

Cryptosporidiosis is one of the leading causes of moderate to severe diarrhea in children in low-resource settings. The therapeutic options for cryptosporidiosis are limited to one drug, nitazoxanide, which unfortunately has poor activity in the most needy populations of malnourished children and HIV-infected persons.
KEYWORDS: Cryptosporidium, diarrhea, drug discovery, pharmacokinetics, tRNA synthetase
ABSTRACT
Cryptosporidiosis is one of the leading causes of moderate to severe diarrhea in children in low-resource settings. The therapeutic options for cryptosporidiosis are limited to one drug, nitazoxanide, which unfortunately has poor activity in the most needy populations of malnourished children and HIV-infected persons. We describe here the discovery and early optimization of a class of imidazopyridine-containing compounds with potential for treating Cryptosporidium infections. The compounds target the Cryptosporidium methionyl-tRNA synthetase (MetRS), an enzyme that is essential for protein synthesis. The most potent compounds inhibited the enzyme with Ki values in the low picomolar range. Cryptosporidium cells in culture were potently inhibited with 50% effective concentrations as low as 7 nM and >1,000-fold selectivity over mammalian cells. A parasite persistence assay indicates that the compounds act by a parasiticidal mechanism. Several compounds were demonstrated to control infection in two murine models of cryptosporidiosis without evidence of toxicity. Pharmacological and physicochemical characteristics of compounds were investigated to determine properties that were associated with higher efficacy. The results indicate that MetRS inhibitors are excellent candidates for development for anticryptosporidiosis therapy.
INTRODUCTION
Although the world has seen substantial progress in the reduction of child mortality in recent decades, much work remains to achieve the United Nations’ Sustainable Development Goals (targeting 2030) for child survival (1). Diarrhea is one of the leading causes of child mortality, responsible for 8% of deaths in children aged 1 to 59 months (2). The Global Enteric Multicenter Study (GEMS) identified Cryptosporidium to be the second leading cause of moderate to severe diarrhea in young children at sites in Africa and Asia (3). The importance of cryptosporidiosis in community diarrhea in developing countries was confirmed in the MAL-ED study (4). Beyond the mortality risk, children who survive cryptosporidiosis suffer from growth and developmental stunting, which contribute to all-cause mortality and disability (5, 6). The recent appreciation of the impact of cryptosporidiosis has drawn attention to the inadequacies in the means to control this infectious disease. No vaccines are in clinical use, and the sole drug for treating cryptosporidiosis (nitazoxanide) has poor efficacy in malnourished children and in patients with human immunodeficiency virus. Vaccine development is likely to be slow due to difficulties raised by antigenic differences within Cryptosporidium, leading to poor cross-protection between species and strains (7). New anticryptosporidial drugs are likely to be the most rapidly developed technology to address the burden of Cryptosporidium infection.
Recent studies suggest that Cryptosporidium is more closely related to gregarine protozoa than to coccidians (8). The genus Cryptosporidium has 27 species that have been identified worldwide that infect four classes of vertebrates (9). The species primarily responsible for human cryptosporidiosis are Cryptosporidium hominis and Cryptosporidium parvum. Water- and foodborne transmission are the major modes of infection, although person-to-person contact is also described (9). The small intestine is the primary site of Cryptosporidium infection in humans, although extraintestinal sites such as the biliary tract, lungs, and pancreas can be involved in immunocompromised and immunocompetent individuals (9–11). The extraintestinal locations may have important implications for developing therapeutics that act at all sites of infection. Cryptosporidium mainly resides in an unusual niche in the intestinal epithelium known as the parasitophorous vacuole, which is insulated from both the intestinal lumen, as well as the host cytoplasm. Therefore, it is not entirely known whether anti-Cryptosporidium drugs should be optimized for luminal or plasma exposure, although a recent study emphasizes the importance of gastrointestinal (luminal) exposure in a murine model (12).
Protein synthesis is a classic antimicrobial drug target, dating back to many of the first antibiotics—such as chloramphenicol, tetracycline, and erythromycin—that inhibit bacterial protein synthesis. Recently, aminoacyl-tRNA synthetase (aaRS) inhibitors have emerged as promising therapeutic candidates for targeting protein synthesis. Using ATP hydrolysis, aaRSs catalyze the formation of tRNAs charged with their cognate amino acids, which serve as the substrates for the formation of new peptides. Mupirocin, a small molecule inhibitor of isoleucyl-tRNA synthetase (13), has been in clinical use for more than 2 decades as a topical treatment for Staphylococcus infections. Tavaborole is a leucyl-tRNA synthetase inhibitor that was approved by the U.S. Food and Drug Administration in 2014 for topical treatment of onychomycosis (14, 15). Halofuginone, a prolyl-tRNA synthetase inhibitor (16), is approved for veterinary use against Cryptosporidium in Europe, although a narrow therapeutic index makes it unsuitable for human use. Three different aaRS inhibitors for systemic use are in clinical trials, demonstrating the potential for safe use beyond topical applications. These include the methionyl-tRNA synthetase (MetRS) inhibitor CRS3213 for Clostridium difficile infections (ClinicalTrials.gov, NCT01551004), the leucyl-tRNA synthetase inhibitor GSK2251052 for Gram-negative bacterial infections (17, 18), and the leucyl-tRNA synthetase inhibitor GSK3936656 for multidrug-resistant tuberculosis (19, 20). Inhibitors of other aaRSs from protozoan parasites, including Cryptosporidium, Plasmodium, Trypanosoma, and Toxoplasma, are also in development (21, 22).
MetRS enzymes fall into two categories: MetRS1 and MetRS2 (23). C. parvum and C. hominis contain a single MetRS gene that aligns with the MetRS1 category, meaning it has close homology to the MetRS of S. aureus, Trypanosoma spp., and the human mitochondrial MetRS. Our group has been developing inhibitors to type 1 MetRS that are shown to have potent activity against trypanosomes and Gram-positive bacteria, including activity in animal models (24–27). Supporting this work are numerous crystal structures of the inhibitors bound to the Trypanosoma brucei MetRS enzyme (28–30), which is 76% identical (19 of 25 residues) to the C. parvum/C. hominis MetRS within the inhibitor binding pocket. Here, the C. parvum MetRS was characterized and MetRS inhibitors were shown to be highly potent with Ki values as low as 0.9 pM. Compound 2093 had the most potent in vitro activity against C. parvum and reduced Cryptosporidium infection to low levels in two murine models without showing signs of toxicity. The physicochemical and pharmacological features associated with in vivo activity are discussed. The research illustrates the potential of target-based drug discovery to develop a novel therapeutic against a formidable eukaryotic pathogen.
RESULTS
Amino acid sequence alignment.
Cryptosporidium parvum (UniProt Q5CVN0) and Cryptosporidium hominis (UPI0000452AB0) have a single MetRS gene in their respective genomes. These sequences were compared to the well-characterized Trypanosoma brucei MetRS by mapping them onto the crystal structures of TbMetRS bound to inhibitors. Table 1 shows the amino acid residues of TbMetRS that form binding pockets that are in direct proximity to MetRS inhibitors (28–30). The C. parvum and C. hominis sequences are identical to each other over the 25 residues forming the compound binding pockets. The Cryptosporidium sequences share 19 identical amino acids with TbMetRS and 18 identical residues with the human mitochondrial MetRS over this region. Only 14 of the 25 amino acids are identical between the Cryptosporidium MetRS and the human cytoplasmic MetRS in this region.
TABLE 1.
Protein sequence analysis of MetRS inhibitor-binding sites from different speciesa
| Organism | Zone, pocket, and amino acid by sequence no.b |
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 247 | 248 | 249 | 250 | 287 | 289 | 290 | 291 | 292 | 456 | 460 | 461 | 470 | 471 | 472 | 473 | 474 | 476 | 477 | 478 | 480 | 481 | 519 | 522 | 523 | |
| b | b | b | l | q | q | q | q | q | q | q | q | q | q | q | q | b | q | b | b | b | b | b | b | b | |
| Trypanosoma brucei | Pro | Ile | Tyr | Tyr | Asp | His | Gly | Gln | Lys | Leu | Ala | Ile | Cys | Val | Tyr | Val | Trp | Asp | Ala | Leu | Asn | Tyr | Ile | Phe | His |
| Cryptosporidium parvum/Cryptosporidium hominis | Ala | Ile | Tyr | Tyr | Asp | His | Gly | Gln | Lys | Ala | Gly | Val | Val | Met | Tyr | Val | Trp | Asp | Ala | Leu | Asn | Tyr | Ile | Phe | His |
| Homo sapiens (mitochondrial) | Pro | Ile | Phe | Tyr | Asp | His | Gly | Leu | Lys | – | Gly | Ile | Thr | Ile | Tyr | Val | Trp | Asp | Ala | Leu | Asn | Tyr | Ile | Phe | His |
| Homo sapiens (cytoplasmic) | Ala | Leu | Pro | Tyr | Asp | Tyr | Gly | Thr | Ala | – | Gly | Thr | Val | Phe | Tyr | Val | Trp | Asp | Ala | Thr | Gly | Tyr | Asn | Phe | His |
Sequence numbers refer to the T. brucei sequence. UniProt accession codes: T. brucei, Q38C91; C. parvum, Q5CVN0; C. hominis, UPI0000452AB0; H. sapiens (mitochondrial), Q96GW9; H. sapiens (cytoplasmic), P56192.
Zones are expressed as follows: l, linker zone; b, benzyl pocket (methionine substrate pocket); q, quinolone pocket (auxiliary pocket formed upon inhibitor binding). –, Ambiguous due to different loop lengths that could be Leu or His.
Enzymology.
The C. parvum MetRS enzyme was overexpressed in Escherichia coli and purified by nickel affinity chromatography, followed by size exclusion chromatography (see Fig. S1 in the supplemental material). The activity was confirmed in the aminoacylation assay which detects the esterification of radiolabeled methionine to the tRNA substrate (Fig. S2). However, in order to measure the Michaelis-Menten constants for the enzyme, an ATP:PPi exchange assay was used, as previously explained (31). The Km for the methionine substrate was comparable to that observed in S. aureus (both in-house and published) (Table 2) but was ∼4-fold higher than the value for the human mitochondrial MetRS (71 versus 18 μM). The Km for the ATP substrate was about 2- to 3-fold higher than that observed in S. aureus (both in-house and published) (Table 2), and ∼10-fold higher than the value for the human mitochondrial MetRS (1040 versus 85 μM).
TABLE 2.
Kinetic parameters of C. parvum MetRS compared to MetRS enzymes from other organisms
MetRS inhibitor activities.
