

An increasing number of multidrug-resistant Acinetobacter baumannii (MDR-AB) infections have been reported worldwide, posing a threat to public health. The establishment of methods to elucidate the mechanism of action (MOA) of A. baumannii-specific antibiotics is needed to develop novel antimicrobial therapeutics with activity against MDR-AB.
KEYWORDS: Acinetobacter baumannii, antibiotic screening, mechanisms of action
ABSTRACT
An increasing number of multidrug-resistant Acinetobacter baumannii (MDR-AB) infections have been reported worldwide, posing a threat to public health. The establishment of methods to elucidate the mechanism of action (MOA) of A. baumannii-specific antibiotics is needed to develop novel antimicrobial therapeutics with activity against MDR-AB. We previously developed bacterial cytological profiling (BCP) to understand the MOA of compounds in Escherichia coli and Bacillus subtilis. Given how distantly related A. baumannii is to these species, it was unclear to what extent it could be applied. Here, we implemented BCP as an antibiotic MOA discovery platform for A. baumannii. We found that the BCP platform can distinguish among six major antibiotic classes and can also subclassify antibiotics that inhibit the same cellular pathway but have different molecular targets. We used BCP to show that the compound NSC145612 inhibits the growth of A. baumannii via targeting RNA transcription. We confirmed this result by isolating and characterizing resistant mutants with mutations in the rpoB gene. Altogether, we conclude that BCP provides a useful tool for MOA studies of antibacterial compounds that are active against A. baumannii.
INTRODUCTION
The discovery of penicillin led to the “golden era” of antibiotic research which lasted for many decades before fading away in the 1970s. Since then, the rate of discovery of novel antibacterial molecules has decreased dramatically, and most of the newly commercialized antibiotics are analogues of existing ones (1–3). Although five new classes of antibiotics acting on Gram-positive bacteria were recently discovered, fewer novel antibiotics against Gram-negative bacteria have been developed (4). The incidence of Gram-negative pathogens that are resistant to almost all existing antibiotics is growing rapidly (5, 6). As a result, the options for treating drug-resistant Gram-negative infections are limited; thus, new antibiotics that act against Gram-negative bacteria are urgently needed (7). Among the Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (ESKAPE) pathogens (5), A. baumannii is of particular concern as it is (8, 9) responsible for a wide range of hospital-acquired infections, including meningitis, bacteremia, and skin infections (10). Apart from their intrinsic resistance, some clinically isolated A. baumannii strains have developed resistance to antibiotics commonly used for treatment, such as β-lactams, aminoglycosides, and tetracyclines (11). Also, a number of cases have been reported of strains that are resistant to colistin (11–13) and tigecycline (11, 14, 15), antibiotics considered to be the last line of defense (16), emphasizing the need for novel antibiotics that are active against the pathogen.
In order to minimize the harmful effects of antibiotics on the microbiome and prevent the spread of antibiotic resistance across various pathogens, narrow spectrum antibiotics may be preferable over broad spectrum ones in some cases (2). Species-specific antibiotic screening platforms have been proposed as a potential approach to discover narrow spectrum antibiotics (17). A mycobacterium-specific screening platform is an example of a successful case of such screening approaches (4, 18). These screens resulted in the discovery of many antibiotics exhibiting both broad spectrum, such as streptomycin (19), and mycobacterium-specific activity, including isoniazid, pyrazinamide, ethionamide, ethambutol (4), and bedaquiline (20). Recently, Gram-specific (21–24) and pathogen-specific (25, 26) antibiotic discovery were also proven to be successful, leading to the identification of narrow spectrum compounds, including some that are active only against A. baumannii (27–29). As more candidate compounds are revealed through screening, there will be a need for better methods to elucidate their mechanism of action (MOA) in A. baumannii.
In recent years, we have developed a method for antibiotic mechanism of action (MOA) study called bacterial cytological profiling (BCP) that can be applied to various bacterial species (30–32). BCP generates reference cytological profiles of bacterial cells upon treatment with different classes of antibiotics. BCP has been proven to be beneficial in MOA studies of antibiotics (30, 31, 33–37) and in a rapid antibiotic susceptibility test (32). Although a BCP-derived method was successfully used in a synergy study between azithromycin and human antimicrobial peptide LL-37 against multidrug-resistant A. baumannii (MDR-AB) (38), reference BCP profiles of A. baumannii treated with various types of antibiotics have not been reported. A. baumannii is very distantly related to Escherichia coli, and it was, therefore, unclear to what extent BCP could be applied. Here, we investigated the utility of BCP for A. baumannii. We showed that BCP is a useful tool for identifying the MOA of antibacterial molecules that inhibit the growth of A. baumannii (see Table S1 in the supplemental material) and used this platform to determine that the compound NSC145612 inhibits transcription in A. baumannii.
RESULTS
BCP in A. baumannii can distinguish different classes of antibiotics.
