

In 2016, we identified a new class A carbapenemase, VCC-1, in a nontoxigenic Vibrio cholerae strain that had been isolated from retail shrimp imported into Canada for human consumption. Shortly thereafter, seven additional VCC-1-producing V. cholerae isolates were recovered along the German coastline.
KEYWORDS: VCC-1, Vibrio cholerae, X-ray structure, avibactam, β-lactamases, carbapenemase, carbapenems
ABSTRACT
In 2016, we identified a new class A carbapenemase, VCC-1, in a nontoxigenic Vibrio cholerae strain that had been isolated from retail shrimp imported into Canada for human consumption. Shortly thereafter, seven additional VCC-1-producing V. cholerae isolates were recovered along the German coastline. These isolates appear to have acquired the VCC-1 gene (blaVCC-1) independently from the Canadian isolate, suggesting that blaVCC-1 is mobile and widely distributed. VCC-1 hydrolyzes penicillins, cephalothin, aztreonam, and carbapenems and, like the broadly disseminated class A carbapenemase KPC-2, is only weakly inhibited by clavulanic acid or tazobactam. Although VCC-1 has yet to be observed in the clinic, its encroachment into aquaculture and other areas with human activity suggests that the enzyme may be emerging as a public health threat. To preemptively address this threat, we examined the structural and functional biology of VCC-1 against the FDA-approved non-β-lactam-based inhibitor avibactam. We found that avibactam restored the in vitro sensitivity of V. cholerae to meropenem, imipenem, and ertapenem. The acylation efficiency was lower for VCC-1 than for KPC-2 and akin to that of Pseudomonas aeruginosa PAO1 AmpC (k2/Ki = 3.0 × 103 M−1 s−1). The tertiary structure of VCC-1 is similar to that of KPC-2, and they bind avibactam similarly; however, our analyses suggest that VCC-1 may be unable to degrade avibactam, as has been found for KPC-2. Based on our prior genomics-based surveillance, we were able to target VCC-1 for detailed molecular studies to gain early insights that could be used to combat this carbapenemase in the future.
INTRODUCTION
Of all clinically available β-lactam antibiotics, carbapenems exhibit the greatest potency and broadest spectrum of activity against Gram-negative and Gram-positive bacteria (1). Once reserved for only the most serious infections, they are now more widely used in hospitals and even outpatient settings (2), and resistance to these once last-resort antibiotics has increased markedly over the last decade (3, 4) as carbapenem-active β-lactamases (carbapenemases) have emerged. β-Lactamases are a large family of β-lactam-degrading hydrolases that can be divided into four molecular classes based on amino acid sequence homology (5). Molecular classes A, C, and D include β-lactamases with a catalytic serine residue in their active sites and are collectively referred to as serine β-lactamases, whereas molecular class B β-lactamases are metalloenzymes that contain catalytic zinc in their active sites (6). A number of serine carbapenemases belonging to classes A and D, as well as metallo-carbapenemases belonging to class B, have emerged.
Class A β-lactamases are generally inhibited by clavulanate, tazobactam, and sulbactam, which are classical β-lactamase inhibitors (BLIs) developed during the 1980s and are based on β-lactam scaffolds that inactivate β-lactamases by forming stable acyl-enzyme adducts with the catalytic serine (7). Notably, however, some class A carbapenemases, such as the widespread family of class A Klebsiella pneumoniae carbapenemase (KPC)-like β-lactamases (8, 9) and other less prevalent class A carbapenemases (e.g., SFC-1 [10, 11], FRI-1 [12], BIC-1 [13], and PenA [14]), are only weakly inhibited by these BLIs. To address the limited spectrum of classical BLIs and increasing prevalence of BLI-resistant β-lactamases, new non-β-lactam-based BLIs are being developed (15). The first to reach the market was avibactam (Fig. 1), which is based on the non-β-lactam scaffold diazobicyclooctanone. Avibactam potently inhibits a broad spectrum of serine β-lactamases, including KPC-like and other inhibitor-resistant class A carbapenemases, as well as class C cephalosporinases and some class D enzymes (16, 17). Avibactam inactivates β-lactamases in a manner distinct from that of classical BLIs. Remarkably, in contrast to the acyl-enzyme linkage formed by classical β-lactam BLIs on the catalytic serine, which is susceptible to hydrolysis (7), the carbamate linkage formed on the serine with avibactam instead undergoes a nonhydrolytic recyclization reaction that regenerates free avibactam (Fig. 1), enabling it to recarbamylate the β-lactamase (18). Notably, in KPC-2 the acyl-enzyme is resolved via a degradative mechanism, where acyl-avibactam is desulfated and dehydrated prior to hydrolysis and release; the degraded molecule is subsequently decarboxylated to yield 5-oxopiperidine-2-carboxamide (19).
FIG 1.
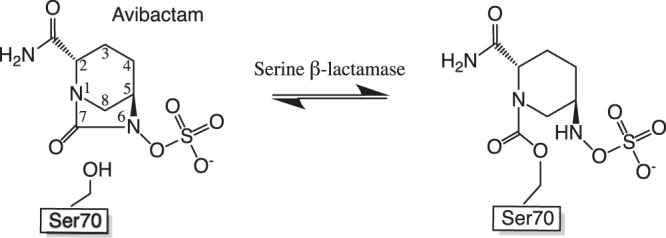
Chemical structure of the recently FDA-approved non-β-lactam-based inhibitor avibactam and its reversible covalent interaction with serine β-lactamases (18).
In 2016, we reported the identification of a new class A carbapenemase, VCC-1, in a nontoxigenic Vibrio cholerae strain (N14-02106) that had been isolated from retail shrimp imported into Canada from India for human consumption (20). In 2017, an additional seven VCC-1 carbapenemase-producing V. cholerae isolates were recovered at different locations along the German coastline (21). Notably, the sequences flanking the VCC-1 gene (blaVCC-1) from the V. cholerae isolates found in Germany differed from those surrounding the blaVCC-1 gene of the Canadian isolate, suggesting that these bacteria independently acquired blaVCC-1 from an unidentified progenitor and that blaVCC-1 may be transferable and widely distributed in the world’s oceans (21). VCC-1 shares the highest amino acid sequence identities with class A carbapenemases IMI (59.1%), NMC-A (59.1%), SFC-1 (58.3%), and KPC-2 (49%). VCC-1 efficiently hydrolyzes penicillins, cephalothin, aztreonam, and carbapenems. Like KPC-2, VCC-1 is only weakly inhibited by clavulanic acid or tazobactam (20). Given its broad spectrum of activity, its poor susceptibility to inhibition by classical BLIs, and its encroachment into aquaculture and other areas with human activity, VCC-1 may be emerging as a new public health threat.
