

Abstract
Low patient compliance and poor bioavailability of ophthalmic medications are the main limitations of topical eye drops. A potential solution to these disadvantages could be provided by thermoresponsive hydrogels, which could be used as the basis for a gelling eye drop for long-term release of therapeutics. We previously reported such a system capable of being retained in the lower fornix of rabbits, continuously releasing an anti-glaucoma drug for one month. Here, we sought to improve the properties of the existing gels as most relevant to patient use without altering the drug release profile. Specifically, we optimized the sol-to-gel transition temperature and de-swelling kinetics of pNIPAAm gels to avoid risk of the gelled drop reverting to liquid during cold or windy weather, and ensure quick gelation upon administration. A reduction of the gel LCST, faster gelation kinetics, and suitable viscosity for the administration as an eye drop were successfully achieved through modification of the poly(ethylene glycol) content in the water phase and its molecular weight. Our data suggest that drug release is not affected by these changes, with representative drug concentration profiles of the previous and new formulations demonstrating comparable anti-glaucoma release kinetics.
TOC
Reduction of the LCST of a pNIPAAm-based thermoresponsive hydrogel through modification of PEG content and molecular weight
Introduction
Topical eye drops are the standard of care for many ocular pathologies, representing around 90% of ophthalmic formulations on the market.1,2 Topical eye drops are easy to apply and generally well-tolerated by patients. However, a major limitation is patient compliance due to the frequency of administration, especially for those affected by chronic pathologies requiring life-long treatment.3,4 In particular, high dosing frequencies are often required because of physiological barriers in the eye that can result in poor bioavailability of the therapeutic agent.5 Specifically, the efficacy of ophthalmic drops can be limited by a short contact time on the surface of the eye due to dilution from tears and lacrimal drainage.6–9 Typically, after the instillation of an eye drop, less than 5% of the active agent is able to reach the target tissues.6,10 The remaining 95% of the drug is absorbed systemically, with possible undesirable side effects.2,10
Over the years, many attempts have been made to improve topical drug delivery systems such as nanosuspensions, ointments, and inserts with the goal of obtaining a more efficient delivery to ocular tissues. Nanosuspensions consist of a sub-micron colloidal dispersion of pure drug particles dispersed in an appropriate medium (mostly water) and stabilized by polymers or surfactants.11,12 Nanosuspensions have been developed to enhance the solubility of hydrophobic drugs, providing prolonged release with improved bioavailability versus traditional eye drops.13 Ointments are another familiar way to administer ophthalmic medications, with advantages over eye drops such as prolonged residence time, reduced dilution from tears and drainage through the nasolacrimal duct, and higher absorbed concentrations.14 The more viscous formulations used in topical ointments can often lead to blurred vision and consequently, administration is typically recommended in the evening only.14,15 Ocular inserts are flexible polymeric materials that are placed in the cul-de-sac of the conjunctiva capable of enhancing the residence time of the drug with reduced systemic absorption.15 An example of such device is the now discontinued Ocusert® system (Alza, Vacaville, CA), a pilocarpine-loaded polymeric reservoir developed for the release of the drug at near-constant rates for 5–7 days, leading to a reduction in the dosing frequency.16 This system was discontinued partially due to patient discomfort, resulting from its 500 μm thickness.17
As a potential solution to the disadvantages of existing topical extended release systems, in situ forming hydrogels have been recently explored as topical eye drops. These gels could be administered as liquid drops at room temperature, and conform as a gel in the lower fornix upon reaching body temperature.18 A known limitation of hydrogels, however, is that release of free drug is limited to the timeframe of hours to days.18–20 However, it may be possible to embed systems capable of providing longer-term release (such as microspheres) into an in situ-forming hydrogel eye drop such that the entire system can be easily administered, comfortably retained, and also provide a longer period of drug release that would be clinically beneficial. We have previously reported such a system containing degradable microspheres made from poly(lactic-co-glycolic) acid (PLGA) that has demonstrated efficacy releasing an anti-glaucoma drug and an antibiotic.4,21 These polymer microparticles were embedded into a thermoresponsive hydrogel based on pNIPAAm and poly(ethylene glycol) (PEG) for administration as an in situ-forming eye drop. This combined polymer microsphere/thermogelling eye drop system has the potential to serve as a long-term release system after one single administration, thus potentially improving the compliance of those patients affected by pathologies requiring high frequency of administration. Further, the adaptable double emulsion fabrication technique can be used to encapsulate and release many types of ocular therapeutics.22,23
Despite the promise of long-term drug release from a topically administered format, the above-mentioned shortcomings of other systems indicate that translation of chronic medications requires a careful consideration of comfort and tolerance to achieve optimal adherence and outcomes. For a thermoresponsive hydrogel eye drop, sol-gel transition temperature and de-swelling kinetics should be optimized to avoid the gelled drop reverting to liquid during cold or windy weather (representing decreased temperatures), and ensure a quick gelation upon administration.
