

Abstract
Lipid-based formulations (LBFs) are a delivery strategy to enhance intestinal absorption of poorly water-soluble drugs. LBF performance is typically evaluated by in vitro lipolysis studies, but these do not accurately predict the in vivo performance. One possible reason is the absence of an absorptive membrane driving sink conditions in the serosal compartment. To explore the impact of absorption under sink conditions on the performance evaluation, we developed a lipolysis-permeation setup that allows simultaneous investigation of intestinal digestion of an LBF and drug absorption. The setup consists of two chambers, an upper one for digestion (luminal), and a lower, receiving one (serosal), separated by a Caco-2 monolayer. Digestions were performed with immobilized lipase, instead of the pancreatic extract typically used during lipolysis, since the latter has proven incompatible with Caco-2 cells. Danazol-loaded LBFs were used to develop the setup, and fenofibrate-loaded LBFs were used to establish an in vitro in vivo correlation. As in regular lipolysis studies, our setup allows for the evaluation of (i) the extent of digestion and (ii) drug distribution in different phases present during lipolysis of drug-loaded LBFs (i.e., oil, aqueous, and solid phase). In addition, our setup can determine drug permeation across Caco-2 monolayers and hence, the absorptive flux of the compound. The presence of the absorptive monolayer and sink conditions tended to reduce aqueous drug concentrations and supersaturation in the digestion chamber. The drug transfer across the Caco-2 membrane accurately reflected in vivo drug exposure upon administration of three different LBFs loaded with fenofibrate, where the traditional lipolysis setup failed to predict in vivo performance. As the new setup reflects the dynamic processes occurring in the gastrointestinal tract, it is a valuable tool that can be used in the development of LBFs prior to in vivo studies.
Keywords: intestinal digestion, absorption, lipid-based formulation, Caco-2, in vitro in vivo correlation
Introduction
Oral administration is the most convenient and least expensive route of drug administration. Unfortunately, oral absorption of an increasing number of drug candidates is limited due to unfavorable biopharmaceutical properties. To permeate the intestinal wall, a drug needs to be in solution, as described by Fick’s first law. However, contemporary drug candidates are often highly lipophilic, and ∼70% of all drug candidates show insufficient solubility to allow complete absorption.1,2 Therefore, a variety of drug delivery strategies have been developed to overcome the solubility issues in oral drug administration.3 Of these, lipid-based formulations (LBFs) promote intestinal absorption by increasing the drug concentration at the absorptive site. Compounds in LBFs are typically predissolved in the formulation, which allows for delivery to the gastrointestinal (GI) tract in solution. The presence of excipients can further increase the solvation capacity (mainly through solubilization) of GI fluids.4,5
Currently, less than 4% of commercial drug products are available as LBFs.6 A complicating factor in the development of LBFs is the selection of appropriate excipients due to poor predictions of their in vivo performance.4,7,8 The standard in vitro lipolysis assay, used to assess the behavior of LBFs in the GI tract, mimics the digestion process in the intestine. During this assay, the LBF is dispersed in simulated intestinal fluids and digested with lipases. Free fatty acids, liberated during the digestion of triglycerides, are titrated with NaOH, which allows for the calculation of the extent of digestion. In addition, aqueous concentrations, considered to be available for absorption, can be determined in the digestion medium.4,9
However, intestinal digestion changes the composition of GI fluids and therefore the intraluminal solvation capacity. In many cases, this generates supersaturation of the drug,3,10 which triggers its precipitation. The in vitro lipolysis assay likely underestimates the in vivo exposure of dissolved drug, as the system does not capture its absorption. The presence of physiologically relevant sink conditions would provide an alternative to the thermodynamically unstable supersaturated state. It therefore has the potential to improve in vitro predictions.4,11
Experiments evaluating in vitro lipolysis and permeation separately but consecutively have been performed to predict intestinal absorption of drugs from LBFs. Predigested LBFs were applied onto either an artificial membrane12 or intestinal rat tissue.13 An alternative to these experiments has been the use of a biopharmaceutical (mathematical) model. This model predicts the effect of an absorption sink, on the basis of data obtained from in vitro lipolysis experiments.14
Despite providing useful information, none of these studies capture the complex dynamics between the release of compounds during digestion and their flux across an absorptive membrane. Therefore, Crum et al. have coupled the in vitro lipolysis assay to an in situ intestinal perfusion in rats, which allows for simultaneous evaluation of digestion and absorption.15 This in-line method has been useful in predicting absorption from different formulations, but is time-consuming and animal-based; therefore, it is primarily suited for mechanistic studies.15,16
The purpose of the present study was to develop a physiologically relevant in vitro method that allows digestion and absorption to occur simultaneously. This would allow real-time observations of rapidly changing intraluminal conditions at a relatively high throughput. Since Caco-2 monolayers are widely used as a predictive tool for the intestinal absorption of drug candidates, these were used in our setup as an absorptive membrane.17 We have previously identified conditions that enable the use of this cell-based system for studies of drug absorption during in vitro lipolysis of LBFs. Importantly, immobilized lipase is used to digest the LBFs instead of the commonly used pancreatic extract, since the latter has proven to be incompatible with cells.18 In vitro data obtained with the new assay using immobilized lipase and a receiver chamber were validated with in vivo literature data of drug absorption after administration of various LBFs to landrace pigs.19
Materials and Methods
Materials
All culture media and supplements were purchased from Invitrogen AB (Sweden). Novozym 435 (immobilized lipase) was obtained from Strem Chemicals (France), and fasted state simulated intestinal fluid (FaSSIF) powder was obtained from biorelevant.com (UK). Maisine 35–1 was a kind gift from Gattefossé (France), and Captex 355 and Capmul MCM were kind gifts from Abitec (WI, USA). Danazol was purchased from Euroasia’s Group of Companies (Mumbai, India). All other compounds were purchased from Sigma-Aldrich (MO, USA).
Lipid-Based Formulations
Six LBFs (herein abbreviated as F1–F6) that (i) represented different classes of the Lipid Formulations Classification System (F1–F3), (ii) were shown to be compatible with Caco-2 cells prior to digestion (F1–F3),18 or (iii) had previously been studied in vivo (F4–F6)19 were prepared as described previously.20 Briefly, excipients were preheated (37 °C) and weighed into glass vials according to predefined fractions (Table 1). Subsequently, vials were sealed, vortexed, and placed on a shaker (300 rpm) at 37 °C for 24 h. Equilibrium solubility of danazol in F1–F3 was determined in triplicate as described previously (Table 1).20 F1–F3 were loaded with 80% of the equilibrium solubility determined for the formulation with the lowest equilibrium solubility (F1, 11.56 mg/g). For the in vitro in vivo correlation (IVIVC) study, the required amount of model compound and LBFs (37 °C) to produce F4–F6 were weighed into glass vials. Subsequently, the vials were sealed, vortexed, and placed on a shaker (300 rpm) at 37 °C for an additional 24 h.
Table 1. Composition and Loading of the LBFsa.
| LBF | type | composition | drug | solubility | Saturation (%) |
|---|---|---|---|---|---|
| (w/w) | mg/g | ||||
| F1 | II-LC | 32.5% soybean oil, 32.5% maisine 35–1, 35% Tween 85 | danazol | 14.45 ± 0.95 | 80.0 |
| F2 | IIIB-LC | 5% soybean oil, 45% Tween 85, 50% carbitol | danazol | 55.26 ± 2.06 | 21.0 |
| F3 | IV | 50% carbitol, 50% cremophor EL | danazol | 50.15 ± 1.15 | 23.1 |
| (w/v) | mg/mL19 | ||||
| F4 | IIIA-MC | 40% miglyol, 20% cremophor RH, 40% Tween 85 | fenofibrate | 143.8 ± 10.9 | 55.6 |
| F5 | IIIA-LC | 40% olive oil, 20% cremophor RH, 40% Tween 85 | fenofibrate | 96.6 ± 3.4 | 82.8 |
| F6 | IV | 33% cremophor RH, 67% Tween 85 | fenofibrate | 104.4 ± 7.7 | 76.6 |
Type indicates the type of formulation according to the Lipid Formulation Classification System.21 Saturation (%) denotes the percentage of the equilibrium solubility the formulation is loaded with. LC, long-chain triglycerides; MC, medium-chain triglycerides.