While VCC-1 has not been observed in the clinic, it is a particularly concerning environmental carbapenem resistance element since it appears to be transferable (21) and has already entered the human food chain on at least one occasion (20). To understand the molecular details of this emerging carbapenemase, we carried out a detailed structural and biochemical analysis of VCC-1 and found that it was potently inhibited by avibactam both in vitro and in V. cholerae. We found that the structure of VCC-1 is similar to that of KPC-2 and that they bind avibactam similarly; however, our analyses suggest that VCC-1 may not readily degrade avibactam, as is found for KPC-2. This work is an example of how current rapid and broad genomics-based surveillance enables targeted molecular studies to gain clinically useful insights into newly emerging antibiotic resistance elements before they reach the clinic.
RESULTS AND DISCUSSION
Avibactam is a potent inhibitor of VCC-1 in vitro and in cell culture.
We previously found that neither clavulanate nor tazobactam is an effective inhibitor of VCC-1 (20). Given the efficacy of avibactam against class A enzymes that are impervious to the classical BLIs, we assessed if VCC-1 could be antagonized by this new inhibitor. Indeed, unlike clavulanic acid or tazobactam, which have 50% inhibitory concentration (IC50) values of 30 ± 1 μM (Ki, 13 ± 3 μM) and 4 ± 0.5 μM (Ki, 2.0 ± 0.4), respectively (20), we found avibactam to be a markedly better inhibitor of VCC-1 with an IC50 of 0.028 ± 0.004 μM (Ki, 0.013 ± 0.002 μM). Moreover, avibactam also rescued the efficacy of carbapenems against the environmental V. cholerae N14-02106 strain, in which the enzyme was discovered. We found that susceptibility testing using the broth microdilution method revealed that avibactam (4 μg/ml) potentiated the activity of carbapenems against the V. cholerae strain. The MICs of three carbapenems (determined in triplicate) were reduced in the presence of avibactam at 4 μg/ml, supporting the effectiveness of avibactam against VCC-1: ertapenem, 64 to 0.5 μg/ml; imipenem, >64 to 16 μg/ml; and meropenem, 64 to 2 μg/ml.
We next carried out enzyme kinetic studies and mass spectrometry (MS) assays using recombinantly expressed and purified VCC-1. We found that avibactam is a covalent, slowly reversible inhibitor of the enzyme, consistent with the inhibition of other class A β-lactamases by avibactam (18, 19). We found the rate constant governing acylation of VCC-1 by avibactam is (3.0 ± 0.5) × 103 M−1 s−1, which is ∼100-fold lower than the reported acylation rate constants measured for the class A enzymes TEM-1 and CTX-M-15, ∼4-fold less than the acylation rate constant measured for KPC-2, and most similar to the acylation rate constant of Pseudomonas aeruginosa PAO1 AmpC (hereafter referred to as AmpC) [(2.9 ± 0.1) × 103 M−1 s−1] (Table 1) (19). Although the VCC-1 acylation rate was measured under the same conditions used to measure that of KPC-2, a portion of the observed differences may be due laboratory-to-laboratory variance. The deacylation half-life governing the dissociation of avibactam from VCC-1, as measured by the return of VCC-1 activity (half-life [t1/2] = 6 ± 1 min) (Table 1; see also Fig. S1 in the supplemental material), is also faster than that reported for TEM-1 (t1/2 = 16 ± 8 min), CTX-M-15 (t1/2 = 40 ± 10 min), and KPC-2 (t1/2 = 82 ± 6 min), as well as for Enterobacter cloacae P99 AmpC (t1/2 = 300 ± 20 min) (19); however, it is similar to that measured for P. aeruginosa AmpC (t1/2 = 6 ± 2 min) (19).
TABLE 1.
Kinetic parameters of avibactam inhibition of VCC-1 versus KPC-2
| Enzyme | Acylation k2/Ki (M−1 s−1) | Deacylation koff (s−1) | Deacylation t1/2 (min) | Kd (μM) |
|---|---|---|---|---|
| VCC-1 | (3.0 ± 0.5) × 103 | (2 ± 0.1) × 10−3 | 6 ± 1 | 0.686 |
| KPC-2a | (1.3 ± 0.1) × 104 | (1.4 ± 0.1) × 10−4 | 82 ± 6 | 0.011 |
| CTX-M-15a | (1.3 ± 0.1 × 105 | (3 ± 1) × 10−4 | 40 ± 10 | 0.002 |
| P. aeruginosa PAO1 AmpCa | (2.9 ± 0.1) × 103 | (1.9 ± 0.6) × 10−3 | 6 ± 2 | 0.66 |
Kinetic parameters are reproduced from reference 19.
To gain insight into the formation and stability of the covalent VCC-1 acyl-enzyme adduct, the enzyme was incubated with an 8:1 molar excess of avibactam for 3 h at 37°C, at which time the enzyme was confirmed to be fully inactivated, as judged by a lack of activity on the chromogenic substrate nitrocefin. To verify formation of a covalent complex, unbound avibactam was removed by ultrafiltration and the remaining complex was analyzed by electrospray ionization (ESI)-MS, which revealed that the mass of the avibactam-inactivated enzyme was 29,455 Da (Fig. 2), whereas the mass determined for the native enzyme was 29,190 Da (expected mass, 29,193 Da) (Fig. S2). This 265-Da mass increase is consistent with formation of a covalent 1:1 complex, where the predicted mass increase stemming from the covalent labeling with avibactam would be 265.24 Da. Monitoring the mass of the inactivated enzyme over time revealed a time-dependent deacylation, resulting in an ∼2:1 ratio of unbound versus bound enzyme after 32 h (Fig. 2). The dissociation constant (Kd) of the VCC-1–avibactam complex (0.686 μM) was found to be ∼40 to 220 times higher than that of other class A enzymes that have been studied in detail with avibactam (19), yet it was similar to a previously determined Kd for avibactam binding to AmpC (0.66 μM) (Table 1) (19). The observation that VCC-1 is still partially acylated, even after removing excess avibactam and incubating for 32 h, is reminiscent of what was observed for AmpC (19). In the case of AmpC, it was proposed that there is a slow approach to an equilibrium between avibactam-acylated and free AmpC that develops due to the nanomolar Kd and relatively fast acylation and deacylation rates for AmpC relative to those for other enzymes that have been studied with avibactam. Given that the rate constants governing the inactivation and reactivation of VCC-1 most resemble those seen for AmpC, we reason a similar scenario holds for VCC-1.
