

Bacteriophage-derived lysins are cell-wall-hydrolytic enzymes that represent a potential new class of antibacterial therapeutics in development to address burgeoning antimicrobial resistance. CF-301, the lead compound in this class, is in clinical development as an adjunctive treatment to potentially improve clinical cure rates of Staphylococcus aureus bacteremia and infective endocarditis (IE) when used in addition to antibiotics.
KEYWORDS: bacteriophage, CF-301, exebacase, human serum, human lysozyme, lysin, Staphylococcus aureus
ABSTRACT
Bacteriophage-derived lysins are cell-wall-hydrolytic enzymes that represent a potential new class of antibacterial therapeutics in development to address burgeoning antimicrobial resistance. CF-301, the lead compound in this class, is in clinical development as an adjunctive treatment to potentially improve clinical cure rates of Staphylococcus aureus bacteremia and infective endocarditis (IE) when used in addition to antibiotics. In order to profile the activity of CF-301 in a clinically relevant milieu, we assessed its in vitro activity in human blood versus in a conventional testing medium (cation-adjusted Mueller-Hinton broth [caMHB]). CF-301 exhibited substantially greater potency (32 to ≥100-fold) in human blood versus caMHB in three standard microbiologic testing formats (e.g., broth dilution MICs, checkerboard synergy, and time-kill assays). We demonstrated that CF-301 acted synergistically with two key human blood factors, human serum lysozyme (HuLYZ) and human serum albumin (HSA), which normally have no nascent antistaphylococcal activity, against a prototypic methicillin-resistant S. aureus (MRSA) strain (MW2). Similar in vitro enhancement of CF-301 activity was also observed in rabbit, horse, and dog (but not rat or mouse) blood. Two well-established MRSA IE models in rabbit and rat were used to validate these findings in vivo by demonstrating comparable synergistic efficacy with standard-of-care anti-MRSA antibiotics at >100-fold lower lysin doses in the rabbit than in the rat model. The unique properties of CF-301 that enable bactericidal potentiation of antimicrobial activity via activation of “latent” host factors in human blood may have important therapeutic implications for durable improvements in clinical outcomes of serious antibiotic-resistant staphylococcal infections.
INTRODUCTION
The emergence and spread of multidrug-resistant bacteria that are not effectively treated by our current antibiotic armamentarium have created an urgent need for novel alternatives to conventional agents. A promising new therapeutic approach to combat resistant bacterial pathogens is the use of recombinantly produced bacteriophage-derived lysins that kill bacteria via enzymatic hydrolysis of their cell walls (1, 2). Lysins have a characteristic modular structure, with catalytic and cell-wall-binding domains, and act to degrade the cell wall peptidoglycan with glycosidase, amidase, endopeptidase, and/or lytic transglycosylase activities. When applied exogenously (as purified recombinant enzymes) to Gram-positive bacteria, lysins elicit rapid osmotic lysis and bacterial cell death in a process referred to as “lysis from without” (3). The potent ability to rapidly kill pathogenic bacteria is the foundation for the development of lysins as powerful new therapeutic agents.
CF-301 is the first agent of the lysin class to enter phase 2 of clinical development in the United States as a systemic treatment for bacteremia and endocarditis due to Staphylococcus aureus (4). Hallmark features of CF-301 include (i) a potent, targeted, and rapid bacteriolytic effect against a broad range of S. aureus isolates, including those resistant to conventional antibiotics; (ii) potent antibiofilm activity; (iii) a low propensity for resistance emergence; and (iv) synergy with conventional antistaphylococcal antibiotics (5, 6). Lysins, including CF-301, are highly effective bacteriolytic agents in the context of standard in vitro assays and in a wide array of animal models of invasive infections (7), yet despite extensive study, there is no full understanding of such lysin activity in complex human physiological contexts, such as blood.
The influence of biologically relevant concentrations of blood matrices (including human) on CF-301 activity is particularly important considering the intended clinical plan to develop CF-301 for systemic use (8). While cation-adjusted Mueller-Hinton broth (caMHB) is an acknowledged reference medium in which to perform standard antimicrobial susceptibility tests (ASTs) (9), it does not replicate the in vivo host environment during infection. Given the well-described reduction in the antimicrobial activity of antimicrobial peptides (AMPs) and certain antibiotics in blood matrices compared to caMHB (10–14), there is an imperative need to understand the impact of blood matrices on CF-301 activity as it pertains to its performance in the context of its target indication. Many antimicrobials exhibit reduced activity in blood; however, some studies have documented enhanced drug activity in such contexts (15), underscoring the need to examine CF-301 in blood matrices.
Here, we report on the results of experiments using multiple different AST formats in both standard artificial medium (caMHB) and a variety of mammalian blood matrices. These studies led to the identification of a novel antimicrobial mechanism by which CF-301 synergizes with and activates latent blood components to facilitate more rapid and potent antistaphylococcal activity in some species (i.e., humans, rabbits, and dogs) but not in others (i.e., mice and rats). These ex vivo study findings were confirmed using the standard infective-endocarditis (IE) model to compare the efficacy of CF-301 in animals in which CF-301 was expected to activate host factors in blood (i.e., rabbits) with that in animals in which this synergistic effect was not predicted by in vitro studies (i.e., rats). The results of this work have important implications for the therapeutic use of CF-301 (and lysins in general) and suggest the potential for enhanced potency when CF-301 is administered systemically to patients, due to the activation of latent antistaphylococcal host factors in human blood.
RESULTS
Hierarchy of CF-301 activities in different mammalian sera.
The initial investigation into the activity of CF-301 in physiological concentrations of blood matrices was undertaken as part of the AST development program. In the course of identifying a robust AST medium for CF-301, we performed time-dependent in vitro bactericidal assays (time-kills) using the prototypical methicillin-resistant S. aureus (MRSA) strain MW2 (USA400; CC1) in the reference AST medium (i.e., caMHB) and compared the results to those determined using sera from various animal species (e.g., rabbits, dogs, cows [calves], rats, and mice). A surprising hierarchy of CF-301 activities was observed based on the minimal sterilizing concentrations of CF-301 in the different species, i.e., 3.2 μg/ml in rabbit and dog sera (Fig. 1A and B), 32 μg/ml in rat and calf sera (Fig. 1C and D), and ≥320 μg/ml in caMHB and mouse serum (Fig. 1E and F).
FIG 1.

CF-301 has bactericidal activity in certain blood matrices superior to that in caMHB. A range of CF-301 concentrations were tested against S. aureus strain MW2 in the following growth matrices: rabbit serum (5 biological replicates) (A); dog serum (8 biological replicates) (B); fetal calf serum (3 biological replicates) (C); rat serum (5 biological replicates) (D); CAMHB (10 replicates) (E); BALB/c mouse serum (5 biological replicates) (F); whole human blood (10 biological replicates) (G); and human serum (20 biological replicates) (H). Mean values (±SEM) are shown for each time point. The limit of detection was 2 log10 CFU per milliliter.
Superiority of CF-301 in human blood matrices.
The hierarchy discovered in the sera of different animals described above led us to determine the activities of CF-301 in both human serum and whole blood. We used the time-kill assay format to demonstrate a rapid bactericidal effect for CF-301 (≥3-log10 CFU/ml reductions) and culture sterilization by 24 h posttreatment using a minimal concentration of 3.2 μg/ml (Fig. 1G and H) in both human serum and whole blood. These findings closely mirror our observations in rabbit and dog sera (Fig. 1E and F).
The time-kill study in human serum was replicated using three additional MRSA strains, three methicillin-sensitive S. aureus (MSSA) strains, and one Streptococcus pyogenes strain. We observed a minimal sterilizing concentration of 3.2 μg/ml for each strain tested (see Fig. S1 in the supplemental material). Compared to the minimal sterilizing concentration of 320 μg/ml observed in caMHB, CF-301 demonstrated a 100-fold increase in potency in human serum, as measured by the ability of CF-301 to sterilize staphylococcal cultures. Vancomycin, which served as a control in these experiments, demonstrated slightly decreased potency in human serum compared to caMHB (see Fig. S2A and B in the supplemental material), which is consistent with previous reports (16).
MICs of CF-301 in human serum.
We employed a second AST method to further validate our observations from time-kill assays. Here, we determined CF-301 MIC values against 74 clinical MSSA and 75 clinical MRSA isolates, as well as 22 additional vancomycin-resistant, linezolid-resistant, and daptomycin-resistant S. aureus strains. A 32-fold decrease in the MIC90 for CF-301 was observed for each staphylococcal strain set tested in human serum compared to caMHB (Table 1). These analyses also demonstrated a narrow range of MIC values in serum (0.25 to 2 μg/ml) compared to the wide range observed with caMHB (0.5 to 128 μg/ml).
TABLE 1.
Comparison of CF-301 MIC values (micrograms per milliliter) determined in caMHB and human serum
| S. aureus type | n | caMHB |
Human serum |
||||
|---|---|---|---|---|---|---|---|
| MIC50 | MIC90 | Range | MIC50 | MIC90 | Range | ||
| MSSA | 74 | 16 | 32 | 8–32 | 0.5 | 1 | 0.25–1 |
| MRSA | 75 | 32 | 32 | 2–128 | 0.5 | 1 | 0.25–2 |
| Othera | 22 | 4 | 32 | 0.5–32 | 0.5 | 1 | 0.25–2 |
Other S. aureus types tested comprised 12 vancomycin-resistant strains, 5 linezolid-resistant strains, and 5 daptomycin-resistant strains.
Potentiation of bactericidal activity is a general feature of CF-301 in human blood matrices.
Having examined CF-301 in human serum against a wide range of staphylococcal isolates, we next examined CF-301 activity against a single MRSA strain, MW2, using a heterogeneous array of 61 different pooled and individual human donor samples of whole blood, serum, and plasma from different commercial sources, featuring variations in age, sex, blood type, and type of anticoagulant used (see Table S1 in the supplemental material). These analyses resulted in a very narrow range of MIC values, from 0.5 to 2 μg/ml, which is consistent with the ability of CF-301 to potentiate high-level activity within the blood matrices of all human sources tested. It was notable that complement-inactivated serum retained high-level CF-301 activity, while delipidated serum did not.
The impact of sample source variation on CF-301 activity against MRSA strain MW2 was also examined in sera from a variety of animal species. We observed the following consistent ranges (n is the number of different commercial sources, strains, and/or lot numbers): mouse, 32 to 64 μg/ml (n = 10); rat, 8 to 16 μg/ml (n = 5); dog, 0.5 to 1 μg/ml (n = 8); and rabbit, 1 μg/ml (n = 2) (see Table S2 in the supplemental material). The hierarchy observed here, with rabbit, dog, and human superior to rat and mouse, is identical to that observed in the time-kill studies described above.
CF-301 synergy with conventional antistaphylococcal antibiotics is observed in human serum.
Synergy between CF-301 and conventional antibiotics was previously demonstrated in caMHB (5). Here, we have extended the synergy study to include human serum in both time-kill and checkerboard assay formats. The goal was to understand the impact of human serum on the ability of CF-301 to synergize with antibiotics.
