

Conspectus

Porous nanostructures and materials based on metal-mediated self-assembly have developed into a vibrantly studied subdiscipline of supramolecular chemistry during the past decades. In principle, two branches of such coordination compounds can be distinguished: Metal–organic frameworks (MOFs) on the one side represent infinite porous networks of metals or metal clusters that are connected via organic ligands to give solid-state materials. On the other hand, metal–organic cages (MOCs) are discrete and soluble systems with only a limited number of pores. Formation of a particular structure type is achieved by carefully balancing the donor site angles within the ligands as well as the nature and coordination geometry of the metal component. Years of research on MOFs and MOCs has yielded numerous types of well-defined porous crystals and complex supramolecular architectures. Since various synthetic routes and postsynthetic modification methods have been established, the focus of recent developments has moved toward the preparation of multifunctional systems that are able to mimic the structural and functional complexity of natural enzymes.
This Account compares different strategies to prepare multifunctional MOFs and heteroleptic MOCs and gives a perspective on where to move forward. While the preparative toolbox for multifunctional MOFs is already quite mature, pore accessibility and substrate diffusion within the crystal have been identified as major challenges yet to be overcome. Only recently have a set of different strategies for the assembly of heteroleptic MOCs been developed. Such multifunctional cages can be formed from either partially protected or “naked” metal cations. Controlled assembly, producing single products rather than statistical mixtures, leans on assembly-dependent approaches making use of either steric effects or shape complementarity between the ligands. Further strategies include coordination-site engineering and hierarchical assembly of preformed components. The main challenge with heteroleptic, functional MOCs is to find a balance between the required dynamic assembly fidelity and the stability of the resulting system under operating conditions. If these limitations can be overcome in the future, chemists will be able to design multifunctional systems of similar activity and complexity as nature’s enzymes from simple and easily accessible synthetic building blocks. Major impacts on chemical sensing, small-molecule recognition and sequestration, drug delivery, and catalysis will be achieved by these materials.
1. Introduction
In recent years, the development of new materials and supramolecular architectures based on biology’s principles of hierarchical assembly, combining covalent and non-covalent interactions and embedding a multitude of orthogonal functionality, has experienced increasing attention.1−3 Natural evolution has tuned proteins to perform highly specific tasks such as molecular recognition, triggered signal transduction, and catalysis with high selectivity and turnover. Proteins constitute complex architectures with discrete pockets for the binding of substrates, chemical signals, and fuels. Enzyme pockets are typically asymmetric and highly functionalized with amino acid residues, giving rise to environments of specific shape, charge, and polarity. These discrete binding sites allow, for example, the selective recognition of small biomolecular signals, triggering consecutive processes. Specific redox and pH conditions significantly deviating from those of the surrounding medium are often established. Enzymatic catalysis is driven by factors such as substrate preorganization, proximity of embedded catalytic sites or cofactors and substrates, and directing effects by the surrounding protein structure. Metalloproteins contain mono- or multinuclear metal centers, usually featuring heteroleptic coordination environments, in a catalytic compartment that is protected by the protein shell.4 Synthetic chemists have been inspired by the structural and functional complexity of biocatalytic systems ever since their molecular features have been stepwise unraveled. Mimicking their capabilities by artificial constructs is regarded as a challenging aim. Thus, in parallel to the breathtaking progress in bioengineering and synthetic biology, fully human-made structures with bioinspired function and dynamics have been developed in the last decades, many of them belonging to the realm of bottom-up supramolecular chemistry (e.g., switchable rotaxanes and catenanes, unidirectional rotors, and molecular machines).5 Modular and dynamic self-assembly, often based on metal cations and organic ligands, has been used extensively in the preparation of these and related structures with relatively moderate synthetic efforts. While approaches toward the preparation of monofunctionalized architectures (containing only one type of bridging organic element) are highly advanced, strategies toward the controlled implementation of multiple functionalities, thus representing a further level of complexity, are still in their infancy. This Account picks up selected examples of two subdisciplines of metallo-supramolecular systems, namely, metal–organic frameworks (MOFs) and metal–organic cages (MOCs, also termed coordination cages), in a comparative manner with a focus on rational assembly strategies toward multifunctional structures and future application potential.
2. Mixed-Ligand Metal–Organic Frameworks
In the past decade, the potential of MOFs as enzyme mimics has been investigated in detail.6 MOFs are highly porous, heterogeneous solid-state materials providing channels and pores of a specific size that are available for the uptake of guests such as gases or small soluble molecules. They are usually built up from organic ligands, often with carboxylate or nitrogen donors, and multinuclear metal clusters. Typically, MOFs are prepared via solvothermal synthesis, where the organic building blocks and metal precursors are heated in a polar solvent such as DMF (Figure 1a). When complex metal clusters, such as Zr6O4(OH)4 in UiO-66 (UiO = University in Oslo; Zr6O4(OH)4(BDC)6, BDC = 1,4-benzenedicarboxylate), are used as building blocks, a modulator (e.g., benzoic acid) that assists in preassembly of the cluster is added to the reaction mixture. After formation of the cluster, the modulator is exchanged with the main ligand, and the MOF crystal grows step by step.7 Homoleptic MOFs have already shown high potential for applications such as gas storage,8 chemical sensing,9 drug delivery,10 and catalysis.11−13 In the context of enzyme mimicry,6 the preparation of mixed-ligand MOFs with multiple functions lining the cavities is emerging as a highly promising approach toward the implementation of fine-tuned reactivity.
Figure 1.