Accordingly, the goal of this work is to improve the properties of the existing gel relevant to patient use without altering the drug release profile. Given variations in external temperature that patients might experience, the properties such as sol-to-gel transition temperature and also the gelation kinetics (to expedite in situ gel formation after administration) could result in a formulation with more desirable characteristics for a self-administered topical gel drop if the desired drug release profile can be maintained. Our data suggest that it is possible to achieve a reduction of the gel LCST, faster gelation kinetics, and suitable viscosity for the administration as an eye drop through modification of PEG content and PEG molecular weight in the water phase. Our data also suggest that drug release is not affected by the adjustments made to the existing formulation, with representative drug concentration profiles of the previous and new formulations demonstrating comparable anti-glaucoma release kinetics.
Experimental
Materials
All materials and reagents were obtained from Sigma (St. Louis, MO) unless otherwise specified.
Microparticle fabrication
Brimonidine tartrate (BT)-loaded poly(lactic-co-glycolic) acid (PLGA) microparticles were fabricated similarly to our previously described methods, using a standard double emulsion procedure.24 Briefly, 200 mg of poly(lactic-co-glycolic) acid (PLGA, lactide:glicolide 50:50, Mw 24–38 kDa) was dissolved in 4 ml of dichloromethane (DCM). 250 μl of a 50 mg/ml of BT aqueous solution was added (Santa Cruz Biotechnologies, Santa Cruz, CA). The suspension was sonicated for 10 seconds at 30% of amplitude (Sonics Vibra- Cell™) and then homogenized in 60 ml 2% poly(vinyl alcohol) (PVA, Polysciences) for 1 minute at 7000 rpm (Silverson L4RT-A homogenizer). The double emulsion was then added to 80 ml 1% PVA solution and allowed to mix for 3 hours to evaporate any remaining DCM. The microparticles were then washed via centrifugation with deionized water and then lyophilized for 48 hours (Virtis Benchtop K Freeze Dryer, Gardiner, NY).
Hydrogel fabrication
Thermoreversible hydrogels were prepared via aqueous free radical polymerization varying the content (100 μl, 200 μl, and 400 μl) and molecular weights (200 Da, and 400 Da) of poly(ethylene glycol) (PEG). The purpose was to tune the lower critical solution temperature (LCST), to ensure stability upon administration. Briefly, the selected amount of PEG was added to 100 mg of NIPAAm. 2 ml of a 0.1 mg/ml ammonium persulfate (APS) aqueous solution was added and vortexed until the solution was homogeneously mixed. To this solution, 5 μl of tetramethylethylenediamine (TEMED) was added and then refrigerated overnight. The resulting polymer was washed 5 times using DI water heated to 45 °C. Four different formulations were fabricated in addition to the original thermoresponsive hydrogel, and the compositions are shown in table 1.
Table 1:
Composition of the different hydrogel formulations.
| Sample ID | NIPAAm | PEG200 | PEG400 | Water content |
|---|---|---|---|---|
| Original formulation | 100 mg | 100 μl | - | 2 ml |
| F1 | 100 mg | 200 μl | - | 2 ml |
| F2 | 100 mg | 400 μl | - | 2 ml |
| F3 | 100 mg | - | 100 μl | 2 ml |
| F4 | 100 mg | - | 400 μl | 2 ml |
Gelling point and transition kinetics
The gelling point was identified via temperature sweep measurements using a rheometer (AR2000, TA Instrument, New Castle, DE). Specifically, the elastic modulus (G’) and viscous modulus (G”) were measured at an angular frequency of 1 rad/s, while the temperature was increased from 10 °C to 40 °C at a rate of 1 °C/min. The temperature at which G’ was equal to G” was assigned as the gelling point.25 In addition, absorbance measurements as a function of the decreasing temperature were carried out at a wavelength of 415 nm using a microplate reader (SoftMax Pro 5 microplate and cuvette reader, Molecular Device, Sunnydale, CA) to assess the reversible nature of the hydrogels. Briefly, the absorbance values of each gel formulation (n=3) were acquired in reverse conditions by cooling the samples from 37 °C to 26 °C.