The initial MetRS inhibitors were synthesized to target trypanosomatid parasites (24–26, 30). The activities of selected compounds on the CpMetRS enzyme, C. parvum parasites cultured in HCT-8 cells, and mammalian cells are shown in Tables 3 and 4. The MetRS inhibitors are characterized by two ring systems tethered by a linker. Table 3 shows four compounds in which the linker is an alkyl chain (“linear linker”). The aminoquinolone (compound 1312) (24) and urea-containing compounds (compound 1433) (25) are weakly active on the CpMetRS enzyme and show little activity on C. parvum cultures. The fluoro-imidazopyridine compounds (compounds 1614 and 1717) (26) display moderate activity on both the enzyme and cultures. Next, a series of 13 compounds containing a ring in the linker region were tested (Table 4). The 1,3-dihydro-2-oxo-imidazole ring was found to be the most active, and thus variants of this scaffold were further investigated. Compound 2093 is the overall most potent compound, with a Ki value on CpMetRS of 0.9 pM and a 50% effective concentration (EC50) between 7 and 36 nM determined in independent laboratories using different readout techniques (luciferase reporter versus fluorescence microscopy) and different C. parvum strains (see Materials and Methods). Importantly, compound 2093 has no cytotoxicity on mammalian cells at the highest (50 μM) concentration tested. Examining the SAR of this scaffold indicates the relative potency at the R1 position as follows: ethyl > CH2CF2H > propyl > H. At the R2 position, Cl is slightly better than F (compare compound 2093 to compound 2114 and compound 2067 to compound 2062). Finally, at R3 the methoxy substitution provides moderately greater potency than chloro (compare compound 2114 to compound 2062, compound 2093 to compound 2067, or compound 2258 to compound 2138). None of the compounds exhibited substantial cytotoxicity on either mammalian cell line tested (CRL-8155 or HepG2).
TABLE 3.
Activities of MetRS inhibitors with linear linkers
| Molecule | Structure | Mean activity ± SD (n)a |
Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C. parvum MetRS Ki (nM) | UW C. parvum EC50 (μM)b | U. VT C. parvum EC50 (μM)c | COX-1 EC50 (μM) | SDH-A EC50 (μM) | CRL-8155 EC50 (μM)* | HepG2 EC50 (μM)* | |||
| 1312 | 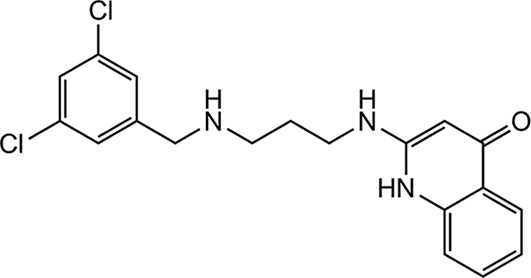 |
0.64 ± 0.01 (2) | 18.55 ± 0.21 (2) | >10.0; >50.0 | 2.43 | >25 | >20.0 | >20.0 | 24 |
| 1433 | 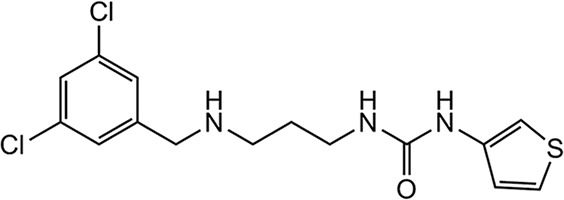 |
>1.33 (2) | >20.00 ± 0.00 (2) | >10.0, 11.2 | 14.7 | 12.2 | 14 ± 2.8 (2) | 16.5 ± 4.9 (2) | 25 |
| 1614 | 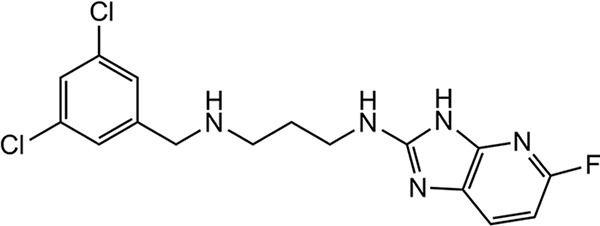 |
0.61 ± 0.06 (2) | 14.80 ± 2.40 (2) | 7.60 ± 0.74 (2) | 4.35 ± 0.92 (2) | 31.5 ± 2.1 (2) | 39.7 ± 0.8 (2) | >50.0 | 26 |
| 1717 | 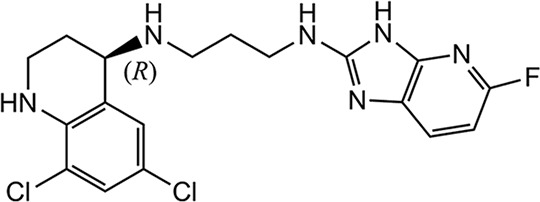 |
0.41 ± 0.06 (2) | 4.10 ± 2.59 (2) | 5.17 ± 0.45 (2) | 1.04 ± 0.51 (2) | >25 (2) | 14.1 ± 9.5 (5) | 25.4 ± 8.4 (4) | 26 |
| 1369d |  |
NA | 2.4 ± 0.7 (2) | ND | NA | NA | >40.0 | >40.0 | 33 |
Values are expressed as means ± the standard deviations, where applicable. *, The cytotoxicity data were previously published for compounds 1312 (24), 1614 (26), 1717 (26), and 1369 (33). NA, not applicable; ND, not done. Reference standards were as follows: CRL-8155 cells, quinacrine EC50 = 3.99 ± 2.27 μM (n = 14); HepG2 cells, quinacrine EC50 = 9.76 ± 2.73 μM (n = 13); COX-1, chloramphenicol EC50 = 6.31 ± 0.91 μM (n = 8); and SDH-A, chloramphenicol EC50 = >50 ± 0 μM (n = 8).
Growth inhibition assay done at University of Washington (UW) (see Materials and Methods).
Growth inhibition assay done at University of Vermont (U. VT) (see Materials and Methods).
Bumped kinase inhibitor (reference standard).
TABLE 4.
Activities of MetRS inhibitors (MetRS inhibitor scaffold and compound 2242) with a single ring in the linker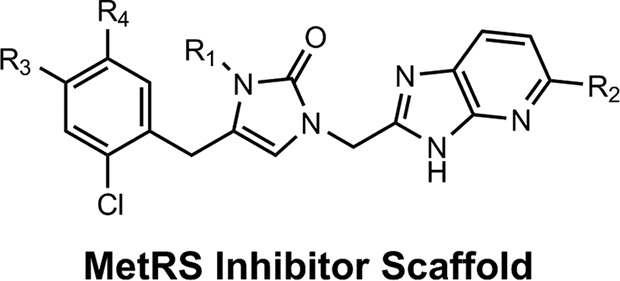
| Molecule | R1 | R2 | R3 | R4 | Mean activity ± SD (n)a |
Source or reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. parvum MetRS Ki (nM) | UW C. parvum EC50 (μM) | U. VT C. parvum EC50 (μM) | COX-1 EC50 (μM) | SDH-A EC50 (μM) | CRL-8155 EC50 (μM)* | Hep G2 EC50 (μM)* | ||||||
| 1962 | H | F | Cl | H | >1.33 (2) | >20.000 ± 0.000 (2) | 8.48 ± 0.81 (2) | >25 | >25 | >50.0 | >50.0 | 27 |
| 2062 | CH2-CH3 | F | Cl | H | 0.0162 ± 0.0017 (2) | 1.865 ± 0.884 (2) | 0.152 (2) | 0.29 | >25 | >50.0 | >50.0 | 27 |
| 2114 | CH2-CH3 | F | OCH3 | H | 0.0023 ± 0.0011 (3) | 0.060 ± 0.011 (2) | 0.075 | >25 | >50.0 | >50.0 | 27 | |
| 2067 | CH2-CH3 | Cl | Cl | H | 0.0093 ± 0.0002 (2) | 0.408 ± 0.286 (2) | 0.076 (2) | 0.042 | >25 | >50.0 | >50.0 | This study |
| 2091 | CH2-CH3 | Br | Cl | H | 0.598 (1) | 0.376 (2) | >50.0 | >50.0 | This study | |||
| 2093 | CH2-CH3 | Cl | OCH3 | H | 0.0009 ± 0.0004 (12) | 0.036 ± 0.004 (2) | 0.007 (2) | 0.039 | >25 | >50.0 | >50.0 | 27 |
| 2069 | CH2-CH2-CH3 | F | Cl | H | 0.016 ± 0.010 (3) | 0.411 ± 0.266 (2) | 0.043 (2) | 0.195 | >25 | >50.0 | >50.0 | This study |
| 2139 | CH2-CF2H | F | Cl | H | 0.066 ± 0.022 (2) | 0.284 ± 0.128 (2) | 0.175 | >25 | 33.3 | >50.0 | This study | |
| 2259 | CH2-CF2H | F | OCH3 | H | 0.0038 ± 0.0014 (3) | 0.134 ± 0.089 (2) | 0.164 | >25 | >50.0 | >50.0 | This study | |
| 2138 | CH2-CF2H | Cl | Cl | H | 0.0187 ± 0.0004 (2) | 0.454 ± 0.252 (2) | 0.416 ± 0.127 (2) | 0.430 | >25 | >50.0 | >50.0 | This study |
| 2258 | CH2-CF2H | Cl | OCH3 | H | 0.0036 ± 0.0019 (3) | 0.19 ± 0.004 (2) | 0.134 | >25 | >50.0 | >50.0 | This study | |
| 2207 | CH2-C(CH3)2OH | Cl | Cl | H | 0.129 (1) | 0.100 ± 0.019 (2) | 0.055 | >25 | >50.0 | >50.0 | This study | |
| 2240 | CH2-C(CH3)2OH | Cl | OCH3 | OCH3 | 0.31 ± 0.170 (2) | >50.0 | >50.0 | This study | ||||
| 2080 | CH(CH3)2 | F | Cl | H | 0.52 ± 0.12 (2) | 0.166 | >25 | >50.0 | >50.0 | This study | ||
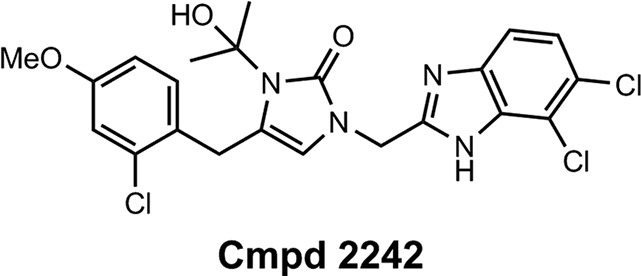
| 2242 | Structure above | 0.014 ± 0.0048 (2) | 0.67 ± 0.19 (2) | 1.57 | >25 | 31,139 | >50.0 | This study | ||||
Values are expressed as means ± the standard deviations, where applicable. *, Cytotoxicity data were previously published for compounds 1962, 2062, 2093, and 2114 (27). The reference standards were as follows: CRL-8155 cells, quinacrine EC50 = 3.99 ± 2.27 μM (n = 14); HepG2 cells, quinacrine EC50 = 9.76 ± 2.73 μM (n = 13); COX-1, chloramphenicol EC50 = 6.31 ± 0.91 μM (n = 8); and SDH-A, chloramphenicol EC50 = >50 ± 0 μM (n = 8).
