

AIDS Clinical Trial Group study A5202 (ClinicalTrials.gov identifier NCT00118898) was a phase 3b, randomized, partially blinded equivalence study of open-label atazanavir/ritonavir or efavirenz, plus either placebo-controlled tenofovir disoproxil fumarate/emtricitabine or abacavir/lamivudine, in treatment-naive adults living with HIV-1, evaluating efficacy, safety, and tolerability. We report an analysis of the contribution of participant characteristics to the disposition of tenofovir plasma concentrations.
KEYWORDS: pharmacokinetics, population pharmacokinetics, tenofovir
ABSTRACT
AIDS Clinical Trial Group study A5202 (ClinicalTrials.gov identifier NCT00118898) was a phase 3b, randomized, partially blinded equivalence study of open-label atazanavir/ritonavir or efavirenz, plus either placebo-controlled tenofovir disoproxil fumarate/emtricitabine or abacavir/lamivudine, in treatment-naive adults living with HIV-1, evaluating efficacy, safety, and tolerability. We report an analysis of the contribution of participant characteristics to the disposition of tenofovir plasma concentrations. Tenofovir concentration data from a total of 817 individuals (88% of the total number of eligible patients randomly assigned to receive treatment in the TDF-containing arms of A5202) were available for analysis. Pharmacokinetic analysis was performed using nonlinear mixed-effects modeling. One- and two-compartment models with first-order absorption and first-order elimination were evaluated. An exponential error model was used for examination of interindividual variability (IIV), and a proportional and mixed-error model was assessed for residual variability. The final structural model contained two compartments with first-order absorption and elimination. IIV was estimated for apparent clearance (CL/F) and the first-order absorption rate constant (ka), and a proportional residual variability model was selected. The final mean parameter estimates were as follows: ka = 2.87 h−1, CL/F = 37.2 liters/h, apparent volumes of the central and peripheral compartments = 127 and 646 liters, respectively, and apparent intercompartmental clearance = 107 liters/h. In addition to race/ethnicity, creatinine clearance and assignment to atazanavir/ritonavir or efavirenz were significantly associated with CL/F (P < 0.001). In conclusion, race/ethnicity is associated with tenofovir oral CL in HIV-1 positive, treatment-naive adults. This covariate relationship raises questions about the possibility of differences in efficacy and risk of adverse events in different patient populations and suggests that examining preexposure prophylaxis regimens and tenofovir exposure in different race/ethnicity groups be considered.
INTRODUCTION
Tenofovir disoproxil fumarate (TDF) is an adenosine-analog nucleotide reverse transcriptase inhibitor (NRTI) prodrug that exhibits activity against HIV-1 and hepatitis B virus infections. TDF is converted to its active intracellular form, tenofovir diphosphate, in a stepwise manner: (i) TDF is converted to tenofovir (TFV) in the intestinal lumen and plasma by diester hydrolysis, and (ii) TFV is internalized intracellularly and subsequently phosphorylated into tenofovir monophosphate and then to its active metabolite, tenofovir diphosphate (1, 2). Tenofovir has been examined in those living with HIV and healthy volunteers and has demonstrated that renal function and concomitant use with ritonavir-boosted HIV-1 protease inhibitors affect plasma TFV concentrations (3–6).
The AIDS Clinical Trials Group study A5202 (ClinicalTrials.gov identifier NCT00118898) randomly assigned 1,857 treatment-naive HIV-1-infected adults to one of the following once-daily regimens: open-label atazanavir and ritonavir (ATV/r; 300/100 mg) or efavirenz (EFV; 600 mg), plus either placebo-controlled tenofovir disoproxil fumarate/emtricitabine (TDF/FTC; 300/200 mg) or abacavir/lamivudine (ABC/3TC; 600/300 mg). Plasma concentration data for each of the antiretrovirals were collected from the majority of study participants for further investigation of differences in the pharmacokinetics of subpopulations. Included in the prespecified pharmacology-related secondary objectives was the exploration of the association of ethnicity and other host factors with the disposition of antiretroviral (ARV) agents. It is critical to determine whether clinically relevant differences in ARV drug exposure exist in potential patient subpopulations, since variability in drug exposure may be associated with differences in ARV drug toxicity and virologic response. In this report, we investigated TFV pharmacokinetics using sparse sampling and population pharmacokinetic modeling to conduct a covariate analysis in exploration of significant host factors that may play a role in the time course of TFV exposure.
RESULTS
Pharmacokinetic data from a total of 817 individuals (88%) were available for analysis. The majority of participants had 3 samples drawn (range, 1 to 5), with a total of 2,166 evaluable plasma concentrations within approximately 6 months of beginning antiretroviral treatment. Information on the previous 3 days of TDF dosing prior to pharmacokinetic sampling was available for 98% of the 2,166 evaluable plasma concentrations, in which 42 and 18 sample collections reported a 2-day difference between the dose prior to sampling and the second dose prior to sampling and between the second and third dose prior to sampling, respectively. One participant reported a 3-day window between the dose prior to sampling and the second dose prior to sampling.
Population pharmacokinetic model.
A two-compartment model with first-order absorption and elimination was ultimately the most appropriate choice to describe the data. A mixture model identified a subpopulation with large residual variability (n = 30), and a total of 78 observations were removed from the data set for model building. The demographics of the remaining participants (n = 787) used in the pharmacokinetic analysis are given in Table 1. The final model included interindividual variability (IIV) terms on apparent clearance (CL/F) and the first-order absorption rate constant (ka), and a proportional model was selected for residual variability. The base model was able to accommodate IIV on ka in addition to CL/F, but the only covariate that was biologically plausible to affect the absorption rate constant (i.e., age) was not significant. Whereas IIV on ka and CL/F during forward selection would not allow estimation of race/ethnicity as a covariate on CL/F with 1 or 2 degrees of freedom, forward selection was continued with IIV on CL/F only, and IIV on ka was added during the multivariable evaluation. This allowed for greater precision of parameter estimates, clinically valuable covariate information to be included in the model, and retention of IIV on ka. In the final model, the IIV variability on CL/F and ka were 18% and 85% (% coefficient of variation [% CV]), with information on covariate model development summarized in Table 2. The eta distribution associated with the absorption rate constant had moderate asymmetrical shrinkage (57%) due to missing data in the absorption phase.
TABLE 1.