In the time-kill assay, synergy between two agents is defined as a ≥2-log10-unit decrease in CFU per milliliter for the combination compared to the CFU reduction provided by the most active single constituent alone (with the second constituent at a largely ineffective concentration) after 24 h (17). We combined each of four different concentrations of CF-301 (from 0.025 to 0.25 μg/ml) with daptomycin (held at the bacteriostatic concentration of 2.5 μg/ml); we demonstrated that all the combinations yielded decreases of ≥2 log10 CFU/ml at 24 h compared to corresponding single-agent values (see Fig. S3 in the supplemental material). These findings are consistent with the enhancement of the activity of CF-301 in addition to antibiotics in human serum. Of note, the minimum concentration of CF-301 demonstrating synergy with daptomycin in human serum (i.e., 0.025 μg/ml) was 160 times lower than the minimum concentration of 4 μg/ml required for synergy with daptomycin in caMHB (5). This suggested a potential link between host factors in human blood and the bactericidal activity of CF-301.
The checkerboard assay (18) was used as a complementary approach to examine synergy between CF-301 and daptomycin or vancomycin. Checkerboards were generated in both caMHB and human serum, with “synergy” defined in the standard manner (19) as an inhibitory activity greater than that predicted by adding the inhibitory activities of the 2 drugs together (i.e., summation fractional inhibitory concentrations [ΣFICMIN {the lowest ΣFIC value obtained among all combinations} or ΣFICAVG {the average ΣFIC value of three consecutive drug combinations}] of ≤0.5). Based on ΣFIC values, CF-301 synergized more effectively with both daptomycin and vancomycin in human serum than in caMHB (see Table S3 in the supplemental material). The lower ΣFIC values observed in serum were particularly significant considering the use of 32 times less CF-301 to generate the serum values than for caMHB. We also extended the checkerboard assays to include 8 additional MRSA strains, each of which demonstrated synergistic ΣFICMIN and ΣFICAVG values in human serum that were superior to those determined in caMHB (see Fig. S4 in the supplemental material).
Identification of latent host factors in human blood which promote synergy with CF-301.
Several key features of human serum potentially required to support the ability of CF-301 to exert high-level activity were examined using a colorimetry-based MIC method. When serum (in the absence of CF-301) was either: (i) pretreated with proteinase K (Fig. 2A), (ii) heated to 75°C for 30 min (Fig. 2B), or (iii) diluted to <6.25% in caMHB (Fig. 2C), it subsequently no longer supported the superior activity of CF-301. These findings led us to posit that CF-301 activates and/or synergizes with serum proteins that are stable upon heating to 75°C and that are abundant. To test this hypothesis, a range of human serum proteins, including many with known antimicrobial activity, were tested for synergy with CF-301 using the same checkerboard assay format detailed above. A critical aspect of this work was the use of a basal medium, referred to as caMHB/HiHuS, comprised of caMHB containing 50% human serum that had been heated to 75°C for 30 min to ablate the potentiation effect. We observed no enhanced bactericidal activity for CF-301 in caMHB/HiHuS unless we supplemented the medium with extrinsically applied blood factors that had not been heat inactivated. While the majority of blood factors tested did not synergize with CF-301, based on ΣFICAVG values of >0.5 (see Table S5 in the supplemental material), we did detect potent synergy using native and recombinant forms of either human lysozyme (HuLYZ) or serum albumin (HSA), with ΣFICAVG values of ≤0.1 in each case. Interestingly, rabbit serum albumin (rabbit SA) also synergized with CF-301 (ΣFICAVG ≤ 0.1) in a manner consistent with the whole-serum data shown in Table S2.
FIG 2.

CF-301 synergizes with human blood components against S. aureus strain MW2. (A) Sensitivity of the synergistic effect to proteinase K. MIC determinations were performed in the presence of alamarBlue, a dye that enabled an assessment of bacterial viability (pink) and death (blue) over the indicated dilution range. (B) Temperature sensitivity of the synergistic effect. (C) Checkerboard analysis of CF-301 and human serum in caMHB. The red squares indicate combinations with diminished synergy. (D and E) Time-kill analysis of CF-301 with a concentration range of HuLYZ (in micrograms per milliliter) and HSA (in milligrams per milliliter), respectively, in CAMHB/HiHuS. Mean values (±SEM) are indicated for three independent technical replicates. (F) Lytic assays examining HuLYZ and HSA without CF-301. (G and H) Lytic activities of 4 μg/ml CF-301 alone and with a concentration range of HuLYZ (in micrograms per milliliter) and HSA (in milligrams per milliliter), respectively. (I) Lytic assays examining CF-301 with HuLYZ and HSA.
The synergy observed in the checkerboard format was confirmed using the time-kill assay in caMHB/HiHuS. We combined CF-301 (at a 0.25× MIC amount determined in caMHB/HiHuS) with a range of HuLYZ concentrations (from 1 to 35 μg/ml) chosen to encompass physiological HuLYZ concentrations between 9.6 and 16.8 μg/ml (20–22). All the combinations of CF-301 tested with HuLYZ at >5 μg/ml resulted in reductions of ≥2 log10 CFU/ml at 24 h (versus combined single-agent values), consistent with synergy (Fig. 2D). HuLYZ alone was completely inactive against S. aureus at all the concentrations tested. Next, the 0.25× MIC amount of CF-301 was combined with a physiological concentration of HSA of 40 mg/ml (10), resulting in a 3-log10 CFU/ml reduction (over CF-301 alone), consistent with synergy (Fig. 2E). HSA alone had no antibacterial activity in the absence of CF-301.
CF-301 potentiates high-level lytic activity with HuLYZ and/or serum albumin.
The impact of HuLYZ and/or HSA on the activity of CF-301 was examined using a “lytic assay” previously developed to follow the loss of optical density in lysin-treated bacterial suspensions (5). Not surprisingly, HuLYZ and HSA had no bacteriolytic activity independent of CF-301 (Fig. 2F). A potent bacteriolytic effect, however, was observed for a single concentration of CF-301 with increasing HuLYZ or HSA concentrations; the combinations reached maximum potency at physiological levels of both HuLYZ and HSA (Fig. 2G and H, respectively). The most potent lytic activity was observed with the combination of HuLYZ and HSA together with CF-301 (Fig. 2I).
The lytic assay is routinely used to determine CF-301 specific activity (5) and, as such, was applied here to study CF-301 in combination with HuLYZ and/or HSA. The combination of either HuLYZ or HSA at physiological concentrations (10 μg/ml and 40 mg/ml, respectively) with CF-301 (4 μg/ml) resulted in 9.8- and 17.8-fold increases in lytic activity, respectively, over CF-301 alone (see Table S6 in the supplemental material). The combination of HuLYZ and HSA together with CF-301 resulted in a significant 25-fold increase in observed activity.
Combinations of CF-301, HuLYZ, and HSA recapitulate the serum effect.
To confirm the ability of CF-301 to potentiate bacteriolysis by leveraging both lysozyme and albumin, we performed a series of “recapitulation” experiments in an MIC format using caMHB and caMHB/HiHuS, each of which has no intrinsic ability (without supplementation) to support high-level CF-301 activity. Recapitulation should be visualized as a decrease in the MIC approaching the 32-fold reduction normally observed for human serum compared to either caMHB or caMHB/HiHuS. As such, supplementation with physiological concentrations of HuLYZ (10 μg/ml) or HSA (40 mg/ml) resulted in either 2-fold or 8- to 16-fold decreases in the CF-301 MIC, respectively (Table 2). The addition of HuLYZ and HSA together had an additive effect, resulting in a 32-fold decrease in the MIC, which effectively recapitulates the 32-fold serum effect.
TABLE 2.
Decreases in CF-301 MIC values using media supplemented with HuLYZ and/or serum albumins
| Supplementation | Fold decrease in CF-301 MIC (μg/ml) compared to base medium with no supplementation |
|
|---|---|---|
| caMHB | caMHB/HiHuS | |
| HuLYZ (10 μg/ml) | 2 | 2 |
| HSA (40 mg/ml) | 16 | 8 |
| HuLYZ (10 μg/ml) + HSA (40 mg/ml) | 32 | 32 |
| Rabbit SA (40 mg/ml) | 16 | 8 |
| Rat SA (20 mg/ml) | 2 | 1 |
| Rat SA (40 mg/ml) | 8 | 16 |
| Mouse SA (20 mg/ml) | 1 | 2 |
| Mouse SA (40 mg/ml) | 8 | 16 |
Recapitulation experiments with animal albumins.
The similar abilities of CF-301 to activate and/or synergize with host factors in human and rabbit sera suggest that the lysozyme and albumin components of each species act in similar manners. To confirm the albumin effect in the MIC assay format, we combined rabbit SA, at the rabbit physiological concentration of 40 mg/ml (23), with CF-301 in either caMHB or caMHB/HiHuS. The combination resulted in an 8- to 16-fold decrease in the CF-301 MIC, identical to that observed for HSA in combination with CF-301 (Table 2). The corresponding experiment using rabbit lysozyme was not performed owing to the lack of a commercial source for the enzyme.
In view of the lack of apparent synergy with rat or mouse serum noted above (Fig. 1), we investigated the effect of combining either rat or mouse SA with CF-301 in caMHB and caMHB/HiHuS. Using each of the SAs at the physiological concentration of 20 mg/ml for each rodent species (24), we observed only 1- to 2-fold decreases in the CF-301 MIC (Table 2). This finding is consistent with the time-kill and MIC studies, in which rat and mouse sera do not yield synergy with CF-301. Interestingly, when we increased the rat or mouse SA to the supraphysiological concentration of 40 mg/ml, we observed 8- to 16-fold decreases in the CF-301 MIC, similar to that observed for HSA. While it is tempting to speculate that albumin (at a sufficiently high concentration) is the missing factor in rodent serum needed for synergy with CF-301, it should be noted that human serum diluted to 20 mg/ml still retains the ability to support high-level CF-301 activity (Fig. 2H).
HSA promotes formation of high-molecular-mass complexes of CF-301.
To understand the mechanism of synergy for CF-301, particularly in light of the high-affinity protein binding capacity of HSA (10), we next examined molecular complex formation by CF-301 and HSA in human serum by Western blot analysis. Specifically, we used polyclonal antibodies specific for CF-301 to examine the supernatants from MIC wells (i.e., only the wells containing the lowest concentration of CF-301 capable of inhibiting growth) with human serum (MIC = 1 μg/ml), caMHB (MIC = 32 μg/ml), or caMHB/HiHuS (MIC = 32 μg/ml). In human serum, which contains the relevant substrates for synergy with CF-301, we detected CF-301 at its expected molecular masses of 26 and 52 kDa (for monomer and dimer forms) and at an unexpected mass of ∼150 kDa (Fig. 3A). In both caMHB and caMHB/HiHuS, which lack the substrates for synergy with CF-301, the ∼150-kDa band was not detected.
FIG 3.
