

Abstract
Liver fibrosis is characterized by excessive deposition of extracellular matrix (ECM) in the liver. Although many fibrogenic cell types may express ECM, the hepatic stellate cell (HSC) is currently considered to be the major effector. HSCs transform into myofibroblast-like cells, also known as hepatic myofibroblasts in a process known as activation; this process is characterized in particular by de novo expression of smooth muscle alpha actin (SM α-actin) and type 1 collagen. The family of actins, which form the cell’s cytoskeleton, are essential in many cellular processes. β-actin and cytoplasmic γ-actin (γ-actin) are ubiquitously expressed, while SM α-actin defines smooth muscle cell and myofibroblast phenotypes. Thus, SM α-actin is tightly associated with multiple functional properties. However, the regulatory mechanisms by which actin isoforms might regulate type 1 collagen remain unclear. In primary HSCs from normal and fibrotic rat liver, we demonstrate that myocardin, a canonical SRF cofactor, is upregulated in hepatic myofibroblasts and differentially regulates SM α-actin, γ-actin and β actins through activation of an ATTA box in the SM α-actin and a CCAAT box in γ-actin and β actin promoters, respectively; moreover, myocardin differentially activated serum response factor (SRF) in CArG boxes of actin promoters. Additionally, myocardin stimulated Smad2 phosphorylation and RhoA expression, leading to increased expression of type 1 collagen in an actin cytoskeleton dependent manner. Myocardin also directly enhanced SRF expression and stimulated collagen 1α1 and 1α2 promoter activities. Additionally, overexpression of myocardin in vivo during carbon tetrachloride-induced liver injury led to increased HSC activation and fibrogenesis. In summary, our data suggest that myocardin plays a critical role in actin cytoskeletal dynamics during HSC activation, in turn, specifically regulating type I collagen expression in hepatic myofibroblasts.
The family of actins are cytoskeletal proteins that play a critical role in a wide array of cellular processes including cell division, migration, contraction and differentiation1–4. Thus, altered expression of actin isoforms has been a focus in multiple physiological and pathological processes5,6. Epithelial-mesenchymal transition (EMT), for example, is a critical biological process in cancer cell metastasis, in which epithelial cancer cells acquire de novo expression of smooth muscle α-actin (SM α-actin), a key biomarker in EMT and differentiate to a motile phenotype7. In wound healing and fibrosis, resident fibroblasts differentiate to the activated myofibroblasts by the virtue of expression of SM α-actin and extracellular matrix8. In liver fibrosis, quiescent hepatic stellate cells (HSCs) differentiate to myofibroblast-like cells, also known as hepatic myofibroblasts, through activation and acquire a number of functional phenotypes related to SM α-actin expression3,9,10.
The six different actin isoforms in mammalian cells are highly conserved and have remarkable amino acid sequence homology11,12. Progress in understanding the functional importance of the relatively minor differences between actin isoforms has been made possible by study of specific actin isoform deficient mice. Deletion of β-actin is lethal13, while cytoplasmic γ-actin deficiency leads to impaired cell growth and survival14. SM α-actin deficiency results in a specific defect of lactation in nursing dams and decreased contractility in activated HSCs3,15. These findings suggest that the cytoplasmic actin isoforms are essential for cell growth and survival, while the more restricted actins play a critical role in tissue specific functions.
Regulation of actin expression has been extensively studied, in particular, for the smooth muscle isoform in vascular smooth muscle cells16. This body of work suggests that actins are serum response factor (SRF) target genes since their promoters contain a 10-base pair cis element CC(A/T)6GG known as the CArG box17, which allows SRF binding and activates transcription. In HSCs, SM α-actin promoter activity appears to be particularly SRF binding dependent since mutation or deletion of the both CArG-B and A boxes leads to nearly complete elimination of the promoter activity18. In contrast, β-actin promoter activity not only requires the CArG box but also an essential CCAAT box19. Little is known about the mechanism underlying the regulation of cytoplasmic γ-actin. In addition to CArG box for SRF binding, SRF-induced transactivation is prominently modulated by its cofactors20. Myocardin, as a restricted expression pattern in cardiac and smooth muscle cells20,21, has been demonstrated to be important SRF cofactors and play a critical role in cardiomyocytes and smooth muscle cell differentiation through activation of muscle specific genes22,23. Interestingly, myocardin was identified in HSCs in an array, linking myocardin with SM α-actin and type 1 collagen expression during HSC activation24. While the molecular pathways underlying the process remains elusive.
In the current study, we explored the molecular mechanisms of actin cytoskeleton regulation by myocardin during HSC activation and hepatic myofibroblast differentiation and the effect of actin cytoskeleton alteration on extracellular matrix protein type 1 collagen expression. Our results demonstrate that myocardin induces a novel actin expression pattern through differential regulation of SM α-actin, cytoplasmic-γ-actin and β-actin isoforms, all of which compose the activated HSC’s cytoskeleton and directly link it to morphological and functional properties in activated HSCs. Further, the data indicate an indirect link to the extracellular matrix protein, type 1 collagen.
MATERIALS AND METHODS
Animal model and cell culture
Sprague Dawley (SD) male rats (400 – 500g) were purchased from Charles’ River Lab and housed in MUSCs animal facility following the NIH and the IACUC guideline. Liver fibrosis was induced by carbon tetrachloride (CCl4) (once a week for 4 ~ 6 weeks) as described25. HSCs were isolated by in situ enzymatic digestion of normal or fibrotic rat liver and purified by Accudenz (Accurate Chemicals, Westbury, NY) gradient as described18. Freshly isolated HSCs were cultured in standard 199OR medium containing 10% calf serum and 10% horse serum (Invitrogen, Grand Island, NY) in a 3% CO2 incubator at 37°C otherwise stated.
Adenovirus and cell infection
A mouse vascular form of myocardin (856 aa) and a dominant negative myocardin (635 aa, without the activation domain) were subcloned from pcDNA3.1-myocardin plasmid construct (a gift from Dr. Olson’s laboratory) into a modified pDC316 shuttle vector (Microbix, Toronto) with pCMV promoter and HA (hemagglutinin) tag. The resulting pDC316-HA-myocardin and pDC316-HA dominant negative myocardin plasmids were cotransfected with pBGHloxΔE1,3-cre viral plasmid into 293HEK cells to generate adenovirus-HA-myocardin (Ad-myocd) and adenovirus-HA-dominant negative myocardin (Ad-myocd-DN). An empty pDC316 shuttle vector with pCMV promoter was cotransfected with pBGHloxΔE1,3-cre viral plasmid into 293HEK cells to generate adenovirus control (Ad-ctr). Viral screening and purification were performed according to manufacture’s instructions (Clontech, Mountain View, CA). Fresh isolated HSCs were cultured for 2 days and then infected with adenoviral vectors (100 MOI) in standard 199OR medium for 3 days. In vivo adenovirus (2.5 × 1011 pfu per rat) administration was performed via portal vein injection as described before26, 27.
Immunoblotting
Immunoblotting was performed as described18. Specific signals were captured by the Syngene G-Box digital imaging system (Syngene, Frederick, MD) and quantitative data obtained from the system’s software. Raw values for control samples were arbitrarily set to 1 (or 100%), and data were presented as fold-increase or percentage of the controls. Antibodies to detect SM α-actin (1A4) and β-actin (AC15), α-tubulin, and Rho kinase inhibitor Y27632 were obtained from Sigma (St. Louis, MO). Antibodies to detect cytoplasmic-γ-actin isoform (AB3265) and total actin (C4), GAPDH (as a loading control) were from Millipore (Temecula, CA). Anti-type 1 collagen (COL.1) antibody was purchased from Rockland (Gilbertsville, PA). Anti-SRF, p-Smad2, total Smad2, p-Erk, and myocardin antibodies were purchased from Cell Signaling (Danvers, MA) and Novus Bio (Littleton, CO) respectively. TGFβ were purchased from Prospec-Tany TechnoGene (Ness Ziona, Israel).
Immunocytochemistry and histology
Immunofluorescence staining was performed as described15. Briefly, HSCs were washed with PBS twice and fixed in 4% buffered formaldehyde solution for 15 minutes. Following exposure to Triton X100 (0.5% in PBS) for 5 minutes and then with 3% BSA in PBS for 30 minutes, cells were incubated with primary antibodies at room temperature for 1 hour. After further washing with PBS, cells were incubated with secondary antibodies for 1 hour. Finally, cells were stained with DAPI (Sigma) for 15 minutes before mounting with FluorSave solution (Calbiochem, San Diego, CA). Anti- SM α-actin antibody (conjugated with Cy3) and anti-β-actin antibody (AC15) were purchased from Sigma (St. Louis, MO). Anti-cytoplasmic-γ-actin isoform (AB3265) antibody was from Millipore (Temecula, CA). Alexa Fluor 488 phalloidin, fluophor 488 donkey anti-sheep, fluophor 555 goat anti-rabbit and mouse antibodies were obtained from Life Technology (Carlsbad, CA). Images were captured with Olympus FV10i LIV confocal microscope (the Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina) and Zeiss Axio Imager M2 (Molecular Morphology, Medical University of South Carolina). For histologic analysis, liver tissue was fixed in 10% buffered formalin (Fisher, NJ). Picrosirius red staining was performed as before28 and collagen content (area %) was quantitated via image-J.