(a) Solvothermal synthesis from mixtures of bridging building blocks leading to mixed-ligand MOFs with statistical positioning of the components. (b) Simplified structure of MOF-5 with five different ligands shown by Yaghi.14
Different strategies have been used to introduce multiple functional ligands. Most commonly, mixed-ligand MOFs have been prepared by solvothermal synthesis from ligand mixtures (Figure 1a). Yaghi and co-workers demonstrated that up to eight BDC ligands with different side functions can be introduced into MOF-5 (Zn4O(BDC)3; Figure 1b).14 All of the functions were distributed statistically over the whole crystal. Another strategy, leading to more ordered structures, involves mixing ligands with different topologies that are incorporated into specific positions. UMCM-1 (UMCM = University of Michigan Crystalline Material; (Zn4O)9(BDC)6(BTB)5, BTB = 1,3,5-benzene-tri-4-carboxyphenyl), for instance, is constructed from tritopic BTB and ditopic BDC that are assembled in a predetermined relation (Figure 2a).15 Further examples have recently been summarized in a review by Yaghi and co-workers.16 A different approach is sequential ligand installation, reported in 2015 by Zhou and co-workers on the example of PCN-700 (PCN = porous coordination network; Zr6O4(OH)8(H2O)4(Me2BPDC)8, Me2BPDC = 2,2′-dimethylbiphenyl-4,4′-dicarboxylate) (Figure 2c).17 First, the MOF is constructed from Zr6O4(OH)8(H2O)4 clusters, each connected to eight Me2-BPDC ligands. The sterically bulky methyl groups force the two phenyl rings to adopt a perpendicular position relative to each other, resulting in a different structure than in the closely related UiO-67 (Zr6O4(OH)4(BPDC)6, BDPC = biphenyl-4,4′-dicarboxylate),7 in which 12 BPDC ligands are connected to each Zr6 cluster (Figure 2b). In PCN-700, two open pockets are formed, which can be postsynthetically filled with BDC and terphenyldicarboxylate (TPDC), respectively. Other approaches include the initial preparation of two-dimensional MOF sheets that are subsequently connected by a second ligand into three-dimensional bulk compounds (Figure 3a). This strategy is similar to the layer-by-layer method, in which a MOF is sequentially grown on a substrate by alternating treatment with the metal precursor and ligands.18 Both strategies allow the controlled introduction of different ligands that in principle can carry different functionalities.
Figure 2.

(a) UMCM-1, a mixed-ligand MOF prepared from tritopic BTB and ditopic BDC ligands. (b) UiO-67, consisting of only one type of ligand. (c) In the related PCN-700, bulky methyl substituents at the 2- and 2′-position lead to perpendicular positioning of the two phenyl rings. As a result, only eight ligands coordinate to each Zr6O6(OH)4 cluster, leaving two open coordination sites in the crystal structure. These open pockets are available for shorter BDC and longer TPDC ligands. Adapted from ref (17). Copyright 2015 American Chemical Society.
Figure 3.

Further examples of methods leading to mixed-ligand MOFs. (a) A 2D MOF sheet is grown first, followed by addition of the second ligand to form a 3D layered compound. (b) Postsynthetic ligand exchange or modification leads to statistical mixtures when diffusion inside the MOF is fast. When diffusion is slower than the exchange or modification process, a core–shell structure is obtained.
While small and robust functionalities such as amine groups can be introduced directly during solvothermal synthesis, more complex and labile functions are added by milder methods such as postsynthetic ligand exchange (PSE) or modification (PSM).19 In both, the balance between diffusion and reaction rate determines the outcome (Figure 3b). Matzger and co-workers tested PSE on three commonly used MOFs that are all based on BDC linkers: MOF-5, UiO-66, and UMCM-8 (Zn4O(BDC)1.5(naphthalene-2,6-dicarboxylate)1.5).20 The authors used the deuterated analogue BDC-d4 for PSE and investigated the resulting samples by Raman spectroscopy. In all three cases, core–shell structures were formed, where the ligand exchange happened at the surface of the particle. Matzger concluded that diffusion of the carboxylic acid is very slow, directing ligand exchange to occur at the outer shell of MOF crystals. In contrast to this, Ott and Primetzhofer used Rutherford backscattering spectrometry to investigate the exchange of BDC-I (I = iodine) within UiO-66.21 They found a homogeneous distribution over the whole crystal even after very short PSE times, indicating fast diffusion of the ligand and comparably slow exchange. The difference in the two observations is attributed to steric and electronic effects of iodine on the ligand exchange.
In recent years, MOFs have been considered as enzyme mimics, as they possess defined pores and channels similar to those of proteins. Pullen et al.22 utilized PSE to functionalize UiO-66 with [FeFe]-(dcbdt)(CO)6 (dcbdt = 1,4-dicarboxylbenzene-2,3-dithiolate), a member of the family of Fe2-hydrogenase active-site mimics that are proton reduction catalysts (Figure 4a). About 14% of the ligands were exchanged in the parent framework, indicating dispersion of the complex over the whole crystal. Incorporation of the catalyst yielded improved performance in photochemical hydrogen production in aqueous buffer solution with Ru(bpy)3Cl as a photosensitizer compared with homogenous [FeFe]-(dcbdt)(CO)6 in solution, which was attributed to stabilization of active catalyst species by the surrounding MOF. In a second study, an analogous complex, [FeFe]-(mcbdt)(CO)6 (mcbdt = 1-monocarboxylbenzene-2,3-dithiolate), was introduced to MIL-101(Cr)-NH2 (MIL = Matériaux de l’Institut Lavoisier; Cr3F(H2O)2O(BDC-NH2)3) via amide coupling at the BDC-NH2 ligands (Figure 4b).23 Improved performance in hydrogen production was observed in this system also. The main difference between UiO-66 and MIL-101 is the pore size (9 vs 29–34 Å, respectively). A direct comparison led to the conclusion that in MIL-101-[FeFe], all of the catalysts are in principle accessible and thus actively participate in hydrogen production, while in UiO-66-[FeFe] only the catalysts on the outer shell were available for reduction by Ru(bpy)3Cl. This study is prominent evidence that pore accessibility plays an important role in the application of MOFs. Accessibility strongly depends on substrate diffusion within the crystal as well as on the pore (window) size. Diffusion pathways increase with grain size, resulting in increasing discrimination of pores that are further inside. Even in mixed-ligand MOFs, functional sites are nonidentical in relation to their position within the crystal (Figure 5a). It should be noted, however, that MOFs are often not perfect crystals and contain defects or cracks, which might influence the pore accessibility. Based on this, a recently developed strategy for improving substrate diffusion within MOFs is the construction of hierarchically porous MOFs, for example, through ligand labilization or use of a modulator.24 Furthermore, a major challenge yet to be overcome is the difficulty of predicting the activity and selectivity of such systems. It is crucial to be able to study and understand the individual steps of these processes. A clear drawback of MOFs in this respect is their insolubility, which complicates the use of traditional solution-based methods such as NMR or advanced (transient) absorption spectroscopy. Both of these shortcomings may be tackled with small-size, soluble coordination cages, which are discussed in the next section.