We first determined, based on cell morphological changes in A. baumannii ATCC 19606, if BCP can distinguish between antibiotics that interfere with six major cellular pathways, namely, protein translation (chloramphenicol), RNA transcription (rifampin), membrane integrity (colistin), lipid synthesis (triclosan), cell wall synthesis (piperacillin), and DNA replication (Ciprofloxacin). After A. baumannii ATCC 19606 was incubated with antibiotics, we found unique cell cytological profiles depending on the class of antibiotics used for treatment (Fig. 1A to F). Chloramphenicol-treated cells had a signature toroidal-shaped chromosome (Fig. 1B), while treatment with the transcription inhibitor rifampin resulted in diffuse 4′,6-diamidino-2-phenylindole (DAPI) staining throughout the cell except for a small rounded region near the cell membrane (Fig. 1C) similar to the BCP profile of actinomycin D-treated E. coli from the previous study (30). Colistin-treated cells were attached together, creating a long chain of small and round cells (Fig. 1D) similar to what we previously reported (38). Triclosan-treated cells were shorter and slightly rounder than untreated cells (Fig. 1E). The cell wall synthesis inhibitor piperacillin resulted in cell elongation without visible cell septation (Fig. 1F). In the case of DNA replication inhibitors, we found that even though DNA replication inhibitors effectively inhibited growth of A. baumannii ATCC 19606 as measured by MIC, only less than 10% of the cells treated with these inhibitors showed a possible DNA replication inhibition phenotype in this strain (Fig. S1). Thus, another well-studied A. baumannii strain, ATCC 17978 (39), was used in a DNA replication experiment (see Fig. S2 and Table S1 in the supplemental material). We found that the cell morphology of A. baumannii ATCC 17978 changed upon ciprofloxacin treatment. The treated cells were elongated and their chromosomes formed a single large nucleoid in the cell center (Fig. 1G). Overall, A. baumannii cytological profiles of cells treated with different antibiotics were similar to those of E. coli shown in our previous study (30). Next, we quantitated 36 different cytological parameters of cells treated with each antibiotic (see Table S2 in the supplemental material) and used principal-component analysis (PCA) to determine if these cell profiles can be used to quantitatively classify the MOAs. The results showed that antibiotics with different MOAs were distinguishable from each other (Fig. 1H) and replicates of each antibiotic treatment were clustered together (Fig. 1I). These results suggest that BCP can be applied to A. baumannii in discriminating antibiotics targeting six major cellular pathways, including protein translation, RNA transcription, membrane integrity, lipid synthesis, cell wall synthesis, and DNA replication.
FIG 1.

A. baumannii cells treated with antibiotics targeting different cellular pathways show distinct morphological changes. (A) Untreated bacterial cells. Bacterial cells were treated with 5× MIC (B, D to F) and 2× MIC (C and G) of each antibiotic for 2 hours and then stained with FM 4-64 (red) and DAPI (blue). Scale bar represents 1 μm. (H) A three-dimensional (3D) PCA graph constructed from PC1 (57.11%), PC2 (17.90%), and PC3 (9.35%) shows antibiotics that are distinguished into different subgroups as coded by colors. Three independent experiments were performed for each antibiotic treatment and cytological parameters (Table S2) measured as described in Materials and Methods. (I) Euclidean cluster map of antibiotics, using values from PC1, PC2, and PC3 of PCA. Ciprofloxacin* indicates that all data for treatment with ciprofloxacin were obtained in A. baumannii ATCC 17978 strain.
In A. baumannii, BCP can subclassify different antibiotics that inhibit the same cellular pathway based on their mechanism of action.
Our previous study in E. coli also showed that BCP can be used to classify subgroups of antibiotics based on their MOA (30). To test if the ability of subclassification by BCP is also observed in A. baumannii, we investigated whether BCP can differentiate various protein translation inhibitors and cell wall synthesis inhibitors. From all protein translation inhibitors tested (Fig. 2A to I), we found that they were classified into 2 groups that correlated with their known MOA, similar to the previous study in E. coli (30), namely, translation inhibition (P1) and aminoglycosides (P2) (Fig. 2J and K). Tetracycline, tigecycline, and minocycline, which are structurally related, were closely clustered in the analysis (Fig. 2K). Protein translation inhibitors belonging to the P1 group bind directly to the ribosome to inhibit translation (40–43), resulting in the formation of toroidal-shaped DNA (Fig. 1B and Fig. 2B to D). In addition to the translation inhibition, aminoglycosides (44) displayed a significant effect on A. baumannii membrane permeability, as indicated by the increase in Sytox green uptake (Fig. 2F to I, right panel) whereas Sytox green signal was not detected in the untreated cells (Fig. 2E, right panel). The increase in Sytox green intensity found in aminoglycoside-treated cells is more than 20 times higher than those of untreated cells (Table S2). This permeability effect of aminoglycosides separated them from the untreated and the others in the P1 group (Fig. 2J and K). We also found that BCP could distinguish between two types of penicillin binding protein (PBP) inhibitors in A. baumannii (Fig. 3), as expected (45, 46). Among all cell wall synthesis inhibitors tested in this study (Table S1), only meropenem, a preferred choice for treating A. baumannii infections (10, 47–49), and piperacillin are active against the strain according to the MIC assay. Meropenem-treated A. baumannii cells were round and bloated compared with control cells, in agreement with its affinity toward PBP2 (50) (Fig. 3A and B). Cells treated with piperacillin were elongated with an average length of 12 μm (Fig. 3C and Table S2), likely due to the affinity of piperacillin toward PBP3 (49), which is required for cell septa formation in A. baumannii. Altogether, these results suggest that BCP in A. baumannii can also subclassify antibiotics based on their MOA (Fig. 2 and 3), similar to what we previously reported in E. coli (30).
FIG 2.