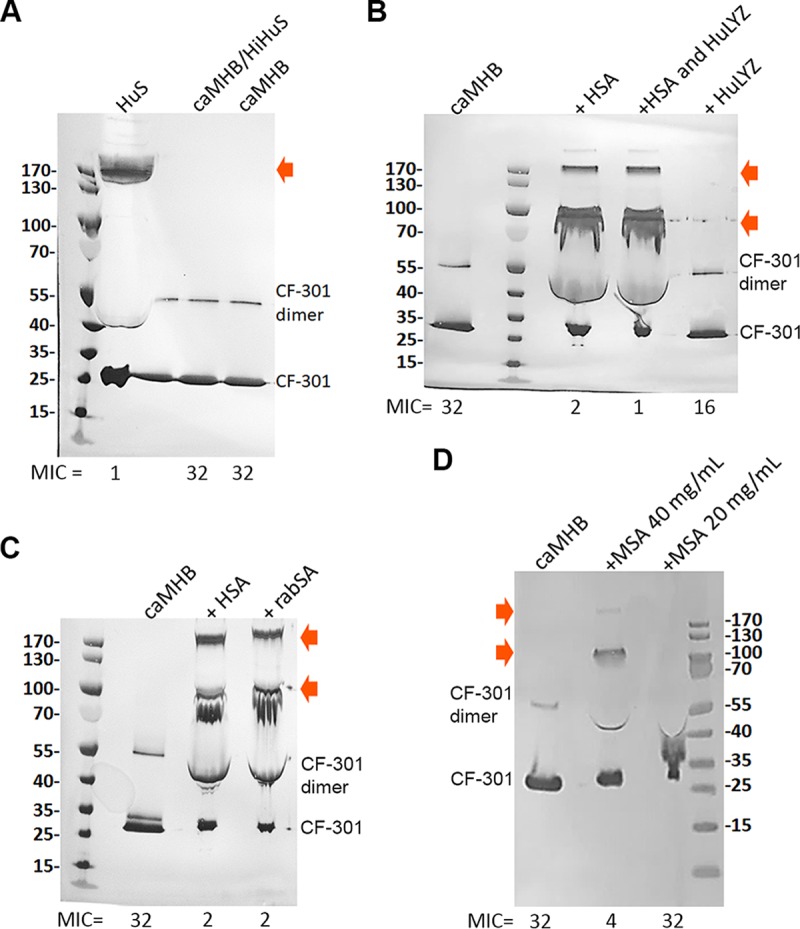
Western blot analyses (using anti-CF-301 antibodies) of MIC well samples under different incubation conditions. High-molecular-mass multimers are indicated by arrows. MIC values are indicated (micrograms per milliliter). (A) Comparison of human serum to caMHB/HiHuS and caMHB. (B) Analysis of caMHB supplemented with HSA (40 mg/ml) and/or HuLYZ (10 μg/ml). (C) Comparison of caMHB supplemented with HSA (40 mg/ml) or rabbit SA (rabSA) (40 mg/ml). (D) Analysis of caMHB supplemented with MSA at 40 mg/ml and 20 mg/ml. Molecular mass standards are indicated (in kDa).
Insight into the nature of the high-molecular-mass band(s) came from the Western blot analysis (as described above) of the supernatant from the MIC well determined in caMHB supplemented with HSA at 40 mg/ml. Here, an MIC shift from 32 μg/ml (in caMHB) to 2 μg/ml (in caMHB with HSA or rabbit SA) was accompanied by the concomitant appearance of both the ∼150-kDa band and an additional ∼95-kDa band (Fig. 3B and C). When we similarly examined caMHB supplemented with HuLYZ (10 μg/ml), no high-molecular-mass bands were observed (Fig. 3B). Likewise, in caMHB supplemented with mouse SA at the mouse physiological concentration of 20 mg/ml (MIC = 32 μg/ml), no high-molecular-mass bands were observed; however, supplementation with mouse SA at the supraphysiological concentration of 40 mg/ml did restore both the upper bands and the low MIC (MIC = 4 μg/ml) (Fig. 3D).
Overall, these findings suggest that HSA promotes and/or participates in the formation of stable CF-301 complexes that potentiate antimicrobial activity. The ability of albumin to form SDS-PAGE-resistant complexes with a variety of other proteins has been described (25), suggesting that CF-301 may form a hetero-oligomeric complex with HSA. In support of this, the ∼150-kDa band was examined by mass spectrometry and sequencing, and the most abundant fragment signals detected were serum albumin and CF-301 (data not shown).
Serum lipids are required for the potentiation effect of CF-301.
Our observation that delipidated human serum does not synergize with CF-301 (see Table S1) raised the notion that fatty acids (FAs) are likely required as part of the mechanism by which CF-301 synergizes with HSA in serum. Most of the free FAs in circulation are bound to HSA (26), and the process of delipidation, while not altering HSA levels, does reduce free-FA levels by approximately one-third (27). To confirm that low FA levels are responsible for the inability of CF-301 to synergize in delipidated serum, we determined the effects of introducing two of the more common FAs in circulation, oleate and palmitate (28), on the performance of delipidated serum in the MIC assay format. The addition of either oleate or palmitate at a physiological concentration of 0.625 mg/ml resulted in 8- and 16-fold decreases in the CF-301 MIC, respectively, with no further decreases associated with the concomitant addition of both lipids (see Table S7 in the supplemental material). Considering that the FAs alone (outside the serum context) have no impact on CF-301 activity (data not shown), it is likely that the effect is mediated through HSA. The effect is also likely to reflect a direct interaction between HSA and CF-301, based on the understanding that FAs bound to high-affinity sites on HSA act to promote additional binding activities to drugs and other compounds (26).
Cell surface-binding studies using CF-301 and HuLYZ in serum.
Fluorescence microscopy was used to test the effect of pretreating S. aureus strain MW2 with different animal sera (or caMHB) on subsequent labeling with rhodamine-conjugated CF-301 (CF-301RHOD). Pretreatment with human or rabbit serum resulted in extensive cell surface labeling, with a short exposure time of 3 ms (Fig. 4A). In contrast, pretreatment with rat serum, mouse serum, or caMHB resulted in poor or no apparent labeling, even with long exposure times of 3 s. Interestingly, high-level labeling with CF-301 was observed in mouse serum after supplementation with 40 mg/ml HSA. In each case, greater surface labeling with CF-301 correlated with low CF-301 MICs.
FIG 4.

Binding and activity of CF-301. (A) Effects of serum and medium pretreatments on CF-301 binding to S. aureus MW2 determined by differential interference contrast (DIC) and fluorescence microscopy. Exposures of 3 ms (human, rabbit, and mouse plus HSA samples) and 3 s (rat, mouse, and caMHB samples) were used to visualize CF-301RHOD. MIC values for each condition are indicated (micrograms per milliliter). (B) Effect of HSA on CF-301 binding to S. aureus ATCC 700699 determined by deconvolution microscopy. Maximum-intensity projections are shown. CF-301RHOD, red; DAPI, blue (scale bar, 2 μm). (C) Effect of CF-301 on HuLYZ binding to S. aureus ATCC 700699 determined by deconvolution microscopy. Maximum-intensity projections are shown. HuLYZAF, green; DAPI, blue (scale bar, 4 μm). (D) Bactericidal activity of CF-301 in human serum and caMHB determined by TEM. CF-301 was tested at 10×, 1×, and 0.1× MIC levels for each environment. Direct magnification values: control, ×13,000; 10× MIC, ×2,600; 1× and 0.1× MIC, ×6,600. Scale bars, 1 μm.
The impact of pretreatment with HSA (at 40 mg/ml) on the surface labeling of S. aureus strain ATCC 700699 with CF-301RHOD was next examined by deconvolution microscopy. Strain ATCC 700699 is a vancomycin intermediate susceptibility (VISA) strain, with a thickened cell wall that enables better visualization of cell surface labeling by deconvolution microscopy. For the HSA pretreatment, but not the phosphate-buffered saline (PBS) control, we observed intense cell surface labeling (Fig. 4B). Interestingly, the HSA pretreatment did not enable binding of lysins specific for either Bacillus anthracis or S. pyogenes (PlyG and PlyC, respectively), which do not have activity against S. aureus (data not shown).
Based on the ability of CF-301 to synergize with HuLYZ, we also used deconvolution microscopy to test the binding of Alexa Fluor-labeled HuLYZ (HuLYZAF) to staphylococci treated with CF-301. Treatments with two physiologically relevant concentrations of CF-301 (1× and 0.5× MIC) for 30 min resulted in extensive surface labeling by HuLYZAF at 25 μg/ml (Fig. 4C). Furthermore, the CF-301 treatments did not facilitate the binding of either the PlyG or PlyC lysin (data not shown), supporting the specificity of the functional interaction between HuLYZ and CF-301 on S. aureus cells.
Visualization of CF-301-mediated bacteriolysis in human serum.
Transmission electron microscopy (TEM) was used to demonstrate the bacteriolytic effect of CF-301 on S. aureus strain MW2 after a short (15-min) treatment in caMHB or human serum. In caMHB, we observed the classic image of lysin-mediated cytoplasmic membrane “bubbling and extrusion” just prior to lysis (29) after treatment with 10× and 1× MIC levels (320 μg/ml and 32 μg/ml, respectively) (Fig. 4D; see Fig. S4). In contrast, treatments in serum, across a range of CF-301 concentrations from 10× to 0.1× MIC (5 to 0.05 μg/ml), resulted in a circumferential dissolution of the electron-dense cell wall material with the staphylococci encased in a proteinaceous sheath possibly consisting of host proteins, including HSA (Fig. 4D; see Fig. S5 in the supplemental material). The bacterial debris in serum also appeared to remain ensheathed after lysis. There was no evidence of cytoplasmic-membrane bubbling after CF-301 treatment in serum.
In vivo efficacy in the IE model further validates ex vivo findings.
We used the gold standard MRSA IE models (30, 31) to compare in vivo efficacy profiles of CF-301 in animals for which in vitro studies predicted CF-301 synergy with host factors in animal serum (i.e., rabbits) versus in animals for which this synergy was not expected (i.e., rats). Readouts from the IE models are based on pharmacokinetic (PK)/pharmacodynamic (PD) relationships as measured by the exposure levels required for efficacy (i.e., reductions in heart valve vegetation CFU burdens). We also evaluated the efficacy of CF-301 treatment in combination with daptomycin to reflect the common use of the agent in serious MRSA infections.
In the rat IE model, a CF-301 dose of 10 mg/kg of body weight administered in addition to the daptomycin human therapeutic dose (HTD) equivalent (40 mg/kg in rats) resulted in an ∼6-log10-unit drop in CFU per gram in heart valve vegetations compared to an ∼3-log10-unit drop in CFU per gram with daptomycin alone (Fig. 5A). The same ∼6-log10-unit decrease in bacterial densities was achieved in the rabbit IE model after administration of total CF-301 doses of only 0.09 to 0.18 mg/kg in addition to a dose of daptomycin lower than the HTD equivalent (Fig. 5B). The sub-HTD equivalent dose of daptomycin in rabbits was chosen, based on the results of a dose-response study (see Fig. S6 in the supplemental material), to match the antistaphylococcal activity previously described using the 40-mg/kg dose in rats (32). The enhanced efficacy of CF-301 in the rabbit IE model (which we attribute to the synergy of CF-301 with rabbit serum components) is thus even more significant, considering the 10-fold higher dose of daptomycin used in the rat than in the rabbit model.