Plasmid constructs, transfection and luciferase activity assay
Rat SM α-actin promoter and the CArG box mutants were described before18. β-actin (Genbank accession number:V01217.1) and cytoplasmic-γ-actin promoters (Genbank accession number:AC095876.6) were cloned from rat genomic DNA by PCR and DNA fragments were ligated into pGL3B luciferase reporter vector (Promega, Madison, WI). CArG and CCAAT boxes in the actin promoters were mutated using site mutagenesis kit (Agilent, Santa Clara, CA). Type 1α1 and 1α2 Collagen promoters were cloned from mouse genomic DNA and ligated into pGL3B luciferase reporter vector (Promega, Madison, WI). The constructs with deletions were created by PCR approach. SRF promoter luciferase reporter constructs were described as before18. All constructs were confirmed by sequencing (Genewiz, South Plainfield, NJ). The myocardin expression construct (pcDNA3.1+myocardin) was obtained from Dr. Eric Olson’s laboratory. Co-transfections were performed in activated rat HSCs with lipofectamine 2000 (Invitrogen, Grand Island, NY) as described before18. Luciferase activity was measured with a dual-luciferase assay kit (Promega, Madison, WI). All transfection experiments were performed in triplicate and repeated twice. Relative light units (RLU) after transfection with a pGL3B reporter vector were arbitrarily set to 1 and data were presented as fold increase relative to pGL3B activity as described29. Primers used for cloning and site mutagenesis are listed in Supplemental Table 1.
Cell contraction assay and wound healing assay
Freshly isolated HSCs were cultured on collagen gels (Advanced Biomatrix, San Diego, CA) as described3. Cells were exposed to adenovirus (100MOI) on the second day of culture and incubated for 3 further days. Then fresh medium containing serum with or without endothelin-1 (ET-1, American peptide company, Sunnyvale, CA) was added. At specified times, gels were released from the plastic substratum, and gel area was measured as described3.
Cell migration was measured using a wound healing assay kit (Cell Biolabs, San Diego, CA). In brief, cells were cultured in 24-well plates containing a specific insert in each well bottom to divide cell cultures to equally. Following 2 days of culture, the insert was removed and formed a clear, sharp demarcation on the plastic substrate. Then HSCs were exposed to adenovirus (100 MOI) in standard medium. Wound closure was subsequently measured over time and the quantitated data were obtained from triplicate experiment according to manufacture’s instructions.
Real time-PCR (RT-PCR) and RNase protection assay (RPA)
RNA from HSCs was isolated using Trizol reagent (Invitrogen) and 1–2 μg of total RNA was used for first-strand cDNA synthesis by SuperScript III (Invitrogen). Resulting cDNA was PCR amplified using gene specific primers (Table S1) and SYBR green supermix (BioRad, Hercules, CA) on a BioRad CFX Connect Real-Time PCR Detection System. For actin isoform RNA expression analysis, 5 μg of total RNA was hybridized with radioactive labeled gene-specific cRNA probes using an RPA III kit (Ambion, Austin, TX) (Supplemental Figure S1) as described before18. Specific signals for SM α-actin, cytoplasmic-γ-actin and β-actin were captured and quantitated using a phosphoimager (Syngene G-Box digital imaging system, Syngene, Frederick, MD). Control signals were arbitrarily set to 1 or 100% and data were presented as fold increase compared to controls.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared and EMSA was performed as described before18. In brief, nuclear extracts (10 μg) were incubated with32P-labeled DNA probes (for probe sequences, see Supplemental materials) at room temperature for 30 minutes. For supershift assays, 2 μl of anti-SRF antibody (Cell Signaling, Danvers, MA) was added to the reaction and incubated for 30 minutes at room temperature prior incubation with labeled probe. DNA-protein complexes were separated by non-denature polyacrylamide gel electrophoresis and specific signals were captured using a phosphoimager (Syngene G-Box digital imaging system (Syngene, Frederick, MD).
Statistical Analysis
Quantitative data were analyzed with Microsoft Excel software. Bar graphs represent mean ± SD (standard deviation). Significance (p < 0.05) was assessed by the Student’s t-test.
RESULTS
Upregulation of myocardin tightly associates with increased expression of actin cytoskeleton.
To explore the basic cell and molecular mechanisms underlying myocardin-mediated HSC activation and myofibroblast-like phenotype differentiation, we first examined myocardin expression in CCl4–induced liver fibrogenesis in vivo. Myocardin mRNA was significantly increased in activated HSCs from fibrotic liver compared to HSCs from normal liver (Figure 1A). Next, since myocardin localizes to the nucleus and physically forms a complex with SRF and potentiates SRF activity30,31, we examined myocardin and SRF protein levels in the nuclear extracts of HSCs from normal and fibrotic livers. We found that myocardin and SRF were upregulated (Figure 1B), suggesting that both of them are required for HSC activation. Since myocardin regulates all smooth muscle specific genes32, we examined the correlation between myocardin and actin isoform expression. As expected, SM α-actin was dramatically upregulated (15–16 fold), and surprisingly, cytoplasmic-γ-actin was also significantly elevated (7–8 fold). Unexpectedly, β-actin, a typical “housekeeping” gene, was also increased (Figure 1C). Further, we examined whether the expression patterns of these proteins in an in vivo liver injury model recapitulated the culture-based in vitro model. Indeed, expression patterns of myocardin, SRF, SM α-actin, cytoplasmic-γ-actin and β-actin in culture-induced HSC activation (Figure 1D, E, F) were similar to that in vivo liver injury (Figure 1B, C). These data strongly suggest that HSC activation is associated with upregulation of myocardin and SRF, which promote actin isoform expression and actin dynamics in response to cellular differentiation signals.
Figure 1.
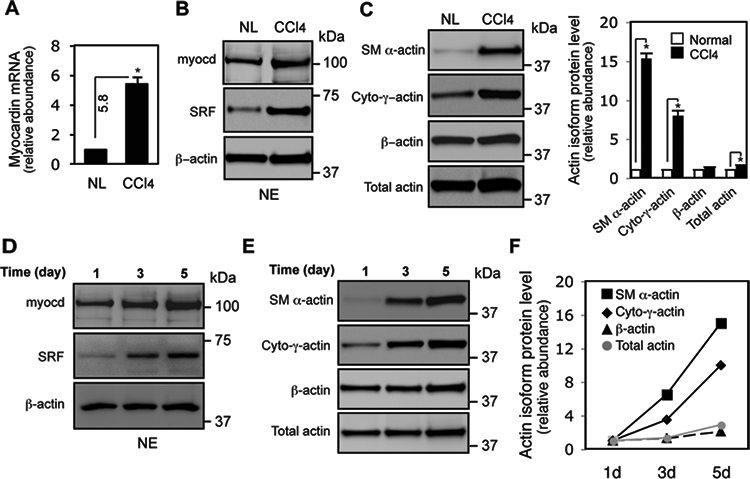
Myocardin is upregulated during HSC activation in vivo and in vitro. HSCs were isolated and in Methods, and in (A), total RNA from freshly isolated HSCs from normal (NL) and fibrotic rat liver (CCl4) was extracted and subjected to RT-PCR to measure myocardin mRNA (n=3, * p < 0.01 for NL vs. CCl4). (B) Myocardin and SRF protein levels were measured using nuclear extracts (NE) from normal (NL) and fibrotic rat liver HSCs (CCl4) (n=3, * p < 0.05 for NL vs. CCl4). (C) Actin isoform protein profiles were measured in whole cell lysates from normal (NL) or fibrotic rat liver HSCs (CCl4). (D) HSCs from normal liver were isolated and grown in culture as in methods and myocardin and SRF levels were measured in nuclear extracts; whole cell lysates were used to assess complete actin isoform profiles (E), and quantitative data are shown graphically (F). β-actin was used as a loading control (an additional Coomassie blue stain is shown in Figure S3). Representative immunoblots from 3 independent experiments were shown.
Myocardin stimulates a HSC myofibroblast-like phenotype differentiation in vitro and in vivo.