A. baumannii cytological profiling differentiating protein translation inhibitors into subgroups by their MOA. Bacterial cells were treated with each antibiotic at 5× MIC for 2 hours and then stained with FM 4-64 (red), DAPI (blue), and Sytox green (green). Scale bar represents 1 μm. (A to D) Cells treated with protein translation inhibitors (P1 group) show distinct cell profiles. (E) Untreated cells. (F to I) Cells treated with aminoglycosides (P2 group) showing altered membrane permeability. Arrows indicate membrane pooling. Sytox green (Right panels) only stains nucleoids in the cells with permeabilized membranes. PCA graph of protein translation inhibitors using PC1 (44.30%), PC2 (26.82%), and PC3 (8.53%) (J) and Euclidean cluster map, using PC1, PC2, and PC3 from PCA (K).
FIG 3.

Cytological profiling of cell wall synthesis inhibitors; meropenem-treated cells showing different profiles to the cells treated with piperacillin. (A to C) Bacterial cells were treated with antibiotics at 5× MIC for 2 hours and then stained with FM 4-64 (red) and DAPI (blue). Scale bar represents 1 μm. PCA graph of cell wall synthesis inhibitors showing only PC1 (64.30%) and PC2 (22.81%) (D) and Euclidean cluster map using PC1, PC2, and PC3 from PCA showing distinct morphological clusters (E).
The compound NSC145612 inhibits the growth of A. baumannii via RNA transcription inhibition.
In this study, we have tested 64 compounds from National Cancer Institute’s Developmental Therapeutics Program library for their antibacterial activities against Gram-negative bacterium E. coli ATCC 25922 and found that 17 compounds were active. Among those Gram-negative active compounds, the compound NSC145612 (Fig. 4A, right panel) showed a promising MIC against A. baumannii ATCC 19606 at 25 μM (Table S1). Although the chemical scaffold of NSC145612 is closely related to rifampin (51), which is an antibiotic inhibiting DNA-dependent RNA polymerase (52), the compound has never been tested for its mechanism of action. In order to investigate if the compound exhibits the same MOA as rifampin, we performed BCP on the compound against A. baumannii. As expected, the result showed that NSC145612-treated A. baumannii cells exhibited a cytological profile identical to rifampin-treated cells (Fig. 4B to D) and grouped together in PCA analysis (Fig. 4E and F), suggesting that NSC145612 inhibits RNA transcription of A. baumannii, similar to rifampin. This conclusion was supported by examining NSC145612 in E. coli ΔtolC, whose growth was inhibited at 30 μM (see Table S3 in the supplemental material). BCP of NSC145612-treated E. coli ΔtolC cells showed decondensed DNA (see Fig. S3 in the supplemental material), which is a hallmark of transcription inhibition in E. coli (30).
FIG 4.

A. baumannii cell treated with NSC145612 show similar profiles to the RNA transcription inhibitor rifampin. (A) Chemical structure of rifamycin, rifampin, and NSC145612. (B) Untreated cells. Bacterial cells were treated with 2× MIC of rifampin (C) or NSC145612 (D) for 2 hours and then stained with FM 4-64 (red) and DAPI (blue). Scale bar represents 1 μm. PCA graph of 6 major classes of representative antibiotics and NSC145612 using PC1 (56.28%), PC2 (18.80%), and PC3 (9.18%) (E) and Euclidean cluster map, using values from PC1, PC2, and PC3 from PCA, showing NSC145612 closely clustered to rifampin (F). Ciprofloxacin* indicates that all data for treatment with Ciprofloxacin was obtained in A. baumannii ATCC 17978 strain.
In order to gain more information regarding the molecular target of the compound NSC145612, we isolated and characterized resistant mutations in both A. baumannii and E. coli ΔtolC. A total of four NSC145612-resistant mutants of E. coli were isolated (Table S3). Whole-genome sequencing of the resistant mutants revealed various mutations in DNA-dependent RNA polymerase subunit B (rpoB gene) (Table S3), a well-known gene responsible for rifampin resistance in E. coli and Mycobacterium tuberculosis (52, 53). Notably, three of our four resistant mutants contain mutations (Table S3) which are located in the rifampin resistance-determining region (RRDR) of the rpoB gene spanning from codon 507 to 533 (54). The rare mutation rpoB(V146F), which is located near the rifampin-binding pocket of the enzyme (55), was also found in one of the NSC145612-resistant mutants (LB143 strain). In accordance with the genetic profiles, the BCP profile of resistant mutants treated with NSC145612 and rifampin showed no cytological changes compared with the untreated controls (Fig. S3), confirming that NSC145612 and rifampin are inactive against the strains containing the rpoB mutation.
To confirm the molecular target of NSC145612 in A. baumannii, two NSC145612-resistant A. baumannii strains were also isolated with MIC above 200 μM (Table 1). As expected, NSC145612-resistant A. baumannii contained a mutation in the rpoB gene, rpoB(G543S) (Table 1), which is located in the RRDR and is known to be responsible for rifampin resistance in A. baumannii (56, 57). BCP results showed that neither NSC145612 nor rifampin treatment resulted in cytological changes of NSC145612-resistant A. baumannii strains compared with the controls (Fig. 5), confirming that NSC145612 and rifampin are inactive against the resistant strains. Overall, these results suggest that NSC145612 inhibits RNA transcription of A. baumannii by targeting its RNA polymerase subunit B.
TABLE 1.