Cell Culture
Caco-2 cells (American Type Culture Collection, VA, USA) were cultivated, as described previously, in an atmosphere of 90% air and 10% CO2.17 Briefly, Caco-2 cells (passage 95 to 105) were seeded on permeable, polycarbonate filter supports (0.45 μm pore size, 75 mm diameter; Transwell Costar, Sigma-Aldrich) at a density of 170 000 cells/cm2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 1% minimum essential medium nonessential amino acids, penicillin (100 U/mL), and streptomycin (100 μg/mL). Monolayers were used for experiments on days 21 to 26 after seeding.
In Vitro Lipolysis
In vitro lipolysis was carried out in a standard glass reaction vessel (Metrohm AG) or in the lipolysis-permeation setup at 37 °C. The standard glass reaction vessel only allows for the evaluation of intestinal digestion but does not capture absorption. The lipolysis-permeation setup consists of two chambers separated by a Caco-2 monolayer. The upper chamber represents the lumen which is used to perform digestion studies. The lower chamber represents the serosal compartment and is used to determine drug absorption across the Caco-2 cells (Figure 1). The receiver chamber (185 mL) contained HBSS supplemented with 4% bovine serum albumin (pH 7.4).
Figure 1.
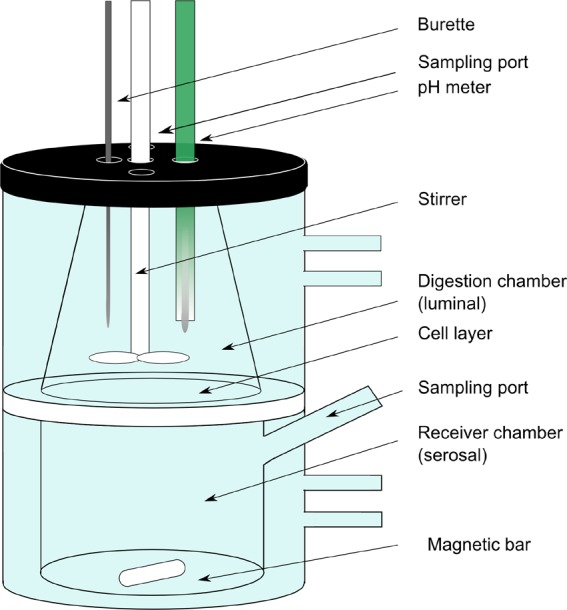
In vitro lipolysis-permeation setup.
Digestion studies, in the standard glass reaction vessel or in the upper chamber of the lipolysis-permeation setup, were performed as described previously with minor modifications.18 Briefly, LBF (1.5 g) was dispersed in 54–60 mL of digestion medium (pH 6.5), comprising 2 mM Tris-maleate, 1.4 CaCl2·2H2O, 150 mM NaCl, and FaSSIF powder (resulting in sodium taurocholate concentrations of 3.0 mM and lecithin concentrations of 0.75 mM). During a 10 min dispersion phase, the pH was manually adjusted to pH 6.5 ± 0.05. Digestion was initiated by addition of pancreatic extract (final concentration 900 USPU/mL) or immobilized lipase (final concentration 125 PLU/mL),18 resulting in a total volume of 60 mL. Pancreatic extract was prepared by mixing 1.6 g of pancreatin powder with 8 mL of lipolysis buffer containing 2 mM Tris-maleate, 1.4 mM CaCl2, and 150 mM NaCl (pH 6.5) followed by centrifugation at 144g and 5 °C for 15 min.22 The right amount of immobilized lipase was weighed and added to the digestion vessel directly. During digestion, a pH-stat (Metrohm 907 Titrando) was used to maintain a pH of 6.5 through titration with 0.2 and 0.6 M NaOH for long-chain/type IV and medium-chain LBFs, respectively.
Caco-2 Monolayer Integrity during in Vitro Lipolysis Studies
To evaluate the membrane integrity of the Caco-2 monolayers during digestion in the upper chamber of the lipolysis-permeation setup, blank LBFs were dispersed and digested in digestion medium spiked with 10 μM Lucifer Yellow. Samples were withdrawn from the receiver chamber at several time points during 3 h. The fluorescence signal in the receiver buffer was measured in a 96-well UV-plate reader (Tecan, Austria).
Drug Distribution during Dispersion and Digestion of Drug-Loaded LBFs
Drug-loaded LBFs were digested in the standard glass reaction vessel and in a lipolysis-permeation setup, i.e., in the presence and absence of the receiver chamber. At several time points (−5, 0, 5, 10, 15, 20, 30, 40, 50, 60, 70, 80, and 90 min), samples were withdrawn from both the digestion (1 mL) and the receiver (50 μL) chamber. Samples taken from the digestion chamber were treated with 5 μL/mL of lipase inhibitor (0.5 M 4-bromophenyl boronic acid in methanol) to inhibit further lipolysis. Subsequently, these samples were vortexed and centrifuged (21 000g at 37 °C for 15 min) to separate the three phases (i.e., oil, aqueous, and solid phases). The three phases were diluted 10–100-fold with acetonitrile, prior to further centrifugation (21 000g at 20 °C for 10 min), dilution (0–10 fold) in mobile phase, and HPLC-UV analysis (see the HPLC Analysis section). Samples withdrawn from the receiver chamber were diluted (1:3) with acetonitrile spiked with 50 nM warfarin as internal standard and quantified using UPLC-MS/MS (see the UPLC-MS/MS Analysis section).
Drug Solubility in the Aqueous Phase
Blank LBFs were dispersed and digested in the standard glass reaction vessel, and after 0, 5, 10, 30, and 60 min of digestion, triplicates of the aqueous phase were collected to determine drug solubility using a small-scale shake-flask method as described before.15 Briefly, an excess of crystalline drug was added, and samples were incubated at 37 °C and 300 rpm. At three time points within 24 h, samples were centrifuged (2300 g at 37 °C for 15 min), and supernatants were diluted 10–100-fold with acetonitrile, prior to further centrifugation (21 000g at 20 °C for 10 min), dilution (0–10 fold) in mobile phase, and HPLC-UV analysis (see the HPLC Analysis section).
HPLC Analysis
Analysis was conducted using an HPLC (Agilent Technologies 1290 Infinity) with a Zorbax Eclipse XDB-C18 column (4.6 mm × 100 mm) at 40 °C. The injection volume was 20 μL. The mobile phase consisted of acetonitrile:sodium acetate buffer (pH 5) 70:30 (v/v) for danazol and 80:20 (v/v) for fenofibrate; an isocratic flow rate was used at 1 mL/min. UV absorbance was monitored at a wavelength of 286 nm for danazol and 287 nm for fenofibrate. The retention times were 2.45 min for danazol and 3.04 min for fenofibrate. Calibration curves were used over a range between 0.78 and 100 μg/mL. Intraday validation with quality control samples (12.5–50 μg/mL) resulted in inaccuracy ranging from 2.44 to 4.79% and 3.59–4.94% and a repeatability (coefficient of variation, CV) of 0.77–1.10% and 0.14–0.37% for danazol and fenofibrate, respectively. The interday inaccuracy for the respective compounds was −2.11–5.99% and −1.79–2.27%, while interassay CV was 3.67–4.57% and 1.04–1.54%.
