

Abstract
Previously, we reported that Y6, a new epigallocatechin gallate derivative, is efficacious in reversing doxorubicin (DOX)--mediated resistance in hepatocellular carcinoma BEL-7404/DOX cells. In this study, we evaluated the efficacy of Y6 in reversing drug resistance both in vitro and in vivo by determining its effect on the adenosine triphosphate-binding cassette protein B1 transporter (ABCB1 or P-glycoprotein, P-gp). Our results showed that Y6 significantly sensitized cells overexpressing the ABCB1 transporter to anticancer drugs that are ABCB1 substrates. Y6 significantly stimulated the adenosine triphosphatase activity of ABCB1. Furthermore, Y6 exhibited a higher docking score as compared with epigallocatechin gallate inside the transmembrane domain of ABCB1. In addition, in the nude mouse tumor xenograft model, Y6 (110 mg/kg, intragastric administration), in combination with doxorubicin (2 mg/kg, intraperitoneal injection), significantly inhibited the growth of BEL-7404/DOX cell xenograft tumors, compared to equivalent epigallocatechin gallate. In conclusion, Y6 significantly reversed ABCB1-mediated multidrug resistance and its mechanisms of action may result from its competitive inhibition of the ABCB1 drug efflux function.
KEY WORDS: Epigallocatechin gallate (EGCG); 5,3′,4′,3″,4″,5″-6-O-ethyl-EGCG (Y6); Drug resistance; Resistance reversal; ABCB1; P-gp
Graphical abstract
Y6, an epigallocatechin gallate derivative, reverses ABCB1-mediated multidrug resistance in vitro and in vivo, and the reversal effect of Y6 is significantly greater than epigallocatechin gallate.
Highlights
-
•
Y6, an epigallocatechin gallate derivative, reverses ABCB1-mediated multidrug resistance in vitro and in vivo, and the reversal effect of Y6 is significantly greater than epigallocatechin gallate.
-
•
Y6 stimulates ABCB1 ATPase activity.
-
•
Y6 has a significantly higher docking score than epigallocatechin gallate, Y6 combined with doxorubicin suppressed the growth of resistant hepatocellular carcinoma tumors.
1. Introduction
The resistance of cancer cells to different structural classes of anticancer drugs is termed multidrug resistance (MDR), which is a major clinical obstacle in the treatment of cancer1., 2.. The mechanisms of MDR are complex and have been extensively discussed previously3., 4., 5.. The most common cause of MDR is the overexpression of adenosine triphosphate (ATP)-binding cassette (ABC) transporters, which use ATP to efflux their substrates (including a wide spectrum of anticancer drugs) and thereby decreasing their intracellular concentrations6., 7., 8.. The ABC transporters superfamily is divided into 7 subfamilies, ABCA to ABCG9., 10., 11.. The human ABCB1 (P-glycoprotein, P-gp), the first identified ABC transporter12., 13., has a molecular weight of 170 kDa. ABCB1 has two nucleotide-binding domains (NBDs) and two transmembrane binding domains (TMDs)14., 15.. The NBDs are responsible for binding and extruding physiological and xenobiotic substrates from the cytoplasm to the extracellular environment. The substrates of ABCB1 include anthracyclines (e.g., doxorubicin and daunorubicin), taxanes (e.g., paclitaxel and docetaxel), vinca alkaloids (e.g., vincristine and vinblastine), epipodophyllotoxins (e.g., teniposide and etoposide), among others1., 2., 11., 14.. The ABCB1 transporter has been reported to have multiple sites that bind and efflux different substrates from cells16., 17., 18., 19., 20..
MDR in cancer cells can be surmounted via a number of mechanisms. This can occur when compounds block the efflux function of certain ABC transporters, and increase the intracellular concentration of certain anticancer drugs21. Epigallocatechin gallate (EGCG), a compound from green tea polyphenols, is unstable due to rapid oxidation, low lipid solubility, low bioavailability, and short duration of action due to its many phenolic hydroxyl groups in the structure (Fig. 1A). Y6 (5,3′,4′,3″,4″,5″-6-O-ethyl-EGCG) is an ethylation product of EGCG with better stability (Fig. 1B). We have reported that Y6 was efficacious in reversing doxorubicin (DOX)-resistance in BEL-7404/DOX cells in vitro via decreasing the expression of the ABCB1 transporter22. Other previous studies have demonstrated that EGCG could reverses resistance to doxorubicin in BEL-7404/DOX hepatocellular cancer cells23, and EGCG could increase the accumulation of rhodamine 123 and daunorubicin via the inhibition of P-gp function in KB-C2 cells24. The results of these studies suggest that EGCG may reverse ABCB1-mediated MDR. However, it is unknown whether Y6 can also reverse MDR mediated by the ABCB1 transporter, and whether the reversal mechanisms are mediated by blocking the efflux function of the ABCB1 transporter.