MIC of NSC145612 and rifampin against A. baumannii strains
| Strain | rpoB mutation | MIC (μM) by compound: |
|
|---|---|---|---|
| NSC145612 | Rifampin | ||
| A. baumannii | 25 | 1.2 | |
| A. baumannii HH1102 | rpoB(G543S) | >200 | 24 |
| A. baumannii HH1105 | rpoB(G543S) | >200 | 24 |
FIG 5.
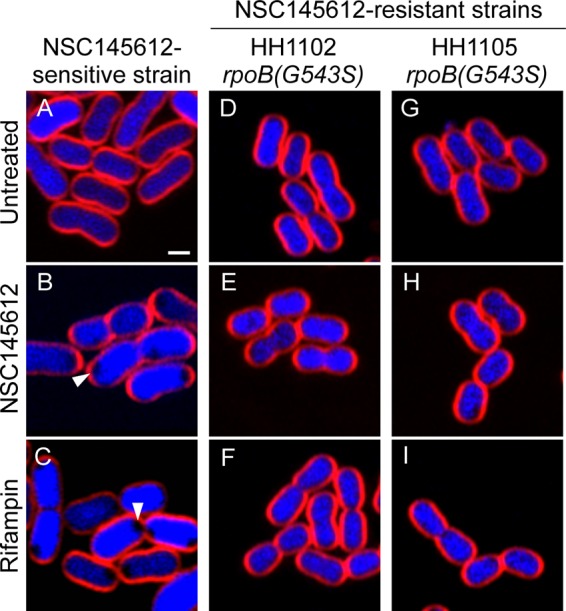
NSC145612-resistant A. baumannii cells show no morphological change upon NSC145612 and rifampin treatment. (A to C) NSC145612-sensitive strain. Arrows indicate signature phenotype of RNA transcription inhibition. (D to I) NSC145612-resistant strains with rpoB mutations indicated. Bacterial cells were treated with 2× MIC of NSC145612 (B, E, and H) or rifampin (C, F, and I) for 2 hours and then stained with FM 4-64 (red) and DAPI (blue). Scale bar represents 1 μm.
DISCUSSION
A recent report from the World Health Organization revealed that among the ESKAPE pathogens, A. baumannii poses a threat to public health and economies worldwide (9). A. baumannii is a successful pathogen due to its ability to survive in desiccated environments and its intrinsic antibiotic resistance (8). As a result, MDR-AB is spreading at an alarming rate (58–60). Multiple approaches exist in order to help mitigate the rise of MDR-AB, including developing more stringent guidelines for antibiotic usage and establishing effective surveillance and containment programs (9). A direct approach to combat MDR-AB is to find new antibiotics that are effective against this pathogen.
BCP has been developed for several species of bacteria, including E. coli, S. aureus, and B. subtilis, but it has not been systematically applied to A. baumannii (31, 32, 61). We studied the cytological profiles of antibiotics commonly used to treat A. baumannii. We show that BCP can be used to identify the MOA of newly discovered compounds to facilitate A. baumannii-specific antibiotic discovery. BCP successfully differentiates A. baumannii cells treated with different antibiotics targeting major cellular pathways, namely, protein translation, RNA transcription, membrane integrity, lipid synthesis, cell wall synthesis, and DNA replication. Similar to E. coli, A. baumannii cytological changes can reveal subgroups of protein translation inhibitors and cell wall synthesis inhibitors, suggesting similar cytological responses across Gram-negative bacterial species.
While A. baumannii ATCC 17978 treated with ciprofloxacin showed a clear cytological profile consistent with inhibiting DNA replication (Fig. S2), treatment of A. baumannii ATCC 19606 with ciprofloxacin did not induce similar cytological changes (Fig. S1). The fact that A. baumannii ATCC 19606 did not respond to DNA replication inhibitors made it unpractical for data from this strain to be used in the analysis. DNA damage and replication inhibition caused by quinolone antibiotics induce SOS responses in E. coli (62–64) and other bacteria (65, 66). In previous studies of E. coli (30, 67), filamentous E. coli observed after quinolone antibiotic treatment was a result of replication-halt-induced SOS response (65). Upon SOS response induction, sulA is derepressed due to the decrease in LexA protein, a master regulator of SOS response genes. SulA then inhibits FtsZ polymerization which leads to cell division inhibition and filamentation (68, 69). However, the SOS response of Acinetobacter spp. is not well understood due to the lack of similar SOS response genes, including lexA and sulA (68, 70–72). Since distinct responses to DNA damage have been observed in different species of Acinetobacter (73, 74), it is possible that A. baumannii ATCC 19606 and ATCC 17978 respond differently to the DNA replication inhibitors. Based on these results, multiple A. baumannii strains should be used to establish a comprehensive database of cytological profiles.
The compound NSC145612 from the National Cancer Institute’s Developmental Therapeutics Program has previously been tested for anticancer and AIDS antiviral activity, all of which gave negative results (51). In this study, its antibacterial activity was tested by BCP and later confirmed by resistant mutant selection and genome sequencing. NSC145612 inhibits the growth of A. baumannii and E. coli via RNA transcription inhibition by targeting the RpoB protein. Altogether, this study proves the utility of BCP as a potential method to reveal the mechanism of action of compounds that are active against A. baumannii.
MATERIALS AND METHODS
Bacteria strains and growth and antibiotics.
Acinetobacter baumannii strain ATCC 19606, strain ATCC 17978, and Escherichia coli strain AD3644 (ΔtolC) were used in this study. The bacteria were grown in LB medium or LB agar at 30°C. A total of 22 antibiotics were tested on A. baumannii from which 15 antibiotics with MIC of less than 112 μg/ml were used in this study (Table S1). The compound NSC145612 was obtained from the National Cancer Institute’s Developmental Therapeutics Program. Preparation of the antibiotics was performed according to the manufacture’s recommendations.