The correlation between enzyme inhibitory activity and C. parvum growth-inhibitory activity was plotted for all of the compounds shown in Tables 3 and 4. A strong correlation was observed (R2 = 0.91) (Fig. 1).
FIG 1.

Correlation between CpMetRS inhibition (Ki) and C. parvum growth inhibition (EC50).
The compounds were also tested for inhibitory activity of mitochondrial protein synthesis in HepG2 cells (COX-1 EC50, Tables 3 and 4). The COX-1 gene is encoded by the mitochondrial genome and expressed in the mitochondrial protein synthesis pathway. The MetRS inhibitors showed a wide range of EC50 values from low micromolar to low nanomolar concentrations. In general, compounds with potent activity on C. parvum cultures also had potent activity on the COX-1 enzyme (e.g., compound 2093). A parallel experiment was performed on the same samples to measure levels of the nuclear encoded SDH-A protein that is imported into the mitochondrion. A reduction in levels of this protein reflects general toxicity to the cells. Except for compound 1433, all compounds exhibited SDH-A EC50 values >25 μM, indicating no general toxicity at this concentration.
The activity of the most potent MetRS inhibitor, compound 2093, was tested against a panel of three different C. parvum strains including a clinical isolate from dairy calves and a C. hominis strain (Table S1). Compound 2093 had EC50 values ranging from 0.006 to 0.029 μM against C. parvum strains and of 0.015 μM against C. hominis. A wide therapeutic index was also documented with 50% cytotoxic concentrations of >25 μM against three mammalian cell lines.
Next, a parasite persistence assay was performed with compound 2093 and control compounds nitazoxanide and MMV665917 (Fig. 2). The persistence curve for compound 2093 closely resembles that of MMV665917, which is believed to have a cidal mechanism of anti-Cryptosporidium activity and is dissimilar to the curve for nitazoxanide which is believed to have a static mechanism (32).
FIG 2.

Parasite persistence assay. Parasite numbers were normalized to numbers of host nuclei over time with increasing concentrations of compound 2093 (A), nitazoxanide (B), or MMV665917 (C). The lower panels show one-phase exponential decay curves for compound 2093 (D), nitazoxanide (E), and MMV665917 (F) using parasite persistence assay data normalized to a percentage of the data for the DMSO control at each time point. Compound 2093 has a curve consistent with a potentially parasiticidal mechanism (akin to MMV665917 and dissimilar to nitazoxanide).
Efficacy in two murine C. parvum infection models.
Compounds with the highest in vitro potency were selected for efficacy studies employing two different mouse models.
In the NOD SCID Gamma (NSG) mouse model (32), adult mice (n = 4 per group) received a challenge dose of 105 oocysts and were treated with study compounds from day 6 to 10 postinfection. Parasites were quantified in stool by PCR on day 5 and day 11 postinfection showing that compound 2093 was associated with 98.6% reduction (P < 0.05) of parasites. Compound 2069 resulted in 83.6% reduction (P < 0.05), whereas compound 2067 had only an 8.9% reduction (not statistically significant) (Fig. S3, experiment 1). A follow-up experiment showed similar results for compound 2093 and about the same level of parasite reduction with compound 2259 (Fig. S3, experiment 2, P < 0.05 for both compounds).
For the gamma interferon (IFN-γ) knockout (KO) mouse model (33), efficacy experiments were also performed using adult IFN-γ KO mice and a luciferase-expressing C. parvum strain. Adult mice (n = 3 per group) received a challenge dose of 103 oocysts and, as above, were treated with study compounds from day 6 to 10 postinfection. Pooled feces from each group were collected daily and the parasite load was quantified by luminometry. Again, compound 2093 was found to be highly efficacious (>4-log10 drop in luminescence) at the oral dose of 50 mg/kg twice daily (BID) (Fig. 3A). Compound 2093 was also tested at lower doses of 50 mg/kg once per day (Fig. 3B) and at 25 mg/kg twice per day (Fig. 3C), and resulted in ∼3-log10 drop in fecal parasite levels in both experiments. At 20 mg/kg once per day, compound 2093 gave ∼1-log10 drop in parasite levels (day 9) that then rebounded to control levels after the treatment was completed (Fig. 3D). Compounds 2114 and 2259 at 50 mg/kg BID gave similar profiles to compound 2093 with a 3- to 4-log10 drop in stool parasite levels (Fig. 3E and F), whereas compound 2258 appeared to be less active (Fig. 3C). Other compounds with modest anticryptosporidial activity (1- to 2-log drop in fecal parasite loads) were compounds 2138 and 2139 (Fig. 3G). Finally, the following compounds had no demonstrable activity: neither compound 2207 at 50 mg/kg perorally once daily, nor compounds 2080, 2240, and 2242 given at 25 mg/kg perorally BID (data not shown). A control compound with strong anti-Cryptosporidium activity, the “bumped kinase inhibitor” compound 1369 (33), was included in some experiments for reference (Fig. 3F and G).
FIG 3.


Efficacy of MetRS inhibitors in murine C. parvum (IFN-γ KO) infection model. Adult IFN-γ KO mice (n = 3 per group) were infected with luciferase-expressing oocysts on day 0 and treated orally with vehicle or test compounds at the indicated doses from days 6 to 10 postinfection. Stools were collected at 24-h intervals, pooled, and quantitated for luminescent parasites (panels on the left). The weights of the mice from the same experiment are shown immediately to the right. Each pair of panels (A to G) represents a separate experiment.
In all experiments, the mice were weighed daily. Weight loss was consistently observed in the mice treated with vehicle control. When weights drop below 20% of the baseline, the mice were euthanized per protocol requirements. (The lines end when all mice were culled.) There was some variability between experiments in the time it took for control mice to reach terminal weight (e.g., compare panel A to panel F in Fig. 3). Mice receiving treatments that led to reduced Cryptosporidium infection had relatively stable weights over the course of the experiment. Once the compound dosing was completed (day 11), a weight gain was sometimes observed (e.g., Fig. 3A and G). Except for compound 2258, the MetRS inhibitors shown in Fig. 3 were well tolerated and led to all mice surviving (without >20% weight loss) until the end of the experiment.
Physicochemical properties, metabolism, and pharmacokinetic studies.
Physicochemical properties of the compounds were calculated or measured (Table 5). The molecular weights for the ring-linker series are in the 400 to 500 g/mol range. The calculated log P values range from 3.25 to 4.75. The solubility of compounds 2093, 2114, and 2259 (the more active compounds in the mouse efficacy model) was higher than for compounds 2067 and 2069. Plasma protein binding is high for all of the tested compounds, ranging from 95% to >99.9%, with the exception of compound 2240, with a plasma protein binding value of 87.7%. Permeability was assessed for a subset of compounds using a parallel artificial membrane permeability assay (PAMPA) method and showed fairly similar permeability values (226 to 319 nm/s) for the tested compounds. For comparison, a highly permeable compound, propranolol, had a permeability value of 600 nm/s (pH 7.4) and a low-permeability compound, methylclothiazide, has a value of 30 nm/s (pH 7.4). The permeability of the compounds was also calculated by computer algorithm, and indicated that the predicted permeability for MetRS inhibitors is more similar to propranolol (known to have high permeability) than methylclothiazide (known to have low permeability). The relationship between the described chemical properties (i.e., the predicted permeability and the solubility/EC50) and in vivo efficacy of the compounds was graphed (Fig. 4). This shows that the most active compounds in vivo cluster at an intermediate level of permeability and have a high ratio of solubility/EC50.
TABLE 5.
Physicochemical properties of ring-linker compoundsa
| Molecule | Mol wt (g/mol) | Log P | H-bond (no.) |
Solubility limit(s) (μM) at: |
Protein binding mouse plasma (% bound) | PAMPA: Pe (nm/s) |
Predicted permeability (cm/s × 104) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Donors | Acceptors | pH 7.4 | pH 2.0 | pH 6.5 | pH 7.4 | pH 5.0 | |||||
| 2114 | 415.85 | 3.47 | 1 | 4 | 69.4 | 78.4 | 53.8 | 98.7 | 319 | 284 | 2.394 |
| 2067 | 436.72 | 4.52 | 1 | 3 | 9.3 | 10.3 | 8.8, 9.8 | 99.7 | 313 | 349 | 3.849 |
| 2091 | 481.18 | 4.67 | 1 | 3 | 49 | <10 | 226 | 238 | 4.500 | ||
| 2093 | 432.31 | 3.76 | 1 | 4 | 26.8 | 60.5, 67.2 | 39, 51.5 | 99.9 | 316 | 336 | 2.333 |
| 2069 | 434.3 | 4.75 | 1 | 3 | 6 | 39.9, 49.1 | 36.4, 29.4 | 99.6 | 286 | 288 | 3.721 |
| 2139 | 456.25 | 4.34 | 1 | 3 | 26.2 | 76.8 | 65 | 99.5 | 269 | 251 | 3.867 |
| 2259 | 451.83 | 3.58 | 1 | 4 | 44.4 | 52 | 80.6 | 95.0 | 2.319 | ||
| 2138 | 472.7 | 4.63 | 1 | 3 | 26.2 | 58.4 | 33 | 100.0 | 235 | 237 | 3.887 |
| 2258 | 468.29 | 3.87 | 1 | 4 | 44.4 | >100 | 39.2 | 98.8 | 2.309 | ||
| 2207 | 476.36 | 3.41 | 2 | 5 | 66.3 | 86.8 | 98.7 | 1.322 | |||
| 2240 | 506.38 | 3.25 | 2 | 6 | 78.1 | 83.2 | 87.7 | 1.118 | |||
| 2080 | 434.3 | 4.65 | 1 | 3 | >100 | 53.6 | 3.853 | ||||
| 2242 | 509.81 | 4.68 | 2 | 4 | 43.1 | <10 | 100.0 | 1.745 | |||
| Propranolol | 259.35 | 2.58 | 2 | 3 | 600 | 200 | 2.449 | ||||
| Methyclothiazide | 360.22 | 0.53 | 2 | 5 | 30 | 30 | 0.646 | ||||
FIG 4.