FIG 2.

Reconstructed spectra, monitoring the reactivation of avibactam-inactivated VCC-1 using ESI-MS. After incubation of VCC-1 with excess avibactam, followed by removal of the inactivator, reactivation of the enzyme was monitored by measuring the amount of covalently labeled VCC-1 at 1 h (top), 10 h (middle), and 32 h (bottom). The peak at 29,190 atomic mass units (amu) corresponds to that of recombinant VCC-1 (expected mass = 29,193 amu), and the peak at 29,455 amu corresponds to that of recombinant VCC-1 covalently modified with 1 molecule of avibactam (expected mass = 29,458 amu). The y axes show ion count.
Molecular structure of VCC-1.
The crystal structure of native VCC-1 was determined by molecular replacement using the structure of the class A carbapenemase NMC-A (PDB accession number 1BUE) (22) as a search model and refined to a 1.7-Å resolution (Table S1). VCC-1 readily crystallizes into a monoclinic (C2) crystal form, with four copies of the enzyme being present per asymmetric unit. Residues Asn6 to Lys265 of each copy in the asymmetric unit could be modeled into density with no significant structural differences between them (root mean square deviation [RMSD], ∼0.3 Å). VCC-1 adopts a two-domain fold that is consistent with other class A enzymes; one domain has an α/β topology, whereas the other is α helical, and the active site is located at the interface of the two domains (Fig. 3A). Structural alignment of VCC-1 against the structure in the Protein Data Bank (PDB) using the Dali server (23) revealed its closest structural neighbors to be the class A β-lactamases NMC-A (RMSD, 1.0 Å; 259 aligned residues), SME-1 (RMSD, 0.9 Å; 259 aligned residues), SFC-1 (RMSD, 0.8 Å; 259 aligned residues), and KPC-2 (RMSD, 0.9 Å; 258 aligned residues).
FIG 3.

X-ray structure of native VCC-1. (A) VCC-1 adopts a two-domain fold typical of other class A enzymes; one has an α/β topology, whereas the other is α helical. Residues comprising the active site are located at the interface of the two domains and shown as sticks. The omega loop containing the catalytic general base Glu166 is colored cyan. (B) Close-up of the VCC-1 active site. The hydrogen-bonding network and water content are consistent with β-lactam acylation occurring via an energetically favorable proton transfer from Lys73 to Glu166 via Ser70 and active-site water W1 (32). Only the dominant His105 side chain conformation is shown.
In keeping with other class A enzymes, the VCC-1 active site is comprised of three conserved motifs that participate in β-lactam hydrolysis, motif I (70SSFK73), motif II (130SDN132), and motif III (234KTG236), as well as an omega loop (residues 164 to 179) containing the conserved Glu residue at position 166. Serine β-lactamases share these three active-site motifs regardless of molecular class (5): the first is Ser-X-X-Lys, which harbors the active-site serine residue that acts as a nucleophile to attack the carbonyl carbon of the β-lactam ring to open it and form a covalent acyl-enzyme adduct (24). The nucleophilicity of the serine is enhanced by the lysine in the motif, which is separated from serine by two variable (X) residues (24). The second and third motifs are (Ser/Tyr)-X-Asn and Lys-(Thr/Ser)-Gly, respectively, which assist in substrate recognition and proton transfer during catalysis (24, 25). Class A β-lactamases also contain a conserved catalytic Glu at position 166 (Ambler [ABL] numbering scheme [26]) within an omega loop (ABL residues 164 to 179 [27]) that serves as a general base that promotes an active-site water to hydrolyze the acyl-enzyme intermediate (24, 28, 29). Glu166 has also been proposed to be the general base in the first step of the reaction, activating the active-site serine to attack the β-lactam carbonyl group (acting in place of the lysine from motif I [Ser-X-X-Lys]) (30, 31). Indeed, ab initio quantum mechanical/molecular mechanical (QM/MM) calculations of the class A TEM-1 β-lactamase suggest that either Glu166 or the lysine from motif I can activate the catalytic serine in class A enzymes (32).
The hydrogen-bonding network and water content that we observed in the native VCC-1 active site (Fig. 3B) are consistent with those from the above-described QM/MM study, which predicts that upon binding a β-lactam, acylation of the enzyme by the antibiotic is initiated through an energetically favorable proton transfer from Lys73 to Glu166 via Ser70 and a water bound within the active site (Fig. 3B, W1) (32). Deprotonation of Lys73 would enable it to act as a general base that facilitates the attack of Ser70 on the carbonyl of the β-lactam ring, leading to formation of a tetrahedral intermediate, as previously proposed (24). Although the Lys73 and Glu166 side chains are close enough to form an ion pair in VCC-1 and other class A enzymes, direct proton transfer between them may be energetically less favorable (32). The active-site geometry of VCC-1 may also allow Glu166 to act as an alternative base to activate Ser70 via water W1 to promote formation of the acyl enzyme, which is consistent with a prediction made for other class A enzymes based on QM/MM calculations (32) and biochemical studies (31, 33). Both mechanisms appear to be feasible (32). We observe an additional water molecule (W2) present in the active site of native VCC-1 that is held in place by hydrogen-bonding interactions with the backbone amides of Ser70 and Thr237. These groups form the oxyanion hole that stabilizes the oxyanion of the tetrahedral intermediate that would occupy the position of W2. We also observed that a disulfide bond between Cys69 and Cys238 adjacent to the oxyanion hole is also present in VCC-1, which is a distinguishing feature of class A carbapenemases (reviewed in reference 11). This disulfide may contribute to altering the overall shape of the carbapenemase active site and help position catalytic residues to accommodate, in particular, the C-6 hydroxyethyl side chain of carbapenems, a unique chemical feature that inhibits noncarbapenemase class A enzymes by aiding in resistance to hydrolysis (1, 34, 35); however, its importance remains controversial (36). In VCC-1, the close proximity of the disulfide bond to the oxyanion hole may stabilize this critical catalytic motif, along with a hydrogen-bonding interaction between the hydroxyl group of Thr237 and Arg220, another feature of carbapenemases (position 237 can vary between a Ser and a Thr [11]) that may underlie substrate selectivity (37). Taken together, VCC-1 possesses all the structural hallmarks of a class A carbapenemase, with particularly close similarity to KPC-2, including the Thr237-Arg220 hydrogen-bonding interaction and Cys69-Cys238 disulfide bond.