UPLC-MS/MS Analysis
UPLC-MS/MS analysis was performed using a Water Xevo TQ MS with electrospray ionization coupled to an Acquity UPLC system (Waters, Milford, MA). A Waters BEH C18 2.1 × 50 mm (1.7 μm) column was used for chromatographic separation. The mobile phase consisted of 5% acetonitrile and 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). Gradient elution was at a constant flow rate of 0.5 mL/min; 95% A decreased linearly to 10% from 0.5 to 1.2 min, followed by a constant flow of 10% A for 0.4 min and a linear increase back to 95% A at 1.7 min until the end of the run (2 min). The injection volume was 10 μL. The column oven and autosampler tray temperature were set at 60 and 10 °C, respectively. The mass spectrometer was operated in the positive electrospray mode for danazol, fenofibrate, and fenofibric acid, and in negative mode for warfarin (used as internal standard). The retention times of these compounds were 1.60, 1.68, 1.47, and 1.40 min, respectively. The precursor–product ion pairs followed were (i) m/z 338 → 148 (cone voltage 28 V and collision energy 22 V) for danazol; (ii) m/z 361 → 233 (cone voltage 20 and collision energy 16 V) for fenofibrate; (iii) m/z 319 → 139 (cone voltage 20 and collision energy 32 V) for fenofibric acid; and (iv) m/z 309 → 163 (cone voltage 22 and collision energy 14 V) for warfarin. Data acquisition and peak integration were performed with MassLynx software (Waters). Since fenofibrate is largely converted to fenofibric acid by hydrolytic enzymes present in Caco-2 cells, permeation for fenofibrate was calculated by including the amount of fenofibric acid appearing in the receiver chamber.23 Calibration curves were used over a range between 0.02 and 10 μg/mL. Methods were validated with quality control samples (0.16–2.50 μg/mL). Intraday and interday validation for danazol resulted in inaccuracy ranging from −2.78 to 8.62% and −9.33–11.25% and repeatability (CV) between 1.15 and 5.49% and 1.93–6.57%. For fenofibrate, intraday and interday validation resulted in inaccuracy ranging from −1.87 to 0.04% and −13.39–10.51% and repeatability (CV) between 2.85 and 5.46% and 3.52–10.9%. For fenofibric acid, intraday and interday validation resulted in inaccuracy ranging from −10.72 to 9.02% and −11.98–13.70% and repeatability (CV) between 1.88 and 6.97% and 4.13–10.16%.
Data Analysis
Data are presented as mean values with standard deviation (n = 3). Statistical analysis was performed in GraphPad Prism 7 (GraphPad Software, USA) using a student’s t-test to evaluate differences between two groups, or an one-way ANOVA followed by a Tukey’s multiple comparison analysis test, to compare differences for more than two groups. p-Values less than 0.05 were considered statistically significant.
The supersaturation ratio was used to evaluate the extent of supersaturation at several time points during the dispersion and digestion phases
To determine the extent of (i) drug distribution to the aqueous phase, (ii) supersaturation, (iii) precipitation, and (iv) permeation, the area under the curves was calculated for aqueous concentration-, supersaturation ratio-, fraction precipitated-, and mass transfer- vs time curves using the trapezoidal rule. In vivo data from the literature were extracted using Plotdigitizer 2.5.0. to evaluate the potential for this method to produce IVIVC.
Results
Conditions Enabling Digestion Studies in the Lipolysis-Permeation Setup
Immobilized Lipase To Digest LBFs
The extent of digestion and the drug distribution across the three phases (aqueous, oil, and solid) of the digestion medium were determined upon lipolysis of F1, F2, and F3 with both pancreatic extract and immobilized lipase. For all three formulations, the pancreatic extract resulted in a more complete digestion than the immobilized lipase (Figure S1). However, the rank order (F1 > F2 > F3) for the total free fatty acids titrated was the same for both enzymes, and it increased with increasing amount of lipids in the LBFs (Table 1).
The less complete digestion with the immobilized lipase was also reflected in the drug distribution across the three digestion phases. In the two lipid-containing formulations (F1 and F2), more danazol resided in the oil phase upon a 90 min digestion with the immobilized lipase than with pancreatic extract (Figure S2). Danazol distribution to the aqueous phase, i.e., the proportion of danazol assumed to be available for absorption, differed after digestion with the two enzymes. The rank order of the proportion of danazol in the aqueous phase after digestion with pancreatic extract was F1 < F2 = F3 and F1 < F3 < F2 with the immobilized lipase. However, since pancreatic extract is not compatible with Caco-2 cells, the immobilized lipase was used in the experiments with the lipolysis-permeation setup.
Membrane Integrity Caco-2 Monolayers
The integrity of the cell membrane over time was assayed from the permeation of Lucifer Yellow across the Caco-2 monolayer during the digestion of blank LBFs in the lipolysis-permeation setup. Figure S3 shows an increase in Lucifer Yellow permeation after approximately 45, 50, and 60 min of digestion of F1, F2, and F3, respectively. Interestingly, the pH of the digestion chamber started to increase around the same time, probably due to mixing of the buffers in the digestion (pH 6.5) and receiver (pH 7.4) chambers. As pH is continuously measured during digestion studies, a stable pH of 6.5 in the digestion chamber was used as an in-line indicator for membrane integrity. Samples withdrawn 15 min prior to an increase in the pH in the digestion chamber were discarded.
Effect of an Absorption Chamber
Danazol Solubilization during Dispersion and Digestion
In vitro lipolysis was performed in both the standard glass reaction vessel and the lipolysis-permeation setup to explore the impact of absorption under sink conditions on the performance evaluation. Aqueous concentrations of danazol in the digestion medium during dispersion and digestion of all test formulations were lower with a receiver chamber (Figure 2A–C), but the AUCs of the total concentration vs time profiles were not significantly affected (Figure 2D). The rank order of danazol exposure in the aqueous phase was F1 < F3 < F2 and F1 < F2 = F3 for experiments performed in the standard vessel and the lipolysis-permeation setup, respectively (Figure 2D).
Figure 2.
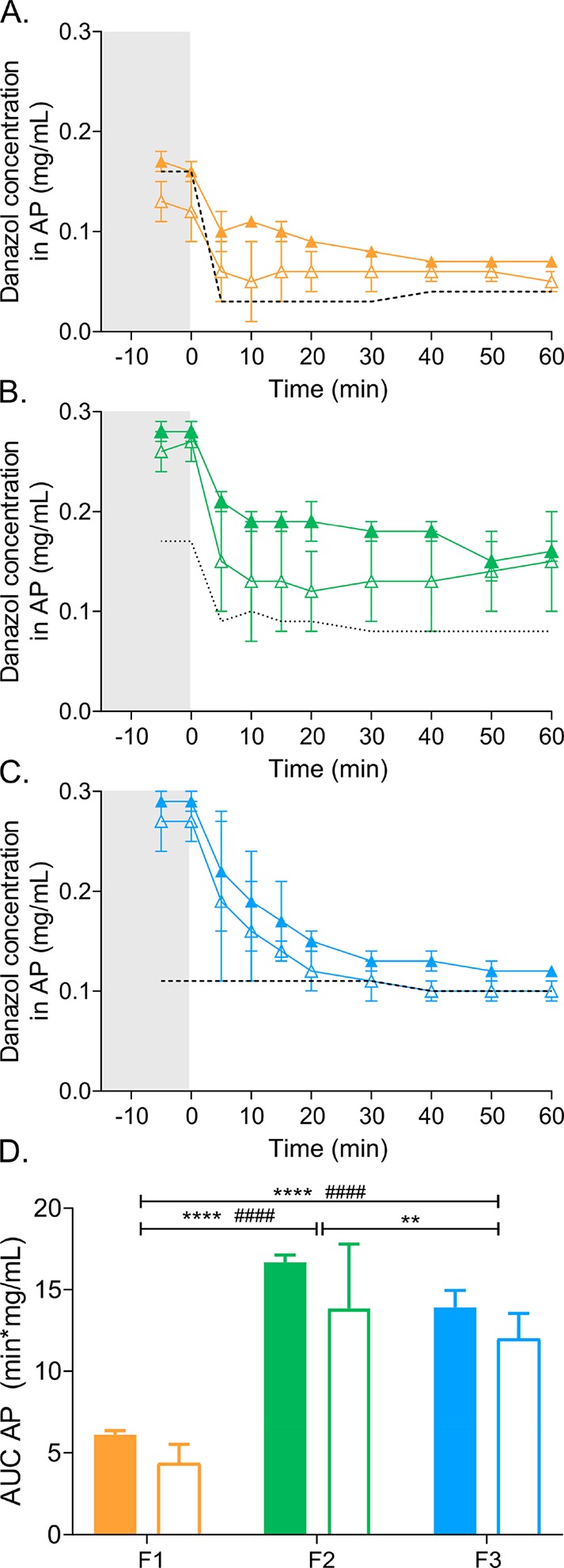
Danazol solubilization in the aqueous phase (AP). (A–C) Danazol concentrations in the aqueous phase determined during dispersion (gray shaded area) and digestion (white area) of F1 (A), F2 (B), and F3 (C), performed in the standard lipolysis vessel (closed symbols) or the lipolysis-permeation setup (open symbols). The dotted line indicates danazol solubility in the AP determined during dispersion and digestion of blank LBFs. (D) AUC of danazol concentrations in the aqueous phase vs time profiles (panel A–C) determined during the dispersion and digestion of F1, F2, and F3. Closed bars represent the AUC determined for lipolysis in the standard vessel and open bars for the lipolysis-permeation setup. Values are expressed as average values ± SD (n = 3). * and # represent significant differences between AUCs determined in the standard lipolysis vessel and the lipolysis-permeation setup, respectively: **** and ####p < 0.0001; ** p < 0.01.