FIG 5.

Evaluation of the blood effect in vivo using pharmacodynamic studies. Shown are efficacies in the rat (A) and rabbit (B) infective-endocarditis model for various CF 301 dosing regimens added to daptomycin. The data are plotted as treatment regimen versus average log10 CFU per gram of tissue for each dose group. Medians and SEM are shown. The insets show the reductions of the bacterial burden in the heart valve vegetation after treatment with a single bolus of the highest concentration of CF-301 (i.e., 10 mg/kg in rat and 1.4 mg/kg in rabbit).
In the rat IE model, the estimated area under the concentration-time curve (AUC)/MIC ratio of ≥0.87 required for efficacy (e.g., an additional ∼3-log10-unit drop in CFU per gram in heart valve vegetation when CF-301 was added to daptomycin) was achieved at the 10-mg/kg dose of CF-301 (Fig. 5; see Table S8 in the supplemental material). A similar AUC/MIC value was obtained in the rabbit model at substantially lower exposures and MICs, using a CF-301 dose of 0.18 mg/kg, in addition to daptomycin. Overall, the in vivo studies demonstrated that a >50-fold higher dose (and >20-fold higher AUC exposure) was required to achieve efficacy in rats (10 mg/kg) similar to that in rabbits (0.18 mg/kg). Importantly, these data further support the selection of the 0.25-mg/kg CF-301 dose as an anticipated efficacious dose of CF-301 in patients with invasive S. aureus infections.
DISCUSSION
CF-301 synergizes with human blood components.
In this report, we describe the substantial ability of CF-301 to activate and synergize with elements of human blood to potentiate an enhanced level of antistaphylococcal activity over that predicted from AST following standardized (e.g., CLSI) procedures using caMHB reference broth. Our findings were initially based on in vitro time-kill assays demonstrating reductions of ≥100-fold in the minimum sterilizing concentration of CF-301 against 11 staphylococcal strains in blood matrices from up to 34 different sources compared to caMHB. We further demonstrated a ≥32-fold reduction in the MIC90 against 171 S. aureus isolates tested in human serum and a consistently low level of MIC variability in the blood, sera, and plasma from 61 different human sources. In combinations with either daptomycin or vancomycin, we observed synergistic activities in human serum at CF-301 concentrations up to 160 times lower than in caMHB. Overall, these results support the potential effectiveness of CF-301 as an intravenously (i.v.) administered antimicrobial agent for the treatment of S. aureus bacteremia and endocarditis.
Furthermore, our findings implicate synergy between CF-301 and two specific components of human blood (i.e., lysozyme and albumin), with no apparent (single-agent) intrinsic antistaphylococcal activity, as key factors associated with enhanced activity and efficacy. The unique ability of CF-301 to activate and synergize with otherwise dormant host “bystanders” (with respect to killing staphylococci) has important implications for the potential therapeutic use of CF-301 and the measurement of its antimicrobial activity. Furthermore, our findings distinctly contrast with the general understanding that, for many systemically delivered conventional small-molecule antibiotics, protein binding in circulation (primarily to albumin) may serve to reduce such drug activity (10, 12, 14, 16, 33–35).
CF-301 leverages the activity of human serum albumin.
There have been no previous descriptions of a role for HSA in promoting antimicrobial activity in the synergistic manner described for CF-301. Our assumptions regarding HSA are based on the following observations: (i) synergy with CF-301 in multiple assay formats, including checkerboards, time-kill assays, and the lytic assay; (ii) the ability of exogenous HSA and rabbit SA to reconstitute the serum effect; (iii) a putative interaction with CF-301 detected by Western blot analysis; and (iv) the promotion of CF-301 binding to the staphylococcal cell surface detected by microscopy. It was particularly notable that rat SA and mouse SA could reconstitute the HSA effect when added at the supraphysiological concentration of 40 mg/ml. The relatively low physiological SA concentrations in rodents of 20 mg/ml, which is half of that observed in rabbits and humans, at least partially explains the inability of rodent sera to serve as substrates for high-level CF-301 activity. While there are undoubtedly other differences in the compositions of rodent and human sera, it is most striking that protein binding, primarily to albumin, is lower in rodents than in humans for most antimicrobials (10, 36, 37). Binding preferences of up to 1,000-fold for HSA over rodent SA have been reported, and there are distinct structural differences in the binding sites of human and rodent albumins to account for differences in the extent of drug binding (38–40). Thus, the reduced capacity of rodent albumins to support high-level CF-301 activity (compared to HSA) may be attributed to both the lower physiological concentration of albumin in rodents. It is also important that a similar hierarchical effect was noted for the antimicrobial peptide polymyxin B nonapeptide, which exhibited superior activity in human serum, intermediate activity in rat serum, and poor activity in mouse serum (41).
Our experiments with delipidated serum further suggest a direct interaction between CF-301 and HSA as part of the mechanism for enhanced activity. The circulating HSA monomer is commonly complexed with lipids, and there are at least 7 high- and intermediate-affinity FA binding sites that can, when bound to FAs, modify and alter interactions with antibiotics and other molecules over the known high-affinity drug and protein binding sites of albumin (including subdomains IIA and IIIA) (26, 42, 43). We hypothesize that the observed interaction with albumin occurs over the SH3b domain of CF-301, which has been shown, in the context of other proteins, to mediate protein-protein interactions (perhaps over a flat, hydrophobic, ligand-binding pocket) (44–46). Such an interaction with albumin is supported by studies showing that albumin may perform chaperone-like functions for client proteins and that the presence of free FAs, at physiological concentrations, stabilizes the chaperone activity (25, 47); furthermore, these interactions lead to the formation of stable high-molecular-weight species observable by SDS-PAGE and similar to that formed between CF-301 and albumin.
S. aureus expresses a range of host matrix binding proteins on its surface in addition to albumin-binding activities (48–50). One of the albumin-binding proteins, Ebh, has been shown to contribute to survival in blood and the overall pathogenesis of staphylococcal infections (49). Albumin-binding proteins are, furthermore, found on a range of pathogenic microorganisms that may adsorb HSA as part of a survival strategy in host tissues (51). Accordingly, we observed rapid accumulation of a dense proteinaceous surface layer on S. aureus in human serum, possibly consisting of albumin and/or other blood components. The layer formed a visible sheath around the staphylococci that modified the visual manifestation of CF-301-mediated bacteriolysis (compared to the event in caMHB); this interaction ultimately resulted in bacterial ghost-like structures (52), often with intact cell envelopes encased in a matrix of possibly host-derived material. The encasement of bacterial debris and fragments (formed postlysis) could help mitigate the potential risk for proinflammatory responses associated with the free release of these fragments into the bloodstream of the host. Furthermore, encasement of bacteria and CF-301 in HSA matrices may also reduce the risk of potentially deleterious immunological reactions. Overall, our findings support the hypothesis that the natural ability of staphylococci to coat themselves with HSA in the bloodstream and thus evade human immune surveillance may be an Achilles’ heel with respect to the enhanced capacity of HSA for binding CF-301, which leads to increased bacteriolysis. In other words, the mechanism of synergy between CF-301 and HSA is based on improved accumulation kinetics for CF-301 at the bacterial cell surface mediated by HSA, resulting in more rapid and efficient bacterial killing by CF-301. While the interaction between CF-301 and HSA was strong, as evidenced by its maintenance in SDS-PAGE gels, the equilibrium binding affinities of lysins for cell wall targets lies in the picomolar to nanomolar range (53, 54) and may thus be favored.
CF-301 leverages the activity of human lysozyme.
The results presented here also allow conclusions to be drawn regarding the ability of CF-301 to activate lysozyme against S. aureus in human serum. First, there is a general understanding that mature S. aureus peptidoglycan is resistant to HuLYZ activity by virtue of O-acetylation at the C-6 position of cell wall N-acetylmuramic acid (21, 55). Accordingly, we observed no antistaphylococcal activity whatsoever for HuLYZ tested alone over a wide range of concentrations in multiple different AST formats. Based on our findings, we propose that CF-301 accumulates at the cell surface in a preferential manner by virtue of interactions with HSA and that cell wall hydrolysis by CF-301 in turn facilitates the activation of HuLYZ. While the exact nature of HuLYZ activity here is unknown, one possible explanation is that CF-301-mediated cleavage of peptidoglycan initiates access of HuLYZ to nascent peptidoglycan formed prior to O-acetylation and that the subsequent hydrolytic activity of HuLYZ promotes more CF-301 binding as bacteriolysis proceeds.
Ex vivo observations translate into in vivo activity.
To understand the in vivo efficacy profile of CF-301 in the intended clinical indication of staphylococcal bacteremia and endocarditis, we conducted experiments in two species (rabbits and rats) with observed differences in the capacity of CF-301 (in addition to daptomycin) to synergize with the respective serum types in the ex vivo formats reported here. The IE model is well established in both rabbits and rats (30, 31) and has a significant biofilm component that is highly relevant with respect to the intended clinical indication for CF-301. In accordance with our ex vivo observations of potent CF-301 synergy in rabbit, but not rat, serum, we observed that a >50-fold higher dose of CF-301 and a >20-fold higher AUC exposure was required to obtain a bactericidal effect in rats (10 mg/kg) similar to that in rabbits (0.18 mg/kg). These models provide further evidence for the potential therapeutic implications of the ability of CF-301 to activate and synergize with HSA and HuLYZ and support the anticipated efficacy of CF-301.
Extension to other lysins.
CF-301 was originally identified as a Streptococcus suis prophage lysin exhibiting a domain arrangement commonly observed in many antistaphylococcal lysins (5, 56) and defined by a catalytic N-terminal cysteine-histidine-dependent amidohydrolase/peptidase (CHAP) domain linked to a cell wall–binding C-terminal domain belonging to the SH3b family of proteins (57, 58). We predicted that lysins of this group, which include enzymes active against both staphylococci and streptococci (like CF-301), as well as enzymes active against only staphylococci (59), would all exhibit the CF-301-like potentiation effect in human blood. As expected, the 32-fold decrease in the MIC observed for CF-301 against S. aureus strain MW2 tested in human serum versus caMHB was similarly observed for a range of antistaphylococcal lytic enzymes, including lysostaphin and LysK (data not shown). These findings suggest a class effect for at least a subset of antistaphylococcal lysins.
Summary and next steps.
Overall, our data demonstrate that the potency of CF-301 can be greatly enhanced as a result of synergistic interactions with the human blood components HSA and lysozyme, which otherwise have no intrinsic antistaphylococcal activity. The proposed mechanism by which CF-301 activates host components to synergize and kill bacteria more efficiently highlights the remarkable potential of the lysin (and possibly the entire lysin class) to serve as an adjunct therapeutic strategy in life-threatening systemic infections in humans. Our findings also distinguish CF-301 from small-molecule antibiotics, which typically exhibit diminished systemic activity because of binding to albumin. Rather than inhibiting activity, CF-301, a biological agent chemically distinct from antibiotics, appears to substantially increase its activity through its interactions with albumin. Our laboratories are currently carrying out investigations to define the exact mechanistic nature of the synergistic interactions between antibiotics and CF-301.