Molecular basis for inhibition of VCC-1 by avibactam.
To capture VCC-1 bound to avibactam for structural analysis, we soaked monoclinic crystals of VCC-1 (the crystal form used to determine the X-ray structure of the native enzyme [Table S1]) in a range of avibactam concentrations (1 to 5 mM). Unfortunately, avibactam caused these crystals to dissolve. However, within the same crystallization drop in which the monoclinic crystal grew, an alternate tetragonal crystal form of VCC-1 appeared ∼2 weeks after the monoclinic crystals appeared, and these crystals could be soaked with up to 5 mM avibactam without any noticeable deterioration. The crystal structure of VCC-1 bound to avibactam was determined by molecular replacement using native VCC-1 (chain D) as the search model and refined to a 1.9-Å resolution (Table S1). Structural alignments of the avibactam-bound VCC-1 structure with the 4 copies of native VCC-1 from the monoclinic crystal form revealed that avibactam binding did not significantly alter the active site or overall conformation of the β-lactamase (RMSD < 0.8 Å) (Fig. 4A).
FIG 4.

X-ray structure of VCC-1 bound to avibactam. (A) Superposition of the avibactam-bound VCC-1 structure with the 4 copies of VCC-1 from the asymmetric unit of the native X-ray structure. Avibactam binding does not significantly alter the active site or overall conformation of the β-lactamase (RMSD < 0.8 Å). (B) Electron density (|Fobs| − |Fcalc| omit map contoured at 4.5 σ) defining avibactam (green carbons) covalently bound to Ser70 (gray carbons) of VCC-1. Positive density is blue, and negative density is red. (C) Avibactam (Avi; green carbon atoms) covalently bound to Ser70 within the VCC-1 active site. VCC-1 carbon atoms are gray, with the exception of residues within the omega loop, which are colored cyan. Dashed yellow lines represent hydrogen-bonding interactions, with their respective distances indicated.
Presently, the crystal structures of six class A β-lactamases (TLA-3 [38], L2 [39], SHV-1 [40], KPC-2 [40], CTX-M-15 [41, 42], BlaC [43]) in complex with avibactam have been reported, and while they share many similarities, interesting differences are also present. For VCC-1, a well-defined electron density can be observed for avibactam covalently bound to the side chain oxygen of Ser70 (Fig. 4B). The six-membered ring of avibactam adopts a chair conformation, while the urea ring has opened across the C-7–N-6 bond to form the carbamoyl linkage with Ser70 of VCC-1. The carbonyl group of the carbamoyl moiety is bound within the oxyanion hole of VCC-1 through 2.7-Å and 3.1-Å hydrogen bonds with the main chain amide nitrogen atoms of residues Ser70 and Thr237, respectively (Fig. 4C). The amide group oxygen of avibactam forms a 3.1-Å hydrogen bond with Asn132. The sulfate moiety of avibactam forms the largest number of hydrogen-bonding interactions with the enzyme. It is situated in a conserved pocket that typically accommodates carboxylate moieties of β-lactams, where it forms hydrogen bonds with the side chains of Ser130 (2.8 Å), Thr235 (2.6 Å), and Thr237 (2.7 Å). The 2.8-Å hydrogen bond observed between Arg220 and Thr237 in the native enzyme is conserved and appears to orient Thr237 within hydrogen-bonding range of the avibactam sulfate moiety. Interestingly, disrupting this hydrogen-bonding network by mutagenesis (Arg220Met) mutation in KPC-2 demonstrates that it plays an important role in avibactam-mediated inhibition of the enzyme, potentially by stabilizing the interaction between Thr237 and the sulfate moiety (44). Alternatively, as seen in SHV-1, which has an Ala at position 237, the loss of the hydrogen bond observed in VCC-1 and KPC-2 between Thr237 and the sulfate of avibactam is compensated for by a direct salt bridge with Arg244.
An aromatic amino acid is conserved among class A carbapenemases at Ambler position 105, where a histidine is a signature of VCC-1 and a number of other related class A enzymes, while a tryptophan is found in KPC-2, and a tyrosine occupies this position in CTX-M-15. His105 of VCC-1 is found at the active-site entrance, where its side chain adopts multiple conformations (Fig. 3B and 4C). In the unliganded structure it is primarily found in a position that is proximal to Ser130 (in 3 of the 4 copies of the asymmetric unit) or rotated ∼90° about the Cα-Cβ bond and pointing toward the solvent (in the remaining copy of the asymmetric unit). When bound to avibactam, His105 adopts the latter conformation (Fig. 4C), which enables the imidazole ring to form weak Van der Waals interactions with C-4 of the avibactam ring, a position reminiscent of the Trp105-avibactam interaction observed in KPC-2 (40). Interestingly, mutational analysis of Trp105 in KPC-2 has demonstrated marked alterations in enzymatic activity for variants of this position against β-lactam antibiotics and up to 10-fold changes in Ki against traditional BLIs, thereby highlighting its importance in binding and/or catalysis (45). In addition, class A enzymes SME-1 and SME-3 differ only in that they contain a histidine and a tryptophan at position 105, respectively; variations at this position can exert a modest 2- to 5-fold difference in catalytic efficiency, and the presence of a tryptophan can broaden the substrate profile of this enzyme (46). Indeed, the presence of the histidine at position 105, in contrast to the tryptophan found in KPC-2, may partially explain the reduced acylation rate of VCC-1 by avibactam compared to that of KPC-2, as discussed below.
Molecular insights into acylation and deacylation of VCC-1 by avibactam.
In addition to direct interactions with avibactam, active-site residues of VCC-1 also maintain a number of hydrogen-bonding interactions with each other (Fig. 4C). In particular, Ser130 retains hydrogen-bonding interactions with epsilon amino groups of Lys73 (3.5 Å) and Lys234 (2.9 Å), suggesting that a nonhydrolytic avibactam deacylation/recyclization process occurs involving the intramolecular attack of N-6 of avibactam on the carbonyl center, aided by the general base catalysis provided by Ser130, which would be assisted by Lys73 (19, 42). Such a recyclization process for avibactam catalyzed by other class A β-lactamases has been proposed (Fig. 4C). A bound water molecule observed in all class A enzymes is thought to be activated by the general base Glu166 to initiate β-lactam deacylation (24, 28, 29), and we consistently observed this water within the active sites of all four monomers of the asymmetric unit in the native VCC-1 and also within the VCC-1–avibactam complex (W1), where it forms a 2.7-Å hydrogen bond with the Glu166 side chain carboxyl group (Fig. 4C). However, consistent with other studies, our kinetic data (Table 1) suggest that avibactam is resistant to hydrolysis by W1, likely due to the stability of the carbamyl linkage and the ability of the five-membered cyclic urea of avibactam, favoring a recyclization pathway (18, 42).