The initiation of digestion (by introducing enzyme into the chamber at time = 0) decreased the aqueous danazol concentrations. The solubilized concentrations of danazol remained above the apparent solubility throughout the 60 min digestion for F1 and F2, indicating supersaturation (Figure 2A,B). During the digestion of F3, precipitation occurred more rapidly, leading to aqueous concentrations close to the apparent solubility, after approximately 30 min (Figure 2C). There was little difference in the apparent solubility of F3 over time, reflecting the low digestibility of this type IV formulation, which contains only surfactant and cosolvent but no glycerides.
The extent of supersaturation was calculated based on concentrations dissolved in the aqueous phase and apparent solubility (Figure 3A–C). Although the presence of a receiver chamber tended to decrease the supersaturation, the total supersaturation, illustrated by the AUCs of the supersaturation ratio vs time curves (Figure 3D), was not significantly lower than in the standard vessel. In the standard vessel, the rank order of total supersaturation was F3 < F1 < F2. There was no significant difference in total supersaturation between formulations in the lipolysis-permeation vessel (Figure 3D).
Figure 3.
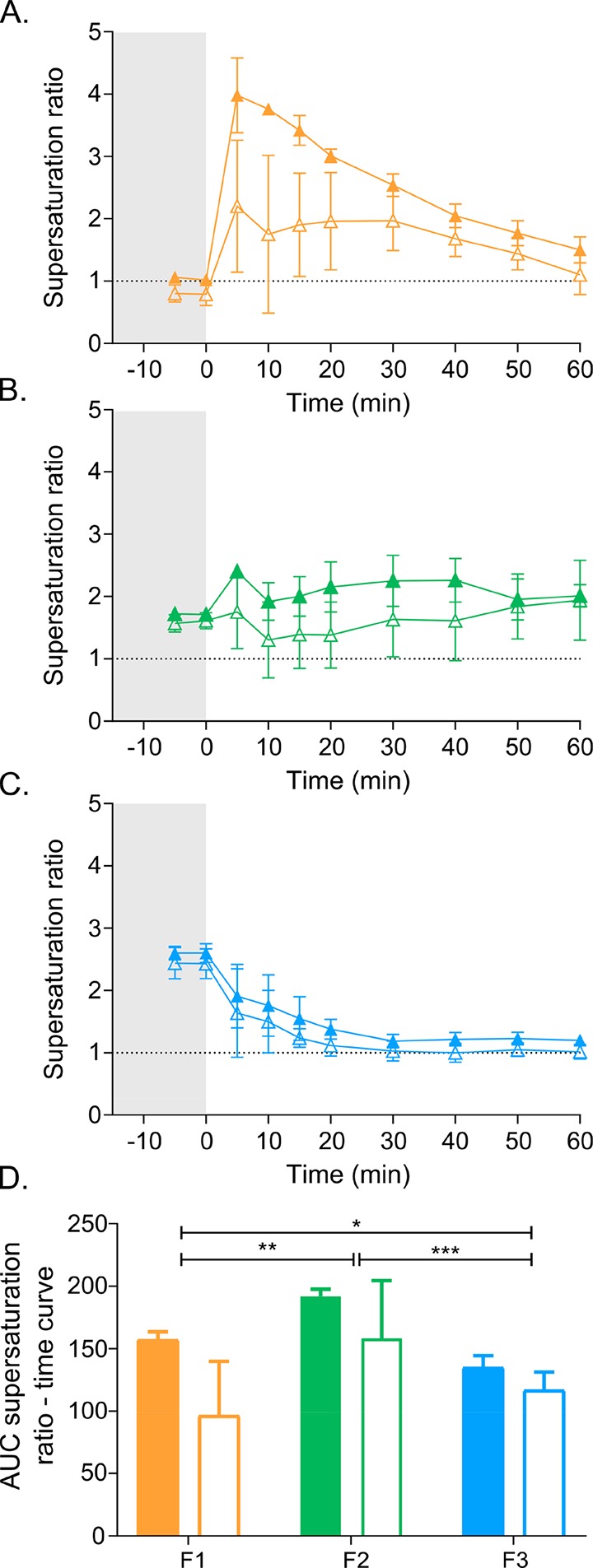
Danazol supersaturation ratios. (A–C) Supersaturation ratio determined during dispersion (gray shaded area) and digestion (white area) of F1 (A), F2 (B), and F3 (C), performed in the standard lipolysis vessel (closed symbols) or the lipolysis-permeation setup (open symbols). The dotted line indicates the SR of 1, the threshold for supersaturation. (D) AUC of the supersaturation ratio vs time profiles (panels A–C) determined during the dispersion and digestion of F1, F2, and F3. Closed bars represent the AUC for lipolysis in the standard vessel and open bars the lipolysis-permeation setup. Values are expressed as average values ± SD (n = 3). * represents a significant difference between AUC’s determined in the standard lipolysis vessel: *** p < 0.001; ** p < 0.01; * p < 0.05.
Danazol Precipitation during Dispersion and Digestion
The initiation of digestion triggered precipitation for all formulations (Figure 4). The total percentage of precipitated danazol was the same for both the standard vessel and the setup with a receiver chamber (Figure 4D). Approximately 15% of the total danazol precipitated during the first 5 min of digestion of F1 and F2 after which no further danazol precipitated (Figure 4A,B). Precipitation was more pronounced for F3 for which 65% danazol precipitated (Figure 4C).
Figure 4.

Danazol precipitation. (A–C) Danazol precipitation determined during dispersion (gray shaded area) and digestion (white area) of F1 (A), F2 (B), and F3 (C), performed in the standard lipolysis vessel (closed symbols) or the lipolysis-permeation setup (open symbols). (D) AUC of danazol precipitation vs time profiles (panel A–C) determined during the dispersion and digestion of F1, F2, and F3. Closed bars represent the AUC determined upon lipolysis in the standard vessel and open bars in the lipolysis-permeation setup. Values are expressed as average values ± SD (n = 3). * and # represent significant differences between AUCs determined in the standard lipolysis vessel and the lipolysis-permeation setup, respectively: **** p < 0.0001; ###p < 0.001; ##p < 0.01; and * p < 0.05.