Figure 4.

(a) Postsynthetic ligand exchange in UiO-66 with a structural mimic of the Fe2-hydrogenase active site. (b) Postsynthetic modification of BDC-NH2 in MIL-101 with a monocarboxylate derivative of the catalyst.
Figure 5.

Comparison of pore accessibility. (a) In MOFs, accessibility is dependent on grain size, pore window size, and diffusion within the MOF crystal. Pores deeply buried inside the crystal (red) are less accessible. Thus, incorporated functionalities in these positions are less likely to contribute to the overall activity of the material. (b) MOCs possess only a few pores, and accessibility depends only on host–guest and solvent interactions. Exchange of guests (e.g., anions, small molecules) is often a very dynamic process. MOCs can be viewed as the smallest possible MOF-like assemblies.
3. Design Principles for Assembly of Heteroleptic Metal–Organic Cages
Metal–organic coordination cages represent the smallest possible MOF-like assemblies featuring a limited number of pores.25 Metal-mediated assembly of homoleptic MOCs has already reached a high level of maturity, and structural characterization by NMR methods and single-crystal X-ray diffraction is straightforward. The preparation of such systems usually proceeds in the following manner: metal precursor and ligands are dissolved and heated until the desired cages have assembled as the thermodynamically most favorable products. Square-planar, diamagnetic palladium(II) has been used extensively, allowing cage assembly to be followed by NMR spectroscopy. In the case of most Pd-mediated assemblies, cage formation with nitrogen-donor ligands is finished after 1–24 h.26 Within the group of Pd-mediated cages, several examples demonstrated the versatility of dynamic ligand exchange. Among others, Yoshizawa and Clever showed cage-to-cage transformations within minute to hour time scales.27,28 In contrast, cages assembled from Pt(II), Ru(II), or Co(III) are kinetically more inert, resulting in substantially slower ligand exchange. The formation of MOFs, on the other hand, is controlled both by kinetic and thermodynamic factors. Reactions times of 24 h or more are common for MOFs because of their extended crystalline structures.
Heteroleptic coordination cages represent a new class of MOCs offering high potential for application in guest recognition, chemical sensing, and catalysis: the combination of a guest binding site with a second function such as chirality, a photosensitizing unit, proton or electron relays, or a catalyst may lead to complexity similar to that present in proteins. All of the components can be brought together in a modular, nonstatistical approach, allowing quick and easy tuning of the chemical environment in the cavity. Such systems not only allow the rational design and detailed examination of an outer coordination sphere around a functionality but also serve as model systems for larger MOFs and, merged with the latter concept, may in the future facilitate exploitation of advantages of both MOF and MOC chemistry. For these reasons, it is highly desirable to advance the methodology for the preparation, examination, and application of functionalized heteroleptic cages.
3.1. General Aspects
Numerous homoleptic cages have been prepared by means of metal-mediated self-assembly over the last decades. Within this overview, we mainly restrict the discussion to the use of banana-shaped ligands to prepare smaller M2L4 cages as well as large M12L24 spheres.29 One successful strategy for obtaining heteroleptic cages is the hierarchical assembly of cis-protected metal centers (e.g., Pd(en) or Pt(PR3)2; en = ethylenediamine) with a suitable set of donor ligands.30 On the other hand, also “naked” metal ions such as square-planar Pd(II) allow the rational formation of mixed-ligand Pd2L2L′2 cages when the right combination of ligands is employed.31 When Pd(II) ions and a mixture of two different bis-monodentate ligands are mixed, three potential outcomes can be expected: (1) narcissistic assembly leading to the formation of coexisting homoleptic cages, (2) formation of statistical mixtures of heteroleptic cages, or (3) assembly of a single heteroleptic species based on rational design. While the former two require further treatment and separation, the latter leads directly to a single desired heteroleptic product. In this context, the principle of integrative self-sorting arises, which is the nonstatistical preparation of a single heteroleptic cage product from a suitable mixture of metal source and ligands (or by mixing of homoleptic cage precursors).32 In the following, different strategies for rational cage design based on integrative self-sorting are discussed (Figure 6).
Figure 6.
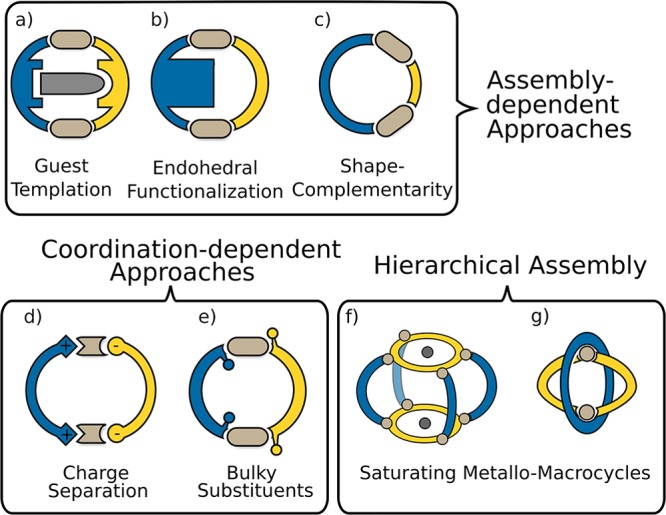
Schematic representation of different approaches for the preparation of heteroleptic MOCs: (a) use of host–guest stabilizing interactions; (b) Endohedral functionalization on one ligand resulting in steric bulk and thus requesting balance with unfunctionalized ligands for assembly; (c) geometric design based on shape complementarity; (d) donor-site engineering using charge separation, e.g., pyridine in combination with carboxylate donors; (e) bulky substituents in proximity to the coordination site; (f) connecting preformed metallo-macrocycles with ligands at open coordination sites; (g) saturating metallo-macrocycles with additional ligands. Strategies (a–c) depend on the shape, length and functionalization of the ligand backbone. Approaches (d) and (e) are based on direct engineering of the coordination sites and their close proximity. Hierarchical assembly of preformed metallo-macrocycles and subsequent introduction of additional ligands leads to structures (f) and (g).