To gain insight into the acylation and deacylation of VCC-1 by avibactam, the structure of the VCC-1–avibactam covalent adduct was superposed with the structures of the avibactam-bound complexes of KPC-2 (PDB accession number 4ZBE; RMSD, 1.0 Å) and CTX-M-15 (PDB accession number 4S2I; RMSD, 1.3 Å) (Fig. 5). VCC-1 is structurally most similar to KPC-2 (RMSD, 1.0 Å), and the acylated avibactam adopts a very similar conformation in both enzymes (Fig. 5A). In contrast, the acylated avibactam in CTX-M-15 adopts a conformation that allows the N-6 atom of the inhibitor to be situated closer to the carbamoyl-carbonyl C-7 (2.9 Å) than these atoms in the KPC-2 and VCC-1 complexes (Fig. 5B). For the VCC-1 and KPC-2 complexes, the N-6 atom faces away from C-7 at a distance of 3.8 Å and 3.9 Å, respectively (Fig. 5A). Further, the N-6 atom of avibactam in the CTX-M-15 complex is within 2.9 Å of Ser130, which is shorter than the comparable distances in the VCC-1 (3.2 Å) and KPC-2 (3.7 Å) complexes. The proximity of N-6 to C-7 and Ser130 in the CTX-M-15 complex suggests that avibactam is well poised to recyclize in CTX-M-15 (41, 42), and this observation may underlie the reason for the slightly higher deacylation rate of avibactam from CTX-M-15 (t1/2 = 40 min) than from KPC-2 (t1/2, 82 min) (19). However, VCC-1 undergoes deacylation with a t1/2 of 6 min (Table 1), which is unexpected, given the remarkable structural similarities between avibactam-bound VCC-1 and KPC-2. Considering also that the acylation rate of VCC-1 [(3.0 ± 0.5) × 103 M−1 s−1] is roughly an order of magnitude lower than that observed for KPC-2 [(1.3 ± 0.1) × 104 M−1 s−1] (19), the structural differences in avibactam bound to CTX-M-15, KPC-2, and VCC-1 cannot fully account for the kinetic variations and indicate that more detailed computational modeling of these enzymes will need to be considered in the future to fully understand their catalytic mechanisms. Further, measuring the above-described kinetic parameters and additional studies of these enzymes performed contemporaneously may better resolve the differences between them and help establish whether VCC-1 is able to degrade avibactam.
FIG 5.

Active-site superpositions of avibactam-bound VCC-1 with avibactam-bound KPC-2 (A) or CTX-M-15 (B). (A) Superposition of VCC-1 and KPC-2 (PDB accession number 4ZBE) bound to avibactam. VCC-1 carbon atoms are colored gray, with bound avibactam carbon atoms colored green. KPC-2 carbon atoms and bound avibactam carbon atoms are colored orange. The hydrogen-bonding interactions in the VCC-1–avibactam complex are represented as dashed yellow lines, whereas they are orange dashed lines in the KPC-2–avibactam complex. The distances between N-6 and C-7 of avibactam in the VCC-1 and KPC-2 complexes are shown as green and orange dashed lines, respectively. (B) Superposition of VCC-1 and CTX-M-15 (PDB accession number 4S2I) bound to avibactam. VCC-1 carbon atoms are colored gray, with bound avibactam carbon atoms being colored green. CTX-M-15 carbon atoms and bound avibactam carbon atoms are colored magenta. Hydrogen-bonding interactions in the VCC-1–avibactam complex are represented as dashed yellow lines, whereas they are magenta dashed lines in the CTX-M-15–avibactam complex. The distances between N-6 and C-7 of avibactam in the VCC-1 and CTX-M-15 complexes are shown as green and magenta dashed lines, respectively.
Concluding remarks.
VCC-1 was the first class A carbapenemase to be identified in members of Vibrionaceae and the first reported class A carbapenemase discovered in food for human consumption; fortunately, it has yet to reach the clinic (20). While public health surveillance efforts revealed the rapid emergence of carbapenemase-mediated resistance elements in human pathogens over the last decade (3), reports of novel carbapenemase-producing bacteria from environmental and food sources have been relatively scarce, even though environmental microbes are considered a major reservoir of antimicrobial resistance (47, 48). The possible overuse of antibiotics in agriculture, aquaculture, and medicine could be selecting for pathogens that have acquired antibiotic resistance mechanisms from this natural reservoir. With the advent of rapid and broad genomics-based surveillance, inclusion of environmental and foodborne sources of antimicrobial resistance in surveillance programs is becoming feasible. Targeting detailed molecular studies based on this information, as presented here for VCC-1, could provide early information about how to combat new and emerging antibiotic resistance elements before they reach the clinic.
MATERIALS AND METHODS
MIC assay.
MICs were performed in either triplicate or quadruplicate using the broth microdilution method (Clinical and Laboratory Standards Institute) in 96-well plates (49). Serial dilutions of meropenem, ertapenem, and imipenem were tested in the presence or absence of 4 μg/ml avibactam in 100 μl cation-adjusted Mueller-Hinton broth (MHB; Becton, Dickinson). Wells were inoculated with 100 μl of 105 cells of Vibrio cholerae N14-02106 grown to an optical density at 600 nm of 0.5. The plates were incubated with shaking at 280 rpm at 37°C for 18 to 20 h. The MIC was determined as the concentration of drug in the well in which there was no growth.
VCC-1 expression and purification.
VCC-1 (GenBank accession number ALU63998.1) expression and purification were determined using pET-28b-VCC-1-transformed Escherichia coli BL21(DE3) cells, as previously described by Mangat et al. (20). Purified VCC-1 was concentrated to 12 mg/ml (the concentration was determined by measurement of the absorption at 280 nm; molecular weight, 29,192 Da; extinction coefficient [ε] value, 30,035 M−1 cm−1). Preparations were judged to be >90% pure, as assessed by SDS-PAGE, and were stored in 10% glycerol at −80°C.
Kinetic analysis.