Other pathogenic staphylococci and streptococci also express albumin-binding proteins on their surfaces that may adsorb HSA in systemic settings and thus may exhibit enhanced susceptibility to lysin-mediated activity. The possibility of targeting microorganisms other than S. aureus with CF-301 can be examined and is supported by the finding, described in this report, that S. pyogenes is susceptible to the synergistic activity of CF-301 in human serum. Other lysins in our research pipeline may also have the ability to synergize with HuLYZ and/or HSA against S. aureus and potentially other pathogens. Finally, we may also consider an expanded array of host environments in which to seek such an effect, including synovial fluid (which has albumin concentrations similar to that of blood) and infected bone, skin, and/or soft tissues that may be infiltrated with such blood components. In this manner, we will continue to develop CF-301, and possibly other, related lysins, to address the issue of widespread antibiotic resistance and emerging infectious threats.
MATERIALS AND METHODS
Bacteria, media, and growth conditions.
A subset of the bacterial strains used in this study are listed in Table S8. The 171 S. aureus strains used in Table 1 were previously described (5), including 74 MSSA and 75 MRSA clinical isolates from 2011 that were obtained by JMI Laboratories (North Liberty, IA) from 66 different hospital sites throughout the United States. The JMI isolates were recovered from a variety of different infection sources (i.e., bloodstream, respiratory, skin/soft tissue, and urinary tract infections) and consist of a genetically heterogeneous group based on sequence analysis (targeting loci including hlgBC, lukSF, and hla) and, for the MRSA strains, SCCmec typing, which identified types I to IV. The bacteria were cultivated on either BBL Trypticase soy agar with 5% sheep blood (TSAB) (Becton, Dickinson & Company [BD]), BBL Mueller-Hinton II broth, caMHB (BD), or brain heart infusion broth (BHI) (BD) unless otherwise indicated. With the exception of HyClone fetal bovine serum (0.1-μm filtered; GE Healthcare Lifesciences), the source, description, and lot number of all human and animal blood matrices tested are listed in Tables S1 and S2. Staphylococci were grown at 37°C with aeration unless otherwise indicated.
Reagents.
Lysin CF-301 was expressed, purified, and stored as previously described (5). Human anti-CF-301 IgG3 monoclonal antibody (expressed and purified by ContraFect Corporation) and mouse anti-human IgG3 heavy chain antibody, AP conjugate (ThermoFisher; 05-3622), were used as primary and secondary antibodies at 1:1,000 dilution for Western blots. Cloned constructs of the human anti-CF-301 IgG3 heavy and light chains were transiently cotransfected in HEK293 cells in serum-free medium. The culture supernatant was harvested 5 days after transfection, and affinity chromatography (Hi Trap Protein G HP column) was used to purify the anti-CF-301 IgG3. The monoclonal antibody (MAb) was selectively bound to the affinity column; washed with 20 mM sodium phosphate, pH 7.0; and eluted in 100 mM glycine-hydrochloric acid, pH 2.7. The fractions of the elution peak were analyzed on a 4 to 12% PAGE gel to confirm the purity of the protein. No degradation or aggregation was observed. A sample was buffer exchanged into 1× Dulbecco's phosphate-buffered saline and concentrated to 1.4 mg/ml. Western blot analysis confirmed the immunological specificity of the monoclonal antibody.
The agents (and vendor sources) tested in combination with CF-301 were as follows: β-defensin-3, human (Anaspec); Leap-1, human (Anaspec); Leap-2, human (GenScript); LL-37, human (Anaspec); LL-18-37 (Anaspec); histatin-5, human (Anaspec); HNP-1, human (Anaspec); HNP-2, human (Anaspec); human platelet factor IV 18 (Anaspec); lactoferrin, human milk (Sigma-Aldrich); lactoferrin, bovine colostrum (Sigma-Aldrich); lactoferricin H, human (Anaspec); human lysozyme, recombinant expressed in rice (Sigma-Aldrich); lysozyme, hen egg (Sigma-Aldrich); lysozyme, human neutrophil derived (Aviva Biosciences); lysozyme, human neutrophil derived (RayBiotech); human serum albumin, recombinant expressed in rice (Sigma-Aldrich); human serum albumin, fraction V (Sigma-Aldrich); human serum albumin, fraction V, fatty acid free, globulin free (Sigma-Aldrich); human serum albumin, recombinant expressed in Saccharomyces cerevisiae (Albumin Bioscience); mouse serum albumin, recombinant expressed in yeast (Albumin Biosciences); rabbit serum albumin (Sigma-Aldrich); and rat serum albumin (Sigma-Aldrich). Sodium oleate, sodium palmitate, vancomycin hydrochloride, daptomycin, and proteinase K-agarose from Tritirachium album were obtained from Sigma-Aldrich. For microscopy, DAPI (4′,6-diamidino-2-phenylindole), Alexa Fluor 488, and NHS-rhodamine were obtained from ThermoFisher Scientific. The labeling and purification of CF-301 conjugated to NHS-rhodamine and HuLYZ conjugated to Alexa Fluor 488 were performed as described by the manufacturer’s protocol. Nonreacted fluorophores were removed using PD-10 desalting columns (GE Healthcare), and labeling efficiencies were determined to be >80% in each case. The activity of CF-301RHOD was confirmed to be equivalent to that of CF-301 using the standard MIC assay. The activity of HuLYZAF was confirmed to be equivalent to that of HuLYZ using a drop dilution assay on a 1% agarose surface impregnated with 1 mg/ml peptidoglycan from Micrococcus luteus (Sigma-Aldrich). The production, purification, and use of green fluorescent protein (GFP)-labeled PlyG (PlyGGFP) and Alexa Fluor 488-labeled PlyC (PlyCAF) were previously described (60, 61).
Time-kill assays.
Bactericidal activities were tested using a standardized time-kill assay format defined by the CLSI (17). The assays were performed in 10 ml of caMHB or the indicated (undiluted) blood matrix with a bacterial inoculum of 5 × 105 CFU/ml in 125-ml glass Erlenmeyer flasks with agitation. The indicated agents were tested across a 10-fold range of concentrations. For daptomycin, caMHB cultures were supplemented with 50 μg/ml Ca2+. Growth controls with buffer alone were always included. Immediately before treatment and at the indicated time intervals thereafter up to 24 h, culture aliquots were removed and diluted in activated charcoal (to impede or halt drug activity). A series of 10-fold dilutions of the inactivated cultures were then plated on TSAB and incubated at 37°C for 24 h prior to colony enumeration. Bactericidal activity was defined as a decrease of ≥3 log10 CFU/ml relative to the initial inoculum. The time-kill curve data were, in every case, provided as arithmetic means and standard deviations from the results of at least 3 independent experiments, exactly according to standard time-kill methodology (17, 62–67). For analyses in serum, blood, and plasma, all replicates were performed using the indicated number (n > 3) of biologically independent samples representing different lot numbers (Tables 1 and 2); thus, if 5 biologically independent replicates are indicated, the analysis was performed in the blood, sera, or plasma of 5 different individuals and/or pooled samples.
MIC assays.
MIC values were determined by broth microdilution using the CLSI reference method (68) in either caMHB or the indicated blood matrices. caMHB/HiHuS was prepared by supplementing caMHB with 50% human serum and then filtering it through a Microcon centrifugal filter unit (Amicon Ultra-15; Millipore) with a 50-kDa cutoff before incubation at 70°C for 20 min to completely inactivate the component(s) responsible for synergy with CF-301. For the supplementation of delipidated serum with fatty acids, 50-mg/ml stock solutions of either oleate in H2O or palmitate in 100% ethanol were prepared and added to the serum to achieve a final concentration of 0.625 mg/ml; the supplemented serum was then incubated at 37°C for 1 h prior to use to promote fatty acid binding to HSA. Colorimetric determination of CF-301 MIC values was performed using alamarBlue (Thermo Fisher Scientific) exactly according to the manufacturer’s protocol. The analysis of CF-301 activity in human serum pretreated for 3 h with proteinase K-agarose beads (Sigma-Aldrich) was performed according to the manufacturer’s protocol. As a control for protease carryover after the removal of proteinase K-agarose beads, the treated serum was diluted 3:4 in untreated serum prior to MIC determination; the addition of untreated serum was expected to restore the synergistic effect only if there was no carryover of either unbound proteinase K or the proteinase K-agarose beads.
Lytic assays.
Overnight cultures of MRSA strain MW2 were diluted 1:100 in BHI and grown for 2.5 h at 37°C with aeration. The exponential-phase cells were washed, concentrated 10-fold in 20 mM phosphate buffer (PB) (pH 7.4), and split into aliquots to which either HSA (Albumin Biosciences) or human neutrophil-derived lysozyme (Aviva Bioscience) over a range of concentrations was added. For each concentration of HSA or lysozyme, 0.1 ml of the mixture was aliquoted in duplicate to a 96-well, flat-bottom, non-tissue-culture-treated microtiter plate (BD). The lytic reaction was then started by adding to all the wells 0.1 ml CF-301 (in phosphate) to a final concentration of 4 μg/ml. Control wells with CF-301, HSA, or HuLYZ alone at appropriate concentrations were included. Samples were mixed, and the optical density at 600 nm (OD600) was followed for 15 min at room temperature in a SpectraMax M5 microplate reader (Molecular Devices).
A variation of the lytic assay was performed to determine CF-301 specific activity. Exponential-phase MW2 cells were prepared as described above and divided into 4-ml aliquots containing either phosphate buffer (reaction mixtures without agents added, with cells alone) or HSA (Albumin Biosciences) and/or human lysozyme (Aviva Bioscience). The CF-301 (starting at 0.5 mg/ml) was 2-fold serially diluted across columns 1 through 11 of a 96-well plate with 20 mM phosphate, pH 7.4, at a volume of 0.1 ml. Column 12 contained no enzyme, only buffer, and was used as an assay control well. Bacterial cells mixed with either buffer, HSA, lysozyme, or HSA combined with HuLYZ were added as 0.1-ml aliquots to each well of three rows of serially diluted CF-301. Each plate was read at 600 nm for 15 min (with shaking for 3 s between reads) at room temperature using a microplate reader as described above. The specific activity in units per milligram of CF-301 was determined based on the CF-301 dilutions displaying curves just above and below the optical density that was 50% of the buffer control at 15 min.
Checkerboard assays.