Demographics of study participants
| Demographic | Value (n = 787)a |
|---|---|
| Treatment arm (3rd drug), no. (%) of participants | |
| ATV/r | 387 (49) |
| EFV | 400d (51) |
| Sex, no. (%) of participants | |
| Female | 127 (16) |
| Male | 660d (84) |
| Race/ethnicity, no. (%) of participants | |
| White (non-Hispanic) | 339d (43) |
| Black (non-Hispanic) | 236 (30) |
| Hispanic | 190 (24) |
| Asian | 14 (2) |
| American Indian/Alaskan | 3 (0.4) |
| Multiracial | 5 (1) |
| Age (yr), median (Q1, Q3)b | 39 (31, 45) |
| Wt (kg), median (Q1, Q3)c | 77.3 (68.2, 88.8) |
| CLCR (ml/min), median (Q1, Q3)c | 113.7 (97.4, 133.6) |
Percentages are rounded to the nearest whole number; Q1 and Q3 represent the 1st and 3rd quartiles, respectively.
Three subjects had no age reported, so the mean was used (38.9 years).
One subject had no CLCR or weight data, so the means were used (117.9 ml/min and 79.5 kg, respectively).
Four subjects had no race/ethnicity reported and three subjects had no sex or third drug reported, so the mode was used for all three categorical covariates.
TABLE 2.
Pharmacokinetic model developmenta
| Phase of development | MVOF | IIV CL (% CV) | IIV ka (% CV) | CCV RV |
|---|---|---|---|---|
| Base | 17,173 | 23.9 | 0.0697 | |
| R1 FS—CLCR | 17,021 | 20.8 | 0.0703 | |
| R2 FS—CLCR/3rd drug | 16,887 | 18.5 | 0.0701 | |
| R3 FS—CLCR/3rd drug/race or ethnicity | 16,860 | 18.1 | 0.07 | |
| Multivariable—IIV ka | 16,839 | 17.7 | 84.5 | 0.0645 |
| R1 BE | All covariates significant on CL, with addition of IIV on ka |
MVOF, minimum value of the objective function; IIV, interindividual variability; CL, apparent tenofovir plasma clearance; ka, absorption rate constant; % CV, % coefficient of variation; CCV RV, constant coefficient of variation, residual variability; R1, R2, and R3, round of covariate analysis; FS, forward selection; CLCR, creatinine clearance; 3rd drug, treatment arm (ATV/r versus EFV); BE, backwards elimination.
The final significant covariates that were identified were creatinine clearance (power model), treatment arm (additive shift; ATV/r as the reference), and race/ethnicity (additive shift; 2 degrees of freedom; white non-Hispanic race as the reference) on apparent clearance (Table 3). The final covariate relationship with apparent clearance was described as follows:
with CL as the apparent total drug clearance, θTVCL as the estimated typical value of clearance, CLCR/113.5 as the creatinine clearance centered to a value of 113.5 ml/min, θCLCR as an estimated factor for CLCR, θ3rd drug as the estimated shift in clearance for those in the efavirenz arm compared to those in the atazanavir/r arm, RACB as an indicator variable for black non-Hispanic race/ethnicity compared to white non-Hispanic race/ethnicity, and RACO as the indicator variable for the combination of Hispanic, Asian, American Indian, Alaskan, and multiracial compared to white non-Hispanic.
TABLE 3.
Final model parameter estimates for tenofovir pharmacokinetics
| Parametere | Model estimate (95% CI) | % RSEf (% shrinkage) | Bootstrap estimateg (95% CI) |
|---|---|---|---|
| ka (h−1) | 2.87 (1.83, 3.91) | 18.5 | 2.87 (2.27, 4.16) |
| CL/F (liters/h) | 37.2 (34.10, 40.29) | 4.2 | 37.40 (33.57, 40.26) |
| Vc/F (liters) | 127 (83.10, 170.90) | 17.6 | 128.73 (86.26, 188.48) |
| Vp/F (liters) | 646 (555.45, 736.55) | 7.2 | 649.55 (547.62, 737.81 |
| Q/F (liters/h) | 107 (70.94, 143.06) | 17.2 | 108.60 (73.26, 144.98) |
| IIV in ka | 0.714 (0.389, 1.039) | 23 (57)d | 0.817 (0.350, 1.497) |
| IIV in CL | 0.0312 (0.025, 0.037) | 10 (17) | 0.0309 (0.025, 0.038) |
| CLCRa,b | 0.442 (0.375, 0.509) | 8 | 0.445 (0.372, 0.511) |
| Treatment arma,c | 8 (6.47, 9.53) | 10 | 7.93 (6.51, 9.55) |
| Black non-Hispanica,c | 3.17 (1.54, 4.80) | 26 | 3.29 (1.65, 4.90) |
| “Other” race/ethnicitya,c | 4.09 (2.40, 5.78) | 21 | 4.11 (2.53, 5.84) |
| CCV RV | 0.0645 (0.0576, 0.0714) | 5 (16) | 0.0641 (0.0575, 0.0712) |
On CL/F.
Power model.
Additive shift.
Significant ETABAR.
ka, absorption rate constant; CL/F, apparent plasma clearance; Vc/F, apparent volume of distribution in the central compartment. Vp/F, apparent volume of distribution in the peripheral compartment; Q/F, apparent intercompartmental clearance; IIV, interindividual variability; CLCR, creatinine clearance; CCV RV, constant coefficient of variation, residual variability.
RSE, relative standard error.
960/1,000 = 96% runs contributed; 36 runs with minimization terminated were skipped when calculating the bootstrap results, and 4 runs with estimates near a boundary were skipped when calculating the bootstrap results.
The final estimated covariate effects showed that there was an average 0.442 change in the natural log of tenofovir plasma CL/F (liters/h) per ml/min in the ln CLCR compared to the centered value (113.5 ml/min) [e.g., for an individual, ln(CL/F) = ln(37.2 liters/h) + 0.442 · ln(CrCl/113.5 ml/min)]. For the median (113.7 ml/min), first quartile (Q1) (97.4 ml/min), third quartile (Q3) (133.6 ml/min), and lowest CLCR (44.4 ml/min) values in the data set, the resultant oral CL values for these individuals would be 37.2, 34.8, 40.0, and 24.6 liters/h, respectively. Those in the efavirenz treatment arm were found to have an average apparent tenofovir clearance that was 8 liters/h greater than that of participants in the atazanavir/ritonavir arm. In comparison to white non-Hispanic individuals, black non-Hispanic participants and those combined into the “other” group (Hispanic, Asian, American Indian/Alaskan, and multiracial) had average apparent tenofovir clearances that were 3.17 liters/h and 4.09 liters/h greater, respectively. A simulation of population predicted values in male participants with median values for weight, CLCR, and age (with no variability) shows the difference in plasma tenofovir exposure between different race/ethnicity groups assigned to either the ATV/r or EFV arms of study A5202 (Fig. 1). Among both treatment arms, those of the “other” and black non-Hispanic race/ethnicity groups were associated with a faster TFV plasma clearance (and therefore reduced plasma exposure) than that of white non-Hispanic participants. A comparison of plasma exposures between racial/ethnic groups per third drug treatment arm is provided in Fig. 2.