All enzyme reactions were performed at 37°C in 0.1 M sodium phosphate buffer, pH 7.0, containing 0.1 mg/ml bovine serum albumin. Nitrocefin hydrolysis was monitored by determination of the change in the absorbance at 490 nm using a iD5 plus plate reader (Molecular Devices, Sunnyvale, CA); the dead time for on- and off-rate experiments was estimated to be 23 s. Determination of Ki for avibactam against VCC-1 was performed under the same conditions previously described for tazobactam and clavulanic acid (20).
On- and off-rate experiments with avibactam against VCC were performed as previously described (19, 50, 51). Avibactam binding to VCC was modeled as a reversible two-step scheme:
where the dissociation constant (Ki) is equal to (k−1/k1) and the overall dissociation constant is K*i = (Ki k−2)/(k2 + k−2). E, I, EI, and EI* represent the free enzyme, free inhibitor, enzyme-inhibitor encounter complex, and enzyme-inhibitor tightly bound complex, respectively. Progress curves were fitted to a general integrated rate equation by nonlinear regression, where v0 is the uninhibited enzyme velocity, vs is the fully inhibited enzyme velocity (estimated using a reaction with no VCC-1), and t is time (measured in seconds).
| (1) |
On-rate experiments were performed using 100 pM VCC-1, 600 μM nitrocefin, and 0.35 to 3.5 μM avibactam in triplicate; standard errors were typically <10%. For the on-rate experiment, the fitted values of the observed rate constant for inactivation (kobs) were plotted against the avibactam concentration, the efficiency of acylation (k2/KI) was determined using the linear relationship in equation 2, and the standard deviation is reported.
| (2) |
where [I] and [S] represent the concentrations of avibactam and nitrocefin, respectively. The Michaelis constant (Km) for nitrocefin against VCC was previously determined to be 264 μM (20).
Off-rate experiments were performed using the jump dilution method. Briefly, 1 μM VCC-1 was incubated for 5 min with 5 and 100 μM avibactam, the mixture was serially diluted 10,000-fold into a final reaction buffer containing 1,800 μM nitrocefin, and the off-rates at each concentration were found to be within 5% of each other. Control reactions were performed in the presence or absence of avibactam postdilution. The resultant progress curves were fit to equation 1 to resolve the off-rate constant (koff); the mean and standard deviation for triplicate values are reported. Kd was calculated by dividing koff by k2/Ki.
MS analysis of acyl-enzyme stability.
Two hundred microliters of VCC-1 (3 μM) was incubated with 25 μM avibactam in phosphate buffer solution (pH 7.15, 130 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) at 37°C for 3 h, at which point inactivation of VCC-1 was confirmed using the chromogenic β-lactamase substrate nitrocefin. Excess avibactam was removed by ultrafiltration using a 10-kDa-molecular-weight-cutoff centrifugation filter unit (Amicon Ultra; Merck Millipore Ltd.). The reaction mixture was then centrifuged at 10,000 × g for 10 min at 4°C, after which 140 μl of phosphate-buffered saline was added to the reaction volume, restoring it to 200 μl. Three more wash steps, each consisting of centrifugation and replenishment of buffer to the retentate, were performed. To monitor the stability of the VCC-1 enzyme-inactivator complex, the resultant 3 μM enzyme product was incubated at 37°C for an additional 1, 10, and 32 h. Samples collected at 1 and 10 h were flash frozen and thawed for analysis at the 32-h time point by liquid chromatography (LC)-MS (Bruker maXis Impact quadrupole time-of-flight LC-MS system) operating in positive electrospray ionization mode. Samples were eluted using a Zorbax-300SB-C8 column under gradient flow (water, 0.1% formic acid and acetonitrile, 0.1% formic acid).
VCC-1 crystallization and structure determination.
VCC-1 crystals were grown at 20°C in 0.7 M NaCl, 0.2 M bis-Tris, pH 5.5, and 22% polyethylene glycol 3350 at a concentration of 12 to 18 mg/ml using the hanging-drop vapor diffusion method, where equal volumes of crystallization buffer and protein were used. Crystals were cryoprotected in crystallization buffer supplemented with 25% glycerol prior to flash cooling to 100 K for X-ray data collection. X-ray data for native VCC-1 were collected from a single crystal (held at 100 K in an N2 gas stream) at beamline 08ID-1 (Canadian Light Source, Saskatoon, Canada).
Crystals of native VCC-1 (monoclinic) dissolved when soaked with avibactam. However, a tetragonal crystal form of the enzyme was found to grow under the same conditions as the monoclinic VCC-1 approximately 3 weeks after the monoclinic crystals had appeared. Fortunately, the tetragonal VCC-1 crystals withstood soaking with avibactam. Tetragonal VCC-1 crystals selected for avibactam soaking had grown at a protein concentration of 10 mg/ml in the presence of an identical buffer which also included 5% glycerol. The crystals were soaked with avibactam at a final concentration of 1 mM for 24 h to obtain the protein-inhibitor complex. Cryoprotection was identical to that of the native protein. Data for the VCC-1–avibactam complex were collected using a Rigaku R-AXIS IV++ detector and a 007HF Microfocus X-ray generator at the University of Manitoba. X-ray data were indexed using the Mosflm program (52) and were then scaled and averaged using the Aimless program (CCP4 suite) (53). The structures of native and avibactam-bound VCC-1 were determined by molecular replacement using the PHASER program (from within the PHENIX package [54]) and the class A carbapenemase NMC-A from E. cloacae (PDB accession number 1BUE) as a search model (from which solvent had been removed). Models of avibactam and its desulfated hydroxylamine form were made using the Jligand program (CCP4 suite) (55), and restraints were generated using the PHENIX eLBOW program (54). Avibactam and its desulfated derivative were manually fit into its ascribed electron densities using the Coot program (24), and geometrical restraints for the covalent carbamyl linkage between avibactam and the catalytic serine of VCC-1 were generated using the PHENIX program (54). Iterative refinement of native and avibactam-bound VCC-1 was carried out using the PHENIX.REFINE (54) and Coot (56) programs. Crystallographic and refinement statistics are presented in Table S1 in the supplemental material. The amino acid residues in the figures with the VCC-1 structure are numbered to be consistent with the Ambler system for class A β-lactamases (26). Determination of root mean square deviations (RMSD) between structurally conserved Cα carbon atoms was carried out using the Dali server (23).
Accession number(s).
The atomic coordinates and structure factors for native VCC-1 and VCC-1 bound to avibactam have been deposited in the Protein Data Bank (http://wwpdb.org/) under accession numbers 6MK6 and 6MKQ, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by Canadian Institutes of Health research grant (CIHR) PJT-148496 to B.L.M. and D.J.V.