The kinetics of the transition was investigated at 37 °C for n=3 gel samples via absorbance measurements at a wavelength of 415 nm to assess the time for phase separation, and consequent transition to gel form. The absorbance values were read every 2 seconds for a total of 10 minutes. The transition time was determined according to the following equation:
where ti is the time at which absorbance value started to increase and te is the time at which the absorbance became stable at the highest value. In addition, the transition kinetics was investigated optically to establish the time needed to the gel to revert to transparency. Briefly, 100 μl of gel samples (n=3) was placed in a 48-well plate and incubated at 37 °C until samples became opaque. Subsequently, the plate containing the samples was transferred into the microplate reader pre-set at room temperature and absorbance was read every 2 seconds for a total of 30 minutes at a wavelength of 415 nm.
Sol fraction and swelling ratio
The degradation rate of the hydrogels, defined as a ratio of change from initial mass at given time points,26 was determined according to the following equation:
where Mi is the initial mass of the gel and Md is the mass of the dry gel after the incubation for the appropriate amount of time in PBS at 37°C. Degradation at each time point was calculated as the average of three samples ± standard deviation and measurements were taken at 0, 7, 14, 21, and 28 days.
The swelling ratio, a comparison of mass change after swelling in PBS, was calculated as the average of five samples for each gel formulation, according to the following equation:
where Ms represents the mass of the gel after equilibrating in PBS at 37 °C, and Md is the mass of the same gel after complete drying.
Viscosity measurements
The viscosity of each formulation was determined at room temperature varying the shear rate from 1s−1 to 10 s−1 using a rheometer (AR2000, TA Instrument, New Castle, DE).
Viability assay
To ensure that the cytocompatibility of the gel has not been affected by the adjustments, the cytotoxicity of the hydrogels was determined by incubating gel samples with Human Conjunctival Epithelial Cells (American Type Culture Collection, Manassas, VA). Briefly, 100 μl of 2·105 cells/ml in growth medium was added to wells in a 96-well plate, while 50 μl of each hydrogel sample was incubated with growth medium for 24 hours in another 96-well plate. After achieving a monolayer of attached cells, 50 μl of the pre-incubated hydrogel samples along with 150 μl of fresh medium was added to wells containing cells. 200 μl of fresh medium was added to cells used as the positive control for viability. Cells were incubated at 37°C with 5% CO2 for 24 hours followed by addition of PrestoBlue® viability agent (Life Science) and additional 2 hours of incubation. Triton X100 was added to negative control wells as a lysis agent and incubated for a total of 15 minutes. Fluorescence was then determined in each well using an emission filter of 500 nm and an excitation filter of 620 nm. The mean and standard deviation of the absorbance values were determined for n=12 samples of each test group. Percent viability was determined according to the following equation:
where F is an average fluorescence value. Moreover, quantitative data on viable cells were obtained using a calibration curve.
In vitro drug release study
In vitro drug release studies were carried out to assess the kinetics of BT from the microparticles embedded into the hydrogels. All gel samples were prepared by dispersing 10 mg microparticles within 100 μl hydrogel. Samples were then placed at 37 °C to ensure their gelation. After gelation occurred, 500 μl PBS was added. At predetermined intervals, the supernatant was removed and replaced with fresh PBS. BT concentration in the supernatant was determined via UV/Vis absorption using a microplate reader set to a wavelength of 320 nm. Background signal from blank microparticles embedded into the gel was subtracted at each time point, and results were reported as the average of three samples ± standard deviation.
Statistics
All the experimental data were expressed as mean ± SD (n=3) One-way analysis of variance (ANOVA) was performed on transition kinetics measurements to determine statistically significant differences, if any, in the transition time. The same method was used to determine statistically significant differences in the time needed to the gel to revert to transparency. Statistically significant differences were designated by a significance criterion (p value) below 0.05.