Figure 1.

Chemical structures of EGCG and Y6.
Therefore, in the present study, we determined whether Y6 is efficacious in reversing drug resistance in ABCB1-transfected HEK293/ABCB1 cells. We also conducted in vitro experiments to ascertain whether the reversal mechanism of Y6 is associated with inhibiting the efflux function of the ABCB1 transporter. In addition, using an in vivo model, we also investigate the therapeutic effect of Y6 in mice xenografted with BEL-7404/DOX cells that overexpress ABCB1 transporters.
2. Materials and methods
2.1. Materials
EGCG (purity 97%) was purchased from Hangzhou Hetian Biotechnology Co., Ltd. (Hangzhou, Zhejiang, China). Y6 (purity 96.87%) was synthesized in our laboratory as previously described22. Doxorubicin, verapamil (VER), dimethylsulfoxide (DMSO) and 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazoliumbromide (MTT) were purchased from Sigma–Aldrich (St. Louis, MO, USA). The ATPase assay kit and membrane vesicles were purchased from BD Biosciences (San Jose, CA, USA). Dulbecco׳s modified Eagle׳s medium (DMEM), fetal bovine serum (FBS), trypsin 0.25% and penicillin/streptomycin were purchased from Hyclone (Thermo Scientific, Logan, UT, USA).
2.2. Cell culture
HEK293/pcDNA3.1 and HEK293/ABCB1 cells were established by transfecting HEK293 with either the empty pcDNA3.1 (HEK293/pcDNA3.1) DNA or vector containing the full length ABCB1 (HEK293/ABCB1), and were cultured in a medium containing 2 mg/mL of G41825. The doxorubicin-selected, ABCB1-overexpressing BEL-7404/DOX cells were kindly provided by the Department of Physiopathology, Guangxi Medical University (Nanning, Guangxi, China), and were cultured in a medium containing 2 mg/mL of doxorubicin22. All cells were cultured in flasks with DMEM supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin at 37 °C in a humidified incubator containing 5% CO2. The cells were harvested using 0.25% trypsin once they reached 80% confluence.
2.3. Cell viability assay
The viability of HEK293/pcDNA3.1 and HEK293/ABCB1 cells to anticancer drugs was measured using the MTT assay. Cells (5000 cells/well) were seeded in 96-well plates (160 μL/well) and cultured for 24 h. For the cytotoxicity experiment of Y6, 40 μL of varying concentrations of Y6 were added into each well to the final concentrations of: 100,30,10,3,1,0.3,0.1,0.03 μmol/L. They were incubated for 48 h at 37 °C. For the reversal experiment, Y6, EGCG, and the control modulator (verapamil) (20 μL/well) were added 1 h prior. Then, twenty microliters of different concentrations of chemotherapeutic drugs (doxorubicin or cisplatin) were added into the designated wells and incubated for 48 h at 37 °C. Subsequently, 20 μL of the MTT solution (4 mg/mL) was added to each well and incubated for an additional 4 h. The solution was discarded and 100 μL of DMSO were added to dissolve the formazan crystals. Finally, light absorbance was determined at 490 nm using the OPSYS microplate reader (Dynex Technology, Chantilly, VA, USA). Verapamil (1 μmol/L) was used as positive control inhibitors of ABCB1.
2.4. ABCB1 ATPase activity assay
The ATPase assay was performed as previously described18. The vanadate-sensitive ATPase activity of ABCB1 was measured in the presence of Y6 by an ATPase assay kit from BD Biosciences (San Jose, CA, USA). The membrane vesicles (20 μg protein/reaction) were incubated in the ATPase buffer with or without 400 μmol/L vanadate, at 37 °C for 5 min. Different concentrations of Y6 or EGCG (0–40 μmol/L) were separately added to the membrane vesicles. The membrane vesicles were incubated at 37 °C for 5 min. Ten microliters of a 25 mmol/L Mg-ATP solution were added to the membrane vesicles. The reaction was incubated at 37 °C for 20 min and then mixed with 100 μL of 5% SDS solution. Two hundred microliters of detection reagent were added to detect and quantify the amount of inorganic phosphate. The mix was incubated for 10 min at room temperature. The light absorption was detected at 880 nm using a spectrophotometer.