3.2. Templating Effects
One approach to obtain multicomponent supramolecular cages can be the addition of guest molecules as templates during cage formation. Early examples of templated heteroleptic cage synthesis were shown by Fujita. In 2000, he utilized cis-protected Pd(II) together with two tritopic pyridine ligands. Assembly into homo- or heteroleptic cages was found to be in an equilibrium that could be influenced by the addition of different guests.33 The same group exploited guest-templated synthesis of a heteroleptic prism from cis-protected Pt(II), tris(pyridine)triazine, and pyrazine. Large aromatic guests such as a triphenylene derivative allowed the selective formation of a multicomponent prism (Figure 7a).34 More recently, Yoshizawa demonstrated the use of fullerene C60 as a template for the formation of a heteroleptic cage (Figure 7b).27 First, two homoleptic cages based on anthracene ligands with phenylene and naphthalene backbones, respectively, were prepared. While the larger cage could host C70 and diethyl malonate-derivatized C60, the smaller cage was unable to host these guests. Mixing the two preformed cages in the absence of guest molecules led to the formation of a statistical mixture of heteroleptic cages. Addition of fullerene led to reorganization into one single species, PdL2L′2 in the cis form. It was concluded that C60 shows the best host–guest interactions with the heteroleptic cage, thus yielding a large energetic contribution to its stabilization. Templating is a powerful strategy to form heteroleptic cages. As a drawback, however, the cavity is already filled with the template.
Figure 7.

(a) Stabilization through guest templation in Fujita’s heteroleptic prism. (b) Templated reorganization of two homoleptic cages into one heteroleptic cage as demonstrated by Yoshizawa. Addition of fullerene C60 leads to energetic stabilization of the heteroleptic structure. Reprinted with permission from ref (27). Copyright 2015 John Wiley and Sons.
3.3. Steric Effects and Ligand Interaction
Hooley investigated the influence of steric bulk in the ligand backbone on the formation of heteroleptic cages with bis(pyridine) ligands (Figure 8a).35 Three ligands with endohedral functions of increasing size were prepared and combined with the unfunctionalized derivative 8c. Unfunctionalized 8c and ligand 8d with the least sterically demanding functional group (NH2) both form homoleptic cages cleanly when Pd(II) is added. Mixing both ligands and Pd(II) showed a complex NMR spectrum, indicating a statistical mixture of heteroleptic cages. Using 8a with bulkier trifluoroacetate in the endohedral position together with the unfunctionalized ligand allowed for the formation of a Pd28a18c3 cage along with homoleptic Pd28c4. Homoleptic cages with 8a were not observed. Crowley examined ligand interaction as a strategy to control heteroleptic assembly. He achieved clean cis-heteroleptic Pd28e28f2 cages by installing amines at the 2-position of the pyridine donor ligands 8f (Figure 8b).36 Formation of only heteroleptic cages was controlled by kinetic effects: hydrogen bonding between amines and α-hydrogens of the unsubstituted ligands stabilized the cis cage. Furthermore, the amines sterically hinder nucleophiles to attack Pd(II) and thus make the heteroleptic cage kinetically most favorable.
Figure 8.

(a) Bulky endohedral function on the backbone of ligand 8a leads to the heteroleptic Pd28a18c3 cage. (b) Postsynthetic ligand exchange promoted by hydrogen-bonding interactions between the ligands 8e and 8f forming a heteroleptic Pd2A2B2 cage.
3.4. Shape Complementarity of Ligands
Li and Zhou37 demonstrated the formation of heteroleptic structures by partial ligand substitution in preformed homoleptic cages. First, homoleptic cages based on dicarboxylic acid ligands and Cu(II) paddlewheel nodes were prepared. Subsequently, the cages were exposed to a dicarboxylate ligand with a longer backbone, leading to a mixed-ligand cage. More recently, Kitagawa showed that such structures can be directly obtained when a mixture of 5-(tert-butyl)isophthalic acid acid and azobenzene-3,3′-dicarboxylic acid is reacted with a Cu(II) source.38
Fujita and co-workers used ligands of different length to study the formation of heteroleptic icosahedral spheres.39 The authors found that the difference in size has to be significant in order to form clean heteroleptic spheres, such as in bis(pyridyl)benzene together with extended bis(pyridylethynylphenyl)benzene. Each ligand individually forms a homoleptic M12L24 cuboctahedral complex when it is reacted with Pd(II). Mixing the ligands 1:1:1 with Pd(II) in one pot results in the clean formation of Pd12L12L′12.
Clever developed a strategy based on geometric complementarity of the ligands. Acridone ligands (A) with inward-bent isoquinoline donors were mixed with phenanthrene-based ligands (P) bearing outward-bent pyridines and Pd(II) to form cis-PdL2L′2 cages (Figure 9).40 The concept could further be expanded to carbazole ligands (C).28 On the basis of these initial results, the Clever lab is currently expanding the ligand scope in order to demonstrate the ubiquitous application of this approach.
Figure 9.

(a) Ligands used for heteroleptic assembly based on shape complementarity. (b) Three different cages have been obtained. Upon addition of Cl–, C1 and C2 partially rearrange into C3.
3.5. Coordination-Site Engineering
Utilization of cis-protected metal centers as building blocks for the hierarchical assembly of heteroleptic cages has been explored extensively. For example, Stang has constructed prisms through the charge separation approach between adjacent carboxylate and pyridine donors.41 Cis-protected Pt(PEt3)2(OTf)2 was reacted with tri- or tetradentate pyridine ligands and sodium terephthalate to obtain multicomponent supramolecular prisms. The formation of heteroleptic structures was attributed to a preference to combine one negatively charged carboxylate and one pyridine at each metal center, leading to charge separation, in contrast to homoleptic assemblies (Figure 10). Interestingly, the authors also showed the transformation of preformed homoleptic supramolecular structures into the heteroleptic form upon mixing. Mukherjee investigated heteroleptic assembly based on cis-protected Pd with a mixture of imidazole and pyridine donors.30b
Figure 10.