Results and discussion
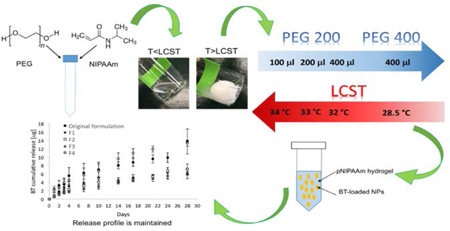
In this study, a previously developed gel formulation was tuned to reduce the LCST and obtain faster gelation kinetics. The results of the characterization of microparticles and thermoresponsive hydrogel can be found in two previously published works 4,24. Briefly, microparticles had a poreless surface morphology and a volume average diameters of 7.46 ± 2.86 μm (via SEM and volume impedance-based sizing). After dispersion, microparticles were confirmed to be homogeneously distributed into the pNIPAAm hydrogel matrix. In addition, the thermoresponsive hydrogel exhibited an LCST of 33.4 °C. Starting from the original formulation of hydrogel, containing 100 μl PEG (200 Da), four new formulations were prepared via aqueous free radical polymerization varying the content (100 μl, 200 μl, and 400 μl) and molecular weights (200 Da, and 400 Da) of PEG. The purpose of the new formulations was to reduce the LCST farther below body temperature, and therefore to improve stability upon administration. The compositions of the new formulations are shown in table 1. The new hydrogels were then characterized to ensure that drug release has not been affected by the adjustments.
Gelling point and transition kinetics
Results shown in Figure 1 represent the gelling point determination via temperature sweep measurements. The rheological analysis of the hydrogels pointed out the typical trend of G’ and G” characterizing thermo-sensitive gels.27 Specifically, G’ and G” values remained only weakly dependent on temperature at low temperature before rising sharply. Such behavior is typical of pNIPAAM-water mixtures and is indicative of gelation induced by phase separation. Specifically, the increased temperature up to the gelling point led to an increase of the thermal motion of the copolymer, with consequent increased flexibility of the gel network and reduced elastic modulus. At temperatures higher than the gelling point, the interaction within the pNIPAAm network was stronger than the hydrogen bonds between pNIPAAm and water. This leads to the elimination of water and transition to a gel state. A reduction of the transition temperature with respect to the original gel was achieved for each of the new formulations. In particular, the use of a higher content of PEG with molecular weight of 400 Da exhibited the lowest gelling point, which was between 28 and 29 °C (Figure 1E). In addition, the same test was carried out on the F4 formulation containing microparticles. The graph in figure 1F shows that the presence of the particles led to a slight decrease of the gelling point. The effect of the PEG content on the LCST can be attributed to the co-nonsolvency of pNIPAAm in water-PEG systems. Briefly, the LCST is the result of a balance between hydrogen bonding and hydrophobic effects in aqueous solutions.28 For this reason, the solvent composition influences the LCST, in particular when a co-solvent or co-solute that modifies water structure and hydrophobic interactions is present.28 The LCST is an indication of the disruption of the hydrogen bonding between pNIPAAm and water upon raising temperature. An additive that competes with those bonds can lower the LCST.28 An example of this phenomenon is the reduction of the LCST of pNIPAAm when methanol is added to water.29,30 Specifically, this is due to the complexation between methanol and water that leads to a reduction in the hydrogen-bonded sites in water molecules. In a same way, it has been demonstrated that PEG interacts through hydrogen bonding with water molecules,31 thus competing with pNIPAAm in the complexation with water molecules. Specifically, the presence of PEG disturbs the hydration layer around pNIPAAm leading to the exposure of some hydrophobic groups, thus causing the collapse and aggregation of pNIPAAm chains and resulting in a decrease of the LCST.32 Accordingly, increasing the content of PEG in the water phase during the optimization of the hydrogels led to the desired reduction in the LCST, potentially ensuring a more stable system that can be worn in a variety of climate conditions.