FIG 1.
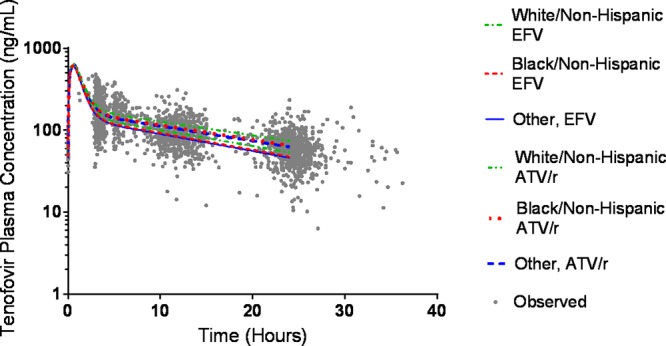
Comparison of predicted TFV concentrations based on race/ethnicity and treatment arm. Simulations are population predictions for males with median values for creatinine clearance, weight, and age. The AUCs (ng · h/ml) were as follows: 2,752 (“other,” EFV), 2,804 (black/non-Hispanic, EFV), 3,001 (white/non-Hispanic, EFV), 3,647 (white/non-Hispanic, ATV/r), 3,361 (black/non-Hispanic, ATV/r), and 3,286 (“other,” ATV/r).
FIG 2.

Comparison of TFV plasma exposures in racial/ethnic groups per third drug. AUC, area under the plasma concentration-time curve; EFV, efavirenz; ATV/r, ritonavir-boosted atazanavir; NG, nanogram; ML, milliliter.
The estimated mean population pharmacokinetic parameters for the final model, with 95% confidence intervals compared to the bootstrap analysis (96% of runs successfully contributed to the bootstrap analysis and met the predefined convergence criteria), are given in Table 3. The prediction-corrected visual predictive check (pcVPC) was generated with model predictions from 2,000 simulations and shows that the observed median (red line) and 5th and 95th percentiles of the data (blue lines) were included within the model-predicted 95% confidence intervals (shaded areas) (Fig. 3). These internal validation techniques show appropriate model specification.
FIG 3.

Prediction-corrected visual predictive check comparing observed and model predicted drug concentrations. Observed concentrations and their 5th, 50th, and 95th percentiles are overlaid on the simulated 95% confidence intervals of the corresponding percentiles.
DISCUSSION
This study has identified CLCR, treatment arm, and race/ethnicity as significant covariates associated with apparent clearance following the recommended TDF dose in treatment-naive adults living with HIV. Tenofovir disoproxil fumarate is recommended as a first-line nucleotide reverse transcriptase inhibitor in national HIV treatment guidelines (7), and its use has increased among non-HIV-infected individuals for preexposure prophylaxis, when used with emtricitabine (8, 9). These findings may provide additional considerations for individualizing TDF-based regimens.
Multiple structural population pharmacokinetic models for plasma tenofovir have been reported as two-compartment models with first-order absorption and elimination (4, 5, 10, 11). Previous population analyses (3–5, 10, 11) have identified plasma tenofovir pharmacokinetic parameters in the following ranges: CL/F, 42 to 66.6 liters/h, apparent volume of distribution in the central compartment (Vc/F), 268 to 1,040 liters; apparent intercompartmental clearance (Q/F), 13.2 to 197 liters/h; apparent volume of distribution in the peripheral compartment (Vp/F), 398 to 1,630 liters; and ka, 0.82 to 1.35 h−1. These values were similar to those estimated in the current analysis: CL/F, 37.2 liters/h; Vc/F, 127 liters; Q/F, 107 liters/h; Vp/F, 646 liters; and ka, 2.87 h−1. The difference noted in the absorption rate constant may be due to differences in population administration of tenofovir in relation to food, as food delays the time to tenofovir maximum concentration in serum (Cmax) (1). Previous pharmacokinetic studies of TFV have also identified renal function and ritonavir-enhanced protease inhibitors as significant covariates in the plasma exposure of TFV (3–5, 10, 11). Our analysis confirms that ATV/r in combination with TDF results in increased TFV plasma concentrations compared to coadministration with EFV. In a study of 12 participants listed in the prescribing information for atazanavir, there was a 37% (90% confidence interval [CI], 1.30 to 1.45) increase in the TFV area under the concentration-time curve (AUC) when coadministered with ATV/r (12).
Study 5202 was a multicenter clinical trial across the United States, including Puerto Rico, which enrolled a large number of participants, including well-represented minority populations. Approximately 23% (n = 429) of the study population were Hispanic, and 33% (n = 615) were black non-Hispanic, allowing for the evaluation of the pharmacokinetics of TFV in these patient populations. Among the three race/ethnicity groups evaluated (white non-Hispanic versus black non-Hispanic and versus “other”), there was a significant difference in TFV oral CL when the white non-Hispanic group was compared to the other two groups. To our knowledge, this is the first report to identify race/ethnicity as a significant covariate for TFV oral CL. The “other” race/ethnicity group in this study consisted predominantly of participants who were Hispanic (190 Hispanic participants out of 212 in this group). Individuals in this race/ethnicity group were associated with a faster TFV oral clearance, compared to white non-Hispanic participants, and therefore reduced plasma exposure (Fig. 1). The question arises if a difference in clearance could affect adverse effects over time. ACTG study A5224s was a substudy of A5202 (n = 269) focused on assessing longer-term changes in metabolic outcomes in participants treated with FTC/TDF or ABC/3TC with either EFV or ATV/r. A5224s compared changes in areas such as limb fat, renal function, glucose, insulin and insulin sensitivity, urine albumin, and urine protein/creatinine ratios between different race/ethnicity groups. However, information comparing these groups within the TDF/FTC arms to the ABC/3TC arms (13–16) was not reported. In the univariate analysis, there was no significant association between race/ethnicity (compared to white non-Hispanic participants) with changes in body mass and bone mineral density adjusted for treatment arm (17).