Charge separation strategy. (a) Homoleptic arrangements of pyridine and carboxylate donors. (b) Combination of the two leads to structures that are energetically more favored because of charge separation. (c) Example of a 3D supramolecular box obtained using charge separation, as reported by Stang. Homoleptic cages from ligand A and B rearrange to the heteroleptic cage when they are mixed. Adapted from ref (41). Copyright 2010 American Chemical Society.
In 2005, Fujita demonstrated the side-chain-directed complementary assembly of a heteroleptic M6L3L′2 prism. A combination of tris(pyridine)triazine and bilutidinyl ligands was reacted with cis-protected Pd(II).42 Sterically demanding methyl groups in proximity to the donor site in the latter ligand led to selective heteroleptic assembly (Figure 11a). A similar approach, but transferred to “naked” Pd(II), was recently utilized by Clever, who prepared acridone- and phenothiazine-based picolyl ligands from 5- or 3-ethynyl-2-picoline.43 The respective ligands featured methyl groups pointing either inward (Ai) or outward (Ao). Using only acridone-based ligands for the formation of homoleptic cages resulted in either complex mixtures or bowl-shaped Pd2L3(CH3CN)2 structures. The formation of clean Pd2L4 cages was less favorable because of the sterically demanding methyl groups. However, a 1:1:1 mixture of Ao, Pi, and Pd(II) led to the distinct formation of one heteroleptic cage. Its identity as the cis-[Pd2Ao2Pi2] stereoisomer was determined by density functional theory calculations and the X-ray structure of a model complex. On the other hand, mixing Ai and Po resulted in a complex mixture of bowl-shaped Pd2Ai3 and an interpenetrated double cage from Pi ligands, containing BF4– and Cl– ions. The main difference between the acridone and phenothiazine backbones is that the former one is flat while the latter has a bent geometry, the two having distinct influences on the steric preference around the metal center (Figure 11c).
Figure 11.

Donor-site engineering to form heteroleptic cages. (a) Bilutidinyl ligand in combination with tris(pyridine)triazine ligand and cis-protected Pd(II). (b) Series of banana-shaped ligands with methyl groups pointing outward or inward. (c) Assembly depends not only on the steric balance at the donor site but also on the angles Φ (related to the flatness of the ligand backbone) and θ (the bend angle of ligand). While acridone has a flat backbone, phenothiazine is bent. Both angles were found to influence the steric preference around the coordination site.
3.6. Hierarchical Assembly
Costas and Ribas prepared A4B2 tetragonal prisms based on hexa-aza macrocyclic Pd complex (A) and a tetra-anionic porphyrin ligand (B). Assembly is driven by the charge separation approach discussed above, using carboxylate donors on the porphyrin to coordinate to the Pd metallacycle. After assembly, the two porphyrins that contain Pd(II) or Zn(II) as the central atom serve as anchors for the encapsulation of functionalized guests. In the first example, Pd-centered porphyrin ligands were used to host a series of anionic π guests.44 In a second study, Zn-centered porphyrin cages allowed the coordinative encapsulation of ligands with further open coordination sites to bind additional metals (Zn, Fe, or Cu) inside the cavity, both in solution and in the solid state.45 Furthermore, together with Reek, Ribas and Costas encapsulated a Rh catalyst inside a molecular cage by coordination to the Zn porphyrins (Figure 12).46 The resulting supramolecular catalyst proved to be highly active and enantioselective for hydroformylation of styrene and its derivatives.
Figure 12.

(a) Schematic of a hierarchically assembled box. (b) Example reported by Ribas and Costas making use of charge separation. The two Zn porphyrin units are available for coordination of a guest. (c) An encapsulated Rh catalyst performed enantioselective hydroformylation of styrene and its derivatives promoted by the confined environment. Reprinted from ref (46). Copyright 2015 American Chemical Society.
4. Summary and Prospectus
In this Account, we have summarized a selection of strategies to access heteroleptic metal–organic systems. First, different approaches for the preparation of MOFs containing more than one type of ligand were examined. Several strategies have already been well-established, such as mixing ligands of different topology during solvothermal synthesis and the utilization of postsynthetic methods. On the other side, the self-assembly of heteroleptic MOCs has revealed a set of synthetic tools based on ligand backbone or donor-site engineering. While MOFs are infinite solid-state materials, MOCs represent finite and soluble coordination compounds. Their different nature results in distinguished promises and challenges for future application. A great strength of MOFs is the combination of molecular building blocks with the properties of a solid-state material. With respect to applications in selective sequestration and catalysis, facile substrate/product separation along with possibilities for systematic molecular-level materials engineering result. This argument has been stressed in almost every recently published article on applications of MOFs. A major concern that is often overlooked in this respect is the limited accessibility of pores that are located deeper inside the crystal. This is especially relevant in catalysis, were substrate diffusion pathways are affected by the grain size. Placement of functional groups within the crystal can be achieved statistically if the ligands have the same topology. Utilizing ligands with different topologies or making use of sequential linker installation enables incorporation of various functions in a controlled fashion. However, when the material is turned into action, diffusion discriminates against pores that are deeply buried. At the same time, it is difficult to distinguish functional sites spectroscopically and to determine their exact location, accessibility, and relative activity in the crystal. Also, for other applications we should raise the question of whether all of the pores are accessible and contribute to the overall function of the material. These drawbacks are clearly invalid for MOCs, which are substantially smaller than MOFs. In MOCs, substrate exchange mostly depends on the tunable kinetics and thermodynamics of the host–guest interaction. Furthermore, most MOCs are soluble, and therefore, solution-based techniques allow their detailed investigation. The main challenge for MOCs in the future will be to find a good balance between control over assembly and stability of the cage under working conditions. For many of the discussed strategies, dynamic assembly plays a paramount role because all of the components coordinate and rearrange until the thermodynamic minimum is reached. When the system is put into action, kinetic stability is highly desired in order to ensure that the components do not disassemble. Future research should be directed toward the development of robust, heteroleptic MOCs and detailed investigations of mechanistic aspects of the assembly and performance of these systems. Ultimately, individual molecular cages could then selectively be transformed into larger MOF-like architectures by linking them postsynthetically.