MICs.
MICs of all antibiotics are shown in Table S1. MICs were determined using the microdilution method (30). Overnight cultures of A. baumannii were diluted 1:100 in LB broth and allowed to grow at 30°C on a roller until exponential phase or until an optical density at 600 (OD600) of 0.2 was obtained. The bacterial culture was further diluted 1:100 into each well of a 96-well plate containing antibiotics in LB medium at appropriate concentrations. Cultures were allowed to grow at 30°C for 24 hours. MICs were determined by observing the concentration of the antibiotic in the well where the bacteria was unable to grow.
Fluorescence microscopy.
Overnight cultures of A. baumannii were diluted 1:500 and those of E. coli at 1:100 in LB broth and grown at 30°C on a roller until exponential phase. Antibiotics were added at concentrations of 0.75 times MIC for colistin; 2 times the MIC for rifampin, NSC145612, and ciprofloxacin; and 5 times the MIC for the rest of the tested antibiotics. Cultures were then grown at 30°C on a roller for 2 hours. A. baumannii cultures were stained with FM 4-64 (2 μg/ml), DAPI (4 μg/ml), and Sytox green (0.5 μM). E. coli cells were stained with FM 4-64 (1 μg/ml), DAPI (2 μg/ml), and Sytox green (0.5 μM). Stained bacterial cultures were harvested by centrifugation at 6,000 × g for 30 seconds and resuspended in 1/10 volume of the same culture medium. Three microliters of this was added to agarose pads (1.2% agarose in 20% LB broth) on concave glass slides. Fluorescence microscopy was performed with consistent imaging parameters throughout all experiments.
Cytological profiling.
Cytological profiles were determined by automated cell analysis using CellProfiler 3.0 (75). Briefly, images were preprocessed on Fiji software (76) and subsequently analyzed on CellProfiler 3.0 software. Cell morphological parameters, such as length, width, area, perimeter, form factor, ferret diameter, radius, compactness, solidity, and eccentricity, of both cell membrane and nucleoid were determined. To obtain the average intensity of Sytox green and DAPI, both the membrane and nucleoid outlines were used and subtracted by background intensity in corresponding images. The fold increase in permeability of aminoglycosides (P2 group) was determined by dividing the Sytox green intensity of aminoglycoside-treated cells with that of the untreated cells (Table S2). Decondensation of the nucleoid was determined by the ratio of the area of the nucleoid to that of the cell membrane.
Statistical analysis.
As described previously (30, 31, 38), the cytological parameters of each antibiotic were obtained from three independent experiments. Profiling data was from automated analysis of the cells in each imaging field. Only images containing more than 20 for long cells and for the rest, more than 30 cells per imaging field were selected into data points. Weighed principal-component analysis (PCA) was performed using statistic tools on MATLAB 2017a. Euclidean cluster analysis was generated from Morpheus (https://software.broadinstitute.org/morpheus).
Isolation of NSC145612-resistant mutants.
In E. coli, resistant mutants were obtained by plating E. coli AD3644 (ΔtolC) onto LB agar plates containing 2× MIC of NSC145612. The plates were incubated at 30°C, and resistant mutants were purified and stabilized on additional 2× MIC of NSC145612 selection plates. In A. baumannii, A. baumannii ATCC 19606 culture was diluted into the LB medium containing NSC145612 starting at 0.5× MIC. This process was repeated with escalating concentrations of NSC145612 until the NSC145612-resistant A. baumannii was obtained. The resistant strains were purified on LB agar plates, and the MICs for NSC145612 and rifampin were determined by the broth dilution method, as mentioned above.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Thailand Research Fund and the Office of the Higher Education Commission (MRG6080081). V.C. was supported by MRG6180027 and Grants for Development of New Faculty Staff, Ratchadaphiseksomphot Endowment Fund.
We thank Naraporn Sirinonthanaweeh and Potchaman Sittipaisankul for fluorescence microscope services and thank Ittipat Meewan for providing chemical structures.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02310-18.