Mass Transfer to the Receiver Chamber
The transfer of danazol across Caco-2 monolayers to the receiver chamber during dispersion and digestion of F1–F3 is depicted in Figure 5A. The total danazol transfer during digestion of F1 and F2 was similar, whereas it was significantly higher for F3 (F1 = F2 < F3, Figure 5B). This rank order was different from both the rank orders of aqueous exposure of danazol in the aqueous phase (F1 < F2 = F3) and total supersaturation (F1 = F2 = F3) in the lipolysis-permeation setup.
Figure 5.

(A) Mass transfer of danazol transfer across Caco-2 monolayers to the receiver chamber during dispersion (gray shaded area) and digestion (white area) of F1 (orange), F2 (green), and F3 (blue) in the lipolysis-permeation setup. (B) AUCs of danazol transfer vs time profiles. Values are expressed as average values ± SD (n = 3). #p < 0.05.
IVIVC
Griffin et al. have previously shown that in vitro digestion of F4–F6 in the standard vessel does not predict in vivo plasma exposure of fenofibrate in landrace pigs.19 They showed that fenofibrate exposure in the aqueous phase predicts a significantly lower exposure to fenofibrate with F6 than with F4 and F5, whereas plasma concentration vs time profiles after administration of the three formulations are similar (Figure 6A).
Figure 6.

Fenofibrate-loaded LBFs in the lipolysis-permeation setup. (A) IVIVC of in vivo plasma exposure and fenofibrate distribution to the aqueous phase in the digestion chamber of the lipolysis-permeation setup during dispersion and digestion. Blue, green, and red symbols represent results for F4, F5, and F6, respectively. The gray symbols represent the data from Griffin et al.19 (B) Fenofibrate transfer across monolayers to the receiver chamber during dispersion (gray shaded area) and digestion (white area) in the lipolysis-permeation setup. (C) IVIVC of in vivo plasma exposure and fenofibrate transfer across monolayers to the receiver chamber during dispersion and digestion. Values are expressed as average values ± SD (n = 3).
In the lipolysis-permeation setup, permeation across the Caco-2 membrane was significantly different for danazol-loaded F1–F3 (Figure 5). Furthermore, there was no correlation between the permeation and the aqueous concentrations of the model drug (Figures 2 and 5). Therefore, we repeated the digestion experiments with F4–F6 to evaluate the capacity of the lipolysis-permeation setup to predict in vivo exposure of fenofibrate. In agreement with findings in the previous study, aqueous fenofibrate concentrations in the digestion chamber were significantly higher upon digestion of F4 and F5 than of F6 (Figure 6A). Furthermore, no correlation was found between in vivo exposure in pigs and supersaturation ratio and precipitation in the digestion chamber (Figure S4). However, mass transfer of fenofibrate was similar for the three formulations (F4, F5, and F6, Figure 6B), thereby indicating that in vivo exposure of fenofibrate would also be similar (Figure 6C).
Discussion
The lack of a biologically relevant in vitro model containing sink conditions is a major reason for the poor prediction of the absorption enhancing capacity of LBFs in vivo.4,8,11,24 In the current study, we therefore developed the first in vitro lipolysis-permeation setup in which intestinal digestion and absorption studies (across a cell-based absorption membrane) are performed simultaneously. The lipolysis-permeation setup allows for the evaluation of digestion of LBFs, as well as solubilization, supersaturation, and permeation of model drugs, in a dynamic environment mimicking the GI tract. Absorption of fenofibrate across the absorptive membrane in this dynamic environment was similar for all three of the drug-loaded LBFs during the digestion, which is in agreement with the in vivo data from landrace pigs after oral administration.19
Caco-2 cells, the gold standard for intestinal permeation studies in vitro,17 were used as the absorptive membrane in this study. We have previously selected digestion conditions that are compatible with Caco-2 monolayers.18 Although pancreatic extract is commonly used in in vitro digestion studies, it is incompatible with Caco-2 cells. Therefore, the LBFs were digested with immobilized lipase (Novozym 435). The activity of the immobilized lipase, in concentrations that allow significant digestion and homogeneous stirring and sampling,18 is slightly lower than that of pancreatic extract (Figure S1). This results in variations in drug distribution across the different phases of the digestion medium (Figure S2). The differences in enzyme activity between immobilized lipase and pancreatic extract may be because immobilized lipase only contains a single enzyme, recombinant lipase B originating from Candida Antarctica. In contrast, pancreatic extract contains a mixture of enzymes.25 Moreover, immobilization of the lipase on polymeric beads possibly limits access of its active site to the triglycerides residing in the oil droplets.18 Nevertheless, the lower activity of immobilized lipase might still resemble in vivo digestion as the specific activity of human pancreatic lipase is up to 8000-fold lower in vivo than under optimized in vitro conditions.26
F1–F3 were used to develop and optimize the lipolysis-permeation setup. The three formulations were selected because the undigested LBFs (in concentrations of 2.5% (w/v) typically used in in vitro digestion studies22) are compatible with Caco-2 cells. In contrast, long exposure (up to 2 h) to digested formulations, containing mixtures of excipients, immobilized lipase, and free fatty acids, are not compatible.18 It was therefore important to identify a marker to evaluate integrity of the Caco-2 monolayer during the lipolysis-permeation experiment. A stable pH in the digestion chamber (pH 6.5) was used as an in situ marker for membrane integrity. Loss of integrity will result in mixing of the buffers in the digestion (pH 6.5) and receiver (pH 7.4) chamber (Figure S3).
Drug distribution across the different phases of the digestion media during the dispersion and digestion of F1–F3 depended on their composition but were largely independent of the presence of a receiver chamber. The concentration of danazol was relatively low in the aqueous phase during dispersion and digestion of F1 (Figure 2A,D), and precipitation was limited (Figure 4A). Most of this highly lipophilic compound (log P 4.9) resided in the oil phase; this is a significant phase during the digestion of the type II LBF because this formulation consists of 65% lipids.20,24 The amount of dissolved drug was higher than the equilibrium solubility during a large fraction of the experiment, indicating supersaturation during digestion of the F1 formulation (Figure 3A). Similarly, supersaturation was maintained during the digestion of F2, a type IIIB formulation containing 5% lipids (Figure 3B). Limited precipitation (Figure 4A,B) occurs during the digestion of these lipid-containing formulations due to the presence of triglycerides and their digestion products that form colloidal structures, enhancing the solubilizing capacity of the digestion media.19 Initial solubilization in the aqueous phase during dispersion of F2, a formulation consisting largely of cosolvent and surfactant, was relatively high and similar to that in the initial solubilization upon dispersion of F3 (Figure 2B–D). However, as F3 is an essentially nondigestible type IV formulation, the solubilization capacity was quickly lost (Figure 2C) due to miscibility of the excipients with the digestion medium. Miscibility induces a high degree of precipitation,9,27 and therefore, only transient supersaturation occurred during the experiments with F3 (Figure 3C).