Relationship of chemical properties to efficacy (Peff) for the MetRS inhibitors. Compounds are color coded as follows: no efficacy in mouse models (<1-log decrease in parasites, red squares [compounds 2067, 2069, 2080, 2169, 2207, 2240, and 2242]), moderate efficacy (1- to 2-log decrease in parasites, orange triangles [compounds 2138, 2139, and 2258]), and strong efficacy (>2-log decrease in parasites, green circles [compounds 2093, 2114, and 2259]). The predicted permeability is plotted against the solubility at pH 6.5/EC50.
In vitro measurements of liver microsome stability showed half-lives ranging from 2 to 29 min (mouse) and 8 to 42 min (human) (Table 6). The blood pharmacokinetics in mice were measured after a single oral dose of 50 mg/kg. Peak blood levels (Cmax) and areas under the curve (AUC) are shown in Table 6. Compound 2067 had the highest Cmax (65 μM) and AUC (17263 min ⋅ μM/liter), whereas compound 2093 had a lower exposure in blood (Cmax [5.8 μM] and AUC [1,863 min ⋅ μM/liter]). The levels of the compounds were also measured in the feces of selected compounds and were observed to be in the 10 to 50 μM range, with the exception of compound 2240, for which the fecal levels were very high at 846 μM.
TABLE 6.
Pharmacological properties of ring-linker compoundsa
| Molecule | Microsome stability (t1/2 [min]) |
Mouse oral PK (mean ± SD)b |
|||
|---|---|---|---|---|---|
| Mice | Humans | Cmax (μM) | AUC (min ⋅ μmol/liter) | Concn (μM) in pooled mouse feces | |
| 2114 | 7.7 | 15.2 | 7.5 ± 2.4 (3) | 1,854 ± 103 (3) | 11.4 ± 4.1 (3) |
| 2067 | 3.1 | 9.3 | 65.2 ± 13.3 (3) | 17,263 ± 5,167 (3) | |
| 2091 | 2 | 9.4 | |||
| 2093 | 3.2 | 8.3 | 5.8 ± 1.54 (3) | 1,863 ± 658 (3) | 31.1 ± 3.5 (3) |
| 2069 | 3.6 | 8.2 | 21.8 ± 8.8 (3) | 3,932 ± 431 (3) | |
| 2139 | 11.7 | 20.1 | 40.7 ± 10.6 (3) | 10,404 ± 2,589 (3) | 36.2 ± 3.7 (3) |
| 2259 | 29.4 | 32.7 | 25.8 ± 16.2 (3) | ||
| 2138 | 5.8 | 15 | 30.8 ± 1.3 (3) | 6,269 ± 1,806 (3) | 43.6 ± 3.8 (3) |
| 2258 | 13.8 | 25.5 | |||
| 2207 | 5.3 | 25.1 | 30.8 ± 6.5 (3) | 3,048 ± 863 (3) | |
| 2240 | 24.2 | 42.3 | 11.1 ± 11.8 (3) | 856 ± 813 (3) | 846 ± 220 (3) |
| 2080 | 1.6 | 8 | 25.4 ± 8.6 (3) | 5,092 ± 1,466 (3) | 17.9 ± 5.4 (3) |
| 2242 | 4.5 | 7.7 | 2.5 ± 0.7 (3) | 512 ± 51 (3) | 52.7 ± 16.2 (3) |
In vitro metabolic stability (t1/2) was measured in mouse and human liver microsomes. Pharmacokinetic studies in mice (n = 3 per compound) were performed with a single peroral dose at 50 mg/kg in MMV vehicle (defined in Materials and Methods). The microsome half-lives for compounds 2093 and 2114 were previously published (27).
n values are indicated in parentheses.
Safety studies.
The compounds with the best in vivo activity (compounds 2093, 2114, and 2259) were tested for inhibition of five human cytochrome P450 (CYP) isoenzymes at a single concentration of 10 μM. Similar results were observed for all three compounds. Only CYP2C8 was inhibited by >50% at this concentration (Table 7). Inhibition of the hERG channels was measured at two concentrations: 10 and 30 μM (Table 8). Again, similar results were observed for three compounds. In the absence of serum, hERG inhibition was approximately 60% at 30 μM, but the inhibition dropped to ∼20% in the presence of bovine serum albumin (BSA). The Ames and in vitro micronucleus tests for genotoxicity were performed on compound 2093, and both results were negative. Detailed results are available in the supplemental material.
TABLE 7.
CYP enzyme inhibition
| Molecule | % inhibitiona |
||||
|---|---|---|---|---|---|
| CYP1A2 | CYP2C8 | CYP2C9 | CYP2D6 | CYP3A4 | |
| 2093 | 4.1 | 87.3 | 13.6 | 36.0 | 25.4 |
| 2114 | 4.0 | 74.7 | –9.20 | 16.6 | 24.9 |
| 2259 | 10.4 | 62.1 | –13.1 | 31.6 | 37.5 |
The values represent the percent inhibition of human CYP isoenzymes obtained with compounds 2093, 2114, and 2259 at 10 μM.
TABLE 8.
hERG inhibition
| Molecule | % inhibitiona |
|||
|---|---|---|---|---|
| –BSA |
+BSA |
|||
| 10 μM | 30 μM | 10 μM | 30 μM | |
| 2093 | 31.6 | 62.1 | 17.9 | 20.9 |
| 2114 | 28.5 | 58.8 | 18.8 | 23.9 |
| 2259 | 23.1 | 64.9 | 12.9 | 19.5 |
The values represent the percent inhibition of the hERG activity for compounds 2093, 2114, and 2259 measured at two concentrations (10 and 30 μM) either without (–) or with (+) bovine serum albumin (BSA).
DISCUSSION
The Cryptosporidium enzyme, methionyl-tRNA synthetase, was targeted for the development of novel drugs to treat cryptosporidiosis. Analysis of the deposited amino acid sequence of the CpMetRS revealed that it clusters with type 1 MetRS enzymes (23) that include trypanosomes, Giardia, and Gram-positive bacteria. In previous work, crystal structures of the T. brucei MetRS revealed the ATP and methionine binding pockets, as well as the binding mode for numerous inhibitors (28, 29). By mapping the C. parvum enzyme to the T. brucei MetRS structure, the analogous binding pocket was identified, demonstrating that 19 of 25 amino acids (76%) forming the surface of the pocket are identical. This indicated that inhibitors of the T. brucei MetRS would be likely to bind the CpMetRS. The binding pocket residues of the C. hominis MetRS are identical to those of C. parvum. The sequence analysis also showed that 18 of 24 residues (75%) were identical in the corresponding human mitochondrial MetRS. The significance of this similarity is discussed below. In contrast, the human cytoplasmic MetRS had a lower degree of identity, i.e., 14 of 24 (58%), which is consistent with the knowledge that the human cytoplasmic MetRS belongs to the MetRS2 category which is not inhibited by the compounds under investigation.
The CpMetRS gene was amplified from genomic DNA, cloned into an expression vector, and overexpressed in E. coli. The purified enzyme was catalytically active in an aminoacylation assay in which [3H]methionine is incorporated into tRNA (Fig. S2). This method uses 100 nM enzyme to provide an acceptable signal to background ratio which constrains the ability to accurately measure the Ki for highly potent inhibitors (since the 50% inhibitory concentration [IC50] can theoretically be no less than half the enzyme concentration). In order to measure the Ki for the MetRS inhibitors, the ATP:PPi exchange assay was adopted (31). In this method, incorporation of [32P]PPi into ATP occurs by the reverse enzyme reaction that can be measured when the receiving substrate, tRNA, is omitted. The Km for the enzyme substrates, methionine and ATP, are within a factor of 2 to 3 of the values measured in this study and reported elsewhere (31) for the S. aureus MetRS. In contrast, the Km values of methionine and ATP for the human mitochondrial MetRS are lower by a factor of 5 to 10.
Since the MetRS inhibitors are competitive with methionine (31), it is possible to accurately measure the IC50s of inhibitors in the reaction by raising the concentration of methionine above its Km (34). The shift in IC50 is proportional to the [substrate]/Km, as described by the Cheng-Prusoff equation for competitive inhibitors (35). Of note, the method of raising the methionine substrate concentration cannot be used in the aminoacylation assay mentioned earlier because the unlabeled methionine competes with the [3H]methionine that is necessary for the readout. The Ki values were determined for selected MetRS inhibitors revealing extraordinary potency of many of these compounds, ranging from 0.0009 to >1.33 nM (Table 4). This is similar to the Ki for antibiotic MetRS inhibitor, REP8839 against the S. aureus MetRS, reported at 0.01 nM (31). The relationship between the Ki and C. parvum EC50 showed a strong correlation (R2 = 0.91; P < 0.0001), which is consistent with the observation that compound 2093 was the most potent compound against the MetRS enzyme and against C. parvum infection of cell cultures. The correlation data support the conclusion that the inhibitors act “on target” to mediate their effects on C. parvum cells.
More than 500 MetRS inhibitors have been developed in our program to optimize their antitrypanosomal and antibacterial activities (24–26, 30). A set of structurally diverse compounds from this library (Tables 3 and 4) was screened against C. parvum, revealing a mix of positive and negative results. The aminoquinolone-containing compounds (exemplified by compound 1312 [24]) had poor activity. The aminoquinolone compounds are in clinical development as antibiotics for Clostridium difficile and S. aureus infections (36, 37). Similarly, compounds with the urea moiety (e.g., compound 1433 [25]) also had poor activity. In contrast, compounds with the fluoro-imidazopyridine (e.g., compounds 1614 and 1717 [26]) demonstrated more potent activity, in the 5 to 10 μM range. Parallel work had indicated that changes to the linker region of the molecule were well tolerated, leading to explorations of various changes, including ring systems at this region of the molecule (30). Compounds with the 1,3-dihydro-2-oxo-imidazole as the linker ring were particularly active (Table 4). Among the compounds containing the 1,3-dihydro-2-oxo-imidazole, various substitutions were explored at the R groups indicated in Table 4. R1 as ethyl (e.g., compound 2062) was more active than R as H (compound 1962) or as propyl (compound 2069). R1 as CH2CF2H appears to be slightly less active than the ethyl version. R2 as F is slightly less active than R2 as Cl, and Br is essentially the same as Cl. Next, substitutions on the benzyl group (on the left side of the structure; Table 4) were explored. It had been previously shown that 3,5 substitutions (such as 3,5-dichlorobenzyl) were particularly potent on the T. brucei MetRS (24). In the orientation created by the ring-linker structures, the 2,4 substitutions (as shown in Table 4) have the greatest potency. Changing R4 from Cl to OCH3, produced the most potent inhibitors in the series (e.g., compounds 2093 and 2114). Compounds with a trisubstituted benzyl group (e.g., 2,4,5 substitutions) had much diminished activity against C. parvum oocysts (data not shown). In summary, compound 2093 was the most potent compound (EC50 = 0.007 μM) and compares very favorably to the published data for the clinical drug, nitazoxanide (EC50 = 3.7 μM) against C. parvum (38).