To evaluate the role of myocardin in HSC activation, primary rat HSCs were used to examine bona fide biological responses to myocardin. Additionally, an adenovirus harboring a smooth muscle form of myocardin (Ad-myocd) and dominant negative myocardin (Ad-myocd-DN) were used to modulate myocardin activity, since HSCs primarily express a smooth muscle form of myocardin (Supplemental Figure S2). HSCs infected with Ad-myocd virus exhibited a more prominent activated appearance - characterized by an enlarged cell size and prominent lamellipodia. In contrast, HSCs infected with dominant negative myocardin had a less activated appearance, characterized by smaller cell size and less prominent cellular processes (Figure 2A). Myocardin induced prominent actin stress fibers, in particular SM α-actin stress fibers (Figure 2B) and cytoplasmic-γ-actin stress fibers (Figure 2C), which were thick and long in the cell cytoplasm. Additionally, cytoplasmic-γ-actin stress fibers appeared to be prominent in filopodia around the membrane (Figure 2C). In contrast, inhibition of myocardin (with the dominant negative construct) appeared to inhibit the activated phenotype - leading to smaller and rounder cells, with stress fibers forming a thick ring (which was mainly composed of SM α-actin and cytoplasmic-γ-actin stress fibers) at the inside of the cell membrane and a disorganized intracellular pattern (Figure 2B, C, lower panel). Dominant negative myocardin also caused corresponding changes in β-actin, characterized by actin spots along thin actin stress fibers (Figure 2D). Additionally, myocardin was localized in both the cytoplasm and nuclei of HSCs, while dominant negative myocardin was primarily localized in the nuclei of HSCs (Figure 2E).
Figure 2.
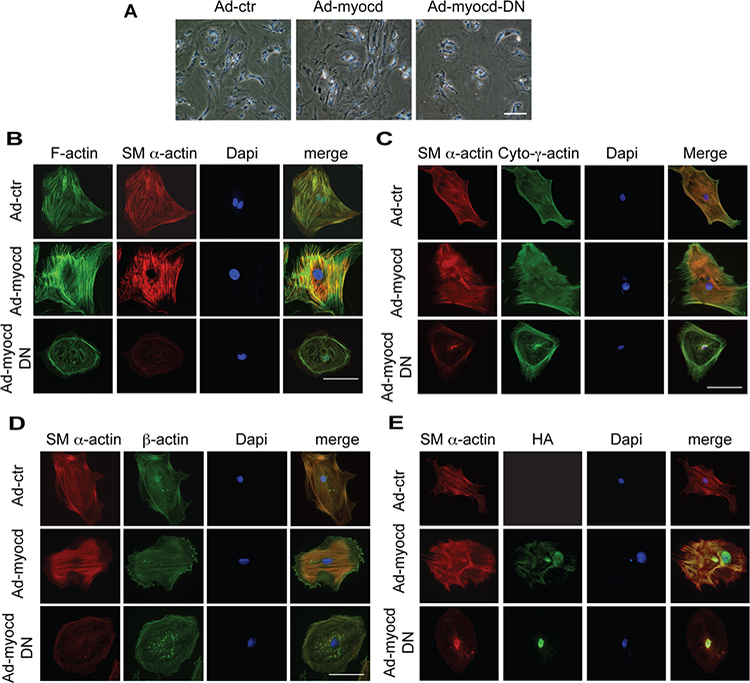
Myocardin induces morphological changes in activated HSCs. (A) HSCs were exposed to adenoviruses (Ad-control (Ad-ctr), Ad-myocd and Ad-myocd-DN, all at 100 MOI) for 3 days and then incubated at 0.5% serum medium for 2 days. Representative images from 3 independent experiments are shown. (B, C, D, E) Cells were grown on glass cover slips and exposed to adenovirus as in (A) and subjected to immunofluorescence labeling as in Methods. Representative images from 3 independent experiments are shown. The scale bar represents 100 μm.
Further, we examined the effect of overexpression of myocardin in CCl4-induced liver fibrogenesis in vivo (Figure 3). Since the myocardin deficient mouse is an embryonic lethal and thus could not be used to examine the effect of myocardin in vivo, we chose to overexpress myocardin in HSCs with an adenovirus expressing myocardin (note that we have previously demonstrated that parenteral administration of adenovirus leads to efficient viral expression in HSCs27). Liver injury was established by administration of CCl4 before administration of adenovirus (Figure 3A, D)27. In the first model, myocardin expression led to a highly activated HSC appearance and increased SM α-actin and type 1α1 mRNA expression compared to control (Figure 3,A–C). In the second CCl4-induced liver fibrosis model, extracellular matrix protein collagen deposition was increased after myocardin overexpression (compared to control, Figure 3, D, E). These data demonstrate the importance of myocardin in regulation of hepatic myofibroblast differentiation in vitro and in vivo.
Figure 3.
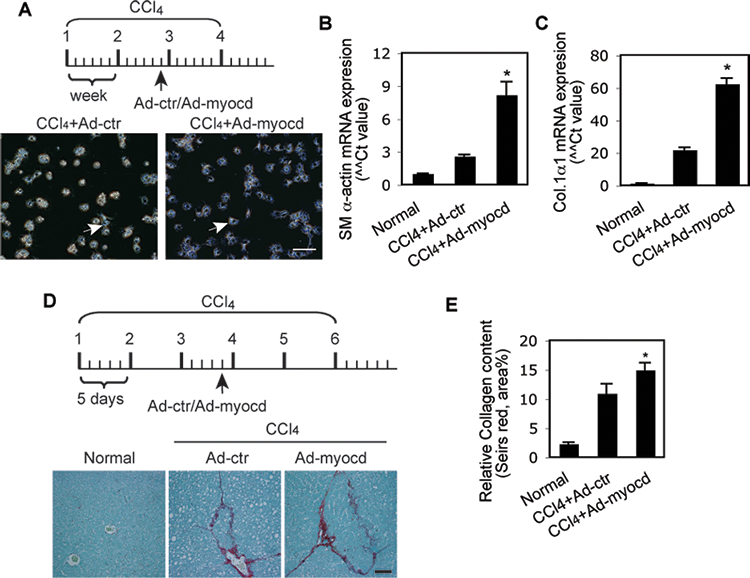
Overexpression of myocardin promotes HSC myofibroblast differentiation and liver fibrosis. (A) A schematic map for overexpression of myocardin with 4 doses of CCl4. 5 days after the final dose of CCl4, HSCs were isolated and cultured overnight; representative phase contrast images are shown (arrow indicates an activated HSC). (B, C) Total RNA was extracted from the same cells and RT-PCR was performed to detect SM α-actin and Col.1α1 mRNA (n=3, p < 0.01 for Ad-control (Ad-ctr) vs. Ad-myocd). (D) A schematic map for overexpression of myocardin with 6 doses of CCl4. 5 days after the final CCl4 dose, whole liver tissues were harvested and subjected to picrosirius red staining as in Methods. Quantitative data is shown graphically (n=3 ~ 4, p < 0.05 for Ad-control (Ad-ctr) vs. Ad-myocd). The scale bar represents 100 μm.
Myocardin mediates functional effects in HSCs.
Prominent functional features of activated HSCs include enhanced contractility and cell motility3. Thus, we examined whether myocardin had functional effects in HSCs. As expected, overexpression of myocardin increased basal HSC contractility, while inhibition of myocardin reduced endothelin-1 (ET-1) mediated HSC contractility (Figure 4A–C). Additionally, inhibition of myocardin activity reduced HSC migration (Figure 4D), a phenotype similar to SM α-actin deficient HSCs3. Notably, exogenous expression of myocardin had minimal effects on cell motility. Taken together, the data demonstrate that myocardin has important functional effects in activated HSCs.
Figure 4.