Biographies
Sonja Pullen, born in 1988, studied chemistry in Münster, Germany (B.Sc., 2010) and Uppsala, Sweden (M.Sc., 2012). She received her Ph.D. in 2017 from Uppsala University, working on MOFs and biomimetic catalysis. She is currently a postdoctoral researcher in the Clever group, holding a Marie Curie Fellowship (MOCCA). Her research interest lies in studying catalysis in confined space.
Guido H. Clever, born in 1976, studied chemistry in Heidelberg, changed to Marburg University, and received his Ph.D. in bioorganic chemistry from LMU Munich in 2006. From 2007–2009 he was an AvH/JSPS postdoctoral researcher and in 2009 became an assistant professor at The University of Tokyo. In 2010 he joined the University of Göttingen as a Junior Professor, where he was appointed as a professor in 2013. In 2015, he became a full professor at TU Dortmund. His ERC-supported research focuses on the redox-, photo-, and host–guest chemistry of functionalized coordination cages. Further interests include metal-mediated DNA and chiral organometallic chromophores.
ERC Consolidator Grant RAMSES, Project 683083 (G.H.C.); Deutsche Forschungsgemeinschaft (GRK 2376, “Confinement-Controlled Chemistry”) (G.H.C.); and ERC Marie-Sklodowska Curie Fellowship, Project 798103 (S.P.).
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Supramolecular Chemistry in Confined Space and Organized Assemblies”.
References
- Wiester M. J.; Ulmann P. A.; Mirkin C. A. Enzyme Mimics Based Upon Supramolecular Coordination Chemistry. Angew. Chem., Int. Ed. 2011, 50, 114–137. 10.1002/anie.201000380. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Gu Z.-Y.; Bosch M.; Perry Z.; Zhou H. C. Biomimicry in Metal–Organic Materials. Coord. Chem. Rev. 2015, 293–294, 327–356. 10.1016/j.ccr.2014.05.031. [DOI] [Google Scholar]
- a Cook T. R.; Stang P. J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. 10.1021/cr5005666. [DOI] [PubMed] [Google Scholar]; b García-Simon C.; Costas M.; Ribas X. Metallosupramolecular receptors for fullerene binding and release. Chem. Soc. Rev. 2016, 45, 40–62. 10.1039/C5CS00315F. [DOI] [PubMed] [Google Scholar]
- Leenders S. H. A. M.; Gramage-Doria R.; de Bruin B.; Reek J. N. H. Transition Metal Catalysis in Confined Spaces. Chem. Soc. Rev. 2015, 44, 433–448. 10.1039/C4CS00192C. [DOI] [PubMed] [Google Scholar]
- Nobel Prize in Chemistry 2016:; a Stoddart J. F. Mechanically Interlocked Molecules (MIMs)—Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56, 11094–11125. 10.1002/anie.201703216. [DOI] [PubMed] [Google Scholar]; b Sauvage J.-P. From Chemical Topology to Molecular Machines (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56, 11080–11093. 10.1002/anie.201702992. [DOI] [PubMed] [Google Scholar]; c Feringa B. L. The Art of Building Small: From Molecular Switches to Motors (Nobel Lecture). Angew. Chem., Int. Ed. 2017, 56, 11060–11078. 10.1002/anie.201702979. [DOI] [PubMed] [Google Scholar]
- a Nath I.; Chakraborty J.; Verpoort F. Metal Organic Frameworks Mimicking Natural Enzymes: A Structural and Functional Analogy. Chem. Soc. Rev. 2016, 45, 4127–4170. 10.1039/C6CS00047A. [DOI] [PubMed] [Google Scholar]; b Cohen S. M.; Zhang Z.; Boissonnault J. A. Toward “metalloMOFzymes”: Metal–Organic Frameworks with Single-Site Metal Catalysts for Small-Molecule Transformations. Inorg. Chem. 2016, 55, 7281–7290. 10.1021/acs.inorgchem.6b00828. [DOI] [PubMed] [Google Scholar]
- Cavka J. H.; Jakobsen S.; Olsbye U.; Guillou N.; Lamberti C.; Bordiga S.; Lillerud K. P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. 10.1021/ja8057953. [DOI] [PubMed] [Google Scholar]
- Li H.; Wang K.; Sun Y.; Lollar C. T.; Li J.; Zhou H.-C. Recent Advances in Gas Storage and Separation Using Metal–Organic Frameworks. Mater. Today 2018, 21, 108–121. 10.1016/j.mattod.2017.07.006. [DOI] [Google Scholar]
- Kreno L. E.; Leong K.; Farha O. K.; Allendorf M.; Van Duyne R. P.; Hupp J. T. Metal–Organic Framework Materials as Chemical Sensors. Chem. Rev. 2012, 112, 1105–1125. 10.1021/cr200324t. [DOI] [PubMed] [Google Scholar]
- Horcajada P.; Gref R.; Baati T.; Allan P. K.; Maurin G.; Couvreur P.; Férey G.; Morris R. E.; Serre C. Metal–Organic Frameworks in Biomedicine. Chem. Rev. 2012, 112, 1232–1268. 10.1021/cr200256v. [DOI] [PubMed] [Google Scholar]
- Yoon M.; Srirambalaji R.; Kim K. Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis. Chem. Rev. 2012, 112, 1196–1231. 10.1021/cr2003147. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Farha O. K.; Roberts J.; Scheidt K. A.; Nguyen S. T.; Hupp J. T. Metal–Organic Framework Materials as Catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. 10.1039/b807080f. [DOI] [PubMed] [Google Scholar]