REFERENCES
- 1.Walsh CT, Wencewicz TA. 2014. Prospects for new antibiotics: a molecule-centered perspective. J Antibiot (Tokyo) 67:7–22. doi: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis K. 2013. Platforms for antibiotic discovery. Nat Rev Drug Discov 12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 5.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 6.Berdy J. 2012. Thoughts and facts about antibiotics: where we are now and where we are heading. J Antibiot 65:385–395. doi: 10.1038/ja.2012.27. [DOI] [PubMed] [Google Scholar]
- 7.Butler MS, Blaskovich MA, Cooper MA. 2013. Antibiotics in the clinical pipeline in 2013. J Antibiot (Tokyo) 66:571–591. doi: 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- 8.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, Ouellette M, Outterson K, Patel J, Cavaleri M, Cox EM, Houchens CR, Grayson ML, Hansen P, Singh N, Theuretzbacher U, Magrini N. 2018. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- 10.Lee C-R, Lee JH, Park M, Park KS, Bae IK, Kim YB, Cha C-J, Jeong BC, Lee SH. 2017. Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol 7:55. doi: 10.3389/fcimb.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doi Y, Murray G, Peleg A. 2015. Acinetobacter baumannii: evolution of antimicrobial resistance—treatment options. Semin Respir Crit Care Med 36:085–098. doi: 10.1055/s-0034-1398388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beceiro A, Moreno A, Fernandez N, Vallejo JA, Aranda J, Adler B, Harper M, Boyce JD, Bou G. 2014. Biological cost of different mechanisms of colistin resistance and their impact on virulence in Acinetobacter baumannii. Antimicrob Agents Chemother 58:518–526. doi: 10.1128/AAC.01597-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rolain J-M, Diene SM, Kempf M, Gimenez G, Robert C, Raoult D. 2013. Real-time sequencing to decipher the molecular mechanism of resistance of a clinical pan-drug-resistant Acinetobacter baumannii isolate from Marseille, France. Antimicrob Agents Chemother 57:592–596. doi: 10.1128/AAC.01314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peleg AY, Potoski BA, Rea R, Adams J, Sethi J, Capitano B, Husain S, Kwak EJ, Bhat SV, Paterson DL. 2007. Acinetobacter baumannii bloodstream infection while receiving tigecycline: a cautionary report. J Antimicrob Chemother 59:128–131. doi: 10.1093/jac/dkl441. [DOI] [PubMed] [Google Scholar]
- 15.Ruzin A, Keeney D, Bradford PA. 2007. AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus–Acinetobacter baumannii complex. J Antimicrob Chemother 59:1001–1004. doi: 10.1093/jac/dkm058. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis 6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 17.Waksman SA, Schatz A, Reynolds DM. 2010. Production of antibiotic substance by actinomycetes. Ann N Y Acad Sci 1213:112–124. doi: 10.1111/j.1749-6632.2010.05861.x. [DOI] [PubMed] [Google Scholar]
- 18.Sneader W. 2005. Drug discovery: a history. John Wiley & Sons Ltd. doi: 10.1002/0470015535. [DOI]
- 19.Schatz A, Bugle E, Waksman SA. 1944. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Proc Soc Exp Biol Med 55:66–69. doi: 10.3181/00379727-55-14461. [DOI] [PubMed] [Google Scholar]
- 20.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Van GJ, Timmerman P, Zhu M, Lee E, Williams P, de CD, Huitric E, Hoffner S, Cambau E, Truffot PC, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 21.Maxson T, Mitchell DA. 2016. Targeted treatment for bacterial infections: prospects for pathogen-specific antibiotics coupled with rapid diagnostics. Tetrahedron 72:3609–3624. doi: 10.1016/j.tet.2015.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swaney SM, Aoki H, Ganoza MC, Shinabarger DL. 1998. The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrob Agents Chemother 42:3251–3255. doi: 10.1128/AAC.42.12.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brickner SJ, Hutchinson DK, Barbachyn MR, Manninen PR, Ulanowicz DA, Garmon SA, Grega KC, Hendges SK, Toops DS, Ford CW, Zurenko GE. 1996. Synthesis and antibacterial activity of U-100592 and U-100766, two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant gram-positive bacterial infections. J Med Chem 39:673–679. doi: 10.1021/jm9509556. [DOI] [PubMed] [Google Scholar]
- 24.Oleson FB Jr, Baltz RH, Eisenstein BI. 2010. Daptomycin: from the mountain to the clinic, with essential help from Francis Tally, MD. Clin Infect Dis 50:S10–S15. doi: 10.1086/647938. [DOI] [PubMed] [Google Scholar]
- 25.Ackermann G, Löffler B, Adler D, Rodloff AC. 2004. In vitro activity of OPT-80 against Clostridium difficile. Antimicrob Agents Chemother 48:2280–2282. doi: 10.1128/AAC.48.6.2280-2282.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melander RJ, Zurawski DV, Melander C. 2018. Narrow-spectrum antibacterial agents. MedChemComm 9:12–21. doi: 10.1039/C7MD00528H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corey BW, Thompson MG, Hittle LE, Jacobs AC, Asafo-Adjei EA, Huggins WM, Melander RJ, Melander C, Ernst RK, Zurawski DV. 2017. 1,2,4-triazolidine-3-thiones have specific activity against Acinetobacter baumannii among common nosocomial pathogens. ACS Infect Dis 3:62–71. doi: 10.1021/acsinfecdis.6b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J-Y, Jeong M-C, Jeon D, Lee Y, Lee WC, Kim Y. 2017. Structure-activity relationship-based screening of antibiotics against Gram-negative Acinetobacter baumannii. Bioorg Med Chem 25:372–380. doi: 10.1016/j.bmc.