2.5. Molecular docking analysis
The human ABCB1 homology model, based on refined mouse ABCB1 (RCSB ID: 4M1M), was kindly provided by S. Aller and prepared as previously described26., 27.. The structures of the ligands Y6 and EGCG were built and prepared using our previous molecular modeling protocols28. The Surflex-dock docking algorithm and scoring functions have been previously described in detail29. Briefly, Surflex-dock generates a ‘Protomol’ using a surface shape-based method. The Protomol consists of a series of molecular fragments that characterizes the surface properties of the target active site, including steric effects and hydrogen bonding interactions. The ligands are aligned to the Protomol and each pose is scored based on hydrophobic and polar interactions between atoms. The best-scored pose of Y6 and EGCG was used for further analysis and graphic representation in Maestro (Schrödinger, Cambridge, MA). All computations were run on a 6-core Intel Xeon processor running the Linux operating system.
2.6. Animal experiments
Male athymic nude mice (BALB/CN-nu/nu, 4–6 weeks old, 16–20 g) were purchased from the experimental animal center of Guangxi Medical University (Nanning, China, license No. SCXK 2014-0002), and maintained under specific-pathogen-free conditions. All animals were maintained on an alternating 12 h light/12 h dark cycle with water and rodent chow available ad libitum. The BEL-7404/DOX cell mouse xenograft model was used, with a slight modification, as reported previously23. Briefly, BEL-7404/DOX cells (1.0×107 cells) were injected into the mice subcutaneously under the armpits. When the size of the subcutaneous tumors reached approximately 5 mm×5 mm, mice were randomized into four experimental groups with 8 mice per group. Group 1 was given Y6, 110 mg/kg p.o., every day, in combination with doxorubicin, 2 mg/kg i.p., every 4 days. Group 2 was given EGCG, 80 mg/kg p.o., every day, in combination with doxorubicin, 2 mg/kg i.p., every 4 days. The other three groups received either doxorubicin, 2 mg/kg i.p., every 4 days, an intragastric dose of Y6, 110 mg/kg, every day or an i.p. injection of a 0.9% solution of NaCl. The two perpendicular diameters (W and L, respectively) of the tumors were measured with a caliper every two days, as well as the weight of each mice. The tumor volume was calculated and determined using the equation:
| (1) |
where L represents the long diameter; W represents the diameter perpendicular to L.
The tumor growth curve was drawn according to the tumor volume and the time of implantation. All the mice were sacrificed by decapitation after 20 days of treatment.
2.7. Statistical analysis
The data were analyzed using a t-test method. All values represent the mean±SD. The a priori P value for significance was P<0.05.
3. Results
3.1. Y6 improves the chemosensitivity of HEK293/ABCB1 cells to doxorubicin treatment
The chemical structure of EGCG and Y6 is shown in Fig. 1. The MTT cytotoxicity assay was performed using HEK293/pcDNA3.1 and ABCB1-transfected HEK293/ABCB1 cells in order to select a non-toxic concentration of Y6 (Fig. 2A). The IC50 value of doxorubicin was 8.80 μmol/L in ABCB1-transfected HEK293/ABCB1 cells, which was significantly higher than the IC50 value for the parental HEK293/pcDNA3.1 cells (0.34 μmol/L, Table 1).
Figure 2.

Cytotoxicity of Y6 in HEK293/pcDNA3.1 and HEK293/ABCB1 cell lines. (A) Cytotoxicity of Y6 was evaluated in pair of parental and MDR cell lines: HEK293/pcDNA3.1 and HEK293/ABCB1. (B) and (C)The concentration–response curves of HEK293/pcDNA3.1 and HEK293/ABCB1 treated with doxorubicin alone and doxorubicin combined with verapamil, EGCG, or Y6. Points with error bars represent the mean ± SD. Three independent experiments were performed in triplicate.
Table 1.
