huang HL, Ko YL. Activin A predicts left ventricular remodeling and mortality in patients with ST‐elevation myocardial infarction. Acta Cardiol Sin 2016; 32: 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang WQ, Yang CY, Li SM, Liu M, Ding M, Liu GH, Yang P. Lipopolysaccharide induced activin A–follistatin imbalance affects cardiac fibrosis. Chin Med J (Engl) 2012; 125: 2205–2212. [PubMed] [Google Scholar]
- 13. Fukushima N, Matsuura K, Akazawa H, Honda A, Nagai T, Takahashi T, Seki A, Murasaki KM, Shimizu T, Okano T, Hagiwara N, Komuro I. A crucial role of activin A‐mediated growth hormone suppression in mouse and human heart failure. PLoS One 2011; 6: e27901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen WJ, Greulich S, van der Meer RW, Rijzewijk LJ, Lamb HJ, de Roos A, Smit JW, Romijn JA, Ruige JB, Lammertsma AA, Lubberink M, Diamant M, Ouwens DM. Activin A is associated with impaired myocardial glucose metabolism and left ventricular remodeling in patients with uncomplicated type 2 diabetes. Cardiovasc Diabetol 2013; 12: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. From AM, Scott CG, Chen HH. The development of heart failure in patients with diabetes mellitus and pre‐clinical diastolic dysfunction a population‐based study. J Am Coll Cardiol 2010; 55: 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aljaroudi W, Halley C, Houghtaling P, Agarwal S, Menon V, Rodriguez L, Grimm RA, Thomas JD, Jaber WA. Impact of body mass index on diastolic function in patients with normal left ventricular ejection fraction. Nutr Diabetes 2012; 2: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rickham PP. Human experimentation: code of ethics of the World Medical Association. Declaration of Helsinki. Br Med J 1964; 2: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. National Cholesterol Education Program . (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106: 3143–3421. [PubMed] [Google Scholar]
- 19. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ, Chamber Quantification Writing Group , American Society of Echocardiography's Guidelines and Standards Committee , European Association of Echocardiography . Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005; 18: 1440–1463. [DOI] [PubMed] [Google Scholar]
- 20. Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics 1989; 45: 255–268. [PubMed] [Google Scholar]
- 21. Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelista A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 2009; 22: 107–133. [DOI] [PubMed] [Google Scholar]
- 22. Paulus WJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Smiseth OA, De Keulenaer G, Leite‐Moreira AF, Borbély A, Edes I, Heymans S, Pezzali N, Pieske B, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 2007; 28: 2539–2550. [DOI] [PubMed] [Google Scholar]
- 23. Greulich S, Maxhera B, Vandenplas G, de Wiza DH, Smiris K, Mueller H, Heinrichs J, Blumensatt M, Cuvelier C, Akhyari P, Ruige JB, Ouwens DM, Eckel J. Secretory products from epicardial adipose tissue of patients with type 2 diabetes mellitus induce cardiomyocyte dysfunction. Circulation 2012; 126: 2324–2334. [DOI] [PubMed] [Google Scholar]
- 24. González N, Moreno‐Villegas Z, González‐Bris A, Egido J, Lorenzo Ó. Regulation of visceral and epicardial adipose tissue for preventing cardiovascular injuries associated to obesity and diabetes. Cardiovasc Diabetol 2017; 16: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahmoudabady M, Mathieu M, Dewachter L, Hadad I, Ray L, Jespers P, Brimioulle S, Naeije R, McEntee K. Activin‐A, transforming growth factor‐beta, and myostatin signaling pathway in experimental dilated cardiomyopathy. J Card Fail 2008; 14: 703–709. [DOI] [PubMed] [Google Scholar]
- 26. Wei Q, Wang YN, Liu HY, Yang J, Yang CY, Liu M, Liu YF, Yang P, Liu ZH. The expression and role of activin A and follistatin in heart failure rats after myocardial infarction. Int J Cardiol 2013; 168: 2994–2997. [DOI] [PubMed] [Google Scholar]
- 27. Shimano M, Ouchi N, Nakamura K, Oshima Y, Higuchi A, Pimentel DR, Panse KD, Lara‐Pezzi E, Lee SJ, Sam F, Walsh K. Cardiac myocyte‐specific ablation of follistatin‐like 3 attenuates stress‐induced myocardial hypertrophy. J Biol Chem 2011; 286: 9840–9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oshima Y, Ouchi N, Shimano M, Pimentel DR, Papanicolaou KN, Panse KD, Tsuchida K, Lara‐Pezzi E, Lee SJ, Walsh K. Activin A and follistatin‐like 3 determine the susceptibility of heart to ischemic injury. Circulation 2009; 120: 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu M, Mao C, Li J, Han F, Yang P. Effects of the activin A–follistatin system on myocardial cell apoptosis through the endoplasmic reticulum stress pathway in heart failure. Int J Mol Sci 2017; 18: 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi Y, Massagué J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- 31. Hu J, Wang X, Wei SM, Tang YH, Zhou Q, Huang CX. Activin A stimulates the proliferation and differentiation of cardiac fibroblasts via the ERK1/2 and p38‐MAPK pathways. Eur J Pharmacol 2016; 789: 319–327. [DOI] [PubMed] [Google Scholar]
- 32. Morishita T, Uzui H, Mitsuke Y, Amaya N, Kaseno K, Ishida K, Fukuoka Y, Ikeda H, Tama N, Yamazaki T, Lee JD, Tada H. Association between matrix metalloproteinase‐9 and worsening heart failure events in patients with chronic heart failure. ESC Heart Fail 2017; 4: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei Q, Liu H, Liu M, Yang C, Yang J, Liu Z, Yang P. Ramipril attenuates left ventricular remodeling by regulating the expression of activin A–follistatin in a rat model of heart failure. Sci Rep 2016; 6: 33677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Activin A levels compared in normal and pathologic metabolic syndrome (MetS) parameters in the obese.