In the iPrEx trial evaluating oral daily doses of TDF/FTC or placebo as preexposure prophylaxis in HIV-seronegative men or transgender women who have sex with men, a relative risk reduction from contracting HIV of 92% was found in those with detectable drug concentrations (quantification range, 10 to 1,500 ng/ml) in the treatment arm (8). The majority of the iPrEx study population was Hispanic (approximately 72%), with the largest number of participants from Peru; however, a separate analysis of ARV exposures by race/ethnicity has not been reported.
Limitations of the current analysis include unknown reasons for variability of drug concentrations, such as other potential drug-drug interactions, effects of comorbid conditions, or other unmeasured confounding factors. In addition, the observations identified by the residual variability mixture model represent apparent nonadherence, since only 2 of the 78 observations identified by the mixture model reported slight deviations from daily dosing (2 days between the second and third dose prior to pharmacokinetic sampling). Unreported noncompliance may be one possible source of the large residual variability seen within the data.
In conclusion, a population pharmacokinetic model with covariate analysis was developed for ACTG study A5202 to analyze the TFV plasma concentration data available from the diverse participant population. The significant covariates associated with apparent TFV clearance included creatinine clearance, treatment arm, and race/ethnicity. As the number of low- and middle-income countries are moving toward antiretroviral regimens containing dolutegravir (DTG) as the preferred first-line treatment, including the fixed-dose combination of TDF/3TC/DTG (18), the findings of this analysis are timely and relevant. These data suggest that additional research examining preexposure prophylaxis regimens and use for treatment may consider possible TFV exposure differences between race/ethnicity groups.
MATERIALS AND METHODS
Participants and study design.
Study A5202 was a phase 3b, randomized, partially blinded equivalence study of four once-daily ARV regimens in treatment-naive adults (≥16 years of age). Participants were randomized 1:1:1:1 and balanced by study site. At screening, study participants were stratified by HIV-1 RNA level (<100,000 or ≥100,000 copies/ml). Participants in the high-screening viral load stratum (based on data safety monitoring board recommendations) and those with toxicity associated with the NRTI backbone who had virologic failure or hepatitis B were unblinded. The primary efficacy, safety, and tolerability results have been reported (19–21).
A sparse sampling strategy for the measurement of plasma drug concentrations was designed to collect three steady-state samples per participant during the first 24 weeks of therapy, including dosing information on the previous 4 doses prior to sampling. Pharmacokinetic drug concentration sampling occurred at week 4, 8, 16, or 24. Three samples were to be collected over two visits, visits A and B. Visit A could occur before or after visit B, or the visits could be combined if medications were regularly scheduled for the morning. At visit A, two plasma samples were obtained, one before a dose and one 3 to 4 h after an observed dose; at visit B, one sample was to be obtained 5 to 15 h (5 to 12 h if all medications were taken in the morning) after a dose (22–24).
Assurances.
The human subject committees of all sites approved the A5202 protocol, and written informed consent was obtained from all participants in compliance with human experimentation guidelines of the U.S. Department of Health and Human Services.
Tenofovir sample processing.
Plasma concentrations of tenofovir were measured at the ACTG Pharmacology Laboratory at the University of Alabama at Birmingham using a high-performance liquid chromatography with tandem mass spectrometry (HPLC-MS) assay (25). After the addition of stably labeled isotope internal standards (IS), samples were processed using a solid-phase extraction method on 30-mg (1-cm3) Oasis MCX cartridges. The eluate was then evaporated under a nitrogen stream and reconstituted in 175 μl of 0.01% trifluoroacetic acid (TFA) for injection. Using a Shimadzu XR HPLC system, reversed phase chromatographic separation of the drug and the internal standard was performed on an Atlantis dC18 analytical column (2.1 by 100 mm, 3.0 μm) under isocratic conditions. A Supelco C18 in-line guard cartridge was used to protect the column from contaminants. A column temperature of 30°C, a rate of 0.2 ml/min, and an injection volume of 10 μl were used. The binary mobile phase consisted of 0.1% formic acid (A)and 0.1% formic acid (B) in acetonitrile with a composition of 90% A to 10% B. Under these conditions, the retention time for tenofovir and its respective internal standard was approximately 2.12 min. A total run time of 5 min was used to ensure complete elution of all peaks of interest. Detection and quantification were accomplished on an API 5500 mass spectrometer by multireaction monitoring. Tenofovir and the IS were detected using the following transitions for protonated products [M + H]+: m/z 288.2 > 176.1 for tenofovir; m/z 293.2 > 181.0 for tenofovir-IS. Mass spectrometer settings were as follows: an API 5500 instrument was used in positive TubolonSpray mode with a source temperature of 500°C. Nitrogen was used as the nebulizer, auxiliary, collision, and curtain gas. The calibration curve was linear over the range of 5 to 1,000 ng/ml for tenofovir using a 50-μl aliquot of human plasma. Validation of the plasma assay showed precision with less than 8% coefficient of variation between calibration standards on different assay runs and accuracy with less than 6% deviation from known concentrations. Plasma quality controls showed overall precision [(SD/mean) × 100%)] with less than 7% and accuracy [(mean − target)/target × 100%] within 10% CV.
Population pharmacokinetic modeling.
An exploratory pharmacokinetic analysis identified significant differences in TFV oral CL when participant characteristics for A5202 were compared (22). With this previously identified structure as a starting base model, the goal of the current analysis was to identify and quantify sources of variability in TFV exposure, while confirming and refining the exploratory base structural model. Pharmacokinetic analysis of TFV (136-mg dose) was performed using nonlinear mixed-effects modeling with NONMEM (version 7.1.0; ICON Development Solutions, Ellicott City, MD, USA) interfaced with Perl-speaks-NONMEM (PsN; https://uupharmacometrics.github.io/PsN/) through Pirana (version 2.9.4; http://www.pirana-software.com/) as the modeling environment (26, 27). The first-order conditional estimation method with interaction was used with a steady-state assumption. One- and two-compartment models with first-order absorption and first-order elimination were evaluated. An exponential error model with log-normal distribution was used for examination of intersubject variability of all pharmacokinetic parameters, adding to the intersubject variability on CL/F which was already contained in the exploratory model. A proportional and mixed (additive and proportional) error model was assessed for residual variability on one- and two-compartment models. In addition, a mixture model of residual variability was used to detect apparent nonadherence during the model building process (28). Goodness-of-fit plots were used to evaluate the base structural model: observations versus individual and population predictions (IPRED and PRED), conditional weighted residuals versus PRED and time, and absolute individual weighted residuals versus IPRED.