Reversal effects of Y6 to ABCB1-mediated MDR in HEK293/pcDNA3.1 and HEK293/ABCB1 cell lines.
| Treatment | HEK293/pcDNA3.1 |
HEK293/ABCB1 |
||
|---|---|---|---|---|
| IC50±SDa (μmol/L) | RFb | IC50±SD (μmol/L) | RF | |
| Doxorubicin | 0.34±0.03 | [1.0] | 8.80±0.32 | [25.9] |
| + EGCG (1 μmol/L) | 0.30±0.08 | [0.9] | 1.43±0.08 | [4.2]* |
| + Y6 (1 μmol/L) | 0.35±0.01 | [1.0] | 1.03±0.05 | [3.0]*,# |
| + Y6 (2 μmol/L) | 0.29±0.05 | [0.9] | 0.62±0.09 | [1.8]* |
| + Verapamil (1 μmol/L) | 0.39±0.05 | [1.1] | 0.77±0.03 | [2.3]* |
| Cisplatin | 1.07±0.07 | [1.0] | 1.05±0.05 | [1.0] |
| + EGCG (1 μmol/L) | 0.92±0.01 | [0.9] | 1.01±0.01 | [1.0] |
| + Y6 (1 μmol/L) | 0.96±0.04 | [0.9] | 1.05±0.06 | [1.0] |
| + Verapamil (1 μmol/L) | 0.91±0.02 | [0.8] | 0.92±0.07 | [0.9] |
P < 0.05 versus no reversal agent group.
P < 0.05 versus doxorubicin–EGCG group
IC50 values are represented as mean ± SD of three independent experiments performed in triplicate.
Values represent the resistance fold (RF) calculated by dividing IC50 values of anticancer drug in HEK293/pcDNA3.1 and HEK293/ABCB1 cells in presence or absence reversal agent by the IC50 value of HEK293/pcDNA3.1 cells without reversal agent.
To determine the reversal effects of Y6 on the ABCB1 transporter, we measured drug cytotoxicity in the HEK293/pcDNA3.1 and ABCB1-overexpressing HEK293/ABCB1 cells, using doxorubicin in the presence of Y6 at the non-toxic doses of 1 and 2 μmol/L, as well as verapamil (1 μmol/L) and EGCG (1 μmol/L). At 1 or 2 μmol/L, Y6 had no significant effect on the viability of HEK293/pcDNA3.1 cells (Fig. 2B). However, 1 or 2 μmol/L of Y6 significantly increased the cytotoxicity of HEK293/ABCB1 cells to doxorubicin, a substrate of the ABCB1 transporter (Fig. 2C). The reversal efficacy of Y6 was similar to verapamil and EGCG and was concentration-dependent (Fig. 2C). Y6 was significantly more efficacious than EGCG in increasing the cytotoxicity of doxorubicin (Fig. 2C). Cisplatin, which is not an ABCB1 substrate, was equally cytotoxic in HEK293/pcDNA3.1 cells and drug-resistant HEK293/ABCB1 cells (Table 1). In contrast, EGCG, Y6, and verapamil did not significantly alter the IC50 values of cisplatin in HEK293/pcDNA3.1 and HEK293/ABCB1cells (Table 1). These results indicate that Y6 significantly reverses ABCB1-mediated drug resistance in HEK293/ABCB1 cells in a concentration-dependent manner, and its efficacy was greater than that of EGCG.
3.2. Y6 stimulates ABCB1 ATPase in a concentration-dependent manner
The ABCB1 transporter effluxes its substrates against a concentration gradient by using energy from ATP hydrolysis and thus, ATP consumption is an indicator of its ATPase activity30. To determine the effect of EGCG and Y6 on the ATPase activity of ABCB1, we measured ABCB1-mediated ATP hydrolysis in the presence of EGCG and Y6 at various concentrations ranging from 0 to 40 μmol/L. The results showed that ATPase activity increased with increasing concentrations of Y6, indicating that Y6 increased the ATPase activity of ABCB1 in a concentration-dependent manner (Fig. 3A). EGCG increased ATPase activity when its concentration changed from 0 to 10 μmol/L, and no further obvious increase in ATPase activity occurred when the concentration of EGCG was >10 μmol/L (Fig. 3B).
Figure 3.

Effect of EGCG and Y6 on the Vi-sensitive ABCB1 ATPase activity. The Vi-sensitive ATPase activities of ABCB1 in membrane vesicles were determined with different concentrations of EGCG and Y6 (0–40 μmol/L). Experiments were repeated for three times.