The checkerboard assay was performed as described previously (18) and was adapted from the CLSI method for broth microdilution (68). Checkerboards were prepared by first aliquoting in each column of a 96-well polystyrene microtiter plate the same amount of CF-301 diluted 2-fold along the x axis. In a separate plate, corresponding rows were prepared in which each well had the same amount of another agent diluted 2-fold along the y axis. The dilutions were then combined, so that each column had a constant amount of CF-301 and doubling dilutions of the second agent, while each row had a constant amount of the second agent and doubling dilutions of CF-301. Each well thus had a unique combination of CF-301 and the second agent. Bacteria were added to each well at concentrations of ∼5 × 105 CFU/ml in caMHB or human serum (pooled male, type AB; sterile filtered; U.S. origin; Sigma-Aldrich). The MIC of each drug, alone and in combination, was recorded after 18 h at 37°C in ambient air. The results are expressed in terms of a ΣFIC index equal to the sum of the FICs for each drug; the FIC for a drug is defined as the MIC of the drug in combination divided by the MIC of the drug used alone. Both the ΣFICMIN and ΣFICAVG are reported for each paired agent. If the ΣFIC index was ≤0.5, the combination was interpreted as being synergistic; between >0.5 and ≤2 was interpreted as additive; and >2 was interpreted as antagonistic (18). Colorimetric determinations of CF-301 MIC values were also performed using alamarBlue (ThermoFisher Scientific) according to the manufacturer’s protocol.
Synergy time-kill assays.
Synergy time-kill curves were performed according to the method described by the CLSI (17). Strain MW2 was suspended in caMHB with 50% HiHuS at a concentration of 5 × 105 CFU/ml and exposed to CF-301 and/or daptomycin, HSA (Albumin Biosciences), and HuLYZ (Aviva Bioscience) for 24 h at 35°C in ambient air with agitation. At timed intervals, culture samples were removed, serially diluted, and plated to determine the number of CFU per milliliter. The resulting kill kinetics determinations are shown graphically by plotting log10 CFU per milliliter versus time. Mean values are indicated (±standard errors of the mean [SEM]) for three independent experiments using the single indicated serum sample. Synergy was defined as a ≥2-log10-unit decrease in the number of CFU per milliliter between the combination and its most active constituent, with the least active constituent tested at an ineffective concentration.
Fluorescence microscopy. (i) Binding of CF-301 to the bacterial cell surface in different serum environments.
Mid-log phase MRSA strain MW2 was suspended at 1 × 107 CFU/ml in either caMHB or 100% serum from either human (Sigma-Aldrich), rabbit (Gibco), rat (BioreclamationIVT), or mouse (BioreclamationIVT) sources and incubated for 30 min at 37°C. An additional mouse serum sample containing 40 mg/ml HSA (Albumin Biosciences) was also tested. After the preincubation, the bacteria were washed with 1× PBS, resuspended in 50 μl of PBS, and attached to the surface of a poly-l-lysine-coated cover glass. The cells were washed and treated with CF-301RHOD (2 μg/ml in PBS) for 10 min before washing and counterstaining with DAPI. Slides were mounted in 50% glycerol and 0.1% p-phenylenediamine in PBS, pH 8. Fluorescence microscopy was performed using a Nikon Eclipse E400 microscope equipped with a Nikon 100×/1.25-numerical-aperture (NA) oil immersion lens and a Retiga EXi fast 1394 camera (QImaging). QCapture Pro version 5.1.1.14 software (QImaging) was used for image capture and processing.
(ii) CF-301 binding in the presence of HSA.
Bacteria were grown, mounted, stained, and visualized as described previously (69). Briefly, 10 μl of exponential-phase VISA strain ATCC 700699 in caMHB was placed on the surface of a poly-l-lysine-coated glass slide for 10 min at room temperature, and unattached cells were removed by washing 5 times with 100 μl PBS. Adherent bacteria were then treated for 30 min with 10 μl PBS containing HSA (Albumin Biosciences) or PBS alone. Duplicate samples were supplemented with 1/10 of the original volume of PBS, or PBS containing NHS-rhodamine-labeled CF-301, to a final concentration of 4 μg/ml (0.25× MIC). The cells were incubated for a further 30 min, washed 5 times with 100 μl PBS, and fixed with 2.6% paraformaldehyde in PBS for 45 min. The slides were then washed 5 times with 100 μl PBS and mounted in PBS, pH 8.0, containing 50% glycerol and 0.1% p-phenylenediamine. DAPI was used as a counterstain under all assay conditions. All the incubation steps were done in a moist environment, and the cells were not allowed to dry. Deconvolution microscopy was performed using a DeltaVision image restoration microscope (Applied Precision/Olympus) equipped with a CoolSnap QE cooled charge-coupled-device (CCD) camera (Photometrics). Imaging was done using an Olympus 100×/1.40-NA UPLS Apo oil immersion objective combined with a 1.5× optovar (magnification enhancer). Z-stacks were taken at 0.15-μm intervals. Images were deconvolved using SoftWoRx software (Applied Precision/DeltaVision), corrected for chromatic aberrations, and presented as maximum-intensity projections combining all relevant z-sections. In a complementary analysis, similar to that described above, the NHS-rhodamine-labeled CF-301 was replaced with either 25 μg/ml PlyGGFP or PlyCAF. After treatment, the samples were visualized by fluorescence microscopy using a Nikon Eclipse E400 microscope equipped with a Nikon 100×/1.25-NA oil immersion lens.
(iii) HuLYZ binding in the presence of CF-301.
Bacteria were prepared and attached to the cover glass as described above. The cells were treated for 30 min at room temperature with 50 μl PBS containing CF-301 at different concentrations or PBS alone. Duplicate samples were then supplemented with 1/10 the original volume of PBS or PBS containing Alexa Fluor 488-labeled HuLYZ to a final concentration of 10 μg/ml. The cells were incubated for a further 30 min at room temperature, washed with PBS, and fixed with 2.6% paraformaldehyde in PBS for 45 min at room temperature. The slides were then washed with PBS and mounted in 20 mM Tris, pH 8.0, 90% glycerol, 0.5% n-propyl gallate. DAPI was used as a counterstain. Deconvolution microscopy was performed as described above. In a complementary analysis, Alexa Fluor 488-labeled CF-301 was replaced with either 25 μg/ml PlyGGFP or PlyCAF. After treatment, the samples were visualized by fluorescence microscopy using a Nikon Eclipse E400 microscope equipped with a Nikon 100×/1.25-NA oil immersion lens.
Electron microscopy.
Mid-log-phase strain MW2 growing in either caMHB or human serum (Sigma-Aldrich) was treated with the indicated concentrations of CF-301 or buffer alone (control) at 37°C for 15 min. The cells were then washed with 1× PB and resuspended in a solution of 4% paraformaldehyde and 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4). The samples were postfixed in 1% osmium tetroxide, block stained with uranyl acetate, and processed according to standard procedures by the Rockefeller University Electron Microscopy Service. Samples were visualized using a Tecnai Spirit BT transmission electron microscope (FEI). The human serum was sterile filtered and obtained from a pooled male population (∼70 subjects) of U.S. origin with type AB blood.
Western blot analysis.
The CF-301 MIC well samples taken from the indicated medium types were analyzed by Western blotting. Sample aliquots of 10 μl each were run on 4 to 12% Tris-glycine minigels (Novex) and then transferred to a polyvinylidene difluoride (PVDF) membrane via electroblotting. The PVDF membranes were incubated with an anti-CF-301-specific protein G affinity-purified human IgG3 recombinant monoclonal antibody, followed by a secondary monoclonal murine anti-IgG3-alkaline phosphatase conjugate detection antibody. Both the primary and secondary antibodies were used at 1:1,000 dilutions. The membrane was stained with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate (NBT-BCIP) chromogenic substrate. The molecular weight of visible bands was determined by comparison to the bands of a molecular weight standard run in the same gel.
Rat infective-endocarditis model.
Sprague-Dawley rats (250 to 275 g), anesthetized with a ketamine (87 mg/kg) and xylazine (13 mg/kg) cocktail via intraperitoneal (i.p.) injection, underwent a standard indwelling transcarotid-transaortic valve to left ventricle catheterization. After 48 h, the animals were challenged with S. aureus strain MW2 (∼1 × 105 CFU/rat i.v.) to induce endocarditis. At 24 h postinfection, a cohort of rats were euthanized and heart valve vegetations were collected to determine initial tissue burdens (control group). The remaining rats were treated with either vehicle (saline; a single i.v. dose) or diaminopimelic acid (DAP) alone (40 mg/kg; subcutaneous [s.c.] injection; once a day [q.d.] for 4 days) or with DAP in addition to CF-301. CF-301 was administered i.v. as a single slow bolus (injection over 5 to 10 min) on the first day of treatment only, just after the initial DAP dose, in four dosing regimens (1, 2.5, 5, and 10 mg/kg). The treated animals were euthanized (sodium pentobarbital at 200 mg/kg by rapid i.p. push) 24 h after the last DAP treatment (5 days postinfection), and the cardiac valve vegetation was removed, weighed, homogenized, and serially diluted in sterile PBS for quantitative culture onto TSAB. All the culture plates were incubated at 37°C for 24 h, and the resulting colonies were enumerated and expressed as log10 CFU per gram of tissue. Data for each organ for different treatment groups were calculated as the median log10 CFU per gram of tissue ± 95% confidence interval (CI).
Population PK modeling of CF-301.
PK data were collected from previous nonclinical toxicology studies that included rats and dogs treated with CF-301 at varying doses (70, 71). Postdose plasma was collected over a time course and analyzed using a validated CF-301 enzyme-linked immunosorbent assay (ELISA). The predicted AUC values for CF-301 at doses of 1, 2.5, 5, and 10 mg/kg as a 10-min intravenous infusion were derived from population PK modeling based on the nonclinical rat and dog PK experiments. For the current study, the previously reported AUC values were divided by the MIC value determined in rat serum (i.e., 16 μg/ml) for MRSA isolate MW2 to yield the calculated AUC/MIC ratios reported in Table S7.
Rabbit infective-endocarditis model.
New Zealand White rabbits (2.2 to 2.5 kg), anesthetized with a ketamine (35 mg/kg) and xylazine (5 mg/kg) cocktail via intramuscular (i.m.) injection, underwent a standard indwelling transcarotid-transaortic valve to left ventricle catheterization (72). At 48 h post-catheter placement, the animals were challenged i.v. with an inoculum of ∼2 × 105 CFU of S. aureus strain MW2 to induce IE. In previous studies, this inoculum has been shown to induce IE in >95% of catheterized animals. At 24 h postinfection, a cohort of rabbits were euthanized, and heart valve vegetations were collected to determine initial tissue burdens (control group). The remaining rabbits were treated with either vehicle (saline; single i.v. dose), DAP alone (4 mg/kg i.v.; q.d. for 4 days), CF-301 alone, or DAP in addition to CF-301. CF-301 was administered i.v. as a single slow bolus (injection over 5 to 10 min) on the first day of treatment only, just after the initial DAP dose, at five dosing regimens (0.09, 0.18, 0.35, 0.70, and 1.4 mg/kg). The treated animals were euthanized (sodium pentobarbital at 200 mg/kg by rapid i.p. push) 24 h after the last DAP treatment (5 days postinfection), and the cardiac valve vegetation was removed, weighed, homogenized, and serially diluted in sterile PBS for quantitative culture onto TSAB. All the culture plates were incubated at 37°C for 24 h, and the resulting colonies were enumerated and expressed as log10 CFU per gram of tissue. The data for each organ for different treatment groups were calculated as the median log10 CFU per gram of tissue ± 95% CI.