Covariate selection.
A covariate analysis was performed to explore the association of several factors with plasma TFV pharmacokinetic parameters. Race/ethnicity, sex, and third drug treatment arm (ATV/r or EFV) were evaluated as categorical covariates using an additive functional form. Race/ethnicity groups available for model building included white non-Hispanic, black non-Hispanic, Hispanic, Asian, American Indian/Alaskan, and multiracial (although the sample size was small for the last three groups). Each group was evaluated individually and combined in stepwise increments starting with those groups with the smallest number of participants. Continuous covariates, which included age at study entry, creatinine clearance (CLCR; calculated by the Cockcroft-Gault equation) (29), and weight in kilograms, were centered and evaluated using power and linear functional forms. Weight and CLCR measurements were obtained at the same time or in close proximity to the time points for tenofovir sampling. Significant covariates were selected using the classical stepwise approach (30). Forward selection was conducted in a univariate manner, with covariate inclusion into the model being determined by a reduction in minimum value of the objective function (MVOF) by >3.84 (P < 0.05; 1 degree of freedom, in a χ2 distribution) and by a reduction in the interindividual variability (IIV) of the pharmacokinetic parameter of interest. For the backward elimination step, covariates were removed separately and considered significant if their removal resulted in an increase in the MVOF of at least 10.83 (α < 0.001; 1 degree of freedom, in a χ2 distribution). Time-varying covariates (CLCR and weight) were included in the data set if CLCR changed to a different stage of renal function (table can be found in the Food and Drug Administration’s “Guidance for Industry on Pharmacokinetics in Patients with Impaired Renal Function” [31]) and weight if it changed by >20 lb. Between forward selection and backward elimination, multivariable model evaluation occurred by exploring the addition of interindividual variability on additional parameters and examination of the residual variability model by precision.
Model evaluation.
The final model was evaluated using 2,000 simulations in a prediction-corrected visual predictive check (pcVPC). Observed concentrations and their 5th, 50th, and 95th percentiles were overlaid on the 95% confidence interval of the corresponding percentiles of the simulations for visual assessment. Prediction correction was used to take into account the differences within a bin coming from the included covariates (32). All the observations and predictions were then normalized using the typical population prediction in relation to the median of typical population predictions per bin across the covariates (32). Nonparametric bootstrap resampling was also performed to evaluate the model (33). One thousand bootstrap data sets were generated to obtain a median and 95% confidence interval for each model parameter for comparison with the original final parameter estimates, with an a priori convergence criterion set at 90% when modeling the bootstrap data sets for evaluation of the convergence rate.
ACKNOWLEDGMENTS
We thank the study participants for their generous donation of time and effort in the successful completion of clinical trial A5202. We also thank Olivia Campagne (University at Buffalo, SUNY) for advice on population modeling and Katie Mollan (University of North Carolina, UNC) for advice and statistical contributions to ACTG A5202. We also acknowledge contributions, either financial and/or drug related, made by GSK/ViiV Healthcare, Gilead, Bristol-Myers Squibb, and Abbott (AbbVie) to the parent study (A5202) and/or substudies of A5202.
Other investigators and contributors included the following: Hector H. Bolivar and Sandra Navarro, University of Miami (site 901), CTU grant AI069477, ACTG grant AI27675, and CFAR grant AI073961; Susan L. Koletar and Diane Gochnour, Ohio State University (site 2301), CTU grant AI069474; Edward Seefried and Julie Hoffman, University of California, San Diego (site 701), CTU grant AI69432; Judith Feinberg and Michelle Saemann, University of Cincinnati (site 2401), CTU grant AI069513; Kristine Patterson, Donna Pittard, and David Currin, University of North Carolina (site 3201), CTU grant AI69423, CFAR grant AI50410, GCRC grant RR00046, and grant RR025747; Kerry Upton and Michael Saag, University of Alabama (site 5801), CTU grant U01 AI069452, CCTS grant 1UL1 RR025777-01; Graham Ray and Steven Johnson, University of Colorado Health Sciences Center (site 6101), CTU grant AI69450, grant AI054907, and grant RR00051; Bartolo Santos and Connie A. Funk, University of Southern California (site 1201), CTU grant 5U01 AI069428; Michael Morgan and Brenda Jackson, Vanderbilt Therapeutics CRS (site 3652), CTU grant AI069439; Pablo Tebas and Aleshia Thomas, University of Pennsylvania, subunit of Children’s Hospital of Philadelphia (site 6201), CTU grant U01 AI069467-03, CFAR grant 5P30 AI045008-10; Ge-Youl Kim and Michael K. Klebert, Washington University (site 2101), CTU grant AI069495; Jorge L. Santana and Santiago Marrero, University of Puerto Rico (site 5401), CTU grant 5U01 AI069415-03; Jane Norris and Sandra Valle, Stanford University (site 501), CTU grant AI69556; Gary Matthew Cox and Martha Silberman, Duke University Medical Center (site 1601), CTU grant 5U01 AI069484-02; Sadia Shaik and Ruben Lopez, Harbor-University of California, Los Angeles Medical Center (site 603), CTU grant AI069424; Margie Vasquez and Demetre Daskalakis, New York