3.3. Docking analysis of Y6 in human ABCB1 homology model
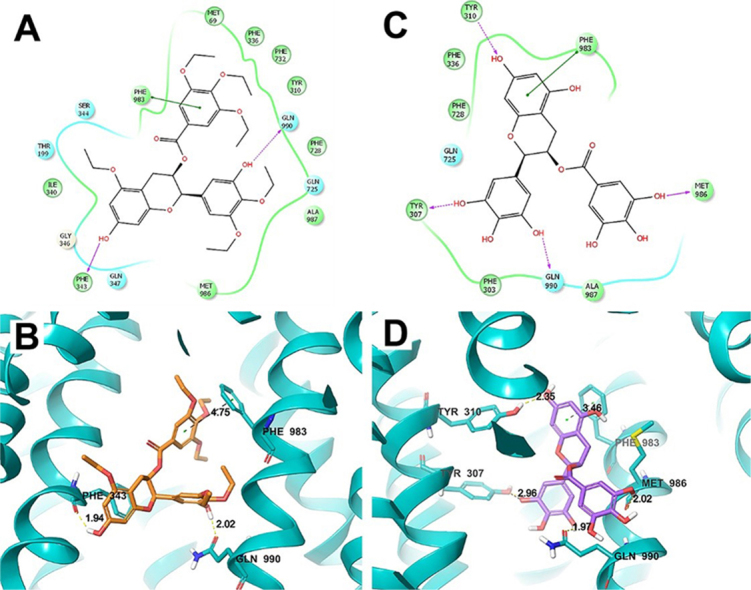
To determine whether Y6 and EGCG directly interact with ABCB1, we performed a molecular docking simulation analysis. The best-scored Y6 exhibited a total Surflex-dock score of –10.545. The docked pose of Y6 into the drug-binding cavity of human ABCB1 is shown in Fig. 4A and B. Y6 was stabilized by the nearby hydrophobic residues Met 69, Tyr310, Phe336, Ile340, Phe343, Phe728, Phe732, Phe983, Met986, and Ala 987. Two hydrogen bonding interactions were observed between the phenolic groups of Y6 and residues Phe 343 and Gln 990 (–OH··OC–Phe 343, 1.94 Å, –OH··CO–Gln 990, 2.02 Å). One phenyl group of Y6 formed a π–π interaction with a side-chain phenyl group of Phe 983 residue.
Figure 4.

Molecular modeling of the binding of Y6 and EGCG to ABCB1 homology. (A) A two-dimensional ligand-receptor interaction diagram with important interactions observed in the docked complex of Y6 with human ABCB1 is shown. The residues within 4 Å are shown as colored bubbles, cyan indicates polar, and green indicates hydrophobic residues. Hydrogen bonds are depicted by purple arrows, while π-stacking aromatic interactions are indicated with green lines. (B) A portion of the transmembrane region of the homology modeled human ABCB1 is shown in a ribbon presentation. Selected residues are depicted as tubes with the CPK coloring except carbon atoms are represented in blue, whereas the ligand is shown with the same color scheme as above except carbon atoms are represented in purple. (C) Two-dimensional ligand–receptor interaction diagram with important interactions observed in the docked complex of EGCG–ABCB1. (D) The docked conformation of EGCG. Color scheme is the same as panel (B) except carbon atoms of EGCG are presented in purple.
The best-scored EGCG exhibited a total Surflex-dock score of –6.707. The docked pose of EGCG into the large-binding cavity of human ABCB1 is shown in Fig. 4C and D. Four hydrogen bonds were formed between the ligand phenolic groups and nearby residues (–O··HO–Tyr 310, 2.35 Å; –OH··Tyr 307, 2.96 Å; –OH··Gln 990, 1.97 Å; –OH··OC–Met 986, 2.02 Å). The same π–π interaction between the ligand and the phenyl ring of Phe 983 with Y6 was formed with EGCG.
3.4. The efficacy of Y6 combined with doxorubicin on the growth inhibition in BEL-7404/DOX cell tumor xenograft mouse model
To determine the effect of Y6 combined with doxorubicin on the reversal of MDR in vivo, we prepared a mouse tumor xenograft using drug-resistant BEL-7404/DOX cells. Based on a previous study and a preliminary experiment23, the intraperitoneal dose of doxorubicin (2 mg/kg) was combined with the gavage dose of Y6 (110 mg/kg) or EGCG (80 mg/kg) in this study. After 20 days of treatment, there was significant no difference in tumor volumes between the control group and the Y6-gavage-treated group (Fig. 5A). However, tumor growth in mice was significantly inhibited by doxorubicin alone or by the combination of doxorubicin and Y6 or EGCG compared to the control (Fig. 5A). The doxorubicin–Y6 combination produced the greatest decrease in tumor growth compared to doxorubicin alone or the combination of doxorubicin and EGCG (#P<0.05, Fig. 5A). The tumor volume was reduced in mice treated with the doxorubicin–Y6 combination based not only in the final average tumor weight at the end of the experiment (Fig. 5B), but also in the appearance of the tumors (Fig. 5C). As shown in Fig. 5B, the average tumor weights of the doxorubicin group, doxorubicin + EGCG and doxorubicin + Y6 were significantly decreased compared to the control group. In addition, the tumor weight in the doxorubicin + Y6 group was significantly lower than that of doxorubicin alone and doxorubicin + EGCG (Fig. 5B). There were no significant differences in the loss of body weight of the treatment groups compared to animals treated with saline. The intergroup comparisons of the body weights are shown in Fig. 5D.