The digestion chamber of the lipolysis-permeation setup was cone shaped (Figure 1) to optimize the absorption-surface-area-to-donor-volume ratio (A/V) while reaching sufficiently high levels of digestion medium to allow pH measurements and titration. Currently available in vitro methods that combine dissolution/release and permeation studies provide (i) small A/V-values (0.04–0.22 cm–1) and (ii) do not allow for evaluation of complex intestinal processes, including digestion.28−30 The A/V (0.47 cm–1) in our setup was relatively high; this allowed for the measurement of the extent of digestion as well as mass transfer while maintaining sink conditions (Figure 5). Precipitation was similar (Figure 4) in either the absence or presence of the receiver chamber. This was despite the fact that permeation to the receiver chamber of the lipolysis-permeation setup provided an alternative to precipitation during the thermodynamically unstable supersaturated state. In addition, the tendency toward lower aqueous concentrations (Figure 2) and supersaturation ratios (Figure 3) in the presence of the receiver chamber was not statistically significant. Despite the relatively large absorption surface area, limited amounts of danazol (<3% of total) permeated across the Caco-2 membrane during the time frame of the study. For highly supersaturated systems, the donor volume should perhaps be reduced. The current volume in the digestion chamber (60 mL) was initially selected to obtain a donor-volume-to-receiver-volume ratio of 1:3, which is what is commonly used in permeation studies.17 However, this volume can easily be decreased to 20 mL. This would result in an A/V of 1.41 cm–1 that more closely reflects that in the human small intestine (1.9–2.3 cm–1).31
The devised lipolysis-permeation model is the first in vitro setup that captures the numerous processes occurring in the continuously evolving environment of the GI tract, including solubilization, supersaturation, precipitation, and permeation. This is of utmost importance, as in vivo exposure upon oral administration of LBFs depends highly on the complex interplay between all of these processes. In vivo exposure depends on permeation, which is correlated to free aqueous concentrations of the compound at the level of the membrane and the apparent permeability of the drug. Removal of compound through permeation triggers the re-equilibration of drug across the different phases present in the intestine, i.e., drug from the oil phase, colloidal structures, or precipitated phase migrates toward the aqueous phase, which again affects the free concentration at the site of absorption and thus permeation.4,24
Aqueous drug concentrations in the digestion chamber did not predict permeation (Figures 2 and 5) because solubilization enhancing effects depend on micellization. During the digestion of F1 and F2, some of the danazol was encapsulated in aggregates formed by lipids and their digestion products. This encapsulation resulted in a solubility-permeability trade-off since the actual free concentration available for absorption is limited, resulting in relatively low permeation (Figure 5).32,33 Similar effects have been observed in vivo in humans.34 In contrast, cosolvents increase solubility without affecting the free fraction driving permeation (F3, Figure 5).35 However, they can affect permeability as their presence (i) reduces the thickness of the unstirred water layer and (ii) increases apparent aqueous solubility; this may decrease membrane permeability due to reduced membrane/aqueous partitioning.35
As an alternative to aqueous concentrations, supersaturation ratios have been used to predict permeation because they provide information on free drug concentrations.15,36 Supersaturation ratios are the ratios between aqueous drug concentrations and equilibrium solubility in the aqueous phase (see the Data Analysis section). In the absence of colloidal structures, increasing ratios indicate increasing free drug concentrations.36 Similarly, free drug concentrations increase with increasing supersaturation in the presence of colloidal structures, provided that colloidal partitioning remains unaltered.15,37 However, dispersion and digestion of different LBFs result in different colloidal structures and colloidal partitioning. Therefore, the supersaturation ratios obtained during digestion of F1–F3 (Figure 3) did not allow for direct comparison between the LBFs and did not predict drug permeation (Figure 5).
Direct measurement (in situ) of free drug concentrations in the dynamic environment of the digestion chamber was not possible. Commonly used separation techniques (e.g., centrifugation or filtration) likely impair the physical integrity of the oily droplets and colloidal structures and therefore alter the drug distribution. Moreover, these separation techniques only allow for the determination of aqueous concentrations and cannot distinguish between free drug and drug encapsulated in micellular/vesicular structures.24 Therefore, the presence of the receiver chamber during digestion was crucial for prediction of permeation based on free drug concentrations.
Indeed, permeation in the lipolysis-permeation setup during digestion of F4–F6 was in accordance with in vivo exposure of fenofibrate upon oral administration in landrace pigs, determined in a previous study by Griffin et al. (Figure 6C).19 Despite some differences in the digestion protocol (e.g., immobilized lipase vs pancreatic extract), the solubilization profiles in our digestion chamber were similar to those in their study, and in agreement with Griffin et al., predicted better in vivo performance for F4 and F5 than for F6 (Figure 6A).
However, the poor prediction of the in vivo exposure by aqueous fenofibrate concentrations can be explained by a solubility-permeability trade-off. Fenofibrate was encapsulated in triglyceride/digestion product-based colloidal structures, present during the digestion of F4 and F5. This encapsulation probably resulted in relatively low free fractions available for permeation, since permeation of fenofibrate to the receiver chamber was similar for all three formulations (Figure 6B). Interestingly, a study coupling an in situ intestinal perfusion in rat to the standard in vitro lipolysis assay produced similar results.15 Their solubilization profiles, obtained during digestion of two triglyceride containing LBFs, predicted higher plasma exposure than the profile obtained during digestion of a type IV formulation. However, fenofibrate absorption and flux into the mesenteric blood was similar for all three formulations.15 Although this in vitro in situ rat model is more biologically relevant (e.g., in terms of A/V and the intestinal microclimate), the complexity of the setup only makes it useful for mechanistic studies, whereas the model devised herein can be used as an in vivo relevant screening tool.
In addition to free drug concentrations, permeation also depends on apparent permeability.24 As mentioned before, cosolvents can have significant effects on apparent permeability.35 Permeation enhancing properties have been reported for other excipients and digestion products (e.g., free fatty acids). Therefore, differences in drug permeation during the digestion of several LBFs can also originate from the effect of digestion medium on the barrier properties of the Caco-2 monolayer.38 As dispersion, digestion, and absorption are occurring simultaneously, permeation effects are inherently studied in our setup.
Despite the accurate prediction of in vivo exposure of fenofibrate with the lipolysis-permeation setup, further research is required to optimize the setup and to study its applicability. For example, compounds other than the model drugs, e.g., bile salts and free fatty acids, are present in the digestion medium. Permeation of these compounds is of interest as their uptake changes the composition of the digestion medium, its solvation capacity, and thereby the free drug concentrations available for permeation. Since many of these compounds are transporter substrates, cell-based systems should be used as an absorptive membrane in studies with them, to accurately capture the continuously changing colloidal structures in the GI tract.39,40 The Caco-2 cell model may not be the most suitable system for these studies since the transporter expression of many transporters (e.g., the apical sodium dependent bile acid transporter) is lower in the model than what is observed in the small intestine.41,42 Other cell-based systems, e.g., MDCK cells, might be more appropriate because they (i) form larger paracellular pores, (ii) mature quicker, and (iii) can be transfected to study the uptake mediated by specific human transporters.43 MDCK cells can also be used to evaluate the inhibition of drug transporters (e.g., P-glycoprotein) by excipients commonly used in LBFs.44
Nevertheless, cell-based membranes in the lipolysis-permeation setup have some disadvantages. Despite their biological relevance, cell-based systems are relatively time-consuming membranes that require 1–3 weeks of culture before experiments can be performed.17,43 In addition, incompatibility of cells with the pancreatic enzyme and some LBFs or digestion products thereof can limit their applicability.18 Artificial membranes may be an alternative that can often be applied instantaneously.12
In addition to lipolysis-permeation studies of LBFs, the proposed setup (Figure 1) can be used to perform dissolution/release-permeation studies of other drug-delivery systems (e.g., amorphous solid dispersions and systems based on different drug carriers).3 As mentioned before, currently available in vitro methods that allow simultaneous evaluation of dissolution and permeation provide smaller A/V-values than the lipolysis-permeation setup. Moreover, the configuration of these systems often does not allow homogeneous stirring. They typically consist of two vertical cylinders connected by a perpendicular fitting that holds a vertically oriented membrane separating the cylinders in two chambers. Unfortunately, solid matter can accumulate near the absorptive membrane, due to the hydrodynamics of the geometry and stirrer configuration of this design. In contrast, our configuration with a horizontally oriented membrane and a cone-shaped digestion chamber (i) improves the hydrodynamics and (ii) allows for the use of a pH-stat to measure changes in pH as a result of digestion. It therefore provides a biologically relevant method that possibly allows for the direct comparison between different formulation strategies. This, in turn, facilitates the selection of the best strategies in an early stage of drug development.