Rabbit pharmacokinetics.
New Zealand White rabbits were dosed with CF-301 (i.v.; slow bolus) at 0.18, 0.35, 0.07, or 1.4 mg/kg. After increasing amounts of time postdose, plasma was collected and analyzed using a validated CF-301 ELISA in the manner described previously (70, 71). The predicted AUC values for CF-301 at doses of 1, 2.5, 5, and 10 mg/kg as a 10-min intravenous infusion were derived using the pharmacokinetic software package WinNonLin (data not shown). These values, divided by the MIC value (1 μg/ml in rabbit serum) of MRSA isolate MW2, resulted in the calculated AUC/MIC ratios reported in Table S8.
Daptomycin dose rationale in rabbits.
Daptomycin dose-response experiments were performed at doses ranging from 1 mg/kg to 10 mg/kg i.v. once daily for 4 days in the rabbit IE model caused by S. aureus strain MW2. While each of the 7 treatment groups used ≥3 animals, the vehicle control used only 2 animals; the vehicle control is also supported by a preestablished understanding of control group data in the rabbit IE model (73). Daptomycin at 4 mg/kg, representing a dose below the human therapeutic dose equivalent, was chosen to explore the benefit of CF-301 therapy in addition to daptomycin. In the rabbit IE model, a daptomycin dose of 4 mg/kg i.v. provided an ∼2- to 3-log10-unit reduction in the bacterial burden compared to vehicle-treated controls. The treated animals still had significant burdens of ∼5 to 7 log10 units, providing a significant dynamic range to observe the added effect of CF-301 on the treatment regimen.
Ethics statement.
Animals were maintained in accordance with the American Association for Accreditation of Laboratory Animal Care criteria and were cared for in accordance with national guidelines. The Animal Research Committee (IACUC) of the LABioMed Research Institute at Harbor-UCLA Medical Center approved the animal IE studies under protocol number 021683.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nadine Soplop at the Electron Microscopy Resource Center at The Rockefeller University for electron microscopy; Roxana Georgescu, Simon Hoffenberg, Jun Oh, Steven M. Jones, and Michael Wittekind for insightful discussions; and Patricia Bradford and Alena Jandourek for help with reviewing the manuscript.
This work was supported by funds from ContraFect Corporation and CDMRP award number W81XWH-16-1-0245. A.S.B., W.A., and Y.Q.X. were supported by research grants from ContraFect Corporation and the above-mentioned CDMRP award.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02291-18.
REFERENCES
- 1.Wittekind M, Schuch R. 2016. Cell wall hydrolases and antibiotics: exploiting synergy to create efficacious new antimicrobial treatments. Curr Opin Microbiol 33:18–24. doi: 10.1016/j.mib.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Love MJ, Bhandari D, Dobson RCJ, Billington C. 2018. Potential for bacteriophage endolysins to supplement or replace antibiotics in food production and clinical care. Antibiotics (Basel) 7:E17. doi: 10.3390/antibiotics7010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischetti VA, Nelson D, Schuch R. 2006. Reinventing phage therapy: are the parts greater than the sum? Nat Biotechnol 24:1508–1511. doi: 10.1038/nbt1206-1508. [DOI] [PubMed] [Google Scholar]
- 4.ClinicalTrials.gov. 2017. Safety, efficacy and pharmacokinetics of cf-301 vs. placebo in addition to antibacterial therapy for treatment of S. aureus bacteremia. https://clinicaltrials.gov/ct2/show/NCT03163446.
- 5.Schuch R, Lee HM, Schneider BC, Sauve KL, Law C, Khan BK, Rotolo JA, Horiuchi Y, Couto DE, Raz A, Fischetti VA, Huang DB, Nowinski RC, Wittekind M. 2014. Combination therapy with lysin CF-301 and antibiotic is superior to antibiotic alone for treating methicillin-resistant Staphylococcus aureus-induced murine bacteremia. J Infect Dis 209:1469–1478. doi: 10.1093/infdis/jit637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuch R, Khan BK, Raz A, Rotolo JA, Wittekind M. 2017. Bacteriophage lysin CF-301, a potent antistaphylococcal biofilm agent. Antimicrob Agents Chemother 61:e02666-16. doi: 10.1128/AAC.02666-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DM. 2012. Endolysins as antimicrobials. Adv Virus Res 83:299–365. doi: 10.1016/B978-0-12-394438-2.00007-4. [DOI] [PubMed] [Google Scholar]
- 8.Cassino C, Murphy G, Boyle J, Rotolo J, Wittekind M. 2016. Results of the first in human study of lysin CF-301 evaluating the safety, tolerability and pharmacokinetic profile in healthy volunteers, poster EVLB62. 26th Eur Congr Clin Microbiol Infect Dis, Amsterdam, Netherlands. [Google Scholar]
- 9.Levison ME, Levison JH. 2009. Pharmacokinetics and Pharmacodynamics of antibacterial agents. Infect Dis Clin North Am 23:791–815. doi: 10.1016/j.idc.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeitlinger MA, Derendorf H, Mouton JW, Cars O, Craig WA, Andes D, Theuretzbacher U. 2011. Protein binding: do we ever learn? Antimicrob Agents Chemother 55:3067–3074. doi: 10.1128/AAC.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt S, Gonzalez D, Derendorf H. 2010. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci 99:1107–1122. doi: 10.1002/jps.21916. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt S, Rock K, Sahre M, Burkhardt O, Brunner M, Lobmeyer MT, Derendorf H. 2008. Effect of protein binding on the pharmacological activity of highly bound antibiotics. Antimicrob Agents Chemother 52:3994–4000. doi: 10.1128/AAC.00427-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeitlinger M, Sauermann R, Fille M, Hausdorfer J, Leitner I, Muller M. 2008. Plasma protein binding of fluoroquinolones affects antimicrobial activity. J Antimicrob Chemother 61:561–567. doi: 10.1093/jac/dkm524. [DOI] [PubMed] [Google Scholar]
- 14.Burian A, Wagner C, Stanek J, Manafi M, Bohmdorfer M, Jager W, Zeitlinger M. 2011. Plasma protein binding may reduce antimicrobial activity by preventing intra-bacterial uptake of antibiotics, for example clindamycin. J Antimicrob Chemother 66:134–137. doi: 10.1093/jac/dkq400. [DOI] [PubMed] [Google Scholar]
- 15.Yeaman MR, Gank KD, Bayer AS, Brass EP. 2002. Synthetic peptides that exert antimicrobial activities in whole blood and blood-derived matrices. Antimicrob Agents Chemother 46:3883–3891. doi: 10.1128/AAC.46.12.3883-3891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stratton CW, Weeks LS. 1990. Effect of human serum on the bactericidal activity of daptomycin and vancomycin against staphylococcal and enterococcal isolates as determined by time-kill kinetic studies. Diagn Microbiol Infect Dis 13:245–252. doi: 10.1016/0732-8893(90)90067-6. [DOI] [PubMed] [Google Scholar]
- 17.CLSI. 1999. Methods for determining bactericidal activity of antimicrobial agents; approved guideline. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 18.Verma P. 2007. Methods for determining bactericidal activity and antimicrobial interactions: synergy testing, time-kill curves, and population analysis, p 275–298. In Schwalbe R, Steele-Moore L, Goodwin AC (ed), Antimicrobial susceptibility testing protocols. CRC Press, Boca Raton, FL. [Google Scholar]
- 19.Moody J. 2010. Synergy testing: broth microdilution checkerboard and broth macrodilution methods, p 5.12.1–5.12.23. In Garcia LS. (ed), Clinical microbiology procedures handbook, vol 2 ASM Press, Washington, DC. [Google Scholar]
- 20.Pruzanski W, Saito S, Ogryzlo MA. 1970. The significance of lysozyme (muramidase) in rheumatoid arthritis. I. Levels in serum and synovial fluid. Arthritis Rheum 13:389–399. doi: 10.1002/art.1780130405. [DOI] [PubMed] [Google Scholar]
- 21.Bera A, Herbert S, Jakob A, Vollmer W, Gotz F. 2005. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol 55:778–787. doi: 10.1111/j.1365-2958.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- 22.Sahin O, Ziaei A, Karaismailoglu E, Taheri N. 2016. The serum angiotensin converting enzyme and lysozyme levels in patients with ocular involvement of autoimmune and infectious diseases. BMC Ophthalmol 16:19. doi: 10.1186/s12886-016-0194-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Georgieva TM, Penchev Georgiev I, Iliev Y, Petrov VS, Vachkov A, Kanelov IN, Tanev SI, Zapryanova D, Pavlov AI, Eckersall D. 2008. Blood serum concentrations of total proteins and main protein fractions in weaning rabbits experimentally infected with E. coli. Rev Med Vet 159:431–436. [Google Scholar]
- 24.Zaias J, Mineau M, Cray C, Yoon D, Altman NH. 2009. Reference values for serum proteins of common laboratory rodent strains. J Am Assoc Lab Anim Sci 48:387–390. [PMC free article] [PubMed] [Google Scholar]
- 25.Finn TE, Nunez AC, Sunde M, Easterbrook-Smith SB. 2012. Serum albumin prevents protein aggregation and amyloid formation and retains chaperone-like activity in the presence of physiological ligands. J Biol Chem 287:21530–21540. doi: 10.1074/jbc.M112.372961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang F, Zhang Y, Liang H. 2014. Interactive association of drugs binding to human serum albumin. Int J Mol Sci 15:3580–3595. doi: 10.3390/ijms15033580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sacks FM, Rudel LL, Conner A, Akeefe H, Kostner G, Baki T, Rothblat G, de la Llera-Moya M, Asztalos B, Perlman T, Zheng C, Alaupovic P, Maltais JA, Brewer HB. 2009. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J Lipid Res 50:894–907. doi: 10.1194/jlr.M800622-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richieri GV, Kleinfeld AM. 1995. Unbound free fatty acid levels in human serum. J Lipid Res 36:229–240. [PubMed] [Google Scholar]
- 29.Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA. 2010. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 54:1603–1612. doi: 10.1128/AAC.01625-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelhady W, Bayer AS, Gonzales R, Li L, Xiong YQ. 2017. Telavancin is active against experimental aortic valve endocarditis caused by daptomycin- and methicillin-resistant Staphylococcus aureus strains. Antimicrob Agents Chemother 61: e01877-16. doi: 10.1128/AAC.01877-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelhady WA, Bayer AS, Xiong YQ. 2012. Experimental endocarditis model of methicillin resistant Staphylococcus aureus (MRSA) in rat. J Vis Exp doi: 10.3791/3863:e3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakoulas G, Eliopoulos GM, Alder J, Eliopoulos CT. 2003. Efficacy of daptomycin in experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 47:1714–1718. doi: 10.1128/AAC.47.5.1714-1718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beer J, Wagner CC, Zeitlinger M. 2009. Protein binding of antimicrobials: methods for quantification and for investigation of its impact on bacterial killing. AAPS J 11:1–12. doi: 10.1208/s12248-008-9072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegde SS, Reyes N, Wiens T, Vanasse N, Skinner R, McCullough J, Kaniga K, Pace J, Thomas R, Shaw JP, Obedencio G, Judice JK. 2004. Pharmacodynamics of telavancin (TD-6424), a novel bactericidal agent, against Gram-positive bacteria. Antimicrob Agents Chemother 48:3043–3050. doi: 10.1128/AAC.48.8.3043-3050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garonzik SM, Lenhard JR, Forrest A, Holden PN, Bulitta JB, Tsuji BT. 2016. Defining the active fraction of daptomycin against methicillin-resistant Staphylococcus aureus (MRSA) using a pharmacokinetic and pharmacodynamic approach. PLoS One 11:e0156131. doi: 10.1371/journal.pone.0156131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig WA, Suh B. 1991. Protein binding and the antimicrobial effects. Methods for determination of protein binding, p 367–402. In Lorian V. (ed), Antibiotics in laboratory medicine, 3rd ed Williams and Wilkins, Baltimore, MD. [Google Scholar]