University/NYC HHC at Bellevue Hospital Center (site 401), CTU grant AI069532; Christina Megill and Todd Stroberg, Cornell Chelsea (site 7804), CTU grant AI69419, CSTC grant RR024996; Jessica Shore and Babafemi Taiwo, Northwestern University CRS (site 2701), CTU grant AI069471; Mitchell Goldman and Molly Boston, Indiana University (site 2601), CTU grant UO1 AI025859; Jeffrey Lennox and Carlos del Rio, Ponce de Leon Center (A5802), CTU grant 5U01 AI069418, CFAR grant P30AI050409; Timothy W. Lane and Kim Epperson, Moses H. Cone Memorial Hospital (site 3203), CTU grant 1U01 A1069423-01; Annie Luetkemeyer and Mary Payne, University of California, San Francisco (site 801), CTU grant 1U01 AI069502-01; Barbara Gripshover and Dawn Antosh, Case Western Reserve University (site 2501), CTU grant AI69501; Jane Reid and Mary Adams, University of Rochester (site 1101), CTU grant U01 AI069511, GCRC grant UL1 RR024160; Sheryl S. Storey and Shelia B. Dunaway, University of Washington (site 1401), CTU grant AI069434; Joel Gallant and Ilene Wiggins, Johns Hopkins University (site 201), CTU grant AI69465; Kimberly Y. Smith and Joan A. Swiatek, Rush University Medical Center (site 2702), CTU grant 5U01 AI069471; Joseph Timpone and Princy Kumar, Georgetown University (site 1008), CTU grant 1U01 AI069494-01; Ardis Moe and Maria Palmer, University of California, Los Angeles Care Center (site 601), CTU grant AI069424; Jon Gothing and Joanne Delaney, Brigham and Women’s Hospital, Boston, MA (site 107), CTU grant AI069472; Kim Whitely and Ann Marie Anderson, Metro Health Center (site 2503), CTU grant AI069501; Scott M. Hammer and Michael T. Yin, HIV Prevention & Treatment (Columbia University) (site 30329), CTU grant 5U01 AI069470, grant 1UL1 RR024156; Mamta Jain and Tianna Petersen, UT Southwestern Medical Center at Dallas (site 3751), CTU grant 3U01 AI046376 05S4; Roberto Corales and Christine Hurley, AIDS Community Health Center (site 1108), CTU grant U01AI069511, GCRC grant UL1 RR024160; Keith Henry and Bette Bordenave, Hennepin County Medical Center (site 1502), grant N01 AI72626; Amanda Youmans and Mary Albrecht, Beth Israel Deaconess (Partners/Harvard) CRS (site 103), CTU grant UOI A106947203; Richard B. Pollard and Abimbola Olusanya, University of California, Davis Medical Center (site 3851), grant AI38858; Paul R. Skolnik and Betsy Adams, Boston Medical Center CRS (site 104), CTU grant AI069472; Karen T. Tashima and Helen Patterson, Miriam Hospital-Brown University (Partners/Harvard) (site 2951), CTU grant 1U01 AI069472-01; Michelle Ukwu and Lauren Rogers, Peabody Health Center (site 31443), CTU grant AI069471; Henry H. Balfour, Jr., and Kathy A. Fox, University of Minnesota (site 1501), CTU grant AI27661; Susan Swindells and Frances Van Meter, University of Nebraska Medical Center (site 1505), CTU grant AI27661; University of Hawaii (site 5201), CTU grant AI34853; Gregory Robbins and Nicole Burgett-Yandow, Massachusetts General Hospital from the Partners/Harvard/BMC ACTU (site 101), CTU grant 1U01 AI069472-01; Charles E. Davis, Jr., and Colleen Boyce, IHV Baltimore Treatment CRS (site 4651), CTU grant 5U01 AI069447 03; William A. O’Brien and Gerianne Casey, University of Texas Medical Branch (site 6301), CTU grant AI032782; Gene D. Morse and Chiu-Bin Hsaio, SUNY-Buffalo (site 1102), CTU grant 5U01 A1027658; San Mateo County AIDS Program (site 505), CTU grant AI27666; Jeffrey L. Meier and Jack T. Stapleton, University of Iowa Healthcare (site 1504), NIAID grant AI27661, grant AI58740; Donna Mildvan and Manuel Revuelta, Beth Israel Medical Center ACTU (site 2851), CTU grant AI46370; David Currin, Wake County HHS (site 30076), CTU grant AI25868; Wafaa El Sadr and Avelino Loquere, Harlem ACTG CRS (site 31483), CTU grant 5U01 AI069470-03; Nyef El-Daher and Tina Johnson, McCree McCuller Wellness Center (site 1107), CTU grant U01 AI069511, GCRC grant UL1 RR024160; Robert Gross and Kathyrn Maffei, University of Pennsylvania Health (site 6206), CTU grant 1U01 AI69467-01; Valery Hughes and Glenn Sturge, Cornell Uptown (site 7803), CTU grant 1U01 AI069419-01; Deborah McMahon and Barbara Rutecki, University of Pittsburgh (site 1001), CTU grant 1UO1 AI069494-01; Michael Wulfsohn, Andrew Cheng, and Norbert Bischofberger, Gilead Sciences; and Lynn Dix and Qiming Liao, GlaxoSmithKline, Inc.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers UM1 AI068634, UM1 AI068636, UM1 AI069424, UM1 AI069511, and UM1 AI106701. Research was supported in part by the University of Rochester Center for AIDS Research under award number P30 AI078498, the University of Washington/Fred Hutchinson Cancer Research Center for AIDS Research under award number UM1 AI27757, the University of California San Francisco Pharmacology Core under award number P30 AI022763, the University of North Carolina Center for AIDS Research under award number P30 AI050410, and the University at Buffalo Pharmacology Specialty Laboratory and the University of Alabama Pharmacology Specialty Laboratory under award number UM1 AI106701. C. S. Venuto and Q. Ma were supported in part by grants K23AI108355 and K08MH098794, respectively.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
E.S.D.’s institution receives grant support from Gilead, Merck, and ViiV, and E.S.D. is a consultant for Bristol Myers Squibb, Gilead, Merck, Janssen, Teva, and ViiV. A.C.C. is a member of a Data and Safety Monitoring Board for a Merck-sponsored study, and her institution has received grant support from Bristol-Myers-Squibb. K.Y.S. is an employee of ViiV Healthcare, beginning December 2013. All other authors have no conflict of interest to declare.