Conclusion
This work introduces the first in vitro lipolysis-permeation setup that allows simultaneous evaluation of intestinal digestion and absorption across Caco-2 monolayers. Unlike the predictions from solubilization profiles, drug permeation across Caco-2 cells into a receiver chamber accurately reproduced in vivo absorption of fenofibrate for three drug-loaded LBFs in landrace pigs. Permeation depends highly on free drug concentrations at the site of absorption. The accurate predictions of in vivo exposure with the permeability data from our lipolysis-permeation setup therefore emphasize the importance of capturing the complex interplay between the multiple in vivo processes affecting free drug concentrations. The proposed setup provides a means to better understand drug absorption from the dynamic environment of the GI tract. Unlike the current empirical and iterative evaluations with suboptimal in vitro models, our model can provide a scientific rationale for the development of advanced drug delivery systems, such as LBFs, prior to in vivo studies.
Acknowledgments
This work has received support from the European Research Council Grant 638965 and the Swedish Research Council Grant 2014-3309.
Glossary
Abbreviations
- A/V
absorption-surface-area-to-donor-volume
- CV
coefficient of variation
- GI
gastrointestinal
- IVIVC
in vitro in vivo correlation
- LBF
lipid-based formulation
- LC
long-chain
- MC
medium-chain
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00811.
Apparent free fatty acid release during in vitro lipolysis of F1, F2, and F3 with pancreatic extract or immobilized lipase; danazol distribution across the aqueous, oil, and pellet phases following 90 min of digestion; pH in the digestion chamber and the lucifer yellow transfer across monolayers to the receiver chamber over time during an in vitro digestion of F1, F2, and F3 performed in the lipolysis-permeation setup; dissolved, supersaturated, precipitated, and permeated fenofibrate during lipolysis-permeation experiments with F4–F5 and the in vitro in vivo correlation between these parameters and plasma exposure in pigs (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Carrière F. Impact of Gastrointestinal Lipolysis on Oral Lipid-Based Formulations and Bioavailability of Lipophilic Drugs. Biochimie 2016, 125, 297–305. 10.1016/j.biochi.2015.11.016. [DOI] [PubMed] [Google Scholar]
- Bergström C. A. S.; Charman W. N.; Porter C. J. H. Computational Prediction of Formulation Strategies for Beyond-Rule-of-5 Compounds. Adv. Drug Delivery Rev. 2016, 101, 6–21. 10.1016/j.addr.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Fong S. Y. K.; Bauer-Brandl A.; Brandl M. Oral Bioavailability Enhancement through Supersaturation: An Update and Meta-Analysis. Expert Opin. Drug Delivery 2017, 14 (3), 403–426. 10.1080/17425247.2016.1218465. [DOI] [PubMed] [Google Scholar]
- Feeney O. M.; Crum M. F.; McEvoy C. L.; Trevaskis N. L.; Williams H. D.; Pouton C. W.; Charman W. N.; Bergström C. A. S.; Porter C. J. H. 50years of Oral Lipid-Based Formulations: Provenance, Progress and Future Perspectives. Adv. Drug Delivery Rev. 2016, 101, 167–194. 10.1016/j.addr.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Porter C. J. H.; Trevaskis N. L.; Charman W. N. Lipids and Lipid-Based Formulations: Optimizing the Oral Delivery of Lipophilic Drugs. Nat. Rev. Drug Discovery 2007, 6 (3), 231–248. 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- Hauss D. J. Oral Lipid-Based Formulations. Adv. Drug Delivery Rev. 2007, 59 (7), 667–676. 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Kollipara S.; Gandhi R. K. Pharmacokinetic Aspects and in Vitro–in Vivo Correlation Potential for Lipid-Based Formulations. Acta Pharm. Sin. B 2014, 4 (5), 333–349. 10.1016/j.apsb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas N.; Richter K.; Pedersen T. B.; Holm R.; Müllertz A.; Rades T. In Vitro Lipolysis Data Does Not Adequately Predict the In Vivo Performance of Lipid-Based Drug Delivery Systems Containing Fenofibrate. AAPS J. 2014, 16 (3), 539–549. 10.1208/s12248-014-9589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams H. D.; Anby M. U.; Sassene P.; Kleberg K.; Bakala-N’Goma J.-C.; Calderone M.; Jannin V.; Igonin A.; Partheil A.; Marchaud D.; et al. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations. 2. The Effect of Bile Salt Concentration and Drug Loading on the Performance of Type I, II, IIIA, IIIB, and IV Formulations during in Vitro Digestion. Mol. Pharmaceutics 2012, 9 (11), 3286–3300. 10.1021/mp300331z. [DOI] [PubMed] [Google Scholar]
- Brouwers J.; Brewster M. E.; Augustijns P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability?. J. Pharm. Sci. 2009, 98 (8), 2549–2572. 10.1002/jps.21650. [DOI] [PubMed] [Google Scholar]
- Gautschi N.; Bergström C. A. S.; Kuentz M. Rapid Determination of Drug Solubilization versus Supersaturation in Natural and Digested Lipids. Int. J. Pharm. 2016, 513 (1–2), 164–174. 10.1016/j.ijpharm.2016.09.015. [DOI] [PubMed] [Google Scholar]
- Bibi H. A.; Holm R.; Bauer-Brandl A. Simultaneous Lipolysis/Permeation in Vitro Model, for the Estimation of Bioavailability of Lipid Based Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2017, 117, 300–307. 10.1016/j.ejpb.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Dahan A.; Hoffman A. The Effect of Different Lipid Based Formulations on the Oral Absorption of Lipophilic Drugs: The Ability of in Vitro Lipolysis and Consecutive Ex Vivo Intestinal Permeability Data to Predict in Vivo Bioavailability in Rats. Eur. J. Pharm. Biopharm. 2007, 67 (1), 96–105. 10.1016/j.ejpb.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Stillhart C.; Imanidis G.; Griffin B. T.; Kuentz M. Biopharmaceutical Modeling of Drug Supersaturation During Lipid-Based Formulation Digestion Considering an Absorption Sink. Pharm. Res. 2014, 31 (12), 3426–3444. 10.1007/s11095-014-1432-1. [DOI] [PubMed] [Google Scholar]
- Crum M. F.; Trevaskis N. L.; Williams H. D.; Pouton C. W.; Porter C. J. H. A New in Vitro Lipid Digestion - in Vivo Absorption Model to Evaluate the Mechanisms of Drug Absorption from Lipid-Based Formulations. Pharm. Res. 2016, 33 (4), 970–982. 10.1007/s11095-015-1843-7. [DOI] [PubMed] [Google Scholar]
- Suys E. J. A.; Chalmers D. K.; Pouton C. W.; Porter C. J. H. Polymeric Precipitation Inhibitors Promote Fenofibrate Supersaturation and Enhance Drug Absorption from a Type IV Lipid-Based Formulation. Mol. Pharmaceutics 2018, 15 (6), 2355. 10.1021/acs.molpharmaceut.8b00206. [DOI] [PubMed] [Google Scholar]