- 37.Colclough N, Ruston L, Wood JM, MacFaul PA. 2014. Species differences in drug plasma binding. Med Chem Commun 5:963–967. doi: 10.1039/C4MD00148F. [DOI] [Google Scholar]
- 38.Johansson MU, Frick IM, Nilsson H, Kraulis PJ, Hober S, Jonasson P, Linhult M, Nygren PA, Uhlen M, Bjorck L, Drakenberg T, Forsen S, Wikstrom M. 2002. Structure, specificity, and mode of interaction for bacterial albumin-binding modules. J Biol Chem 277:8114–8120. doi: 10.1074/jbc.M109943200. [DOI] [PubMed] [Google Scholar]
- 39.Aubry AF, Markoglou N, McGann A. 1995. Comparison of drug binding interactions on human, rat and rabbit serum albumin using high-performance displacement chromatography. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 112:257–266. doi: 10.1016/0742-8413(95)02019-5. [DOI] [PubMed] [Google Scholar]
- 40.Massolini G, De Lorenzi E, Ponci MC, Caccialanza G. 1996. Comparison of drug binding sites on rat and human serum albumins using immobilized-protein stationary phases as a tool for the selection of suitable animal models in pharmacological studies. Boll Chim Farm 135:382. [PubMed] [Google Scholar]
- 41.Vaara M, Viljanen P, Vaara T, Makela PH. 1984. An outer membrane-disorganizing peptide PMBN sensitizes E. coli strains to serum bactericidal action. J Immunol 132:2582–2589. [PubMed] [Google Scholar]
- 42.Huang BX, Dass C, Kim HY. 2005. Probing conformational changes of human serum albumin due to unsaturated fatty acid binding by chemical cross-linking and mass spectrometry. Biochem J 387:695–702. doi: 10.1042/BJ20041624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. 2005. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol 353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 44.Lu JZ, Fujiwara T, Komatsuzawa H, Sugai M, Sakon J. 2006. Cell wall-targeting domain of glycylglycine endopeptidase distinguishes among peptidoglycan cross-bridges. J Biol Chem 281:549–558. doi: 10.1074/jbc.M509691200. [DOI] [PubMed] [Google Scholar]
- 45.Tossavainen H, Raulinaitis V, Kauppinen L, Pentikainen U, Maaheimo H, Permi P. 2018. Structural and functional insights into lysostaphin-substrate interaction. Front Mol Biosci 5:60. doi: 10.3389/fmolb.2018.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whisstock JC, Lesk AM. 1999. SH3 domains in prokaryotes. Trends Biochem Sci 24:132–133. doi: 10.1016/S0968-0004(99)01366-3. [DOI] [PubMed] [Google Scholar]
- 47.Marini I, Moschini R, Del Corso A, Mura U. 2005. Chaperone-like features of bovine serum albumin: a comparison with alpha-crystallin. Cell Mol Life Sci 62:3092–3099. doi: 10.1007/s00018-005-5397-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Josse J, Laurent F, Diot A. 2017. Staphylococcal adhesion and host cell invasion: fibronectin-binding and other mechanisms. Front Microbiol 8:2433. doi: 10.3389/fmicb.2017.02433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng AG, Missiakas D, Schneewind O. 2014. The giant protein Ebh is a determinant of Staphylococcus aureus cell size and complement resistance. J Bacteriol 196:971–981. doi: 10.1128/JB.01366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crosby HA, Kwiecinski J, Horswill AR. 2016. Staphylococcus aureus aggregation and coagulation mechanisms, and their function in host-pathogen interactions. Adv Appl Microbiol 96:1–41. doi: 10.1016/bs.aambs.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Egesten A, Frick IM, Morgelin M, Olin AI, Bjorck L. 2011. Binding of albumin promotes bacterial survival at the epithelial surface. J Biol Chem 286:2469–2476. doi: 10.1074/jbc.M110.148171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu X, Ju X, Du L, Yuan J, Wang L, He R, Chen Z. 2017. Production of bacterial ghosts from Gram-positive pathogen Listeria monocytogenes. Foodborne Pathog Dis 14:1–7. doi: 10.1089/fpd.2016.2184. [DOI] [PubMed] [Google Scholar]
- 53.Schmelcher M, Shabarova T, Eugster MR, Eichenseher F, Tchang VS, Banz M, Loessner MJ. 2010. Rapid multiplex detection and differentiation of listeria cells by use of fluorescent phage endolysin cell wall binding domains. Appl Environ Microbiol 76:5745–5756. doi: 10.1128/AEM.00801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmelcher M, Tchang VS, Loessner MJ. 2011. Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb Biotechnol 4:651–662. doi: 10.1111/j.1751-7915.2011.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bera A, Biswas R, Herbert S, Gotz F. 2006. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect Immun 74:4598–4604. doi: 10.1128/IAI.00301-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilmer DB, Schmitz JE, Euler CW, Fischetti VA. 2013. Novel bacteriophage lysin with broad lytic activity protects against mixed infection by Streptococcus pyogenes and methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:2743–2750. doi: 10.1128/AAC.02526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haddad Kashani H, Schmelcher M, Sabzalipoor H, Seyed Hosseini E, Moniri R. 2018. Recombinant endolysins as potential therapeutics against antibiotic-resistant Staphylococcus aureus: current status of research and novel delivery strategies. Clin Microbiol Rev 31:e00071-17. doi: 10.1128/CMR.00071-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verbree CT, Datwyler SM, Meile S, Eichenseher F, Donovan DM, Loessner MJ, Schmelcher M. 2017. Identification of peptidoglycan hydrolase constructs with synergistic staphylolytic activity in cow's milk. Appl Environ Microbiol 83: e03445-16. doi: 10.1128/AEM.03445-16. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Becker SC, Foster-Frey J, Stodola AJ, Anacker D, Donovan DM. 2009. Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene 443:32–41. doi: 10.1016/j.gene.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 60.Nelson D, Schuch R, Chahales P, Zhu S, Fischetti VA. 2006. PlyC: a multimeric bacteriophage lysin. Proc Natl Acad Sci U S A 103:10765–10770. doi: 10.1073/pnas.0604521103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuch R, Pelzek AJ, Kan S, Fischetti VA. 2010. Prevalence of Bacillus anthracis-like organisms and bacteriophages in the intestinal tract of Eisenia fetida earthworms. Appl Environ Microbiol 76:2286–2294. doi: 10.1128/AEM.02518-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rybak MJ, Hershberger E, Moldovan T, Grucz RG. 2000. In vitro activities of daptomycin, vancomycin, linezolid, and quinupristin-dalfopristin against staphylococci and enterococci, including vancomycin-intermediate and -resistant strains. Antimicrob Agents Chemother 44:1062–1066. doi: 10.1128/AAC.44.4.1062-1066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith JR, Barber KE, Raut A, Aboutaleb M, Sakoulas G, Rybak MJ. 2015. β-Lactam combinations with daptomycin provide synergy against vancomycin-resistant Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother 70:1738–1743. doi: 10.1093/jac/dkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McKay GA, Beaulieu S, Arhin FF, Belley A, Sarmiento I, Parr T Jr, Moeck G. 2009. Time-kill kinetics of oritavancin and comparator agents against Staphylococcus aureus, Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother 63:1191–1199. doi: 10.1093/jac/dkp126. [DOI] [PubMed] [Google Scholar]
- 65.Mishra NM, Stolarzewicz I, Cannaerts D, Schuermans J, Lavigne R, Looz Y, Landuyt B, Schoofs L, Schols D, Paeshuyse J, Hickenbotham P, Clokie M, Luyten W, Van der Eycken EV, Briers Y. 2018. Iterative chemical engineering of vancomycin leads to novel vancomycin analogs with a high in vitro therapeutic index. Front Microbiol 9:1175. doi: 10.3389/fmicb.2018.01175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foerster S, Unemo M, Hathaway LJ, Low N, Althaus CL. 2016. Time-kill curve analysis and pharmacodynamic modelling for in vitro evaluation of antimicrobials against Neisseria gonorrhoeae. BMC Microbiol 16:216. doi: 10.1186/s12866-016-0838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerits E, Blommaert E, Lippell A, O’Neill AJ, Weytjens B, De Maeyer D, Fierro AC, Marchal K, Marchand A, Chaltin P, Spincemaille P, De Brucker K, Thevissen K, Cammue BPA, Swings T, Liebens V, Fauvart M, Verstraeten N, Michiels J. 2016. Elucidation of the mode of action of a new antibacterial compound active against Staphylococcus aureus and Pseudomonas aeruginosa. PLoS One 11:e0155139. doi: 10.1371/journal.pone.0155139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.CLSI. 2018. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 11th ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 69.Raz A. 2017. Detection of intracellular proteins by high-resolution immunofluorescence microscopy in Streptococcus pyogenes. Methods Mol Biol 1535:219–228. doi: 10.1007/978-1-4939-6673-8_14. [DOI] [PubMed] [Google Scholar]
- 70.Ghahramani P, Khariton T, Schuch R, Rotolo JA, Ramirez RA, Wittekind M. 2016. Pharmacokinetic indices driving antibacterial efficacy of cf-301—a novel first-in-class lysin, abstr W-059. Am Conf Pharmacometrics, Bellevue, Washington. [Google Scholar]
- 71.Chiu J, Khariton T, Ghahramani P, Lapinskas P, Cassino C, Carabeo T. 2016. Interspecies scaling in pre-clinical population pharmacokinetics of CF-301, abstr W-039. Am Conf Pharmacometrics, Bellevue, Washington. [Google Scholar]
- 72.Weidenmaier C, Peschel A, Kempf VA, Lucindo N, Yeaman MR, Bayer AS. 2005. DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun 73:8033–8038. doi: 10.1128/IAI.73.12.8033-8038.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiong YQ, Abdelhady W, Tang C, Bayer AS. 2016. Comparative efficacy of telavancin and daptomycin in experimental endocarditis due to multi-clonotype MRSA strains. J Antimicrob Chemother 71:2890–2894. doi: 10.1093/jac/dkw249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.