The activity of the MetRS inhibitors against C. parvum infection was tested in two different murine models. The NOD SCID gamma mouse model was performed with a PCR-based readout comparing pretreatment (day 5) to posttreatment (day 11) fecal parasite levels. Fecal oocysts were significantly reduced for compound 2093 (tested twice), as well as for compounds 2069 and 2259, but not for compound 2067. The activities were then retested in the Cryptosporidium infection model using adult IFN-γ KO mice. In this model, the mice were monitored for 20 days postinfection. Quantitation of stool parasite loads again showed that compound 2093 was highly efficacious (>4-log10 drop in luminescence) at an oral dose of 50 mg/kg BID (Fig. 3A). The greater magnitude of parasite reduction observed in the IFN-γ KO mice may be due to differences in the models and readout methods. In addition, the challenge dose was lower in the IFN-γ KO mice (103) compared to the NSG model (105), which may account for the differences if there is an inoculum effect as seen with some bacterial infections (39). A persistence of a low luminescence signal above background levels (log10 RLU of 2.5) was detected during the remainder of the monitoring period in the IFN-γ model, the significance of which is unclear. Importantly, the parasite signal did not rebound to the high levels observed with the controls. Also, the mice maintained their body weight and survived to the end of the 20-day observation period, unlike the vehicle-treated mice that needed to be euthanized on day 17 due to the loss of >20% body weight (Fig. 3A). Several follow-up experiments confirmed the in vivo activity of compound 2093 at lower doses (e.g., 50 mg/kg/day divided into one or two doses), but the activity was substantially diminished at the dose of 20 mg/kg once per day. The other compounds with excellent in vivo activity were compounds 2114 and 2259. The mice appeared to tolerate the treatments without any observed side effects and did not experience the weight loss that was observed in the vehicle-treated mice. The one exception was for compound 2258, where the mice lost weight and were euthanized on day 13 (before the control mice needed to be euthanized). For the other compounds, it was encouraging to observe that potent anticryptosporidial activity was associated with good clinical outcomes in the mice.
The compounds with the greatest in vivo efficacy (compounds 2093, 2114, and 2259) were among the most potent in vitro compounds. To further understand the reasons for the more potent in vivo activity, various physicochemical properties were assessed. Some general characteristics of these potent in vivo compounds are molecular weights in the 400 to 450 range, log P values in the 3.5 to 3.75 range, and a relatively high solubility (>25 μM) at pH levels reflective of the gastrointestinal tract and plasma. The MetRS inhibitors are all highly protein-bound in the range of 95 to 99.9% (except for compound 2240 with 87.7% protein binding). Apparently, high protein binding is not detrimental to activity since compound 2093 is 99.9% bound to mouse plasma proteins. Figure 4 allows for visualization of the chemical properties as they relate to in vivo efficacy. It shows that the most active compounds in vivo have relatively high solubility/EC50, apparently reflecting the importance of in vitro potency (EC50) and, at least, moderate solubility. Interestingly, the most effective compounds had intermediate levels of predicated permeability perhaps, suggesting that overly permeable compounds may be completely absorbed into the blood and thus unavailable in the gut for local antiparasitic activity and that compounds with low permeability may not cross membranes sufficiently to exert effects on the parasites either.
The liver microsome metabolic half-lives for the MetRS inhibitors were relatively low (<15 min for most compounds tested; Table 6). In fact, the lead compound 2093 has half-lives of 3.2 and 8.3 min in murine and human microsomes, respectively. This short in vitro half-life does not directly translate to low plasma exposure in vivo, probably because of the protective effects of the high plasma protein binding.
The pharmacokinetic (PK) properties in mice of several of the MetRS inhibitors were also assessed. The two most potent compounds (2093 and 2114) had similar PK profiles, with a Cmax in the 6 to 8 μM range and an AUC of ∼2,000 min ⋅ μmol/liter. The fecal levels of compounds 2093 and 2114 were 31 and 11 μM, respectively. The fecal levels suggest that sufficient amounts of intact compound (>100 × EC50) are available in the fecal stream to exert anticryptosporidiosis effects. Previous studies with “bumped kinase inhibitors” indicate that intestinal levels of compound better correlate with antcryptosporidiosis activity than do plasma levels (12). Both fecal levels and plasma levels were similar enough among the tested CpMetRS inhibitors; thus, it is not possible to make firm conclusions about the most favorable properties. One exception is that compound 2240 had very high average fecal levels (846 μM) and yet had no in vivo anti-Cryptosporidium activity. We speculate that this compound may have passed through the gastrointestinal tract in an insoluble form that was not available for local anticryptosporidiosis activity. If this is true, then merely delivering insoluble compound to the gut is not sufficient. Since the pathogen lives in an intracellular niche, it is necessary for the compound to be in solution and sufficiently permeable to reach that niche either directly from the intestinal lumen or via the bloodstream.
The safety of the compounds is of great importance given that the target population for anticryptosporidiosis treatment will include very young children and other vulnerable groups. A potential concern for MetRS inhibitors is cross-activity on the human mitochondrial MetRS enzyme that could lead to mitochondrial dysfunction. The human mitochondrial MetRS and the CpMetRS are identical at 18 of 25 residues in the compound binding pocket (see Table 1), indicating moderate similarity. An assay was performed to quantify cytochrome oxidase 1 (COX-1) enzyme levels in human liver cells (HepG2) after a 6-day incubation with MetRS inhibitors. This enzyme is encoded and expressed in the mitochondrion, whereas the control protein (SDH-A) is encoded in the nucleus and expressed in the cytoplasm. The single ring-linker MetRS inhibitors (Table 4), in fact, demonstrated substantial inhibition of COX-1 expression levels with EC50 values as low as 0.039 μM in the case of compound 2093. This is only slightly above the EC50 value against C. parvum (0.007 to 0.036 μM). The other single ring-linker compounds generally had EC50 values of <0.5 μM in this assay. The in vivo effects that may result from mitochondrial protein synthesis inhibition will require further investigation. It is worth noting that many antibiotics that work by inhibiting prokaryotic protein synthesis also inhibit mitochondrial protein synthesis (40). For example, the commonly used antibiotics such as doxycycline (COX-1 EC50 = 6.6 μM) and linezolid (COX-1 EC50 = 15 μM) are used at plasma levels that approximate or exceed the EC50 determined in the COX-1 assay (41, 42). In the case of linezolid, toxicity due to mitochondrial inhibition can be observed during normal use of the drug, although this typically does not become serious until after 4 weeks of treatment (43). With anticipated treatment courses for cryptosporidiosis being relatively short (ideally no more than 3 days), it is likely that brief exposures to MetRS inhibitors would be well tolerated, although clearly this will require careful investigation. If mitochondrial inhibition is a problem, then additional effort to identify more selective compounds will be pursued in future work.
The compounds with the best in vivo activity (compounds 2093, 2114, and 2259) were also tested for inhibition of the hERG channels and CYP450 enzymes. The hERG inhibition assay screens for potential of the compound to promote dangerous cardiac dysrhythmias. In the presence of plasma protein (BSA), the percent inhibition was only ∼20% at concentrations of 30 μM, which is reassuring. The concentration that substantially inhibits hERG channels is likely to be several multiples above the peak concentrations that would occur during treatment. This same compound had little effect on CYP450 enzymes except for CYP2C8 (causing 62 to 87% inhibition at 10 μM). Common drugs that are metabolized by CYP2C8 include rosiglitazone (antidiabetic), montelukast (asthma), cerivastatin (statin), and amodiaquine (antimalarial). The coadministration of these MetRS inhibitors with the listed drugs could potentially lead to drug-drug interactions. Finally, Ames and in vitro micronucleus tests were done on compound 2093 and were negative. This provides reassurance that compound 2093 is not genotoxic.
In summary, the imidazopyridine compounds described here have potent activity against the C. parvum MetRS enzyme, as well as against cultures of C. parvum and C. hominis. Parasite persistence assays suggest the compounds have parasiticidal effects on the parasites. Most importantly, the MetRS inhibitors controlled C. parvum infection in two murine models without producing side effects. The active compounds demonstrated substantial plasma exposures, as well as fecal levels, but it is not entirely clear from these data which of these pharmacological parameters is most relevant. The MetRS inhibitors are capable of inhibiting the human mitochondrial MetRS enzyme as determined by reduced levels of a mitochondrial protein (COX-1) in cultured HepG2 cells; however, clinical toxicity may be unlikely if the duration of treatment is kept to short durations (e.g., <1 week). Future studies with compound 2093 and other MetRS inhibitors in the calf Cryptosporidium infection model, as well as in vivo toxicology studies, will help establish the potential for developing these compounds for the treatment of human cryptosporidiosis.
MATERIALS AND METHODS
Protein sequence alignments.
Global pairwise amino acid sequence alignments were generated with the NCBI alignment tool Clustal Omega (44).
CpMetRS cloning, expression, and purification.
The CpMetRS gene (UniProtKB accession number Q5CVN0) was PCR amplified from genomic DNA isolated from the C. parvum Iowa II strain. The PCR product was then cloned into the AVA0421 plasmid (45), and the sequence was verified. The expression of the recombinant protein was performed as previously described (45). The protein was purified by nickel affinity chromatography, followed by size exclusion chromatography.
Enzyme assays.
Inhibition of CpMetRS was measured using an ATP:PPi exchange assay as previously described (31, 46) with some modifications. The compounds were preincubated for 5 min at room temperature in a 96 well-plate with 30 nM CpMetRS, 125 mM l-methionine, 25 μM NaPPi, ∼2 μCi of [32P]tetrasodium pyrophosphate (NEX019001MC; Perkin-Elmer), 2.5 mM dithiothreitol, 100 mM Tris-HCl (pH 8.0), 10 mM magnesium acetate, 80 mM KCl, and 2% dimethyl sulfoxide (DMSO). The reaction was started with the addition of 25 μM ATP; after a 10-min incubation at room temperature, 5 μl of the reaction mixture (in duplicate) was quenched into a MultiScreenHTS Durapore 96-well filter plate (MSHVN4B50; Millipore Sigma) containing a mixture of 200 μl of 10% charcoal with 0.5% HCl and 50 μl of 1 M HCl with 200 mM sodium pyrophosphate. The filter plates were washed three times with 200 μl of 1 M HCl with 200 mM sodium pyrophosphate on a vacuum manifold. The plates were dried for 30 min at room temperature, and then 25 μl of scintillation cocktail was added. Plates were incubated at room temperature for ∼1.5 h before the counts per minute (cpm) were quantified on a MicroBeta2 scintillation counter (Perkin-Elmer). The percent inhibition was calculated by subtracting the background wells (containing all assay reagents except ATP and compound) and comparing this value to the high control wells (containing all assay reagents without compound). The IC50s were calculated by nonlinear regression methods using the Collaborative Drug Database (Burlingame, CA; www.collaborativedrug.com). The Ki values were calculated from the IC50s that were shifted above the enzyme concentration (30 nM) using the Cheng-Prusoff equation as follows: IC50 = (1 + [Met]/KmMet)(1 + KmATP/[ATP])Ki (31). The Km values for l-methionine were determined using the assay conditions described above with either SaMetRS or CpMetRS (30 nM) without compounds and with ∼5 μCi of [32P]tetrasodium pyrophosphate, 2.5 mM ATP, and 2.5 mM NaPPi while titrating the l-methionine. The reaction was quenched as described above at different time intervals, typically 0, 4, 8, 12, 16, and 20 min. Similarly, the Km values for ATP were measured by using 1 mM l-methionine and 2.5 mM NaPPi while titrating the ATP. The Km and Vmax values were calculated using Prism (v3.0) software. The kcat is equal to the Vmax divided by the enzyme concentration (30 nM).