- Pullen S.; Ott S. Photochemical Hydrogen Production with Metal–Organic Frameworks. Top. Catal. 2016, 59, 1712–1721. 10.1007/s11244-016-0690-z. [DOI] [Google Scholar]
- Deng H.; Doonan C. J.; Furukawa H.; Ferreira R. B.; Towne J.; Knobler C. B.; Wang B.; Yaghi O. K. Multiple Functional Groups of Varying Ratios in Metal–Organic Frameworks. Science 2010, 327, 846–850. 10.1126/science.1181761. [DOI] [PubMed] [Google Scholar]
- Koh K.; Wong-Foy A. G.; Matzger A. J. A Crystalline Mesoporous Coordination Copolymer with High Microporosity. Angew. Chem., Int. Ed. 2008, 47, 677–680. 10.1002/anie.200705020. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; Müller U.; Yaghi O. M. “Heterogeneity within Order” in Metal–Organic Frameworks. Angew. Chem., Int. Ed. 2015, 54, 3417–3430. 10.1002/anie.201410252. [DOI] [PubMed] [Google Scholar]
- Yuan S.; Lu W.; Chen Y.-P.; Zhang Q.; Liu T.-F.; Feng D.; Wang X.; Qin J.; Zhou H.-C. Sequential Linker Installation: Precise Placement of Functional Groups in Multivariate Metal–Organic Frameworks. J. Am. Chem. Soc. 2015, 137, 3177–3180. 10.1021/ja512762r. [DOI] [PubMed] [Google Scholar]
- Shekhah O. Layer-By-Layer Method for the Synthesis and Growth of Surface Mounted Metal–Organic Frameworks (SURMOFs). Materials 2010, 3, 1302–1315. 10.3390/ma3021302. [DOI] [Google Scholar]
- Cohen S. M. The Postsynthetic Renaissance in Porous Solids. J. Am. Chem. Soc. 2017, 139, 2855–2863. 10.1021/jacs.6b11259. [DOI] [PubMed] [Google Scholar]
- Boissonnault J. A.; Wong-Foy A. G.; Matzger A. J. Core-Shell Structures Arise Naturally During Ligand Exchange in Metal–Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 14841–14844. 10.1021/jacs.7b08349. [DOI] [PubMed] [Google Scholar]
- Fluch U.; Paneta V.; Primetzhofer D.; Ott S. Uniform Distribution of Post-Synthetic Linker Exchange in Metal–Organic Frameworks Revealed by Rutherford Backscattering Spectrometry. Chem. Commun. 2017, 53, 6516–6519. 10.1039/C7CC02631E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen S.; Fei H.; Orthaber A.; Cohen S. M.; Ott S. Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal–Organic Framework. J. Am. Chem. Soc. 2013, 135, 16997–17003. 10.1021/ja407176p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S.; Pascanu V.; Pullen S.; González-Miera G.; Martín-Matute B.; Ott S. Catalyst Accessibility to Chemical Reductants in Metal–Organic Frameworks. Chem. Commun. 2017, 53, 3257–3260. 10.1039/C7CC00022G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yuan S.; Zou L.; Qin J.-S.; Li J.; Huang L.; Feng L.; Wang X.; Bosch M.; Alsalme A.; Cagin T.; Zhou H.-C. Construction of hierarchically porous metal–organic frameworks through linker labilization. Nat. Commun. 2017, 8, 15356. 10.1038/ncomms15356. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cai G.; Jiang H.-L. A Modulator-Induced Defect-Formation Strategy to Hierarchically Porous Metal–Organic Frameworks with High Stability. Angew. Chem. 2017, 129, 578–582. 10.1002/ange.201610914. [DOI] [PubMed] [Google Scholar]
- Cook T. R.; Zheng Y.-R.; Stang P. J Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials. Chem. Rev. 2013, 113, 734–777. 10.1021/cr3002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Debata N. B.; Tripathy D.; Chand D. K. Self-Assembled Coordination Complexes From Various Palladium(II) Components and Bidentate or Polydentate Ligands. Coord. Chem. Rev. 2012, 256, 1831–1945. 10.1016/j.ccr.2012.04.001. [DOI] [Google Scholar]; b Harris K.; Fujita D.; Fujita M. Giant Hollow MnL2n Spherical Complexes: Structure, Functionalisation and Applications. Chem. Commun. 2013, 49, 6703–6712. 10.1039/c3cc43191f. [DOI] [PubMed] [Google Scholar]; c Han M.; Engelhard D. M.; Clever G. H. Self-Assembled Coordination Cages Based on Banana-Shaped Ligands. Chem. Soc. Rev. 2014, 43, 1848–1860. 10.1039/C3CS60473J. [DOI] [PubMed] [Google Scholar]; d Clever G. H.; Punt P. Cation-Anion Arrangement Patterns in Self-Assembled Pd2L4 and Pd4L8 Coordination Cages. Acc. Chem. Res. 2017, 50, 2233–2243. 10.1021/acs.accounts.7b00231. [DOI] [PubMed] [Google Scholar]; e Vasdev R. A. S.; Preston D.; Crowley J. D. Multicavity Metallosupramolecular Architectures. Chem. - Asian J. 2017, 12, 2513–2523. 10.1002/asia.201700948. [DOI] [PubMed] [Google Scholar]
- Yamashina M.; Yuki T.; Sei Y.; Akita M.; Yoshizawa M. Anisotropic Expansion of an M2L4 Coordination Capsule: Host Capability and Frame Rearrangement. Chem. - Eur. J. 2015, 21, 4200–4204. 10.1002/chem.201406445. [DOI] [PubMed] [Google Scholar]
- Bloch W. M.; Holstein J. J.; Hiller W.; Clever G. H. Morphological Control of Heteroleptic cis- and trans-Pd2L2L′2 Cages. Angew. Chem., Int. Ed. 2017, 56, 8285–8289. 10.1002/anie.201702573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Regeni I.; Clever G. H. Structure Relationships Between Bis-Monodentate Ligands and Coordination Driven Self-Assemblies. Coord. Chem. Rev. 2018, 374, 1–14. 10.1016/j.ccr.2018.06.010. [DOI] [Google Scholar]