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Cheng Y-S, Sun W, Xu M, Shen M, Khraiwesh M, Sciotti RJ, Zheng W. 2018. Repurposing screen identifies unconventional drugs with activity against multidrug resistant Acinetobacter baumannii. Front Cell Infect Microbiol 8:438. doi: 10.3389/fcimb.2018.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nonejuie P, Burkart M, Pogliano K, Pogliano J. 2013. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A 110:16169–16174. doi: 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamsa A, Lopez-Garrido J, Quach D, Riley EP, Pogliano J, Pogliano K. 2016. Rapid inhibition profiling in Bacillus subtilis to identify the mechanism of action of new antimicrobials. ACS Chem Biol 11:2222–2231. doi: 10.1021/acschembio.5b01050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quach DT, Sakoulas G, Nizet V, Pogliano J, Pogliano K. 2016. Bacterial cytological profiling (BCP) as a rapid and accurate antimicrobial susceptibility testing method for Staphylococcus aureus. EBioMedicine 4:95–103. doi: 10.1016/j.ebiom.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alpha CJ, Campos M, Jacobs-Wagner C, Strobel SA. 2015. Mycofumigation by the volatile organic compound-producing fungus Muscodor albus induces bacterial cell death through DNA damage. Appl Environ Microbiol 81:1147–1156. doi: 10.1128/AEM.03294-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nayar AS, Dougherty TJ, Ferguson KE, Granger BA, McWilliams L, Stacey C, Leach LJ, Narita S, Tokuda H, Miller AA, Brown DG, McLeod SM. 2015. Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay. J Bacteriol 197:1726–1734. doi: 10.1128/JB.02552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson MZ, Wang R, Gitai Z, Seyedsayamdost MR. 2016. Mode of action and resistance studies unveil new roles for tropodithietic acid as an anticancer agent and the γ-glutamyl cycle as a proton sink. Proc Natl Acad Sci U S A 113:1630–1635. doi: 10.1073/pnas.1518034113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurley KA, Santos TMA, Nepomuceno GM, Huynh V, Shaw JT, Weibel DB. 2016. Targeting the bacterial division protein FtsZ. J Med Chem 59:6975–6998. doi: 10.1021/acs.jmedchem.5b01098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jayamani E, Rajamuthiah R, Larkins-Ford J, Fuchs BB, Conery AL, Vilcinskas A, Ausubel FM, Mylonakis E. 2015. Insect-derived cecropins display activity against Acinetobacter baumannii in a whole-animal high-throughput Caenorhabditis elegans model. Antimicrob Agents Chemother 59:1728–1737. doi: 10.1128/AAC.04198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin L, Nonejuie P, Munguia J, Hollands A, Olson J, Dam Q, Kumaraswamy M, Rivera H, Corriden R, Rohde M, Hensler ME, Burkart MD, Pogliano J, Sakoulas G, Nizet V. 2015. Azithromycin synergizes with cationic antimicrobial peptides to exert bactericidal and therapeutic activity against highly multidrug-resistant gram-negative bacterial pathogens. EBioMedicine 2:690–698. doi: 10.1016/j.ebiom.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baumann P, Doudoroff M, Stanier RY. 1968. A study of the Moraxella group. II. Oxidative-negative species (genus Acinetobacter). J Bacteriol 95:1520–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson DN. 2011. On the specificity of antibiotics targeting the large ribosomal subunit. Ann N Y Acad Sci 1241:1–16. doi: 10.1111/j.1749-6632.2011.06192.x. [DOI] [PubMed] [Google Scholar]
- 41.Parnham MJ, Haber VE, Giamarellos-Bourboulis EJ, Perletti G, Verleden GM, Vos R. 2014. Azithromycin: mechanisms of action and their relevance for clinical applications. Pharmacol Ther 143:225–245. doi: 10.1016/j.pharmthera.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Chopra I, Roberts M. 2001. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazzei T, Mini E, Novelli A, Periti P. 1993. Chemistry and mode of action of macrolides. J Antimicrob Chemother 31:1–9. doi: 10.1093/jac/31.suppl_C.1. [DOI] [PubMed] [Google Scholar]
- 44.Jana S, Deb JK. 2006. Molecular understanding of aminoglycoside action and resistance. Appl Microbiol Biotechnol 70:140–150. doi: 10.1007/s00253-005-0279-0. [DOI] [PubMed] [Google Scholar]
- 45.Typas A, Banzhaf M, Gross CA, Vollmer W. 2011. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spratt BG. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A 72:2999–3003. doi: 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies TA, Shang W, Bush K, Flamm RK. 2008. Affinity of doripenem and comparators to penicillin-binding proteins in Escherichia coli and Pseudomonas aeruginosa. Antimicrob Agents Chemother 52:1510–1512. doi: 10.1128/AAC.01529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cayô R, Rodríguez M-C, Espinal P, Fernández-Cuenca F, Ocampo-Sosa AA, Pascual Á, Ayala JA, Vila J, Martínez-Martínez L. 2011. Analysis of genes encoding penicillin-binding proteins in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother 55:5907–5913. doi: 10.1128/AAC.00459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kocaoglu O, Carlson EE. 2015. Profiling of β-lactam selectivity for penicillin-binding proteins in Escherichia coli strain DC2. Antimicrob Agents Chemother 59:2785–2790. doi: 10.1128/AAC.04552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sumita Y, Fukasawa M. 1995. Potent activity of meropenem against Escherichia coli arising from its simultaneous binding to penicillin-binding proteins 2 and 3. J Antimicrob Chemother 36:53–64. doi: 10.1093/jac/36.1.53. [DOI] [PubMed] [Google Scholar]
- 51.National Center for Biotechnology Information. 2008. PubChem Compound Database: CID 24192962. National Center for Biotechnology Information, Bethesda, MD. https://pubchem.ncbi.nlm.nih.gov/compound/24192962.