Contributor Information
Collaborators: Hector H. Bolivar, Sandra Navarro, Susan L. Koletar, Diane Gochnour, Edward Seefried, Julie Hoffman, Judith Feinberg, Michelle Saemann, Kristine Patterson, Donna Pittard, David Currin, Kerry Upton, Michael Saag, Graham Ray, Steven Johnson, Bartolo Santos, Connie A. Funk, Michael Morgan, Brenda Jackson, Pablo Tebas, Aleshia Thomas, Ge-Youl Kim, Michael K. Klebert, Jorge L. Santana, Santiago Marrero, Jane Norris, Sandra Valle, Gary Matthew Cox, Martha Silberman, Sadia Shaik, Ruben Lopez, Margie Vasquez, Demetre Daskalakis, Christina Megill, Todd Stroberg, Jessica Shore, Babafemi Taiwo, Mitchell Goldman, Molly Boston, Jeffrey Lennox, Carlos del Rio, Timothy W. Lane, Kim Epperson, Annie Luetkemeyer, Mary Payne, Barbara Gripshover, Dawn Antosh, Jane Reid, Mary Adams, Sheryl S. Storey, Shelia B. Dunaway, Joel Gallant, Ilene Wiggins, Kimberly Y. Smith, Joan A. Swiatek, Joseph Timpone, Princy Kumar, Ardis Moe, Maria Palmer, Jon Gothing, Joanne Delaney, Kim Whitely, Ann Marie Anderson, Scott M. Hammer, Michael T. Yin, Mamta Jain, Tianna Petersen, Roberto Corales, Christine Hurley, Keith Henry, Bette Bordenave, Amanda Youmans, Mary Albrecht, Richard B. Pollard, Abimbola Olusanya, Paul R. Skolnik, Betsy Adams, Karen T. Tashima, Helen Patterson, Michelle Ukwu, Lauren Rogers, Henry H. Balfour, Jr., Kathy A. Fox, Susan Swindells, Frances Van Meter, Gregory Robbins, Nicole Burgett-Yandow, Charles E. Davis, Jr., Colleen Boyce, William A. O’Brien, Gerianne Casey, Gene D. Morse, Chiu-Bin Hsaio, Jeffrey L. Meier, Jack T. Stapleton, Donna Mildvan, Manuel Revuelta, David Currin, Wafaa El Sadr, Avelino Loquere, Nyef El-Daher, Tina Johnson, Robert Gross, Kathyrn Maffei, Valery Hughes, Glenn Sturge, Deborah McMahon, Barbara Rutecki, Michael Wulfsohn, Andrew Cheng, Norbert Bischofberger, Lynn Dix, and Qiming Liao
REFERENCES
- 1.Gilead Sciences, Inc. April 2017. Viread: tenofovir disoproxil fumarate. Prescribing information. Gilead Sciences, Inc., Foster City, CA. [Google Scholar]
- 2.Geboers S, Haenen S, Mols R, Brouwers J, Tack J, Annaert P, Augustijns P. 2015. Intestinal behavior of the ester prodrug tenofovir DF in humans. Int J Pharm 485:131–137. doi: 10.1016/j.ijpharm.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Jullien V, Treluyer JM, Rey E, Jaffray P, Krivine A, Moachon L, Lillo-Le Louet A, Lescoat A, Dupin N, Salmon D, Pons G, Urien S. 2005. Population pharmacokinetics of tenofovir in human immunodeficiency virus-infected patients taking highly active antiretroviral therapy. Antimicrob Agents Chemother 49:3361–3366. doi: 10.1128/AAC.49.8.3361-3366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Punyawudho B, Thammajaruk N, Thongpeang P, Matthews G, Lewin SR, Burger D, Ruxrungtham K, Avihingsanon A. 2015. Population pharmacokinetics of tenofovir in HIV/HBV co-infected patients. Int J Clin Pharmacol Ther 53:947–954. doi: 10.5414/CP202386. [DOI] [PubMed] [Google Scholar]
- 5.Baheti G, Kiser JJ, Havens PL, Fletcher CV. 2011. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV-1-infected patients. Antimicrob Agents Chemother 55:5294–5299. doi: 10.1128/AAC.05317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, Goti V, Chaturvedula A, Haberer JE, Fossler MJ, Sale ME, Bangsberg D, Baeten JM, Celum CL, Hendrix CW. 2016. Population pharmacokinetics of tenofovir in HIV-1-uninfected members of serodiscordant couples and effect of dose reporting methods. Antimicrob Agents Chemother 60:5379–5386. doi: 10.1128/AAC.00559-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.U.S. Department of Health and Human Services. 2017. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. What to start: initial combination regimens for the antiretroviral-naive patient. https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv/11/what-to-start. Accessed 21 December 2017.
- 8.Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapia M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernandez T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker LG, Mayer KH, Kallas EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang F, McConnell JJ, Zheng JH, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV, iPrEx Study Team. 2010. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson PL, Glidden DV, Liu A, Buchbinder S, Lama JR, Guanira JV, McMahan V, Bushman LR, Casapia M, Montoya-Herrera O, Veloso VG, Mayer KH, Chariyalertsak S, Schechter M, Bekker LG, Kallas EG, Grant RM, iPrEx Study Team. 2012. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med 4:151ra125. doi: 10.1126/scitranslmed.3004006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson L, Yapa HM, Jackson A, Moyle G, Else L, Amara A, Khoo S, Back D, Karolia Z, Higgs C, Boffito M. 2015. Plasma tenofovir, emtricitabine, and rilpivirine and intracellular tenofovir diphosphate and emtricitabine triphosphate pharmacokinetics following drug intake cessation. Antimicrob Agents Chemother 59:6080–6086. doi: 10.1128/AAC.01441-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valade E, Bouazza N, Lui G, Illamola SM, Benaboud S, Treluyer JM, Cobat A, Foissac F, De Sousa Mendes M, Chenevier-Gobeaux C, Suzan-Monti M, Rouzioux C, Assoumou L, Viard JP, Urien S, Ghosn J, Hirt D. 2017. Population pharmacokinetic modeling of tenofovir in the genital tract of male HIV-infected patients. Antimicrob Agents Chemother 61:e02062-16. doi: 10.1128/AAC.02062-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bristol-Myers Squibb Company. September 2016. Prescribing information. Reyataz (atazanavir). Bristol-Myers Squibb Company, Princeton, NJ.