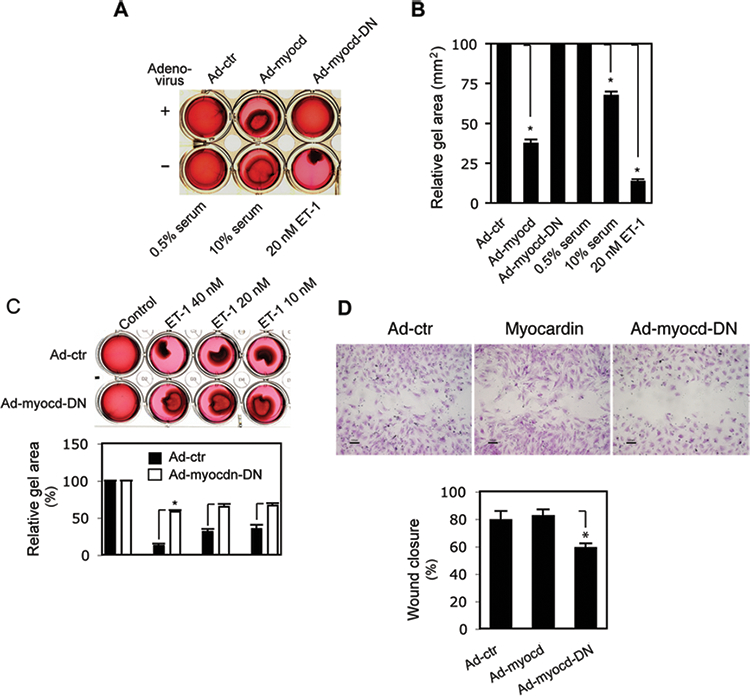
Myocardin-mediated functional effects. HSCs were isolated and grown on collagen lattices for 2 days and exposed to adenovirus as indicated for 3 days. Lattice contraction was as in Methods and images were taken 12 hours after gel lattice release; (A) 10% serum and 20 nM ET-1 were used as positive controls and 0.5% serum as a negative control. A representative image from 3 independent experiments is shown. In (B), quantitative data are presented graphically (the stronger the contraction, the smaller the gel area. n=3, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-myocd, 10% serum and 20nM ET-1, respectively). (C) Cells were isolated, grown in 0.5% serum containing medium, and exposed to different concentrations of ET-1 as indicated at the time of lattice release; images were taken after gel lattice release (6 hours and a representative image from 3 independent experiments is shown). In the panel below the image, quantitative data are shown (n=3, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-myocd-DN). (D) HSCs were grown in a 24-well wound healing assay plate for 2 days and then exposed to the indicated adenovirus at the same time as application of the scratch wound (images were taken after further 3 days of culture; representative images from 3 independent experiments are shown. In the graph below the images, the area of wound closure was measured as in Methods and quantitative data are shown graphically (n=3, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-myocd-DN).
Myocardin differentially regulates actin isoform expression.
To explore the molecular basis of myocardin mediated morphological and functional effects on HSCs, we examined actin isoform expression following exogenous expression of myocardin or dominant negative myocardin in activated HSCs. To measure the bona fide target actin isoform mRNA expression, we used unique probes specific to nucleotides in the amino termini of the actins (with RNase protection assay (RPA), Supplemental Figure S1). SM α-actin and cytoplasmic-γ-actin mRNA levels were upregulated by myocardin (6–7 fold and almost 2-fold vs. control, respectively) (Figure 5A, B). β-actin mRNA was also increased compared to the control. In contrast, inhibition of myocardin led to significant decreases in SM α-actin and cytoplasmic-γ-actin mRNAs, while β-actin mRNA was only modestly affected (Figure 5A, C).
Figure 5.
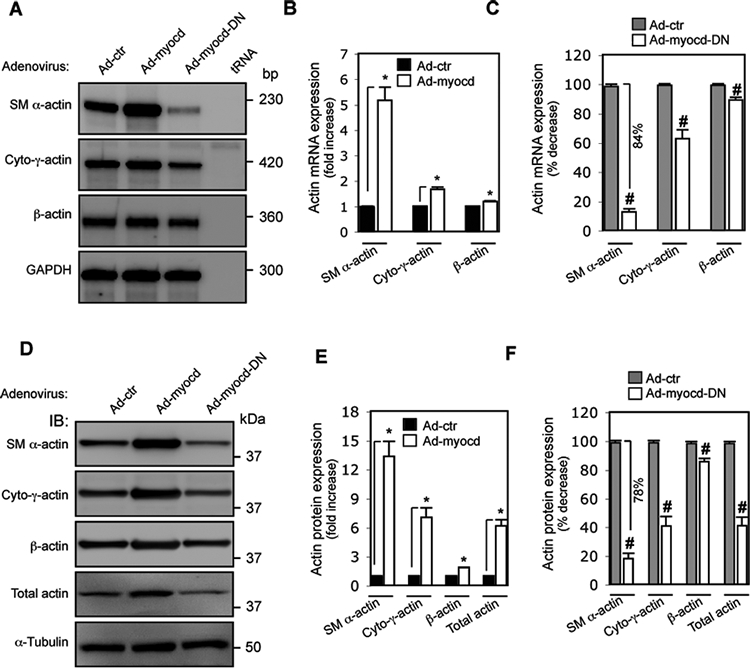
Myocardin induces differential expression of actin isoforms. HSCs were isolated as above and exposed to adenovirus as indicated for 3 days and then incubated in 0.5% serum medium for 2 days. (A) Actin isoform mRNA profiles were assessed by RNase protection assay (RPA). Representative images from 3 independent experiments are shown. (B) Quantitative data depicting changes in actin isoform mRNA expression induced by myocardin, or dominant negative myocardin (C) are depicted graphically. (D) Whole HSC lysates were subjected to immunoblotting to analyze actin isoform profiles (representative immunoblots from 3 independent experiments are shown) and (E) quantitative data depicting changes in actin isoform expression induced by myocardin, or dominant negative myocardin (F) are presented graphically. (n=4, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-myocd; # p < 0.05 for Ad-control vs. Ad-myocd-DN).
Next, we examined regulation of actin protein isoforms by myocardin. Overexpression of myocardin caused dramatic upregulation of SM α-actin. Cytoplasmic-γ-actin and β-actin were also significantly increased (7–8 fold and 0.8–1 fold vs. control, respectively), which all composed of the increased total actin (Figure 5D, E). These results were similar to the actin expression patterns in activated HSCs induced by culture or in vivo liver injury (Figure 1 C, E, F), suggesting a bona fide biological effect of myocardin on actin isoform expression. In contrast, after blocking myocardin function (Ad-myocd-DN), both SM α-actin and cytoplasmic-γ-actin were significantly decreased (by 78%, 56%, respectively); β-actin was also reduced, which all together led to more than 50% reduction in the total actin (Figure 5D, F). In aggregate, these data suggest that myocardin differentially regulates actin isoform expression, which appears to be primarily via transcriptional regulation.
CArG box and ATTA, CCAAT boxes are specifically required for myocardin-induced differential regulation of actin isoform transcription.
The CArG box, found in the promoter region of many genes coding for structural proteins, has been established as the core SRF binding sequence16,17. We next examined whether CArG boxes in the different actin promoters (Figure 6A) might have differential activity in HSCs. We found that SRF binding activity in the SM α-actin promoter was lower than in the cytoplasmic-γ-actin or β-actin promoter at baseline (Figure 6B, black columns 1 vs. 5 and 9). However, myocardin potently stimulated SRF binding to the SM α-actin promoter, which was greater than for the cytoplasmic-γ-actin or β-actin promoter (Figure 6B, open columns 2 vs. 1, 6 vs. 5 and 10 vs. 9). Mutation of CArG boxes (Figure 6A) in the SM α-actin promoter nearly eliminated SM α-actin promoter activity and almost completely abrogated myocardin-induced promoter activity (Figure 6B, columns 3 and 4). In contrast, mutation of CArG boxes in cytoplasmic-γ-actin or β-actin promoters had a less prominent effect, particularly in the cytoplasmic-γ-actin promoter. CArG box mutation had a substantial effect on both cytoplasmic-γ-actin and β-actin promoter activities with myocardin stimulation (Figure 6B, columns 7 vs. 5, 8 vs. 6 and 11 vs. 9, 12 vs. 10). These data indicate that the CArG boxes in actin promoters are required for basal and myocardin-induced transcriptional activation, but that there is a differential effect for the different actin CArG boxes.
Figure 6.