- 52.Floss HG, Yu T-W. 2005. Rifamycin mode of action, resistance, and biosynthesis. Chem Rev 105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- 53.Goldstein BP. 2014. Resistance to rifampicin: a review. J Antibiot (Tokyo) 67:625–630. doi: 10.1038/ja.2014.107. [DOI] [PubMed] [Google Scholar]
- 54.Jamieson FB, Guthrie JL, Neemuchwala A, Lastovetska O, Melano RG, Mehaffy C. 2014. Profiling of rpoB mutations and MICs for rifampin and rifabutin in Mycobacterium tuberculosis. J Clin Microbiol 52:2157–2162. doi: 10.1128/JCM.00691-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siu GKH, Zhang Y, Lau TCK, Lau RWT, Ho P-L, Yew W-W, Tsui SKW, Cheng VCC, Yuen K-Y, Yam W-C. 2011. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 66:730–733. doi: 10.1093/jac/dkq519. [DOI] [PubMed] [Google Scholar]
- 56.Giannouli M, Di Popolo A, Durante-Mangoni E, Bernardo M, Cuccurullo S, Amato G, Tripodi M-F, Triassi M, Utili R, Zarrilli R. 2012. Molecular epidemiology and mechanisms of rifampicin resistance in Acinetobacter baumannii isolates from Italy. Int J Antimicrob Agents 39:58–63. doi: 10.1016/j.ijantimicag.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 57.Tupin A, Gualtieri M, Roquet-Banères F, Morichaud Z, Brodolin K, Leonetti J-P. 2010. Resistance to rifampicin: at the crossroads between ecological, genomic and medical concerns. Int J Antimicrob Agents 35:519–523. doi: 10.1016/j.ijantimicag.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 58.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maragakis LL, Perl TM. 2008. Antimicrobial resistance: Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clin Infect Dis 46:1254–1263. doi: 10.1086/529198. [DOI] [PubMed] [Google Scholar]
- 60.Lin M-F, Lan C-Y. 2014. Antimicrobial resistance in Acinetobacter baumannii: from bench to bedside. World J Clin Cases 2:787–814. doi: 10.12998/wjcc.v2.i12.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumaraswamy M, Lin L, Olson J, Sun C-F, Nonejuie P, Corriden R, Döhrmann S, Ali SR, Amaro D, Rohde M, Pogliano J, Sakoulas G, Nizet V. 2016. Standard susceptibility testing overlooks potent azithromycin activity and cationic peptide synergy against MDR Stenotrophomonas maltophilia. J Antimicrob Chemother 71:1264–1269. doi: 10.1093/jac/dkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Little JW, Mount DW. 1982. The SOS regulatory system of Escherichia coli. Cell 29:11–22. doi: 10.1016/0092-8674(82)90085-X. [DOI] [PubMed] [Google Scholar]
- 63.Ysern P, Clerch B, Castańo M, Gibert I, Barbé J, Llagostera M. 1990. Induction of SOS genes in Escherichia coli and mutagenesis in Salmonella typhimurium by fluoroquinolones. Mutagenesis 5:63–66. doi: 10.1093/mutage/5.1.63. [DOI] [PubMed] [Google Scholar]
- 64.Piddock LJ, Walters RN, Diver JM. 1990. Correlation of quinolone MIC and inhibition of DNA, RNA, and protein synthesis and induction of the SOS response in Escherichia coli. Antimicrob Agents Chemother 34:2331–2336. doi: 10.1128/AAC.34.12.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drlica K, Malik M, Kerns RJ, Zhao X. 2008. Quinolone-mediated bacterial death. Antimicrob Agents Chemother 52:385–392. doi: 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelley WL. 2006. Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Mol Microbiol 62:1228–1238. doi: 10.1111/j.1365-2958.2006.05444.x. [DOI] [PubMed] [Google Scholar]
- 67.López E, Elez M, Matic I, Blázquez J. 2007. Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli: antibiotic-stimulated genetic recombination. Mol Microbiol 64:83–93. doi: 10.1111/j.1365-2958.2007.05642.x. [DOI] [PubMed] [Google Scholar]
- 68.Robinson A, Brzoska AJ, Turner KM, Withers R, Harry EJ, Lewis PJ, Dixon NE. 2010. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol Mol Biol Rev 74:273–297. doi: 10.1128/MMBR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cordell SC, Robinson EJH, Lowe J. 2003. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc Natl Acad Sci U S A 100:7889–7894. doi: 10.1073/pnas.1330742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aranda J, Poza M, Shingu-Vázquez M, Cortés P, Boyce JD, Adler B, Barbé J, Bou G. 2013. Identification of a DNA-damage-inducible regulon in Acinetobacter baumannii. J Bacteriol 195:5577–5582. doi: 10.1128/JB.00853-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Norton MD, Spilkia AJ, Godoy VG. 2013. Antibiotic resistance acquired through a DNA damage-inducible response in Acinetobacter baumannii. J Bacteriol 195:1335–1345. doi: 10.1128/JB.02176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aranda J, Bardina C, Beceiro A, Rumbo S, Cabral MP, Barbe J, Bou G. 2011. Acinetobacter baumannii RecA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J Bacteriol 193:3740–3747. doi: 10.1128/JB.00389-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hare JM, Ferrell JC, Witkowski TA, Grice AN. 2014. Prophage induction and differential RecA and UmuDAb transcriptome regulation in the DNA damage responses of Acinetobacter baumannii and Acinetobacter baylyi. PLoS One 9:e93861. doi: 10.1371/journal.pone.0093861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hare JM, Bradley JA, Lin C.-l, Elam TJ. 2012. Diverse responses to UV light exposure in Acinetobacter include the capacity for DNA damage-induced mutagenesis in the opportunistic pathogens Acinetobacter baumannii and Acinetobacter ursingii. Microbiology 158:601–611. doi: 10.1099/mic.0.054668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang I, Friman O, Guertin DA, Chang J, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.