Chemistry.
The synthesis of compounds 1312 (24), 1433 (25), 1614 (26), 1717 (26), 1962 (27), 2062 (27), 2093 (27), and 2114 (27) was as reported previously. The synthesis of compounds 2067, 2069, 2080, 2091, 2138, 2139, 2207, 2240, 2242, 2258, and 2259 is described in the supplemental material.
Propagation methods and growth inhibition assays for C. parvum.
Genetically modified nanoluciferase (Nluc)-expressing C. parvum Iowa strain oocysts were propagated in female IFN-γ KO mice (C57BL/6 IFN-c-deficient mice B6.129S7-Ifngtm1Ts/J; The Jackson Laboratory, Bar Harbor, ME) (47). Mice were infected by oral gavage with 1,000 oocysts in 0.1 ml of DPBS (Sigma, St. Louis, MO). Fecal samples were collected starting 3 to 5 days after infection. Multiple times per week for 2 to 3 weeks, mice were transferred to a clean cage for 1 to 2 h, and feces were collected and stored in a 2.5% potassium dichromate solution at 4°C. Oocysts were purified from feces using sucrose flotation, followed by cesium chloride gradient as previously described (48).
Growth inhibition assays at University of Washington were performed as follows. HCT-8 cells were added to a 96-well plate and allowed to grow for 72 h to reach 90 to 100% confluence. The medium was removed, and test compounds were added in serial dilutions prior to the addition of 1,000 oocysts per well in 0.1 ml of RPMI 1640 medium supplemented with 10% horse serum and 1% penicillin-streptomycin. Plates were incubated for 72 h; Nano-Glo luciferase reagent (Promega, Madison, WI) was then added, and the plates were read on an EnVision multilabel plate reader (Perkin-Elmer, Waltham, MA). EC50 curves were calculated as previously described using Prism v6.07 (GraphPad Software, La Jolla, CA) (33).
C. parvum growth inhibition assays completed at the University of Vermont were performed as described previously using wild-type C. parvum Iowa strain oocysts freshly isolated from calves (purchased from Bunch Grass Farm, Deary, ID) and high-content microscopy (32, 38). Excystation of oocysts was induced by treatment with 10 mM hydrochloric acid (10 min at 37°C), followed by exposure to 2 mM sodium taurocholate (Sigma-Aldrich) in phosphate-buffered saline (PBS) for 10 min at 16°C. Excysted oocysts were then added to 95% confluent HCT-8 cell monolayers in 384-well plates (∼5,500 oocysts per well). Compounds were added 3 h after infection, and assay plates were incubated for 48 h postinfection at 37°C under 5% CO2. The cell monolayers were then washed three times with PBS containing 111 mM d-galactose, fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, and blocked overnight with 4% BSA in PBS. Parasitophorous vacuoles were stained with 1.33 μg/ml of fluorescein-labeled Vicia villosa lectin (Vector Laboratories) diluted in 1% BSA in PBS with 0.1% Tween 20 for 1 h at 37°C, followed by the addition of Hoechst 33258 (AnaSpec) at a final concentration of 0.09 mM diluted in water for another 15 min at 37°C. Wells were then washed five times with PBS containing 0.1% Tween 20. A Nikon Eclipse TE2000 epifluorescence microscope with an automated stage was programmed to focus on the center of each well and take a 3-by-3 composite image using an EXi Blue fluorescence microscopy camera (QImaging, Canada) with a 20× objective (numerical aperture, 0.45). Nucleus and parasite images were exported separately as tiff files and analyzed on an ImageJ platform (National Institutes of Health) using previously developed macros (38).
The methods for screening C. hominis and clinical isolates of C. parvum from dairy calves (reported in the supplemental material) were the same as those reported previously (49). The strains were collected by C. W. McNamara at CALIBR (32).
Parasite persistence assays.
The procedures were previously published (32). Briefly, HCT-8 cell monolayer infections were established as described above in 384-well culture plates using wild-type C. parvum Iowa strain oocysts. After approximately 24 h, compounds were added at various concentrations as labeled, and then parasites and host cells were enumerated at multiple time points using immunofluorescence microscopy. Data points are means and standard deviations for four culture wells per time point and are representative of three independent experiments. The P value is the replicates test result (note that a P value of ≥0.05 indicates a valid curve fit).
Mammalian cell growth inhibition assays.
Compounds were tested against CRL-8155 human lymphocytic cells and HepG2 human hepatocellular cells, as previously described (50). Briefly, compounds were incubated with CRL-8155 cells (30,000/well) or HepG2 cells (25,000/well) for 48 h in 96-well plates and then developed using alamarBlue (Thermo Fisher Scientific). The EC50 values were calculated from the percent inhibition by nonlinear regression methods using the Collaborative Drug Discovery database (Burlingame, CA) as previously described (50).
MitoBiogenesis In-Cell ELISA colorimetric assay.
At 24 h before the addition of the compounds, 6,000 HepG2 (human hepatocellular) cells were seeded per well into Gibco collagen I-coated plates (96 well, A1142803; Thermo Fisher Scientific) in culture media (50). The next day, the medium was removed, and the compounds were added in culture media. Every 48 h thereafter during the 6-day incubation period, the media with compounds were replaced with freshly diluted compounds in media. The plates were then fixed with 4% paraformaldehyde and developed by using a MitoBiogenesis In-Cell ELISA kit (colorimetric; ab110217; Abcam) according to the manufacturer’s instructions. The EC50 values were calculated from the normalized data by nonlinear regression methods using the Collaborative Drug Discovery database (Burlingame, CA) or Prism (v3.0) software.
Mouse efficacy models.
The efficacy in adult IFN-γ KO mice determined at the University of Washington was carried out as previously described (33) and approved by the University of Washington Institutional Animal Care and Use Committee. Briefly, female IFN-γ KO mice (B6.129S7-Ifngtm1Ts/J; Jackson Laboratories) aged 8 to 10 weeks were infected by oral gavage with 1,000 Nluc-tagged C. parvum UGA1 oocysts in 0.1 ml of Dulbecco PBS. Each experimental group included three mice. On day 6 postinfection, the mice were administered compounds orally in a formulation of 3% ethanol, 7% Tween 80, and 90% saline for 5 days. The fecal samples were collected out to day 20 postinfection and the luminescence was quantified in relative light units and normalized as previously described (33).
All NOD SCID gamma mouse studies were performed in compliance with animal care guidelines and were approved by the University of Vermont Institutional Animal Care and Use Committee. Male NOD SCID gamma mice with normal flora (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) (51) were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed for at least a week for acclimatization. At the age of 4 to 5 weeks, mice were infected by oral gavage with 105 C. parvum Iowa strain oocysts. Treatment was started on day 6 after infection. Mice (four per experimental group) were treated perorally with test compounds at the indicated doses from days 6 to 10 postinfection. Oocyst shedding in feces was monitored using a previously validated quantitative PCR assay and primers (52).
Measurements of compound solubility.
Portions (2 μl) of 20 mM DMSO stock solution of compound were added to 398 μl at pH 7.4, 2.0, or 6.5 in PBS buffer. The mixture was vigorously shaken to mix the sample thoroughly, and then the sample was incubated at 25°C overnight. The mixture was centrifuged at 25°C for 20 min at 15,000 × g. The supernatant (100 μl) was transferred into another vial and diluted with 100 μl of CH3CN. The 2-fold-diluted supernatant (100 μl) was analyzed by using a high-pressure liquid chromatography/UV system.
Measurements of plasma protein binding.
Compound binding to mouse plasma proteins was determined by using rapid equilibrium dialysis device (catalog no. 89809; Thermo Fisher Scientific), 96-well equilibrium dialyzer plates (catalog no. SDIS 9610EN; Nest Group, Inc.), or in-house prepared microdialysis plates (53) according to published methods (27, 53) and the manufacturer’s instructions (54).
Parallel artificial membrane permeability assay.
The donor well was filled with 200 μl of PRISMA HT buffer (pH 5.0 or 7.4; pION, Inc.) containing 10 μM test compound. The filter on the bottom of each acceptor well was coated with 4 μl of a GIT-0 lipid solution (pION, Inc.) and filled with 200 μl of acceptor sink buffer (pION, Inc.). The acceptor filter plate was put on the donor plate and incubated for 3 h. After the incubation, the amount of test compound in both the donor and acceptor wells was measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to calculate the permeability rate.
Microsome stability assays.
Liver microsomes were purchased from Sekisui XenoTech, LLC (Kansas City, KS). The microsomes (0.2 mg of protein/ml) and the compound (1 μM) were mixed in phosphate buffer (pH 7.4). The reactions were initiated by adding an NADPH-generating system (a mixture of MgCl2, β-NADP+, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase) to the mixtures before incubation. Incubations were conducted at 37°C and terminated by adding acetonitrile. The zero-time incubations, which served as the controls, were terminated by adding acetonitrile before adding an NADPH-generating system. After the samples were mixed and centrifuged, the compound concentration in the supernatant fractions were measured by LC-MS/MS. Control compounds demonstrated the following in vitro clearance rates: flutamide, 161 μl/min/mg (human) and 304 μl/min/mg (mouse); quinidine, 4 μl/min/mg (human) and 27 μl/min/mg (mouse); and cilostazol, 28 μl/min/mg (human) and 100 μl/min/mg (mouse).
CYP450 inhibition assays.