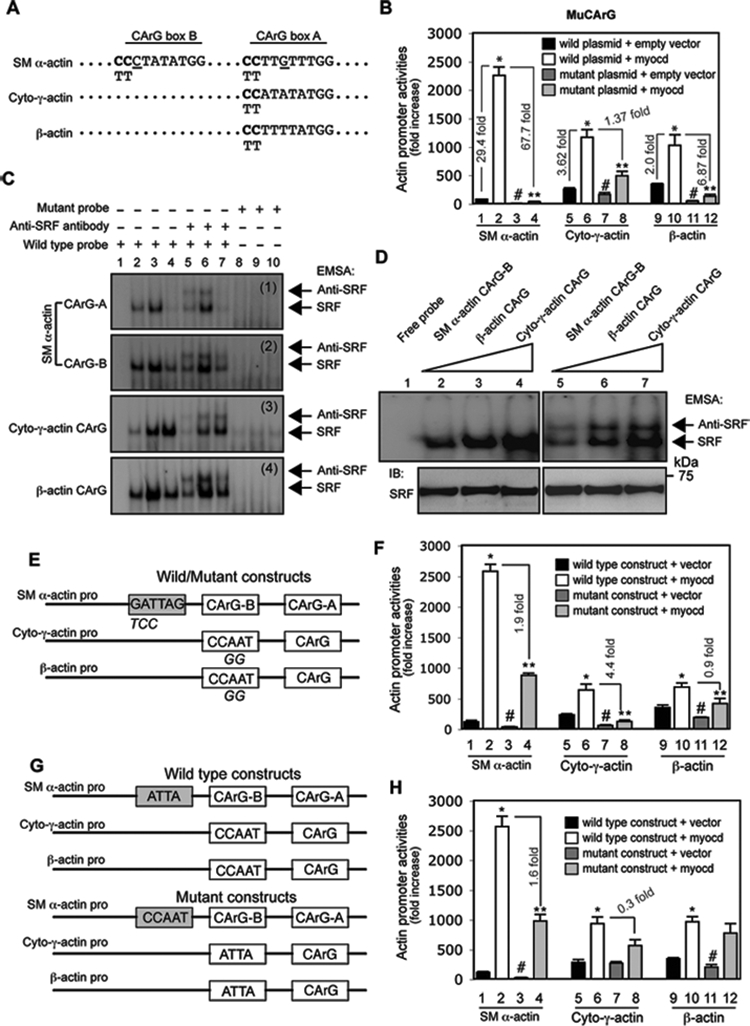
Myocardin-induced differential regulation of actin isoforms is CArG box and ATTA, CCAAT box dependent. (A) CArG box sequences from rat SM α-actin, cytoplasmic-γ-actin (Cyto-γ-actin) and β-actin promoters are aligned. Site mutations are shown in bold letters (CC in CArG boxes were replaced by TT). (B) Activated HSCs were cotransfected with luciferase reporter constructs containing wild type or the CArG box mutation as in (A) and a myocardin expression plasmid (Myocd) or empty vector. Cells were harvested 2 days later to detect for promoter activity. (C) After growth for 2 days after isolation, HSCs were exposed to the indicated adenoviral vectors for 3 days and 2 days later, they were subjected to EMSA to measure the effect of myocardin on SRF binding activity. SRF binding and supershifted bands are highlighted by arrows (lane 1, 2, 5, 8: nuclear extracts from HSCs infected with Ad-control virus; lane 3, 6, 9: nuclear extracts from HSCs infected with Ad-myocd virus; lane 4, 7, 10: nuclear extracts from HSCs infected with Ad-myocd-DN virus). Representative data from 3 independent experiments are shown. (D) EMSA was performed using nuclear extracts (10 μg) from activated HSCs and the same amount of different actin CArG box probes (1 × 105 cpm) as indicated. SRF binding and supershifted complexes were indicated by arrows (upper panel). Nuclear extracts were probed by anti-SRF antibody as loading control (bottom panel). Representative data from 3 independent experiments are shown. (E) A schematic diagram of wild type and mutant actin isoform promoters is shown (mutated nucleotides were indicated below the consensus sequences); (F) luciferase assays were performed as in (B). (G) A schematic diagram of exchanged elements (ATTA and CCAAT boxes) among SM α-actin, cytoplasmic-γ-actin (cyto-γ-actin) and β-actin promoters is shown; (H) luciferase activity assay was performed as in (B). (n=3, * p < 0.01 for wild plasmid + empty vector vs. wild plasmid + myocardin; n=3, # p < 0.05 for wild plasmid + empty vector vs. mutant plasmid + empty vector; n=3, ** p < 0.05 for wild plasmid + myocardin vs. mutant plasmid + myocardin).
We further examined whether SRF binding activity could be associated with the observed differential (myocardin-mediated) actin promoter activities. As expected, SRF binding activity to all of the actin CArG boxes was increased in nuclear extracts after expression of myocardin (Figure 6C, lane 3 vs. lane 2), indicating that myocardin enhanced SRF binding activity, consistent with previous data in smooth muscle cells33. Interestingly, SRF binding activity to the CArG boxes in the SM α-actin promoter was significantly reduced by a dominant negative myocardin construct (Figure 6C, panel 1 and 2; lane 4 vs. lane 2), but SRF binding activity to CArG boxes in cytoplasmic-γ-actin and β-actin promoters were minimally affected (Figure 6C, panel 3, 4; lane 4 vs. lane 2). We hypothesized that such differential SRF binding activity among the CArG boxes might be due to different A/T content, since CArG boxes in the SM α-actin promoter contain single C or G substitutions (Figure 6A). By using identical amounts of nuclear extract from activated HSCs (Figure 6D, lower panel) and the same amount of probe harboring individual CArG box sequences (Figure 6D), we found that the SM α-actin CArG-B box exhibited lower SRF binding activity (Figure 6D, lane 2) than cytoplasmic β- or γ-actin CArG boxes (Figure 6D, lane 4). Interestingly, although the CArG boxes in cytoplasmic-γ-actin and β-actin promoters are both conserved (Figure 6A), more SRF binding activity was present in the CArG box of cytoplasmic-γ-actin than in the β-actin CArG box (Figure 6D, lane 4 vs. 3). Taken together, these results revealed that CArG boxes with different A/T content led to differential SRF binding. Further, the SM α-actin promoter appeared to be more sensitive to myocardin stimulation than the other two actin promoters, suggesting a reverse relationship between SRF binding and myocardin sensitivity.
Previous studies revealed that a conserved ATTA box in SM α-actin promoter33 and a CCAAT box19 in the cytoplasmic β-actin promoter play an important role in actin mRNA expression. Therefore we examined whether these boxes have functional effects on myocardin-induced promoter activity among the actin isoforms (Figure 6E). Myocardin-induced SM α-actin promoter activity was prominently reduced in the ATTA box mutant construct (1.9 fold reduction) compared to the wild type construct (Figure 6F, column 4 vs. 2). Similarly, the CCAAT box mutation significantly abrogated myocardin-induced cytoplasmic-γ-actin and β-actin promoter activity (4.4 and 0.9 fold reduction, respectively) (Figure 6F, columns 8 vs. 6; 12 vs. 10). These results suggest that the ATTA (SM α-actin promoter) and CCAAT boxes (cytoplasmic-γ-actin and β-actin promoter) are required for full myocardin-mediated transactivation.
Next, we examined whether the ATTA box was able to stimulate cytoplasmic-γ-actin and β-actin promoter activity since it appeared to be critical for myocardin-induced promoter activity in the SM α-actin promoter (Figure 6G). Interestingly, substitution of the CCAAT box in the cytoplasmic-γ-actin promoter with the ATTA box did not change the promoter activity as that in the CCAAT box mutant construct (Figure 6F), but such substitution prominently restored myocardin-induced cytoplasmic-γ-actin promoter activity (Figure 6H, column 8 vs. 6). Similar to the β-actin promoter, replacement CCAAT box with the ATTA box also significantly restored myocardin-mediated promoter activity (Figure 5H, column 12 vs. 10). In contrast, substitution of the ATTA box in the SM α-actin promoter with a CCAAT box led to the same result as that in the ATTA box mutant SM α-actin promoter construct (Figure 6H, column 4 vs. 2). These results suggest that the ATTA box is an indispensable element in SM α-actin promoter (i.e. to mediate myocardin-induced full promoter activity). However, the CCAAT box is specifically required for myocardin-induced cytoplasmic-γ-actin and β-actin actin promoter activity.
Myocardin mediates type 1 collagen expression in HSCs.
Type 1 collagen expression is regulated via multiple factors, including MRTF/A34. Myocardin, as a major member in this family35, may also play a role in regulation of type 1 collagen expression in HSCs. Indeed, type 1 collagen expression was increased by wild type myocardin but decreased by dominant negative myocardin (Fig. 7A). To explore the molecular pathways underlying this process, we examined the effect of myocardin on TGFβ-induced type 1 collagen expression. As expected, TGFβ enhanced type 1 collagen expression in control HSCs. SM α-actin and cytoplasmic γ-actin were also upregulated by TGFβ. However, TGFβ-induced type 1 collagen expression was abrogated by dominant negative myocardin, which was accompanied by reduced expression of actin isoforms (Fig. 7B). Additionally, myocardin prominently enhanced Smad2 phosphorylation (p-Smad2), but dominant negative myocardin did not significantly reduce Smad2 phosphorylation compared to the control (Fig. 7C). The results suggest that myocardin is able to stimulate TGFβ signaling through myocardin-mediated actin expression, thereby regulates type 1 collagen expression36.
Figure 7.