figure 1:
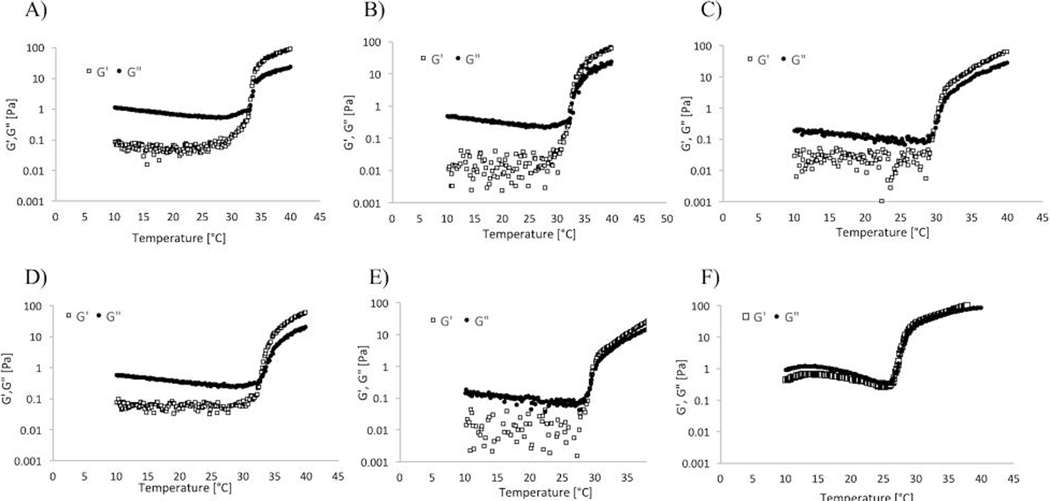
elastic modulus (g’) and storage modulus (g”) during temperature ramps for a) original formulation, b) f1, c) f2, d) f3, e) f4, and f) f4 containing microparticles.
It is well known that the LCST, phase change kinetics, and swelling of pNIPAAm-based hydrogels can be tuned by adding co-monomers or crosslinkers.33 Interestingly, the choice of using PEG as an additive instead of adding a co-monomer or crosslinking agent, allows obtaining an opaque drop that can be easily seen with the naked eye.34 Moreover, the lack of additional monomers favors maintaining the LCST near values suitable for physiological applications, even when the content of PEG is high.29,35 To confirm the accuracy of our results in terms of sol-to-gel transition temperature, we tested the LCST of formulations prepared by adding PEG after the polymerization of NIPAAm. Interestingly, the values of the LCST obtained on these hydrogels were the same as the ones of the hydrogels fabricated adding PEG at the same time along with NIPAAm (data not shown), confirming that PEG can be added either during or after the polymerization. This suggests that even if PEG is not participating in the reaction, a sufficient amount is still present in the final formulation after the purification procedure to allow the modulation of the LCST.
Absorbance measurements as a function of the decreasing temperature were carried out to assess the reversible nature of the hydrogels. Specifically, Figure 2 shows that each of the formulations reverted to transparency below the LCST, suggesting that the gel could be manually removed once drug release is exhausted by irrigating the fornix with liquid below the LCST. Moreover, absorbance measurements were carried out to assess the transition kinetics. Results shown in Figure 3A suggest that the transition was faster as the amount and/or molecular weight of PEG was increased. A transition in the order of seconds might be useful in the case of a gelling eye drop, since it is reported that such a kind of formulation should turn into a gel immediately after contact with the eye in order to increase bioavailability.36 In this work, the F4 formulation had the fastest sol-to-gel transition, with a transition time significantly faster (p<0.01) than the one of the original formulation.
figure 2:
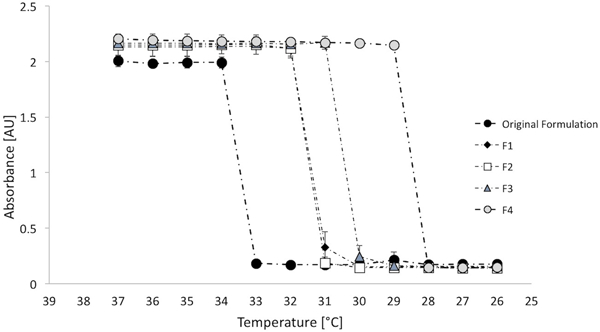
absorbance measurements at 415 nm as a function of decreasing temperature for n=3 gel samples for each formulation to assess the reversible nature of the hydrogels.
figure 3:
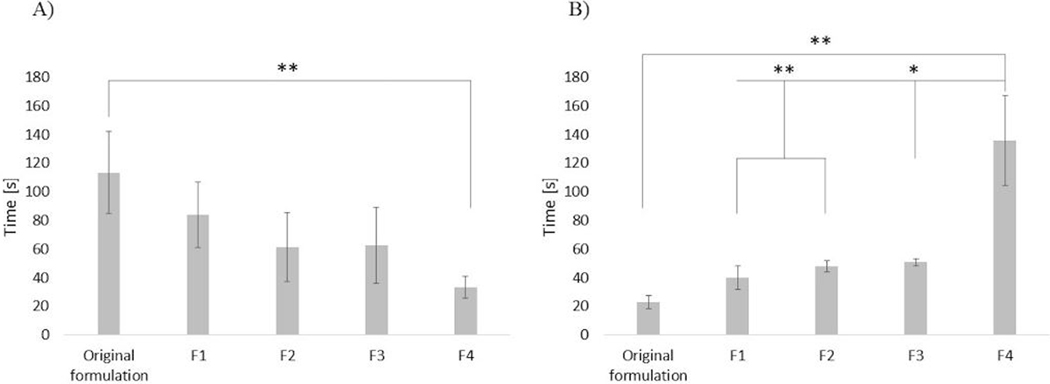
a) transition time for n=3 samples of each hydrogel formulation and b) reverse conditions. statistical significance determined using one-way analysis of variance (anova) with *p < 0.05 and **p < 0.01.