- Hubatsch I.; Ragnarsson E. G. E.; Artursson P. Determination of Drug Permeability and Prediction of Drug Absorption in Caco-2 Monolayers. Nat. Protoc. 2007, 2 (9), 2111–2119. 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- Keemink J.; Bergström C. A. S. Caco-2 Cell Conditions Enabling Studies of Drug Absorption from Digestible Lipid-Based Formulations. Pharm. Res. 2018, 35 (4), 74. 10.1007/s11095-017-2327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin B. T.; Kuentz M.; Vertzoni M.; Kostewicz E. S.; Fei Y.; Faisal W.; Stillhart C.; O’Driscoll C. M.; Reppas C.; Dressman J. B. Comparison of in Vitro Tests at Various Levels of Complexity for the Prediction of in Vivo Performance of Lipid-Based Formulations: Case Studies with Fenofibrate. Eur. J. Pharm. Biopharm. 2014, 86 (3), 427–437. 10.1016/j.ejpb.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Alskär L. C.; Porter C. J. H.; Bergström C. A. S. Tools for Early Prediction of Drug Loading in Lipid-Based Formulations. Mol. Pharmaceutics 2016, 13 (1), 251–261. 10.1021/acs.molpharmaceut.5b00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouton C. W. Formulation of Poorly Water-Soluble Drugs for Oral Administration: Physicochemical and Physiological Issues and the Lipid Formulation Classification System. Eur. J. Pharm. Sci. 2006, 29 (3–4), 278–287. 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Williams H. D.; Sassene P.; Kleberg K.; Bakala-N’Goma J.-C.; Calderone M.; Jannin V.; Igonin A.; Partheil A.; Marchaud D.; Jule E.; et al. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of in Vitro Digestion Profiles across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101 (9), 3360–3380. 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- Linn M.; Collnot E.-M.; Djuric D.; Hempel K.; Fabian E.; Kolter K.; Lehr C.-M. Soluplus® as an Effective Absorption Enhancer of Poorly Soluble Drugs in Vitro and in Vivo. Eur. J. Pharm. Sci. 2012, 45 (3), 336–343. 10.1016/j.ejps.2011.11.025. [DOI] [PubMed] [Google Scholar]
- Bernkop-Schnürch A.; Jalil A. Do Drug Release Studies from SEDDS Make Any Sense?. J. Controlled Release 2018, 271, 55–59. 10.1016/j.jconrel.2017.12.027. [DOI] [PubMed] [Google Scholar]
- Phan S.; Salentinig S.; Hawley A.; Boyd B. J. Immobilised Lipase for in Vitro Lipolysis Experiments. J. Pharm. Sci. 2015, 104 (4), 1311–1318. 10.1002/jps.24327. [DOI] [PubMed] [Google Scholar]
- Carrière F.; Renou C.; Lopez V.; De Caro J.; Ferrato F.; Lengsfeld H.; De Caro A.; Laugier R.; Verger R. The Specific Activities of Human Digestive Lipases Measured from the in Vivo and in Vitro Lipolysis of Test Meals. Gastroenterology 2000, 119 (4), 949–960. 10.1053/gast.2000.18140. [DOI] [PubMed] [Google Scholar]
- Cuiné J. F.; McEvoy C. L.; Charman W. N.; Pouton C. W.; Edwards G. A.; Benameur H.; Porter C. J. H. Evaluation of the Impact of Surfactant Digestion on the Bioavailability of Danazol after Oral Administration of Lipidic Self-Emulsifying Formulations to Dogs. J. Pharm. Sci. 2008, 97 (2), 995–1012. 10.1002/jps.21246. [DOI] [PubMed] [Google Scholar]
- Kataoka M.; Masaoka Y.; Yamazaki Y.; Sakane T.; Sezaki H.; Yamashita S. In Vitro System to Evaluate Oral Absorption of Poorly Water-Soluble Drugs: Simultaneous Analysis on Dissolution and Permeation of Drugs. Pharm. Res. 2003, 20 (10), 1674–1680. 10.1023/A:1026107906191. [DOI] [PubMed] [Google Scholar]
- Mols R.; Deferme S.; Augustijns P. Sulfasalazine Transport in In-Vitro, Ex-Vivo and in-Vivo Absorption Models: Contribution of Efflux Carriers and Their Modulation by Co-Administration of Synthetic Nature-Identical Fruit Extracts. J. Pharm. Pharmacol. 2005, 57 (12), 1565–1573. 10.1211/jpp.57.12.0006. [DOI] [PubMed] [Google Scholar]
- Tsinman O.; Tsinman K.. Degree and Extent of Supersaturation of Amorphous Pharmaceuticals and Their Flux through Lipophilic Membranes. AAPS abstract, 2016.
- Mudie D. M.; Shi Y.; Ping H.; Gao P.; Amidon G. L.; Amidon G. E. Mechanistic Analysis of Solute Transport in an in Vitro Physiological Two-Phase Dissolution Apparatus. Biopharm. Drug Dispos. 2012, 33 (7), 378–402. 10.1002/bdd.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amidon G. E.; Higuchi W. I.; Ho N. F. H. Theoretical and Experimental Studies of Transport of Micelle-Solubilized Solutes. J. Pharm. Sci. 1982, 71 (1), 77–84. 10.1002/jps.2600710120. [DOI] [PubMed] [Google Scholar]
- Bevernage J.; Brouwers J.; Brewster M. E.; Augustijns P. Evaluation of Gastrointestinal Drug Supersaturation and Precipitation: Strategies and Issues. Int. J. Pharm. 2013, 453 (1), 25–35. 10.1016/j.ijpharm.2012.11.026. [DOI] [PubMed] [Google Scholar]
- Hens B.; Brouwers J.; Corsetti M.; Augustijns P. Gastrointestinal Behavior of Nano- and Microsized Fenofibrate: In Vivo Evaluation in Man and in Vitro Simulation by Assessment of the Permeation Potential. Eur. J. Pharm. Sci. 2015, 77, 40–47. 10.1016/j.ejps.2015.05.023. [DOI] [PubMed] [Google Scholar]
- Miller J. M.; Beig A.; Carr R. A.; Webster G. K.; Dahan A. The Solubility-Permeability Interplay When Using Cosolvents for Solubilization: Revising the Way We Use Solubility-Enabling Formulations. Mol. Pharmaceutics 2012, 9 (3), 581–590. 10.1021/mp200460u. [DOI] [PubMed] [Google Scholar]
- Bevernage J.; Brouwers J.; Annaert P.; Augustijns P. Drug Precipitation-Permeation Interplay: Supersaturation in an Absorptive Environment. Eur. J. Pharm. Biopharm. 2012, 82 (2), 424–428. 10.1016/j.ejpb.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Boyd B. J.; Porter C. J. H.; Charman W. N. Using the Polymer Partitioning Method to Probe the Thermodynamic Activity of Poorly Water soluble Drugs Solubilized in Model Lipid Digestion Products. J. Pharm. Sci. 2003, 92 (6), 1262–1271. 10.1002/jps.10390. [DOI] [PubMed] [Google Scholar]
- Aguirre T. A. S.; Teijeiro-Osorio D.; Rosa M.; Coulter I. S.; Alonso M. J.; Brayden D. J. Current Status of Selected Oral Peptide Technologies in Advanced Preclinical Development and in Clinical Trials. Adv. Drug Delivery Rev. 2016, 106, 223–241. 10.1016/j.addr.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Anderson C. M.; Stahl A. SLC27 Fatty Acid Transport Proteins. Mol. Aspects Med. 2013, 34 (2–3), 516–528. 10.1016/j.mam.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini K. M.; Huang S.-M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L. R.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discovery 2010, 9 (3), 215–236. 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ölander M.; Wisniewski J. R.; Matsson P.; Lundquist P.; Artursson P. The Proteome of Filter-Grown Caco-2 Cells With a Focus on Proteins Involved in Drug Disposition. J. Pharm. Sci. 2016, 105 (2), 817–827. 10.1016/j.xphs.2015.10.030. [DOI] [PubMed] [Google Scholar]
- Gröer C.; Brück S.; Lai Y.; Paulick A.; Busemann A.; Heidecke C. D.; Siegmund W.; Oswald S. LC-MS/MS-Based Quantification of Clinically Relevant Intestinal Uptake and Efflux Transporter Proteins. J. Pharm. Biomed. Anal. 2013, 85, 253–261. 10.1016/j.jpba.2013.07.031. [DOI] [PubMed] [Google Scholar]
- Volpe D. A. Drug-Permeability and Transporter Assays in Caco-2 and MDCK Cell Lines. Future Med. Chem. 2011, 3 (16), 2063–2077. 10.4155/fmc.11.149. [DOI] [PubMed] [Google Scholar]
- Fatouros D. G.; Mullertz A. In Vitro Lipid Digestion Models in Design of Drug Delivery Systems for Enhancing Oral Bioavailability. Expert Opin. Drug Metab. Toxicol. 2008, 4 (1), 65–76. 10.1517/17425255.4.1.65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.