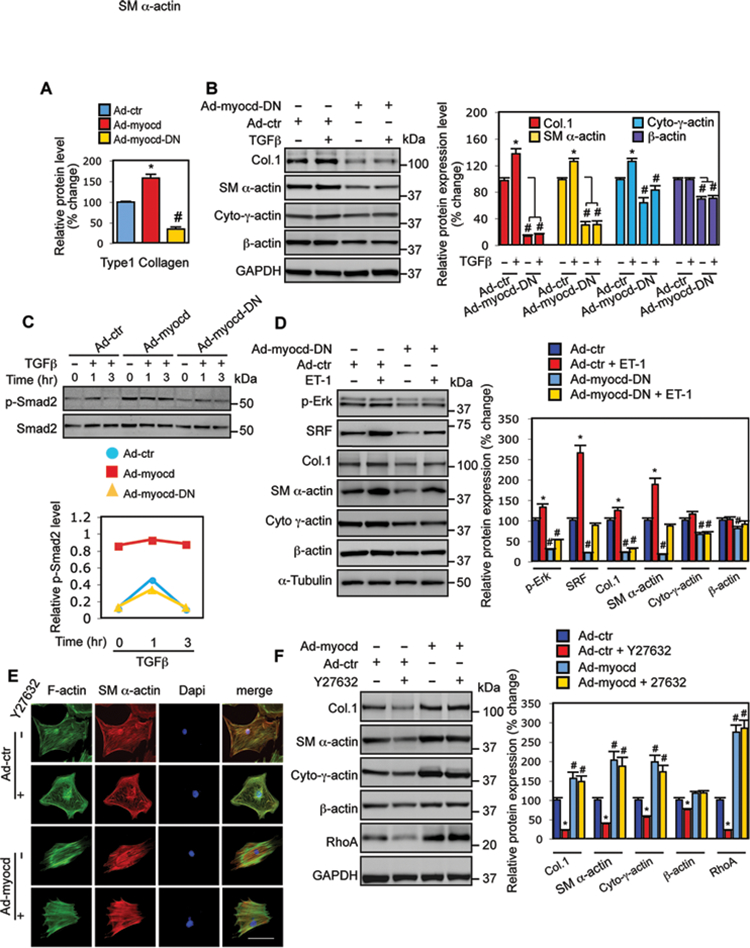
Myocardin modulates type 1 collagen expression. (A) HSCs were exposed to adenoviruses as indicated for 3 days and then incubated in 0.5% serum medium for 2 further days. Type 1 collagen (Col.1) was measured by immunoblotting and quantitative data are shown (n=3, * p < 0.01 for Ad-control (Ad-ctr) vs. Ad-myocd; # p < 0.05 for Ad-ctr vs. Ad-myocd-DN). (B) HSCs were exposed to Ad-control or Ad-myocd-DN for 3 days and then incubated in 0.5% serum medium with or without TGFβ (10 μg) for 2 further days. Type 1 collagen and actin isoform profiling were performed by immunoblotting. Representative images from 3 independent experiments were shown and quantitative data are shown in the graph to the right (n=3, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-ctr + TGFβ; #p < 0.05 for Ad-ctr vs. Ad-myocd-DN with or without TGFβ). (C) HSCs were as in (B); HSCs were exposed to TGFβ (10 μg) for 0, 1, and 3 hours, respectively. Whole cell lysates were assayed for phospho-Smad2 (p-Smad2) and total Smad2 as indicated. Representative immunoblots from 3 independent experiments are shown. Quantitative data are shown in the graph below. (D) HSCs were as in (B), and were exposed to ET-1 (20 nM) for 2 days; cell lysates were assayed for p-Erk, SRF, Col.1 and actin isoform by immunoblotting; representative images from 3 independent experiments are shown and quantitative data are shown in the graph to the right (n=3, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-ctr + ET-1; #p < 0.05 for Ad-ctr vs. Ad-myocd-DN with or without ET-1). (E, F) HSCs were isolated and grown on either glass cover slips as in Figure (2B) or standard culture dishes as above. Following virus infection for 3 days, cells were incubated in 0.5% serum medium with or without Y27632 (10 μM) for 1 day. Cells on glass cover slips were fixed and immunostained as in Methods and whole cell lysates from the same cell preparations were subjected to immunoblotting to detect type 1 collagen, RhoA and actin isoforms (left panel). Representative images from 3 independent experiments are shown and quantitative data are shown in the graph to the right (n=3, * p < 0.05 for Ad-control (Ad-ctr) vs. Ad-ctr with Y27632; # p < 0.05 for Ad-ctr vs. Ad-myocd with or without Y27632).
We next examined the role of myocardin in endothelin-1 (ET-1)-mediated type 1 collagen expression, since ET-1 also increases type 1 collagen expression (via Erk and AP-1 pathways37). Similar to TGFβ, ET-1 upregulated type 1 collagen expression, accompanied by increased expression of SM α-actin, cytoplasmic γ-actin, SRF and Erk-phosphorylation. These effects were prominently inhibited by dominant negative myocardin (Fig. 7D), suggesting that myocardin is able to regulate type 1 collagen expression via endothelin-1-mediated actin cytoskeleton proteins, SRF and Erk pathways.
Finally, since Rho signaling mediates actin stress fiber formation and disruption of Rho pathway suppresses collagen accumulation in activated HSCs38, we examined the effect of Y-27632, a specific Rho kinase inhibitor, on myocardin-induced type 1 collagen expression. As expected, Y-27632 disrupted actin stress fibers and led to a disorganized actin cytoskeleton in control HSCs (Fig. 7E). The effect of Y27632 was abrogated by exogenous expression of myocardin (Fig. 7E, lower panel). Type 1 collagen expression was significantly decreased by Y-27632 in control HSCs, but myocardin rescued the effect of Y-27632 (Fig. 7F, top panel). Simultaneously, Rho inhibition resulted in decreased SM α-actin and cytoplasmic γ-actin expression, and was also rescued by myocardin. These findings provide further support for the concept that myocardin mediates actin and actin dependent regulation of type 1 collagen expression.
Myocardin targets SRF expression and stimulates type 1 collagen transcription.
In activated HSCs, since the SRF gene promoter has CArG boxes18,39, and since upregulation of myocardin coincided with increased SRF expression (Fig. 1B, D), we postulated that myocardin might directly regulate SRF. Indeed, exogenous expression of myocardin increased both SRF mRNA and SRF protein levels in HSCs, while dominant negative myocardin decreased SRF mRNA and protein expression (Fig. 8A, B). To further examine the transcriptional regulation of SRF, wild type (WT) and CArG mutant SRF promoter constructs were cotransfected with myocardin expression plasmid in activated HSCs18. Myocardin dramatically upregulated SRF promoter activity (8.8 fold increase), while myocardin failed to fully elevate SRF promoter activity in the CArG box mutant SRF promoter construct (Fig. 8C). The data suggest that myocardin positively stimulates an SRF autoregulatory loop.
Figure. 8.
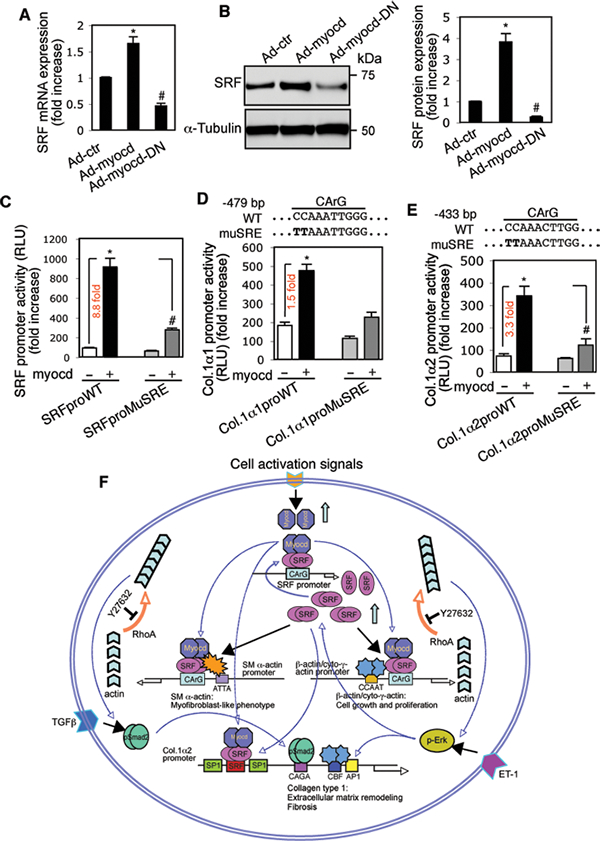
Myocardin activates SRF and type 1 collagen transcription via CArG boxes in actin promoters. HSCs were cultured and infected with adenoviruses as in Figure 7A. (A) SRF mRNA expression was measured by RT-PCR and (B) SRF was measured by immunoblotting; representative images from 3 independent experiments are shown and quantitative data are shown in the graph to the right (n=5, * p < 0.01 for Ad-control (Ad-ctr) vs. Ad-myocd; # p < 0.05 for Ad-ctr vs. Ad-myocd-DN). (C) Activated HSCs were cotransfected with a wild type (WT) SRF promoter (SRFproWT) or CArG box mutant SRF promoter (SRFproMuSRE) and a myocardin expression plasmid (myocd) as in Fig. 6B. (D, E) Activated HSCs were cotransfected with wild type 1α1 and 1α2 collagen promoters (Col.1α1proWT, Col.1α2proWT) or CArG box mutant type 1α1 and 2 collagen promoters (Col.1α1proMuSRE, Col.1α2proMuSRE) and a myocardin expression plasmid as in Fig. 6B. Quantitative data are depicted graphically (n=3, * p < 0.01 for WT construct with empty vector vs. with myocardin expression plasmid; #p < 0.01 for WT construct with myocardin expression plasmid vs. SRE mutant construct with a myocardin expression plasmid). (F) A schematic diagram providing a conceptual overview of myocardin-mediated hepatic stellate cell activation and myofibroblast differentiation is shown.