C.S.M., G.V., and B.L.M. contributed to the writing of the manuscript. MICs were determined by V.L. Enzyme kinetics were performed by C.S.M. Crystallographic data were generated by G.V., V.L., and B.L.M., and mass spectrometry was performed by V.H. and D.J.V.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02112-18.
REFERENCES
- 1.Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. 2011. Carbapenems: past, present, and future. Antimicrob Agents Chemother 55:4943–4960. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush K. 2013. Carbapenemases: partners in crime. J Glob Antimicrob Resist 1:7–16. doi: 10.1016/j.jgar.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Nordmann P, Poirel L. 2014. The difficult-to-control spread of carbapenemase producers among Enterobacteriaceae worldwide. Clin Microbiol Infect 20:821–830. doi: 10.1111/1469-0691.12719. [DOI] [PubMed] [Google Scholar]
- 4.Jean SS, Lee WS, Lam C, Hsu CW, Chen RJ, Hsueh PR. 2015. Carbapenemase-producing Gram-negative bacteria: current epidemics, antimicrobial susceptibility and treatment options. Future Microbiol 10:407–425. doi: 10.2217/fmb.14.135. [DOI] [PubMed] [Google Scholar]
- 5.Bush K. 2013. The ABCD's of beta-lactamase nomenclature. J Infect Chemother 19:549–559. doi: 10.1007/s10156-013-0640-7. [DOI] [PubMed] [Google Scholar]
- 6.Palzkill T. 2013. Metallo-beta-lactamase structure and function. Ann N Y Acad Sci 1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drawz SM, Bonomo RA. 2010. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordmann P, Cuzon G, Naas T. 2009. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis 9:228–236. doi: 10.1016/S1473-3099(09)70054-4. [DOI] [PubMed] [Google Scholar]
- 9.Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, Bonomo RA. 2010. Inhibitor resistance in the KPC-2 beta-lactamase, a preeminent property of this class A beta-lactamase. Antimicrob Agents Chemother 54:890–897. doi: 10.1128/AAC.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonseca F, Sarmento AC, Henriques I, Samyn B, van Beeumen J, Domingues P, Domingues MR, Saavedra MJ, Correia A. 2007. Biochemical characterization of SFC-1, a class A carbapenem-hydrolyzing beta-lactamase. Antimicrob Agents Chemother 51:4512–4514. doi: 10.1128/AAC.00491-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naas T, Dortet L, Iorga BI. 2016. Structural and functional aspects of class A carbapenemases. Curr Drug Targets 17:1006–1028. doi: 10.2174/1389450117666160310144501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dortet L, Poirel L, Abbas S, Oueslati S, Nordmann P. 2015. Genetic and biochemical characterization of FRI-1, a carbapenem-hydrolyzing class A beta-lactamase from Enterobacter cloacae. Antimicrob Agents Chemother 59:7420–7425. doi: 10.1128/AAC.01636-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girlich D, Poirel L, Nordmann P. 2010. Novel Ambler class A carbapenem-hydrolyzing beta-lactamase from a Pseudomonas fluorescens isolate from the Seine River, Paris, France. Antimicrob Agents Chemother 54:328–332. doi: 10.1128/AAC.00961-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papp-Wallace KM, Taracila MA, Gatta JA, Ohuchi N, Bonomo RA, Nukaga M. 2013. Insights into beta-lactamases from Burkholderia species, two phylogenetically related yet distinct resistance determinants. J Biol Chem 288:19090–19102. doi: 10.1074/jbc.M113.458315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Docquier JD, Mangani S. 2018. An update on beta-lactamase inhibitor discovery and development. Drug Resist Updat 36:13–29. doi: 10.1016/j.drup.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Bonnefoy A, Dupuis-Hamelin C, Steier V, Delachaume C, Seys C, Stachyra T, Fairley M, Guitton M, Lampilas M. 2004. In vitro activity of AVE1330A, an innovative broad-spectrum non-beta-lactam beta-lactamase inhibitor. J Antimicrob Chemother 54:410–417. doi: 10.1093/jac/dkh358. [DOI] [PubMed] [Google Scholar]
- 17.Wang DY, Abboud MI, Markoulides MS, Brem J, Schofield CJ. 2016. The road to avibactam: the first clinically useful non-beta-lactam working somewhat like a beta-lactam. Future Med Chem 8:1063–1084. doi: 10.4155/fmc-2016-0078. [DOI] [PubMed] [Google Scholar]
- 18.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-beta-lactam beta-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D beta-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangat CS, Boyd D, Janecko N, Martz S-L, Desruisseau A, Carpenter M, Reid-Smith RJ, Mulvey MR. 2016. Characterization of VCC-1, a novel Ambler class A carbapenemase from Vibrio cholerae isolated from imported retail shrimp sold in Canada. Antimicrob Agents Chemother 60:1819–1825. doi: 10.1128/AAC.02812-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammerl JA, Jackel C, Bortolaia V, Schwartz K, Bier N, Hendriksen RS, Guerra B, Strauch E. 2017. Carbapenemase VCC-1-producing Vibrio cholerae in coastal waters of Germany. Emerg Infect Dis 23:1735–1737. doi: 10.3201/eid2310.161625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swarén P, Maveyraud L, Raquet X, Cabantous S, Duez C, Pédelacq J-D, Mariotte-Boyer S, Mourey L, Labia R, Nicolas-Chanoine M-H, Nordmann P, Frère J-M, Samama J-P. 1998. X-ray analysis of the NMC-A β-lactamase at 1.64-Å resolution, a class A carbapenemase with broad substrate specificity. J Biol Chem 273:26714–26721. doi: 10.1074/jbc.273.41.26714. [DOI] [PubMed] [Google Scholar]
- 23.Holm L, Rosenström P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. 1992. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 Å resolution. Nature 359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 25.Lamotte-Brasseur J, Knox J, Kelly JA, Charlier P, Fonzé E, Dideberg O, Frére JM. 1994. The structures and catalytic mechanisms of active-site serine β-lactamases. Biotechnol Genet Eng Rev 12:189–230. doi: 10.1080/02648725.1994.10647912. [DOI] [PubMed] [Google Scholar]