Despite the faster sol-gel transition time, the gel-sol transition for F4 was significantly slower (p<0.01) than all other formulations. This may in fact be an advantage of F4, as the gel depot could withstand longer exposure times to ambient temperature without undergoing phase transition.
Sol fraction and swelling ratio
The gels were also examined to determine that the newly-formulated gels retained the desirable property of non-biodegradability. Degradation of all gel formulations (that were maintained at 37°C over a period of 28 days) was negligible as demonstrated by the constant solid fractions (Figure 4), defined as the ratio of change from initial mass to mass given at certain time points.
figure 4:
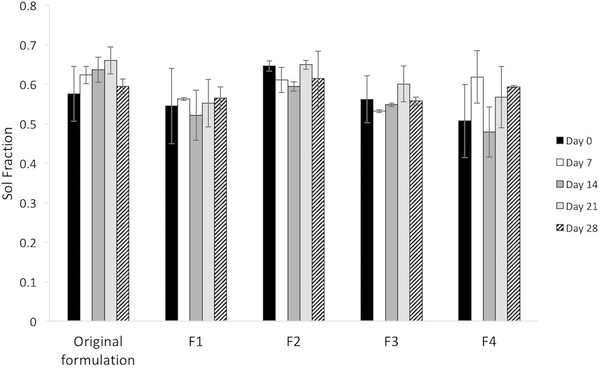
solid fraction of new hydrogel formulations at 37°c over time, representing degradation and comparison with the solid fraction of the original formulation.
Swelling ratios were also measured, with Figure 5 representing a comparison of mass change upon swelling in PBS, calculated as the average of five samples for each gel formulation. The result suggests a slight decrease in the swelling ratio as a function of the increase both in the density and in the molecular weight of the PEG.
figure 5:
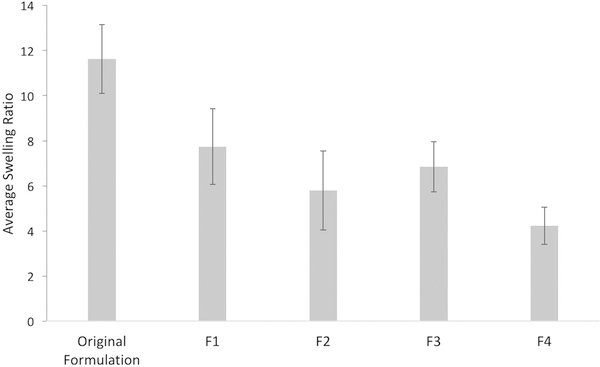
swelling ratio calculated as the average n=5 samples for each new formulation and comparison with the swelling ratio of the original formulation.
Viscosity measurements
Rheological measurements were carried out to evaluate the viscosity of all the formulations prior to gelation. A viscosity that allows an ease of administration into the eye as a liquid is another main pre-requisite for a gelling eye drop.
The viscosity of each formulation was independent of shear rate in the range of 1 to 10 s−1, suggesting that each formulation behaves as a Newtonian liquid when the temperature is below the gelling point (Figure 6). The increase of PEG content led to a decrease in the viscosity values that might be due to an overall dilution of the pNIPAAm when PEG content is increased. Moreover, the molecular weight of PEG seems to have no effect. In fact, hydrogel formulations with the same amount of PEG but different molecular weights exhibited similar viscosity values. Viscosity values ± standard deviations are reported in table 2. In addition, the viscosity of the F4 formulation containing microparticles was tested in the same conditions. The presence of the particles led to an increase of the viscosity of the hydrogel with average value rising from 0.076 ± 0.003 to 0.112 ± 0.008 Pa·s. It has been reported that before gelling, the formulation should have a viscosity in the range of 5–1000 mPa·s.36 The variability in the viscosity values may be due to a slight change in the PEG content due to its extraction by water washings. However, each of the hydrogel formulations tested had viscosity values within the aforementioned range, suggesting that these proposed hydrogels could be easily administered as a traditional eye drop. Future studies could include GPC analysis of the water collected after purification to measure PEG washout to better control this property. Notably, the addition of the microparticles in the F4 formulation led to an increase in the viscosity of the gel, however the value was still in the range required for a gelling eye drop, indicating that the presence of the particles does not affect its potential applicability. In addition, the presence of microparticles led to an increase in G’ and G” values at low temperature. However, there are no large differences in the modulus values when the temperature is above the LCST, suggesting that the presence of the microparticles does not affect the properties of the hydrogel when it is eventually placed at body temperature.