Since the type 1α1 and 1α2 collagen promoters also contain a CArG box40, we also examined whether myocardin might potentiate type 1α1 and 1α2 collagen promoter activity. Exogenous expression of myocardin led to a significant increase in both type 1α1 and 1α2 collagen promoter activities (Fig. 8D, E), while the myocardin-induced effect was inhibited by mutation of the CArG box in both promoters. These data suggest that myocardin may directly activate type 1α1 and 1α2 collagen transcription via SRF.
DISCUSSION
A defining property of the HSC to myofibroblast differentiation is the de novo expression of SM α-actin, which associates with multiple functional features of activated HSCs9, although the fundamental molecular mechanisms underlying hepatic myofibroblast differentiation remain poorly understood. In the present study, our results demonstrate that myocardin is an important factor to promote HSC activation process. Myocardin positively targets SRF autoregulatory loop to increase SRF expression. Myocardin and SRF appear to serve as a core regulatory platform to differentially regulate actin isoform expression through myocardin’s different sensitivity to differential SRF binding activity on the CArG boxes as well as the ATTA and CCAAT boxes in different actin promoters. Myocardin-induced upregulation of actin expression not only contributes to morphological and functional features in activated HSCs, but also indirectly regulates type 1 collagen expression (Fig. 8F).
HSC activation toward myofibroblast differentiation is a complicated molecular process in which multiple factors are involved, including MRTF/A34 and myocardin (Fig. 1). Our results suggest that HSCs may have unique myofibroblastic features since HSCs contain vitamin A lipid droplets10 and express myocardin (Fig. 1)24, 41, which are different from the myofibroblasts of other tissue sources such as myofibroblasts in kidney and lung fibrosis42, 43, suggesting that different mechanisms may exist among different tissue types during wound healing process. Myocardin is located primarily in the nucleus (Fig. 2)35 and readily activates gene transcription via interaction with SRF. In contrast, MRTF/A is an actin binding protein and its nuclear translocation requires the release from actin binding in response to serum stimulation and other signals that promote actin polymerization20. It is likely that myocardin stimulates HSC activation via upregulation of smooth muscle specific genes, while MRTF/A may potentiate the effect of myocardin in response to increased actin polymerization during HSC activation and myofibroblast differentiation.
SM α-actin is a well-known molecular marker for myofibroblast differentiation8–10. Our results revealed that in addition to SM α-actin, cytoplasmic γ-actin was significantly upregulated in activated HSCs (Fig. 1). The finding suggests that cytoplasmic γ-actin may also play an important role in promotion of HSC activation, which was not reported in previous studies regarding to HSC activation. Noticeably, cytoplasmic β-actin was also increased, which together with SM α-actin and cytoplasmic γ-actin consisted of the total actin pool in activated HSCs. We also found that myocardin enhanced actin stress fiber formation (Fig. 2), presumably due to increased RhoA expression induced by myocardin (Fig. 7F). Taken together, increased expression of actin isoforms and actin stress fibers are likely critical for morphological and functional attributes of hepatic myofibroblasts (Fig. 2, 3, 4).
The molecular mechanism underlying differential regulation of the actin isoforms during HSC activation remains poorly understood. Our results revealed that an opposite phenomenon between myocardin-induced actin promoter activity and the SRF binding activity to the CArG box of the actin promoters − the higher promoter activity was induced by myocardin with the lower SRF binding activity. The nature under this phenomenon was largely due to the A/T content and maybe the ratio of A to T in the CArG boxes. Our findings were consistent with a previous study in smooth muscle cells44, in which the conserved c-fos CArG box had more SRF binding activity than the CArG boxes of SM α-actin promoter, while myocardin-induced promoter activity was prominently reduced in the CArG boxes of SM α-actin promoter with c-fos CArG box substitution. Noticeably, our results also revealed that conserved cis elements - ATTA in the SM α-actin promoter and CCAAT box in the cytoplasmic γ-/β-actin actin promoters mediated myocardin-induced actin promoter activity since mutation of these conserved elements prominently damped myocardin-induced promoter activity in all three actin promoter activity (Fig. 6E, F). Thus, the CArG-A and B boxes as well as the ATTA box provided myocardin-induced complete SM α-actin promoter activity. Unlike SM α-actin as a myocardin regulatory gene was reported in previous studies32, myocardin-induced upregulation of cytoplasmic γ-actin and β-actin were not previously described. Our results showed that the CCAAT box was likely more important than the CArG box for cytoplasmic γ-actin promoter activity; while the CArG box appeared to be more important than the CCAAT box for β-actin promoter activity (Fig. 6B, E). Interestingly, myocardin-induced promoter activity between cytoplasmic γ-actin and β-actin promoters was similar (Fig. 6B), but more prominently increased expression of cytoplasmic γ-actin mRNA and protein was observed compared to β-actin (Fig. 1, 5). Our data highlight the complicated nature of myocardin-induced regulation of actin isoforms and further raise the possibility that posttranscriptional regulation might be important in actin isoform expression in activated HSCs.
Previous investigation revealed that MRTF/A was involved in regulation of type 1 collagen expression in liver wound healing34. Our results demonstrated that myocardin also plays an important role in regulation of type 1 collagen expression in activated HSCs in vitro (Fig. 7) and in vivo (Fig. 3). TGFβ signaling is critical to mediate type 1 collagen expression in various fibrotic conditions45. Unlike MRTF/A, which did not increase p-Smad246, our data revealed that myocardin prominently elevated p-Smad2 level (Fig. 7C), suggesting that myocardin was able to signal to the TGFβ pathway. Notably, a dominant negative myocardin decreased type 1 collagen expression and all three actin isoforms without an effect on p-Smad2, suggesting an indirect effect of TGFβ signaling through myocardin-mediated actin expression (Fig. 7B, C). Furthermore, our results suggested that myocardin was also able to affect ET-1 signaling via regulating Erk phosphorylation and SRF expression (Fig. 7D), which is another important pathway in type 1 collagen expression since COL1α2 promoter has both AP-1 and SRF binding sites47.
The Rho pathway plays an important role in actin polymerization5,36. Our results indicated that myocardin stimulates RhoA expression (Fig. 7F), which is likely the molecular mechanism for myocardin-induced actin stress fiber formation (Fig. 2, 7E) and functional features such as enhanced contractility in HSCs (Fig. 4)36. Indeed, basal Rho activity was required to maintain a normal actin dynamics and type 1 collagen expression, which was documented by suppression of Rho signaling with Y27632 (Fig. 7E, F). However, exogenous expression of myocardin upregulated type 1 collagen production and all actin isoforms as well as RhoA levels in presence of Y27632, suggesting a feedback pathway from myocardin to RhoA via actin and actin polymerization36. It is likely that myocardin-induced Rho/actin signals may be resulted from heterodimerization with MRTF-A through its conserved leucine zipper (LZ) domain at least in part20 since HSCs express both myocardin and MRTF-A.
Additionally, our results showed that myocardin was able to directly regulate type lα1 and 1α2 collagen transactivation via CArG boxes in the promoters (Fig. 8D, E). Although myocardin, as an SRF cofactor, was previously reported to regulate SRF target genes23,32, our results for the first time showed that myocardin was able to regulate SRF expression (Fig. 8A,B), which provided a positive feed back regulatory loop from myocardin to SRF, and SRF to SRF target genes (Fig. 8F).
In summary, we have demonstrated that myocardin promotes HSC activation and myofibroblast differentiation. The CArG box, together with ATTA and/or CCAAT boxes, in the actin promoters primarily mediate myocardin-induced differential expression of actin isoforms, which make up the activated HSC cytoskeleton. We further show that myocardin also stimulates type 1 collagen transcription, enhances SRF expression, which appears to be a critical mechanism to rapidly enrich SRF expression in activated HSCs. Thus, myocardin is responsible for morphological and functional changes that occur during HSC activation and myofibroblast differentiation, and raise the possibility that this pathway could be targeted therapeutically.
Supplementary Material
Acknowledgments
We thank Eric Olson (UT Southwestern, TX) for myocardin expression plasmids (dominant active and negative version). We also thank Yingyu Ren for hepatic stellate cell preparation and Mudan Ren for assistance with immunofluorescence staining.