- 13.McComsey GA, Kitch D, Sax PE, Tebas P, Tierney C, Jahed NC, Myers L, Melbourne K, Ha B, Daar ES. 2011. Peripheral and central fat changes in subjects randomized to abacavir-lamivudine or tenofovir-emtricitabine with atazanavir-ritonavir or efavirenz: ACTG Study A5224s. Clin Infect Dis 53:185–196. doi: 10.1093/cid/cir324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta SK, Kitch D, Tierney C, Daar ES, Sax PE, Melbourne K, Ha B, McComsey GA, AIDS Clinical Trials Group Study A5224s Team. 2014. Cystatin C-based renal function changes after antiretroviral initiation: a substudy of a randomized trial. Open Forum Infect Dis 1:ofu003. doi: 10.1093/ofid/ofu003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erlandson KM, Kitch D, Tierney C, Sax PE, Daar ES, Melbourne KM, Ha B, McComsey GA. 2014. Impact of randomized antiretroviral therapy initiation on glucose metabolism. AIDS 28:1451–1461. doi: 10.1097/QAD.0000000000000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyatt CM, Kitch D, Gupta SK, Tierney C, Daar ES, Sax PE, Ha B, Melbourne K, McComsey GA, AIDS Clinical Trials Group Study A5224s Team. 2014. Changes in proteinuria and albuminuria with initiation of antiretroviral therapy: data from a randomized trial comparing tenofovir disoproxil fumarate/emtricitabine versus abacavir/lamivudine. J Acquir Immune Defic Syndr 67:36–44. doi: 10.1097/QAI.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erlandson KM, Kitch D, Tierney C, Sax PE, Daar ES, Tebas P, Melbourne K, Ha B, Jahed NC, McComsey GA. 2013. Weight and lean body mass change with antiretroviral initiation and impact on bone mineral density. AIDS 27:2069–2079. doi: 10.1097/QAD.0b013e328361d25d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.World Health Organization. 2018. Dolutegravir (DTG) and the fixed dose combination (FDC) of tenofovir/lamivudine/dolutegravir (TLD). Briefing note. Accessed on 15 November 2018. http://www.who.int/hiv/pub/arv/DTG-TLD-arv_briefing_2018.pdf?ua=1.
- 19.Sax PE, Tierney C, Collier AC, Fischl MA, Mollan K, Peeples L, Godfrey C, Jahed NC, Myers L, Katzenstein D, Farajallah A, Rooney JF, Ha B, Woodward WC, Koletar SL, Johnson VA, Geiseler PJ, Daar ES, AIDS Clinical Trials Group Study A5202 Team. 2009. Abacavir-lamivudine versus tenofovir-emtricitabine for initial HIV-1 therapy. N Engl J Med 361:2230–2240. doi: 10.1056/NEJMoa0906768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daar ES, Tierney C, Fischl MA, Sax PE, Mollan K, Budhathoki C, Godfrey C, Jahed NC, Myers L, Katzenstein D, Farajallah A, Rooney JF, Pappa KA, Woodward WC, Patterson K, Bolivar H, Benson CA, Collier AC, AIDS Clinical Trials Group Study A5202 Team. 2011. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann Intern Med 154:445–456. doi: 10.7326/0003-4819-154-7-201104050-00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sax PE, Tierney C, Collier AC, Daar ES, Mollan K, Budhathoki C, Godfrey C, Jahed NC, Myers L, Katzenstein D, Farajallah A, Rooney JF, Ha B, Woodward WC, Feinberg J, Tashima K, Murphy RL, Fischl MA, AIDS Clinical Trials Group Study A5202 Team. 2011. Abacavir/lamivudine versus tenofovir DF/emtricitabine as part of combination regimens for initial treatment of HIV: final results. J Infect Dis 204:1191–1201. doi: 10.1093/infdis/jir505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venuto CS, Ma Q, Daar ES, Sax PE, Fischl M, Collier AC, Mollan K, Smith KY, Tierney C, Morse GD. 2012. Tenofovir plasma pharmacokinetics in AIDS Clinical Trials Group Study A5202, poster TUPE058. AIDS 2012, Washington, DC, 22 to 27 July 2012. [Google Scholar]
- 23.Venuto CS, Mollan K, Ma Q, Daar ES, Sax PE, Fischl M, Collier AC, Smith KY, Tierney C, Morse GD, Bolivar HH, Navarro S, Koletar SL, Gochnour D, Seefried E, Hoffman J, Feinberg J, Saemann M, Patterson K, Pittard D, Currin D, Upton K, Saag M, Ray G, Johnson S, Santos B, Funk CA, Morgan M, Jackson B, Tebas P, Thomas A, Kim G-Y, Klebert MK, Santana JL, Marrero S, Norris J, Valle S, Cox GM, Silberman M, Shaik S, Lopez R, Vasquez M, Daskalakis D, Megill C, Stroberg T, Shore J, Taiwo B, Goldman M, Boston M, Lennox J, et al. 2014. Sex differences in atazanavir pharmacokinetics and associations with time to clinical events: AIDS Clinical Trials Group Study A5202. J Antimicrob Chemother 69:3300–3310. doi: 10.1093/jac/dku303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bednasz CJ, Venuto CS, Ma Q, Daar ES, Sax PE, Fischl MA, Collier AC, Smith KY, Tierney C, Yang Y, Wilding GE, Morse GD. 2017. Efavirenz therapeutic range in HIV-1 treatment naïve participants. Ther Drug Monit 39:596–603. doi: 10.1097/FTD.0000000000000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James AMKJ, Ofotokun I, Sheth A, Acosta EP. 2013. Tenofovir and emtricitabine uptake into non-monocytic female genital tract cells with and without hormonal contraceptives. J Exp Pharmacol 5:55–64. doi: 10.2147/JEP.S45308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindbom L, Ribbing J, Jonsson EN. 2004. Perl-speaks-NONMEM (PsN)–a Perl module for NONMEM related programming. Comput Methods Programs Biomed 75:85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Keizer RJ, Karlsson MO, Hooker A. 2013. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol 2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibiansky L, Gibiansky E, Cosson V, Frey N, Stark FS. 2014. Methods to detect non-compliance and reduce its impact on population PK parameter estimates. J Pharmacokinet Pharmacodyn 41:279–289. doi: 10.1007/s10928-014-9364-2. [DOI] [PubMed] [Google Scholar]
- 29.Cockcroft DW, Gault MH. 1976. Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 30.Boekmann AJ, Sheiner LB, Beal SL. 1992. NONMEM user’s guide: part V. Introductory guide. University of California, San Francisco, CA. [Google Scholar]
- 31.U.S. Food and Drug Administration. 2010. Guidance for industry on pharmacokinetics in patients with impaired renal function−study design, data analysis, and impact on dosing and labeling. U.S. Food and Drug Administration, Washington, DC. [Google Scholar]
- 32.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. 2011. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parke J, Holford NH, Charles BG. 1999. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Methods Programs Biomed 59:19–29. doi: 10.1016/S0169-2607(98)00098-4. [DOI] [PubMed] [Google Scholar]