Figure 5.

Antitumor effect of doxorubicin combined with Y6 on BEL-7404/DOX cell tumor xenograft in nude mice. (A) Tumor volume change in the 20 days of treatment. Each data point represents a mean±SD (n=8). *P<0.05 versus control group; #P<0.05 versus doxorubicin-treated group. (B) Tumor weights after 20 days of treatment. Error bars, SD. *P<0.05 versus control group, #P<0.05 versus doxorubicin-treated group. (C) The tumor tissues from the nude mice after 20 days of treatment. (D) The mean body weight of mice (n=8) before and after 20 days of treatment. Error bars, SD. *P<0.05 versus the body weight before subcutaneous injection of BEL-7404/DOX cells.
4. Discussion
Our previous study showed that Y6 was effective in inhibiting cell proliferation on doxorubicin resistant BEL-7404/DOX cells22. In this study, we showed that the non-toxic concentrations of Y6 resensitized the treatment of doxorubicin in ABCB1-transfected HEK293/ABCB1 cells, indicating that Y6 was effective in reversing resistance on HEK293/ABCB1 cells. In addition, the reversal effect of Y6 was stronger than that of EGCG.
The mechanisms of drug resistance are complex in tumor cells not only down-regulating expression of ABCB1 but also inhibiting the transport function of ABCB1 can reverse the resistance to chemotherapeutic drugs. In the previous experiment, Y6 was proven to down-regulate the expression of ABCB122. But no published data reported whether Y6 affects efflux of drug substrates from cytoplasm to extracellular matrix by utilizing ATP-driven energy. In the present study, we investigated the effect of Y6 on the ATPase activity and affinity to ABCB1 transporter.
The substrate transport process of ABCB1 was described in previous studies. Substrate enters into the internal drug-binding pocket, then ATP binding and hydrolysis at the NBDs causes ABCB1 conformational change and discharges the substrate and the drug-binding sites to the outer leaflet20., 31., 32.. In this process, the anticancer drugs or reversing agents, such as ATP modulators, could bind to ATP-binding in NBD as a non-competitive inhibitor or bind to the drug-binding pocket in TMD as a competitive inhibitor. ATP modulators are divided into three classes: Class I compounds stimulate ATPase activity at low concentrations but inhibit the activity at high concentrations; Class II agents stimulate ATPase activity in a concentration-dependent manner; Class III agents inhibit ATPase activity33. Interestingly, Y6, as a derivative of EGCG, had a distinct stimulatory effect on ATPase activity from that of EGCG. Y6 strongly stimulated the ATPase activity of ABCB1 in a dose-dependent manner and therefore is a Class II agent. Meanwhile, in the case of EGCG, the ATPase activity was stimulated by EGCG in a dose-dependent manner at a low concentration, and then leveled off when EGCG reached a certain concentration. This result indicated that Y6 had a stronger stimulatory efficacy on ATPase activity of ABCB1 than EGCG. When drugs bind to the drug-substrate-binding site in TMD of the ABCB1 transporter, it would stimulate ATPase hydrolytic activity to support energy. A stronger stimulatory efficacy was likely to mean strong competitive substrate bind to drug-substrate-binding site.
Molecular docking analysis is a tool used to calculate the binding affinity of a protein--ligand complex. In the molecular docking study, Y6 exhibited a much higher docking score (–10.545) than that of EGCG (–6.707). The binding cavity in the transmembrane domain of human ABCB1 consists of many hydrophobic and aromatic residues. A positive correlation between ABCB1 inhibitory activity and lipophilicity of the compounds has already been suggested34. Therefore, although more hydrogen bonds can be formed between EGCG and ABCB1, an excess of polar phenolic groups in EGCG may hinder the binding of ligand inside the binding cavity. Based on the above, Y6, unlike EGCG, was a competitive substrate, as it interacted with drug--substrate-binding site of the ABCB1 transporter and stimulated the ATPase activity of ABCB1. This could explain why the reversal effect of Y6 was significantly greater than EGCG in vitro. Overall, the results demonstrate that reversal of MDR by Y6 is likely due to its interactions with the ABCB1 transporter, similar to other known typical competitive substrates, such as doxorubicin, that reverse resistance.