Disclosure/Conflict of Interest
This work was supported by NIH grant R01 DK098819. The authors declare no competing or financial interests.
Abbreviations
- HSC
hepatic stellate cell
- SM α-actin
smooth muscle α-actin
- Cyto-γ-actin
cytoplasmic gamma actin
- Col.1
type 1 collagen
- Myocd
myocardin
- SRF
serum response factor
- Ad
adenovirus
- Ad-ctr
adenovirus control
- DN
dominant negative
- HA
hemagglutinin
- TGFβ
transformation growth factor beta
- ET-1
endothelin-1
- RhoA
Ras homolog gene family, member A
- Smad2
Similarity to the Drosophila gene Mothers Against Decapentaplegic (Mad)2
- Erk
extracellular-signal-regulated kinase
- AP-1
activator protein 1
- RPA
RNase protection assay
- CCl4
carbon tetrachloride
- EMSA
electrophoretic mobility shift assay
- RT-PCR
real-time PCR
REFERENCES
- 1.Heng YW, Koh CG. Actin cytoskeleton dynamics and the cell division cycle. The international journal of biochemistry & cell biology 2010;42(10):1622–1633. [DOI] [PubMed] [Google Scholar]
- 2.Gardel ML, Schneider IC, Aratyn-Schaus Y, et al. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol 2010;26:315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rockey DC, Weymouth N, Shi Z. Smooth muscle alpha actin (Acta2) and myofibroblast function during hepatic wound healing. PloS one 2013;8(10):e77166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arteriosclerosis, thrombosis, and vascular biology 2011;31(7):1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandbo N, Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Translational research : the journal of laboratory and clinical medicine 2011;158(4):181–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochimica et biophysica acta 2007;1773(5):642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology 2014;15(3):178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinz B, Phan SH, Thannickal VJ, et al. The myofibroblast: one function, multiple origins. The American journal of pathology 2007;170(6):1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brenner DA, Kisseleva T, Scholten D, et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis & tissue repair 2012;5(Suppl 1):S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct 2003;28(2):105–112. [DOI] [PubMed] [Google Scholar]
- 11.Perrin BJ, Ervasti JM. The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 2010;67(10):630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunnell TM, Ervasti JM. Structural and functional properties of the actin gene family. Critical reviews in eukaryotic gene expression 2011;21(3):255–266. [DOI] [PubMed] [Google Scholar]
- 13.Bunnell TM, Burbach BJ, Shimizu Y, et al. beta-Actin specifically controls cell growth, migration, and the G-actin pool. Molecular biology of the cell 2011;22(21):4047–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunnell TM, Ervasti JM. Delayed embryonic development and impaired cell growth and survival in Actg1 null mice. Cytoskeleton (Hoboken) 2010;67(9):564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weymouth N, Shi Z, Rockey DC. Smooth muscle alpha actin is specifically required for the maintenance of lactation. Developmental biology 2012;363(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological reviews 2004;84(3):767–801. [DOI] [PubMed] [Google Scholar]
- 17.Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. American journal of physiology Cell physiology 2007;292(1):C70–81. [DOI] [PubMed] [Google Scholar]
- 18.Shi Z, Rockey DC. Interferon-gamma-mediated inhibition of serum response factor-dependent smooth muscle-specific gene expression. J Biol Chem 2010;285(42):32415–32424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danilition SL, Frederickson RM, Taylor CY, et al. Transcription factor binding and spacing constraints in the human beta-actin proximal promoter. Nucleic acids research 1991;19(24):6913–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parmacek MS. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circulation research 2007;100(5):633–644. [DOI] [PubMed] [Google Scholar]
- 21.Huang J, Wang T, Wright AC, et al. Myocardin is required for maintenance of vascular and visceral smooth muscle homeostasis during postnatal development. Proceedings of the National Academy of Sciences of the United States of America 2015;112(14):4447–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Wang DZ, Wang Z, et al. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proceedings of the National Academy of Sciences of the United States of America 2003;100(16):9366–9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Wang DZ, Pipes GC, et al. Myocardin is a master regulator of smooth muscle gene expression. Proceedings of the National Academy of Sciences of the United States of America 2003;100(12):7129–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada H, Ochi T, Imasato A, et al. Gene expression profiling and functional assays of activated hepatic stellate cells suggest that myocardin has a role in activation. Liver international : official journal of the International Association for the Study of the Liver 2010;30(1):42–54. [DOI] [PubMed] [Google Scholar]
- 25.Rockey DC, Boyles JK, Gabbiani G, et al. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. Journal of submicroscopic cytology and pathology 1992;24(2):193–203. [PubMed] [Google Scholar]
- 26.Gerolami R, Cardoso J, Bralet MP, et al. Enhanced in vivo adenovirus-mediated gene transfer to rat hepatocarcinomas by selective administration into the hepatic artery. Gene Ther 1998;5(7):896–904. [DOI] [PubMed] [Google Scholar]
- 27.Yu Q, Que LG, Rockey DC. Adenovirus-mediated gene transfer to nonparenchymal cells in normal and injured liver. American journal of physiology Gastrointestinal and liver physiology 2002;282(3):G565–572. [DOI] [PubMed] [Google Scholar]
- 28.Rehman H, Liu Q, Krishnasamy Y, et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. International journal of physiology, pathophysiology and pharmacology 2016;8(1):14–27. [PMC free article] [PubMed] [Google Scholar]
- 29.Li T, Shi Z, Rockey DC. Preproendothelin-1 expression is negatively regulated by IFNgamma during hepatic stellate cell activation. American journal of physiology Gastrointestinal and liver physiology 2012;302(9):G948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang D, Chang PS, Wang Z, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001;105(7):851–862. [DOI] [PubMed] [Google Scholar]
- 31.Mack CP, Hinson JS. Regulation of smooth muscle differentiation by the myocardin family of serum response factor co-factors. Journal of thrombosis and haemostasis : JTH 2005;3(9):1976–1984. [DOI] [PubMed] [Google Scholar]
- 32.Du KL, Ip HS, Li J, et al. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Molecular and cellular biology 2003;23(7):2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida T, Hoofnagle MH, Owens GK. Myocardin and Prx1 contribute to angiotensin II-induced expression of smooth muscle alpha-actin. Circulation research 2004;94(8):1075–1082. [DOI] [PubMed] [Google Scholar]
- 34.Tian W, Hao C, Fan Z, et al. Myocardin related transcription factor A programs epigenetic activation of hepatic stellate cells. Journal of hepatology 2015;62(1):165–174. [DOI] [PubMed] [Google Scholar]
- 35.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes & development 2006;20(12):1545–1556. [DOI] [PubMed] [Google Scholar]
- 36.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nature reviews Molecular cell biology 2010;11(5):353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. Journal of molecular and cellular cardiology 2014;70:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murata T, Arii S, Nakamura T, et al. Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of rat liver fibrosis in association with inactivation of hepatic stellate cells. Journal of hepatology 2001;35(4):474–481. [DOI] [PubMed] [Google Scholar]
- 39.Belaguli NS, Schildmeyer LA, Schwartz RJ. Organization and myogenic restricted expression of the murine serum response factor gene. A role for autoregulation. The Journal of biological chemistry 1997;272(29):18222–18231. [DOI] [PubMed] [Google Scholar]
- 40.Luchsinger LL, Patenaude CA, Smith BD, et al. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J Biol Chem 2011;286(51):44116–44125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wirz W, Antoine M, Tag CG, et al. Hepatic stellate cells display a functional vascular smooth muscle cell phenotype in a three-dimensional co-culture model with endothelial cells. Differentiation 2008;76(7):784–794. [DOI] [PubMed] [Google Scholar]
- 42.Sun KH, Chang Y, Reed NI, et al. alpha-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFbeta activation or collagen production across multiple models of organ fibrosis. American journal of physiology Lung cellular and molecular physiology 2016;310(9):L824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeji M, Moriyama T, Oseto S, et al. Smooth muscle alpha-actin deficiency in myofibroblasts leads to enhanced renal tissue fibrosis. J Biol Chem 2006;281(52):40193–40200. [DOI] [PubMed] [Google Scholar]
- 44.Hendrix JA, Wamhoff BR, McDonald OG, et al. 5’ CArG degeneracy in smooth muscle alpha-actin is required for injury-induced gene suppression in vivo. The Journal of clinical investigation 2005;115(2):418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2004;18(7):816–827. [DOI] [PubMed] [Google Scholar]
- 46.Velasquez LS, Sutherland LB, Liu Z, et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proceedings of the National Academy of Sciences of the United States of America 2013;110(42):16850–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Small EM, Thatcher JE, Sutherland LB, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circulation research 2010;107(2):294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.