Human liver microsomes were purchased from Sekisui XenoTech, LLC (Kansas City, KS). The microsomes (0.1 mg of protein/ml), substrates (tacrine, paclitaxel, tolbutamide, dextromethorphan, and midazolam) and the compound (10 μM) were mixed in phosphate buffer (pH 7.4). The reactions were initiated by adding an NADPH-generating system (a mixture of MgCl2, β-NADP+, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase) to the mixtures before incubation. Incubations were conducted at 37°C for 10 min and terminated by adding acetonitrile. The activities of CYP1A2, CYP2C8, CYP2C9, CYP2D6, and CYP3A4 were determined by the peak of 1-hydroxytacrine, 6α-hydroxypaclitaxel, 4-hydroxytolbutamide, dextrorphan, and 1′-hydroxymidazolam, respectively. The activities of the test samples were expressed as the percentage of activity remaining compared to a control sample containing no inhibitor.
hERG inhibition assays.
hERG/CHO cells stably expressing the hERG channel were purchased from Millipore, Ltd. (UK). Cells were cultured at 32°C and 5% CO2 in Ham’s F-12 medium supplemented with 10% fetal bovine serum and 500 μg/ml Geneticin. The hERG inhibition assay was performed on the IonWorks Quattro (Molecular Devices) system in population patch clamp mode. The extracellular solution was PBS with calcium and magnesium. The intracellular solution contained 120 μM amphotericin B, 140 mM KCl, 2 mM MgCl2, 1 mM EGTA, and 20 mM HEPES (pH 7.3). The hERG current was measured under the potential-clamp protocol (holding potential, –80 mV; first voltage, 40 mV for 2 s; second voltage, –50 mV for 2 s). After patch perforation, the peak-tail current before the addition of the compounds was measured as the pre-hERG current. Test compounds were incubated on the cells for a period of 5 min. The peak-tail current after addition of the compounds was measured as the post-hERG current. The hERG inhibition assays were performed in triplicate in two kinds of extracellular solutions which contained 1% BSA or none.
Mouse pharmacokinetics.
Nonfasted female Swiss-Webster mice (n = 3) were administered 50 mg/kg of compound by oral gavage in a vehicle consisting of 7% Tween 80, 3% ethanol, 5% DMSO, and 0.9% saline. Tail blood was collected at 30, 60, 120, 240, 360, 480, and 1,440 min into heparinized capillary tubes and spotted onto Whatman gel blotting paper as previously described (50). Whole-blood samples were extracted with acetonitrile and analyzed by LC-MS/MS. The values of the pharmacokinetic parameters were calculated using Phoenix WinNonlin (v6.3) software (Certara, Princeton, NJ).
Feces were collected and pooled from all three mice from the entire duration of the pharmacokinetic experiment described above (0 to 1,440 min). The pellets were suspended in 4× water by weight and homogenized. To extract compounds of interest, acetonitrile (8× by volume) was added to the homogenate containing 100 mg of feces, followed by the addition of internal standard solution (90% acetonitrile, 10% water). After centrifuging the homogenate solution, the supernatant was dried in a Speed-Vac. The dried samples were reconstituted in a LC-MS sample solution (50% Milli-Q water:50% acetonitrile). The solution was centrifuged, and the supernatant was transferred to a liquid chromatography insert. Each compound was tested in triplicate. Similarly, fecal homogenates from vehicle mice were prepared to make calibration standards. The compound concentrations for the calibration standards were 0 nM, 10 nM, 100 nM, 1 μM, 5 μM, 10 μM, 20 μM, and 40 μM. The compound concentrations for each homogenate from the treated mice group were calculated from the calibration curves using Microsoft Excel software.
Supplementary Material
ACKNOWLEDGMENTS
Support for this project was provided by PATH with funding from the UK government (grant 300341-111) and by the National Institutes of Health (grants AI097177 and HHSN272201700059C). The purified recombinant C. parvum MetRS protein (UniProt Q5CVN0) described here was provided by the Seattle Structural Genomics Center for Infectious Disease (www.SSGCID.org) supported by Federal Contract HHSN272201700059C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services.
We acknowledge Nora Molasky, Uyen Nguyen, and Omeed Faghih for performing mammalian cell cytotoxicity assays; Molly McCloskey and Grant Whitman for assisting with in vivo efficacy experiments; Zack Herbst for analytical chemistry on pharmacokinetic assays; and Hannah Udell for cloning and purifying the C. parvum MetRS enzyme. We acknowledge the contributions of project oversight by Eugenio de Hostos (PATH). We also acknowledge the valuable contributions made by collaborating scientists at Takeda (Yasushi Miyazaki) and CALIBR (Melissa Love and Case McNamara).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02061-18.
REFERENCES
- 1.United Nations. 2018. Sustainable development goals. United Nations, New York, NY: https://www.un.org/sustainabledevelopment/sustainable-development-goals/. [Google Scholar]
- 2.World Health Organization. 2018. Global health estimates 2016: deaths by cause, age, sex, by country and by region, 2000–2016. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 4.Platts-Mills JA, Babji S, Bodhidatta L, Gratz J, Haque R, Havt A, McCormick BJ, McGrath M, Olortegui MP, Samie A, Shakoor S, Mondal D, Lima IF, Hariraju D, Rayamajhi BB, Qureshi S, Kabir F, Yori PP, Mufamadi B, Amour C, Carreon JD, Richard SA, Lang D, Bessong P, Mduma E, Ahmed T, Lima AA, Mason CJ, Zaidi AK, Bhutta ZA, Kosek M, Guerrant RL, Gottlieb M, Miller M, Kang G, Houpt ER. 2015. Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED). Lancet Glob Health 3:e564–e575. doi: 10.1016/S2214-109X(15)00151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korpe PS, Haque R, Gilchrist C, Valencia C, Niu F, Lu M, Ma JZ, Petri SE, Reichman D, Kabir M, Duggal P, Petri WA Jr. 2016. Natural history of cryptosporidiosis in a longitudinal study of slum-dwelling Bangladeshi children: association with severe malnutrition. PLoS Negl Trop Dis 10:e0004564. doi: 10.1371/journal.pntd.0004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalil IA, Troeger C, Rao PC, Blacker BF, Brown A, Brewer TG, Colombara DV, De Hostos EL, Engmann C, Guerrant RL, Haque R, Houpt ER, Kang G, Korpe PS, Kotloff KL, Lima AAM, Petri WA, Platts-Mills JA, Shoultz DA, Forouzanfar MH, Hay SI, Reiner RC, Mokdad AH. 2018. Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: a meta-analyses study. Lancet Glob Health 6:e758–e768. doi: 10.1016/S2214-109X(18)30283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Checkley W, White AC Jr, Jaganath D, Arrowood MJ, Chalmers RM, Chen XM, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, Huston CD, Kotloff KL, Kang G, Mead JR, Miller M, Petri WA Jr, Priest JW, Roos DS, Striepen B, Thompson RC, Ward HD, Van Voorhis WA, Xiao L, Zhu G, Houpt ER. 2015. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis 15:85–94. doi: 10.1016/S1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clode PL, Koh WH, Thompson RCA. 2015. Life without a host cell: what is Cryptosporidium? Trends Parasitol 31:614–624. doi: 10.1016/j.pt.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Shrivastava AK, Kumar S, Smith WA, Sahu PS. 2017. Revisiting the global problem of cryptosporidiosis and recommendations. Trop Parasitol 7:8–17. doi: 10.4103/2229-5070.202290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mor SM, Tumwine JK, Ndeezi G, Srinivasan MG, Kaddu-Mulindwa DH, Tzipori S, Griffiths JK. 2010. Respiratory cryptosporidiosis in HIV-seronegative children in Uganda: potential for respiratory transmission. Clin Infect Dis 50:1366–1372. doi: 10.1086/652140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mor SM, Ascolillo LR, Nakato R, Ndeezi G, Tumwine JK, Okwera A, Sponseller JK, Tzipori S, Griffiths JK. 2018. Expectoration of Cryptosporidium parasites in sputum of human immunodeficiency virus-positive and -negative adults. Am J Trop Med Hyg 98:1086–1090. doi: 10.4269/ajtmh.17-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnold SLM, Choi R, Hulverson MA, Schaefer DA, Vinayak S, Vidadala RSR, McCloskey MC, Whitman GR, Huang W, Barrett LK, Ojo KK, Fan E, Maly DJ, Riggs MW, Striepen B, Van Voorhis WC. 2017. Necessity of bumped kinase inhibitor gastrointestinal exposure in treating Cryptosporidium infection. J Infect Dis 216:55–63. doi: 10.1093/infdis/jix247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakama T, Nureki O, Yokoyama S. 2001. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J Biol Chem 276:47387–47393. doi: 10.1074/jbc.M109089200. [DOI] [PubMed] [Google Scholar]
- 14.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crepin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MR. 2007. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 15.Elewski BE, Aly R, Baldwin SL, Gonzalez Soto RF, Rich P, Weisfeld M, Wiltz H, Zane LT, Pollak R. 2015. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol 73:62–69. doi: 10.1016/j.jaad.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo CY, Mazitschek R, Whitman M. 2012. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Dwyer K, Spivak AT, Ingraham K, Min S, Holmes DJ, Jakielaszek C, Rittenhouse S, Kwan AL, Livi GP, Sathe G, Thomas E, Van HS, Miller LA, Twynholm M, Tomayko J, Dalessandro M, Caltabiano M, Scangarella-Oman NE, Brown JR. 2015. Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob Agents Chemother 59:289–298. doi: 10.1128/AAC.03774-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez V, Crepin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang YK, Zhou Y, Nieman JA, Anugula MR, Keramane el M, Savariraj K, Reddy DS, Sharma R, Subedi R, Singh R, O’Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MR, Plattner JJ. 2013. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother 57:1394–1403. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Hernandez V, Rock FL, Choi W, Mak YSL, Mohan M, Mao W, Zhou Y, Easom EE, Plattner JJ, Zou W, Perez-Herran E, Giordano I, Mendoza-Losana A, Alemparte C, Rullas J, Angulo-Barturen I, Crouch S, Ortega F, Barros D, Alley MRK. 2017. Discovery of a potent and specific Mycobacterium tuberculosis leucyl-tRNA synthetase inhibitor: (S)-3-(aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J Med Chem 60:8011–8026. doi: 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed] [Google Scholar]