In the xenograft mice implanted with BEL-7404/DOX cells, the in vivo anti-MDR efficacy of Y6 was determined by measuring the volume and weight of the tumors. The results indicated that Y6, in combination with doxorubicin, significantly decreased tumor growth compared to treatment with saline, doxorubicin, or EGCG plus doxorubicin. Furthermore, along with the increased weight in mice after finishing treatment, Y6 alone, or in combination with doxorubicin, did not produce significant observable toxicity or body weight loss during the experimental period. This result indicated that the effect of Y6 in reversing MDR also works in vivo. This result provides the rationale for using Y6 in future clinical trials.
In conclusion, this study demonstrated that Y6 reversed ABCB1-mediated MDR both in vitro and in vivo. The mechanisms of Y6 in reversing MDR involved the inhibition efflux function of ABCB1 and being a typical competitive substrate of ABCB1. These results suggest that Y6 may have the potential to be a reversal agent when combine with conventional anticancer drugs to re-sensitize tumor chemotherapy.
Acknowledgments
The authors would like to acknowledge Dr. Xiaocong Kuang (College of Basic Medicine, Guangxi Medical University, Nanning, China) for providing the BEL-7404/DOX cells. We thank Dr. Stephen Aller (The University of Alabama at Birmingham, Birmingham, US) for kindly providing human ABCB1 homology model. We thank Dr. Tanaji T. Talele (St. John׳s University, New York, NY, US) for providing the computational resources for molecular modeling, and Drs. Charles R. Ashby Jr. (St. John׳s University, Queens, NY, US) and Yangmin Chen (MediMedia Managed Markets, an ICON company, Yardley, PA, US) for reviewing and editing the paper. This work was supported by the National Natural Science Foundation of China (No. 81160532), the Open Project of Guangxi Colleges and Universities Key Laboratory of Biological Molecular Medicine Research (No. GXBMR201602, China), the Young and Middle-aged Teachers Foundation Ability Enhancement Project of Guangxi Colleges and Universities (No. 2018KY0102, China), and US NIH (No. 1R15CA143701).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Zhe-Sheng Chen, Email: chenz@stjohns.edu.
Gang Liang, Email: lianggang22@aliyun.com.
References
- 1.Gottesman M.M., Fojo T., Bates S.E. Multidrug resistance in cancer. Role of ATP-dependent transporters. Nat Rev Cancer. 2002;92:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 2.Sodani K., Patel A., Kathawala R.J., Chen Z.S. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer. 2012;31:58–72. doi: 10.5732/cjc.011.10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottesman M.M. Mechanisms of cancer drug resistance. Ann Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 4.Fan Y.F., Zhang W., Zeng L., Lei Z.N., Cai C.Y., Gupta P. Dacomitinib antagonizes multidrug resistance (MDR) in cancer cells by inhibiting the efflux activity of ABCB1 and ABCG2 transporters. Cancer Lett. 2018;421:186–198. doi: 10.1016/j.canlet.2018.01.021. [DOI] [PubMed] [Google Scholar]
- 5.An X., Sarmiento C., Tan T., Zhu H. Regulation of multidrug resistance by microRNAs in anti-cancer therapy. Acta Pharm Sin B. 2017;7:38–51. doi: 10.1016/j.apsb.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borths E.L., Locher K.P., Lee A.T., Rees D.C. The structure of Escherichia coli BtuF and binding to its cognate ATP binding cassette transporter. Proc Nati Acad Sci U S A. 2002;99:16642–16647. doi: 10.1073/pnas.262659699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Locher K.P. Structure and mechanism of ABC transporters. Curr Opin Struct Biol. 2004;14:426–431. doi: 10.1016/j.sbi.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Locher K.P., Borths E. ABC transporter architecture and mechanism: implications from the crystal structures of BtuCD and BtuF. FEBS Lett. 2004;564:264–268. doi: 10.1016/S0014-5793(04)00289-3. [DOI] [PubMed] [Google Scholar]
- 9.Dean M., Allikmets R. Complete characterization of the human ABC gene family. J Bioenerg Biomembr. 2001;33:475–479. doi: 10.1023/a:1012823120935. [DOI] [PubMed] [Google Scholar]
- 10.Dean M., Annilo T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu Rev Genom Hum Genet. 2005;6:123–142. doi: 10.1146/annurev.genom.6.080604.162122. [DOI] [PubMed] [Google Scholar]
- 11.Schinkel A.H., Jonker J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 12.Juliano R.L., Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 13.Ueda K., Cornwell M.M., Gottesman M.M., Pastan I., Roninson I.B., Ling V. The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem Biophys Res Commun. 1986;141:956–962. doi: 10.1016/s0006-291x(86)80136-x. [DOI] [PubMed] [Google Scholar]