figure 6:
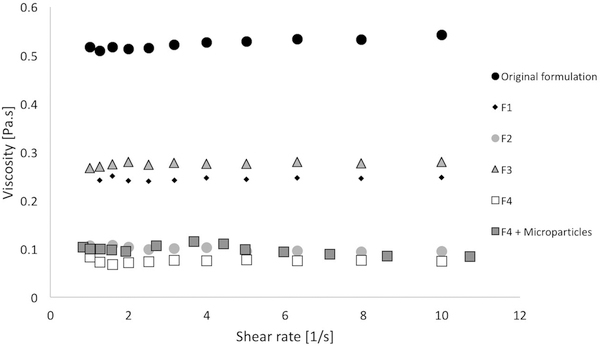
viscosity at room temperature as a function of the shear rate for each gel formulation.
Table 2:
Average viscosity values ± standard deviations for each formulation of hydrogel.
| Sample ID | Viscosity [Pa·s] |
|---|---|
| Original formulation | 0.523 ± 0.010 |
| F1 | 0.244 ± 0.003 |
| F2 | 0.100 ± 0.004 |
| F3 | 0.276 ± 0.004 |
| F4 | 0.076 ± 0.003 |
| F4 containing MPs | 0.112 ± 0.008 |
Viability assay
Human Conjunctival Epithelial Cells were chosen as the representative cell line to test the cytotoxicity of all the formulations. Results of n=12 samples of each gel are shown in Figure 7. The percent viability was determined to be around 80% for each formulation, suggesting that the modifications on the original hydrogel did not affect the cytocompatibility of the material. Notably, although low molecular weight PEG can show some toxicity, viability values reassure on its use and potential translational applications. In fact, considering the theoretical composition of each formulation, and the theoretical volume of each eye drop, the amount of PEG that will be administered only ranges between 2.5 and 10 μl. The same amount was tested in vitro, with results showing viability values above the minimum threshold recommended for medical devices, which is fixed at 70%.37 Importantly, similar low molecular weight PEGs are approved by USFDA for pharmaceutical applications, including ophthalmic ones, supporting the safety of using such component.38
figure 7:
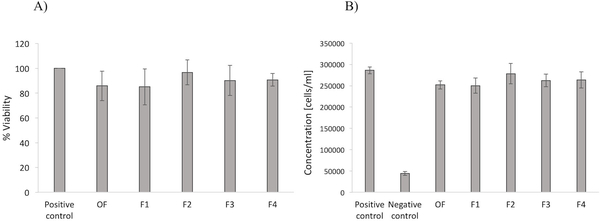
a) human conjunctival epithelial cell viability for each formulation of hydrogel (n=12) compared to positive control (no gel). b) concentration of cells after 24 h incubation with each formulation of hydrogel compared to positive (no gel) and negative (lysis agent) controls.
In vitro drug release study
In vitro drug release assays were carried out to determine if changes in PEG content and/or molecular weight affected the release kinetics. Results shown in Figure 8A represent a comparison between brimonidine released from BT-loaded microparticles embedded in different gel formulations over a period of 4 weeks. All the hydrogel formulations exhibited similar release kinetics over a period of 28 days. These results suggest that the modulation of the properties of the existing hydrogel relevant to patient use did not affect drug release, with representative drug concentration profiles demonstrating comparable release kinetics. Moreover, for all the hydrogel formulations tested, the amount of the anti-glaucoma drug released over the entire length of the study was above the minimum limit of absorption reported in the literature (Figure 8B).39 Among the formulations developed in this work, F4 presented the lowest LCST with the fastest transition kinetics, lowest viscosity, and an amount of anti-glaucoma drug released closely matching the amount released from the original formulation of gel. Potentially, this formulation might be the most suitable candidate to be used as a topical depot for a prolonged release of ophthalmic medications, with more desirable characteristics than traditional eye drops.