- 26.Ambler RP, Coulson AF, Frère JM, Ghuysen JM, Joris B, Forsman M, Levesque RC, Tiraby G, Waley SG. 1991. Biochem J 276:269–270. doi: 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levitt PS, Papp-Wallace KM, Taracila MA, Hujer AM, Winkler ML, Smith KM, Xu Y, Harris ME, Bonomo RA. 2012. Exploring the role of a conserved class A residue in the omega-loop of KPC-2 beta-lactamase: a mechanism for ceftazidime hydrolysis. J Biol Chem 287:31783–31793. doi: 10.1074/jbc.M112.348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adachi H, Ohta T, Matsuzawa H. 1991. Site-directed mutants, at position 166, of RTEM-1 beta-lactamase that form a stable acyl-enzyme intermediate with penicillin. J Biol Chem 266:3186–3191. [PubMed] [Google Scholar]
- 29.Herzberg O, Moult J. 1987. Bacterial resistance to beta-lactam antibiotics: crystal structure of beta-lactamase from Staphylococcus aureus PC1 at 2.5 Å resolution. Science 236:694–701. doi: 10.1126/science.3107125. [DOI] [PubMed] [Google Scholar]
- 30.Minasov G, Wang X, Shoichet BK. 2002. An ultrahigh resolution structure of TEM-1 beta-lactamase suggests a role for Glu166 as the general base in acylation. J Am Chem Soc 124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 31.Damblon C, Raquet X, Lian LY, Lamotte-Brasseur J, Fonze E, Charlier P, Roberts GC, Frère JM. 1996. The catalytic mechanism of beta-lactamases: NMR titration of an active-site lysine residue of the TEM-1 enzyme. Proc Natl Acad Sci U S A 93:1747–1752. doi: 10.1073/pnas.93.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. 2005. Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc 127:15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 33.Guillaume G, Vanhove M, Lamotte-Brasseur J, Ledent P, Jamin M, Joris B, Frère J-M. 1997. Site-directed mutagenesis of glutamate 166 in two β-lactamases: kinetic and molecular modeling studies. J Biol Chem 272:5438–5444. doi: 10.1074/jbc.272.9.5438. [DOI] [PubMed] [Google Scholar]
- 34.Fonseca F, Chudyk EI, van der Kamp MW, Correia A, Mulholland AJ, Spencer J. 2012. The basis for carbapenem hydrolysis by class A beta-lactamases: a combined investigation using crystallography and simulations. J Am Chem Soc 134:18275–18285. doi: 10.1021/ja304460j. [DOI] [PubMed] [Google Scholar]
- 35.Smith CA, Frase H, Toth M, Kumarasiri M, Wiafe K, Munoz J, Mobashery S, Vakulenko SB. 2012. Structural basis for progression toward the carbapenemase activity in the GES family of beta-lactamases. J Am Chem Soc 134:19512–19515. doi: 10.1021/ja308197j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CA, Nossoni Z, Toth M, Stewart NK, Frase H, Vakulenko SB. 2016. Role of the conserved disulfide bridge in class A carbapenemases. J Biol Chem 291:22196–22206. doi: 10.1074/jbc.M116.749648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papp-Wallace KM, Taracila M, Hornick JM, Hujer AM, Hujer KM, Distler AM, Endimiani A, Bonomo RA. 2010. Substrate selectivity and a novel role in inhibitor discrimination by residue 237 in the KPC-2 beta-lactamase. Antimicrob Agents Chemother 54:2867–2877. doi: 10.1128/AAC.00197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin W, Wachino JI, Yamaguchi Y, Kimura K, Kumar A, Yamada M, Morinaka A, Sakamaki Y, Yonezawa M, Kurosaki H, Arakawa Y. 2017. Structural insights into the TLA-3 extended-spectrum beta-lactamase and its inhibition by avibactam and OP0595. Antimicrob Agents Chemother 61:e00501-17. doi: 10.1128/AAC.00501-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvopina K, Hinchliffe P, Brem J, Heesom KJ, Johnson S, Cain R, Lohans CT, Fishwick CWG, Schofield CJ, Spencer J, Avison MB. 2017. Structural/mechanistic insights into the efficacy of nonclassical beta-lactamase inhibitors against extensively drug resistant Stenotrophomonas maltophilia clinical isolates. Mol Microbiol 106:492–504. doi: 10.1111/mmi.13831. [DOI] [PubMed] [Google Scholar]
- 40.Krishnan NP, Nguyen NQ, Papp-Wallace KM, Bonomo RA, van den Akker F. 2015. Inhibition of Klebsiella β-lactamases (SHV-1 and KPC-2) by avibactam: a structural study. PLoS One 10:e0136813. doi: 10.1371/journal.pone.0136813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier JD. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC beta-lactamases. Antimicrob Agents Chemother 57:2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King DT, King AM, Lal SM, Wright GD, Strynadka NCJ. 2015. Molecular mechanism of avibactam-mediated β-lactamase inhibition. ACS Infect Dis 1:175–184. doi: 10.1021/acsinfecdis.5b00007. [DOI] [PubMed] [Google Scholar]
- 43.Xu H, Hazra S, Blanchard JS. 2012. NXL104 irreversibly inhibits the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 51:4551–4557. doi: 10.1021/bi300508r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papp-Wallace KM, Winkler ML, Taracila MA, Bonomo RA. 2015. Variants of beta-lactamase KPC-2 that are resistant to inhibition by avibactam. Antimicrob Agents Chemother 59:3710–3717. doi: 10.1128/AAC.04406-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papp-Wallace KM, Taracila M, Wallace CJ, Hujer KM, Bethel CR, Hornick JM, Bonomo RA. 2010. Elucidating the role of Trp105 in the KPC-2 beta-lactamase. Protein Sci 19:1714–1727. doi: 10.1002/pro.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walther-Rasmussen J, Høiby N. 2007. Class A carbapenemases. J Antimicrob Chemother 60:470–482. doi: 10.1093/jac/dkm226. [DOI] [PubMed] [Google Scholar]
- 47.Costa VM, McGrann KM, Hughes DW, Wright GD. 2006. Sampling the antibiotic resistome. Science 311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 48.Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. 2010. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 8:251–259. doi: 10.1038/nrmicro2312. [DOI] [PubMed] [Google Scholar]
- 49.Clinical and Laboratory Standards Institute. 2017. Performance standards for antimicrobial susceptibility testing, 27th ed CLSI supplement M100. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 50.Copeland RA, Basavapathruni A, Moyer M, Scott MP. 2011. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal Biochem 416:206–210. doi: 10.1016/j.ab.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 51.Morrison JF, Walsh CT. 1988. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Rel Areas Mol Biol 61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 52.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans PR, Murshudov GN. 2013. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. 2012. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr 68:431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.