- 14.Tiwari A.K., Sodani K., Dai C.L., Ashby C.R., Jr, Chen Z.S. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol. 2011;12:570–594. doi: 10.2174/138920111795164048. [DOI] [PubMed] [Google Scholar]
- 15.Wu C.P., Hsieh C.H., Wu Y.S. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8:1996–2011. doi: 10.1021/mp200261n. [DOI] [PubMed] [Google Scholar]
- 16.Roninson I.B., Chin J.E., Choi K.G., Gros P., Housman D.E., Fojo A. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc Natl Acad Sci U S A. 1986;83:4538–4542. doi: 10.1073/pnas.83.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Bliek A.M., Baas F., Ten Houte de Lange T., Kooiman P.M., van der Velde-Koerts T., Borst P. The human mdr3 gene encodes a novel P-glycoprotein homologue and gives rise to alternatively spliced mRNAs in liver. EMBO J. 1987;6:3325–3331. doi: 10.1002/j.1460-2075.1987.tb02653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Bruijn M.H., Van der Bliek A.M., Biedler J.L., Borst P. Differential amplification and disproportionate expression of five genes in three multidrug-resistant Chinese hamster lung cell lines. Mol Cell Biol. 1986;6:4717–4722. doi: 10.1128/mcb.6.12.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gros P., Raymond M., Bell J., Housman D. Cloning and characterization of a second member of the mouse mdr gene family. Mol Cell Biol. 1988;8:2770–2778. doi: 10.1128/mcb.8.7.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aller S.G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C.P., Calcagno A.M., Ambudkar S.V. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr Mol Pharmacol. 2008;1:93–105. doi: 10.2174/1874467210801020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen Y., Zhao R.Q., Zhang Y.K., Gupta P., Fu L.X., Tang A.Z. Effect of Y6, an epigallocatechingallate derivative, on reversing doxorubicin drug resistance in human hepatocellular carcinoma cells. Oncotarget. 2017;8:29760–29770. doi: 10.18632/oncotarget.15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang G., Tang A., Lin X., Li L., Zhang S., Huang Z. Green tea catechins augment the antitumor activity of doxorubicin in an in vivo mouse model for chemoresistant liver cancer. Int J Oncol. 2010;37:111–123. [PubMed] [Google Scholar]
- 24.Kitagawa S., Nabekura T., Kamiyama S. Inhibition of P-glycoprotein function by tea catechins in KB-C2 cells. J Pharm Pharmacol. 2004;56:1001–1005. doi: 10.1211/0022357044003. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H., Zhang Y.K., Wang Y.J., Kathawala R.J., Patel A., Zhu H. WHI-P154 enhances the chemotherapeutic effect of anticancer agents in ABCG2-overexpressing cells. Cancer Sci. 2014;105:1071–1078. doi: 10.1111/cas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J., Jaimes K.F., Aller S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y.K., Zhang G.N., Wang Y.J., Patel B.A., Talele T.T., Yang D.H. Bafetinib (INNO-406) reverses multidrug resistance by inhibiting the efflux function of ABCB1 and ABCG2 transporters. Sci Rep. 2016;6:25694. doi: 10.1038/srep25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y.K., Zhang H., Zhang G.N., Wang Y.J., Kathawala R.J., Si R. Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents. Oncotarget. 2015;6:24277–24290. doi: 10.18632/oncotarget.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain A.N. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y.J., Kathawala R.J., Zhang Y.K., Patel A., Kumar P., Shukla S. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem Pharmacol. 2014;90:367–378. doi: 10.1016/j.bcp.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauna Z.E., Ambudkar S.V. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A. 2000;97:2515–2520. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sauna Z.E., Ambudkar S.V. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein the two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J Biol Chem. 2001;276:11653–11661. doi: 10.1074/jbc.M011294200. [DOI] [PubMed] [Google Scholar]
- 33.Ambudkar S.V., Dey S., Hrycyna C.A., Ramachandra M., Pastan I., Gottesman M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter 1. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 34.Pajeva I.K., Globisch C., Wiese M. Combined pharmacophore modeling, docking, and 3D QSAR studies of ABCB1 and ABCC1 transporter inhibitors. Chem Med Chem. 2009;4:1883–1896. doi: 10.1002/cmdc.200900282. [DOI] [PubMed] [Google Scholar]