- 20.Palencia A, Li X, Bu W, Choi W, Ding CZ, Easom EE, Feng L, Hernandez V, Houston P, Liu L, Meewan M, Mohan M, Rock FL, Sexton H, Zhang S, Zhou Y, Wan B, Wang Y, Franzblau SG, Woolhiser L, Gruppo V, Lenaerts AJ, O’Malley T, Parish T, Cooper CB, Waters MG, Ma Z, Ioerger TR, Sacchettini JC, Rullas J, Angulo-Barturen I, Perez-Herran E, Mendoza A, Barros D, Cusack S, Plattner JJ, Alley MR. 2016. Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase. Antimicrob Agents Chemother 60:6271–6280. doi: 10.1128/AAC.01339-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pham JS, Dawson KL, Jackson KE, Lim EE, Pasaje CF, Turner KE, Ralph SA. 2014. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int J Parasitol Drugs Drug Resist 4:1–13. doi: 10.1016/j.ijpddr.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palencia A, Liu RJ, Lukarska M, Gut J, Bougdour A, Touquet B, Wang ED, Li X, Alley MR, Freund YR, Rosenthal PJ, Hakimi MA, Cusack S. 2016. Cryptosporidium and Toxoplasma parasites are inhibited by a benzoxaborole targeting leucyl-tRNA synthetase. Antimicrob Agents Chemother 60:5817–5827. doi: 10.1128/AAC.00873-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gentry DR, Ingraham KA, Stanhope MJ, Rittenhouse S, Jarvest RL, O’Hanlon PJ, Brown JR, Holmes DJ. 2003. Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae with two distinct methionyl-tRNA synthetase genes. Antimicrob Agents Chemother 47:1784–1789. doi: 10.1128/AAC.47.6.1784-1789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata S, Gillespie JR, Kelley AM, Napuli AJ, Zhang Z, Kovzun KV, Pefley RM, Lam J, Zucker FH, Van Voorhis WC, Merritt EA, Hol WG, Verlinde CL, Fan E, Buckner FS. 2011. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei infection in mice. Antimicrob Agents Chemother 55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibata S, Gillespie JR, Ranade RM, Koh CY, Kim JE, Laydbak JU, Zucker FH, Hol WG, Verlinde CL, Buckner FS, Fan E. 2012. Urea-based inhibitors of Trypanosoma brucei methionyl-tRNA synthetase: selectivity and in vivo characterization. J Med Chem 55:6342–6351. doi: 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Koh CY, Ranade RM, Shibata S, Gillespie JR, Hulverson MA, Huang W, Nguyen J, Pendem N, Gelb MH, Verlinde CLMJ, Hol WGJ, Buckner FS, Fan E. 2016. 5-Fluoro-imidazo[4,5-b]pyridine is a privileged fragment that conveys bioavailability to potent trypanosomal methionyl-tRNA synthetase inhibitors. ACS Infect Dis 2:399–404. doi: 10.1021/acsinfecdis.6b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faghih O, Zhang Z, Ranade RM, Gillespie JR, Creason SA, Huang W, Shibata S, Barros-Alvarez X, Verlinde C, Hol WGJ, Fan E, Buckner FS. 2017. Development of methionyl-tRNA synthetase inhibitors as antibiotics for Gram-positive bacterial infections. Antimicrob Agents Chemother 61:e00999-17. doi: 10.1128/AAC.00999-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh CY, Kim JE, Shibata S, Ranade RM, Yu M, Liu J, Gillespie JR, Buckner FS, Verlinde CL, Fan E, Hol WG. 2012. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 20:1681–1691. doi: 10.1016/j.str.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koh CY, Kim JE, Wetzel AB, de van der Schueren WJ, Shibata S, Ranade RM, Liu J, Zhang Z, Gillespie JR, Buckner FS, Verlinde CLMJ, Fan E, Hol WGJ. 2014. Structures of Trypanosoma brucei methionyl-tRNA synthetase with urea-based inhibitors provide guidance for drug design against sleeping sickness. PLoS Negl Trop Dis 8:e2775. doi: 10.1371/journal.pntd.0002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang W, Zhang Z, Barros-Alvarez X, Koh CY, Ranade RM, Gillespie JR, Creason SA, Shibata S, Verlinde CL, Hol WG, Buckner FS, Fan E. 2016. Structure-guided design of novel Trypanosoma brucei methionyl-tRNA synthetase inhibitors. Eur J Med Chem 124:1081–1092. doi: 10.1016/j.ejmech.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green LS, Bullard JM, Ribble W, Dean F, Ayers DF, Ochsner UA, Janjic N, Jarvis TC. 2009. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob Agents Chemother 53:86–94. doi: 10.1128/AAC.00275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jumani RS, Bessoff K, Love MS, Miller P, Stebbins EE, Teixeira JE, Campbell MA, Meyers MJ, Zambriski JA, Nunez V, Woods AK, McNamara CW, Huston CD. 2018. A novel piperazine-based drug lead for cryptosporidiosis from the medicines for malaria venture open-access malaria box. Antimicrob Agents Chemother 62:e01505-17. doi: 10.1128/AAC.01505-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hulverson MA, Vinayak S, Choi R, Schaefer DA, Castellanos-Gonzalez A, Vidadala RSR, Brooks CF, Herbert GT, Betzer DP, Whitman GR, Sparks HN, Arnold SLM, Rivas KL, Barrett LK, White AC Jr, Maly DJ, Riggs MW, Striepen B, Van Voorhis WC, Ojo KK. 2017. Bumped-kinase inhibitors for cryptosporidiosis therapy. J Infect Dis 215:1275–1284. doi: 10.1093/infdis/jix120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pope AJ, Moore KJ, McVey M, Mensah L, Benson N, Osbourne N, Broom N, Brown MJ, O’Hanlon P. 1998. Characterization of isoleucyl-tRNA synthetase from Staphylococcus aureus. II. Mechanism of inhibition by reaction intermediate and pseudomonic acid analogues studied using transient and steady-state kinetics. J Biol Chem 273:31691–31701. doi: 10.1074/jbc.273.48.31691. [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y, Prusoff WH. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- 36.Critchley IA, Ochsner UA. 2008. Recent advances in the preclinical evaluation of the topical antibacterial agent REP8839. Curr Opin Chem Biol 12:409–417. doi: 10.1016/j.cbpa.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 37.Critchley IA, Green LS, Young CL, Bullard JM, Evans RJ, Price M, Jarvis TC, Guiles JW, Janjic N, Ochsner UA. 2009. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J Antimicrob Chemother 63:954–963. doi: 10.1093/jac/dkp041. [DOI] [PubMed] [Google Scholar]
- 38.Bessoff K, Sateriale A, Lee KK, Huston CD. 2013. Drug repurposing screen reveals FDA-approved inhibitors of human HMG-CoA reductase and isoprenoid synthesis that block Cryptosporidium parvum growth. Antimicrob Agents Chemother 57:1804–1814. doi: 10.1128/AAC.02460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soriano F, Santamaria M, Ponte C, Castilla C, Fernandez-Roblas R. 1988. In vivo significance of the inoculum effect of antibiotics on Escherichia coli. Eur J Clin Microbiol Infect Dis 7:410–412. doi: 10.1007/BF01962350. [DOI] [PubMed] [Google Scholar]
- 40.Cohen BH, Saneto RP. 2012. Mitochondrial translational inhibitors in the pharmacopeia. Biochim Biophys Acta 1819:1067–1074. doi: 10.1016/j.bbagrm.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 41.Ribes S, Pachon-Ibanez ME, Dominguez MA, Fernandez R, Tubau F, Ariza J, Gudiol F, Cabellos C. 2010. In vitro and in vivo activities of linezolid alone and combined with vancomycin and imipenem against Staphylococcus aureus with reduced susceptibility to glycopeptides. Eur J Clin Microbiol Infect Dis 29:1361–1367. doi: 10.1007/s10096-010-1007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newton PN, Chaulet JF, Brockman A, Chierakul W, Dondorp A, Ruangveerayuth R, Looareesuwan S, Mounier C, White NJ. 2005. Pharmacokinetics of oral doxycycline during combination treatment of severe falciparum malaria. Antimicrob Agents Chemother 49:1622–1625. doi: 10.1128/AAC.49.4.1622-1625.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garrabou G, Soriano A, Lopez S, Guallar JP, Giralt M, Villarroya F, Martinez JA, Casademont J, Cardellach F, Mensa J, Miro O. 2007. Reversible inhibition of mitochondrial protein synthesis during linezolid-related hyperlactatemia. Antimicrob Agents Chemother 51:962–967. doi: 10.1128/AAC.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi R, Kelley A, Leibly D, Hewitt SN, Napuli A, Van Voorhis W. 2011. Immobilized metal-affinity chromatography protein-recovery screening is predictive of crystallographic structure success. Acta Crystallogr Sect F Struct Biol Crystallogr Commun 67:998–1005. doi: 10.1107/S1744309111017374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beebe K, Waas W, Druzina Z, Guo M, Schimmel P. 2007. A universal plate format for increased throughput of assays that monitor multiple aminoacyl transfer RNA synthetase activities. Anal Biochem 368:111–121. doi: 10.1016/j.ab.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Cipriano MJ, Striepen B. 2015. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 523:477–480. doi: 10.1038/nature14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arrowood MJ, Donaldson K. 1996. Improved purification methods for calf-derived Cryptosporidium parvum oocysts using discontinuous sucrose and cesium chloride gradients. J Eukaryot Microbiol 43:89S. doi: 10.1111/j.1550-7408.1996.tb05015.x. [DOI] [PubMed] [Google Scholar]
- 49.Love MS, Beasley FC, Jumani RS, Wright TM, Chatterjee AK, Huston CD, Schultz PG, McNamara CW. 2017. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl Trop Dis 11:e0005373. doi: 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silva DG, Gillespie JR, Ranade RM, Herbst ZM, Nguyen UTT, Buckner FS, Montanari CA, Gelb MH. 2017. A new class of antitrypanosomal agents based on imidazopyridines. ACS Med Chem Lett 8:766–770. doi: 10.1021/acsmedchemlett.7b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R. 2005. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγ-null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 52.Parr JB, Sevilleja JE, Samie A, Amidou S, Alcantara C, Stroup SE, Kohli A, Fayer R, Lima AAM, Houpt ER, Guerrant RL. 2007. Detection and quantification of Cryptosporidium in HCT-8 cells and human fecal specimens using real-time polymerase chain reaction. Am J Trop Med Hyg 76:938–942. doi: 10.4269/ajtmh.2007.76.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herbst ZM, Shibata S, Fan E, Gelb MH. 2018. An inexpensive, in-house-made, microdialysis device for measuring drug-protein binding. ACS Med Chem Lett 9:279–282. doi: 10.1021/acsmedchemlett.7b00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thermo Fisher Scientific. 2017. Instructions: single-use RED plate with inserts. Pierce Biotechnology, Rockford, IL: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0011619_SgleUse_RED_Plate_Insert_UG.pdf. [Google Scholar]
- 55.Spencer AC, Heck A, Takeuchi N, Watanabe K, Spremulli LL. 2004. Characterization of the human mitochondrial methionyl-tRNA synthetase. Biochemistry 43:9743–9754. doi: 10.1021/bi049639w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.