- a Saha M. L.; Yan X.; Stang P. J. Photophysical Properties of Organoplatinum(II) Compounds and Derived Self-Assembled Metallacycles and Metallacages: Fluorescence and Its Applications. Acc. Chem. Res. 2016, 49, 2527–2539. 10.1021/acs.accounts.6b00416. [DOI] [PubMed] [Google Scholar]; b Mukherjee S.; Mukherjee P. S. Template-Free Multicomponent Coordination-Driven Self-Assembly of Pd(II)/Pt(II) Molecular Cages. Chem. Commun. 2014, 50, 2239–2248. 10.1039/C3CC49192G. [DOI] [PubMed] [Google Scholar]
- Bloch W. M.; Clever G. H. Integrative Self-Sorting of Coordination Cages Based on ‘Naked’ Metal Ions. Chem. Commun. 2017, 53, 8506–8516. 10.1039/C7CC03379F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z.; Jiang W.; Schalley C. A. Integrative Self-Sorting: A Versatile Strategy for the Construction of Complex Supramolecular Architecture. Chem. Soc. Rev. 2015, 44, 779–798. 10.1039/C4CS00305E. [DOI] [PubMed] [Google Scholar]
- Hiraoka S.; Kubota Y.; Fujita M. Self- and Hetero-Recognition in the Guest-Controlled Assembly of Pd(II)-linked Cages from Two Different Ligands. Chem. Commun. 2000, 1509–1510. 10.1039/b004271o. [DOI] [Google Scholar]
- Kumazawa K.; Biradha K.; Kusukawa T.; Okano T.; Fujita M. Multicomponent Assembly of a Pyrazine-Pillared Coordination Cage That Selectively Binds Planar Guests by Intercalation. Angew. Chem., Int. Ed. 2003, 42, 3909–3913. 10.1002/anie.200351797. [DOI] [PubMed] [Google Scholar]
- Johnson A. M.; Hooley R. J. Steric Effects Control Self-Sorting in Self-Assembled Clusters. Inorg. Chem. 2011, 50, 4671–4673. 10.1021/ic2001688. [DOI] [PubMed] [Google Scholar]
- Preston D.; Barnsley J. E.; Gordon K. C.; Crowley J. D. Controlled Formation of Heteroleptic [Pd2(La)2(Lb)2]4+ Cages. J. Am. Chem. Soc. 2016, 138, 10578–10585. 10.1021/jacs.6b05629. [DOI] [PubMed] [Google Scholar]
- Li J.-R.; Zhou H.-C. Bridging-Ligand-Substitution Strategy for the Preparation of Metal–Organic Polyhedra. Nat. Chem. 2010, 2, 893–898. 10.1038/nchem.803. [DOI] [PubMed] [Google Scholar]
- Hosono N.; Omoto K.; Kitagawa S. Anisotropic Coordination Star Polymer Realized by Self-Sorting Core Modulation. Chem. Commun. 2017, 53, 8180–8183. 10.1039/C7CC04381C. [DOI] [PubMed] [Google Scholar]
- Sun Q.-F.; Sato S.; Fujita M. An M12(L1)12(L2)12 Cantellated Tetrahedron: A Case Study on Mixed-Ligand Self-Assembly. Angew. Chem., Int. Ed. 2014, 53, 13510–13513. 10.1002/anie.201408652. [DOI] [PubMed] [Google Scholar]
- Bloch W. M.; Abe Y.; Holstein J. J.; Wandtke C. M.; Dittrich B.; Clever G. H. Geometric Complementarity in Assembly and Guest Recognition of a Bent Heteroleptic cis-[Pd2La2Lb2] Coordination Cage. J. Am. Chem. Soc. 2016, 138, 13750–13755. 10.1021/jacs.6b08694. [DOI] [PubMed] [Google Scholar]
- Zheng Y.-R.; Zhao Z.; Wang M.; Ghosh K.; Pollock J. B.; Cook T. R.; Stang P. J. A Facile Approach Toward Multicomponent Supramolecular Structures: Selective Self-Assembly via Charge Separation. J. Am. Chem. Soc. 2010, 132, 16873–16882. 10.1021/ja106251f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa M.; Nagao M.; Kumazawa K.; Fujita M. Side Chain-Directed Complementary cis-Coordination of Two Pyridines on Pd(II): Selective Multicomponent Assembly of Square-, Rectangular-, and Trigonal Prism-Shaped Molecules. J. Organomet. Chem. 2005, 690, 5383–5388. 10.1016/j.jorganchem.2005.06.022. [DOI] [Google Scholar]
- Zhu R.; Bloch W. M.; Holstein J. J.; Mandal A.; Schäfer L. V.; Clever G. H. Donor-Site-Directed Rational Assembly of Heteroleptic cis-[Pd2L2L’2] Coordination Cages From Picolyl Ligands. Chem. - Eur. J. 2018, 24, 12976–12982. 10.1002/chem.201802188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Simón C.; Garcia-Borràs M.; Gómez L.; Garcia-Bosch I.; Osuna S.; Swart M.; Luis J. M.; Rovira C.; Almeida M.; Imaz I.; Maspoch D.; Costas M.; Ribas X. Self-Assembled Tetragonal Prismatic Molecular Cage Highly Selective for Anionic π Guests. Chem. - Eur. J. 2013, 19, 1445–1456. 10.1002/chem.201203376. [DOI] [PubMed] [Google Scholar]
- Colomban C.; Martin-Diaconescu V.; Parella T.; Goeb S.; García-Simón C.; Lloret-Fillol J.; Costas M.; Ribas X. Design of Zn-, Cu-, and Fe-Coordination Complexes Confined in a Self-Assembled Nanocage. Inorg. Chem. 2018, 57, 3529–3539. 10.1021/acs.inorgchem.7b02852. [DOI] [PubMed] [Google Scholar]
- García-Simón C.; Gramage-Doria R.; Raoufmoghaddam S.; Parella T.; Costas M.; Ribas X.; Reek J. N. H. Enantioselective Hydroformylation by a Rh-Catalyst Entrapped in a Supramolecular Metallocage. J. Am. Chem. Soc. 2015, 137, 2680–2687. 10.1021/ja512637k. [DOI] [PubMed] [Google Scholar]