figure 8:
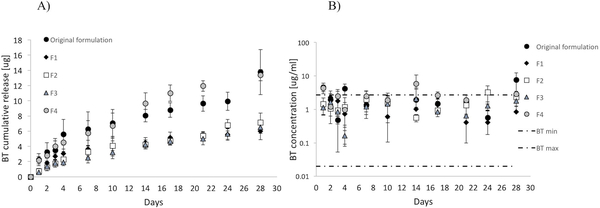
a) cumulative in vitro brimonidine tartrate release from bt-loaded microparticles embedded in the original and new formulations of hydrogel. b) daily release of brimonidine tartrate compared to the minimum and maximum levels of topical bt absorbed into the anterior chamber.
Conclusions
Adjustments to the existing thermoresponsive hydrogel presented in this work led to an improved formulation with a lower LCST and gelation temperature, faster sol-to-gel transition kinetics, and suitable viscosity to be used as a gelling eye drop with the potential of improving patient outcomes. This formulation appears to be most suitable to provide the necessary stability to be worn in a variety of climatic conditions without the risk of being removed. Ultimately, testing the new formulations in expanded preclinical toxicity studies and in humans will be important in the future in order to actually know whether these physical changes result in a better clinical formulation. In addition, the modulation of such properties could potentially lead to better patient tolerability and adherence rates. Importantly, drug release was not affected by the adjustments made to the existing formulation, with representative drug concentration profiles before and after the formulation change demonstrating comparable anti-glaucoma release kinetics. Future studies will be focused on the utilization of the improved formulation for the release of other drugs and/or combined therapeutics, to extend its potential applicability to the treatment of different kinds of ocular pathologies.
Acknowledgements
We acknowledge support from NIH CORE Grant P30 EY08098 to the Department of Ophthalmology, from the Eye and Ear Foundation of Pittsburgh, and from an unrestricted grant from Research to Prevent Blindness, New York, NY.
Footnotes
Conflicts of interest
The authors report no conflict of interest.
References
- 1.Asasutjarit R, Thanasanchokpibull S, Fuongfuchat A and Veeranondha S, Int. J. Pharm, 2011, 411, 128–135. [DOI] [PubMed] [Google Scholar]
- 2.Yu S, Zhang X, Tan G, Tian L, Liu D, Liu Y, Yang X and Pan W, Carbohydr. Polym, 2017, 155, 208–217. [DOI] [PubMed] [Google Scholar]
- 3.Ratay ML, Bellotti E, Gottardi R and Little SR, Adv. Healthc. Mater, 2017, 1700733, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fedorchak MV, Conner IP, Cugini A, Schuman JS and Little SR, Sci. Rep, 2017, 7, 8639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cholkar K, Patel SP, Vadlapudi AD and Mitra AK, J. Ocul. Pharmacol. Ther, 2013, 29, 106–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jünemann AGM, Chorągiewicz T, Ozimek M, Grieb P and Rejdak R, Ophthalmol. J, 2016, 1, 29–35. [Google Scholar]
- 7.Cholkar AKMK, Patel A, A Dutt Vadlapudi, Recent Pat. Nanomed., 2014, 2, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison PW and V Khutoryanskiy V, Ther. Deliv, 2014, 5, 1297–1315. [DOI] [PubMed] [Google Scholar]
- 9.Sahoo SK, Dilnawaz F and Krishnakumar S, Drug Discov. Today, 2008, 13, 144–151. [DOI] [PubMed] [Google Scholar]
- 10.Peng CC, Kim J and Chauhan A, Biomaterials, 2010, 31, 4032–4047. [DOI] [PubMed] [Google Scholar]
- 11.Kassem MA, Abdel Rahman AA, Ghorab MM, Ahmed MB and Khalil RM, Int. J. Pharm, 2007, 340, 126–133. [DOI] [PubMed] [Google Scholar]
- 12.Ali HSM, York P, Ali AMA and Blagden N, J. Control. Release, 2011, 149, 175–181. [DOI] [PubMed] [Google Scholar]
- 13.Ameeduzzafar A, Ali J, Fazil M, Qumbar M, Khan N and Ali A, Drug Deliv, 2016, 23, 710–726. [DOI] [PubMed] [Google Scholar]
- 14.Yellepeddi VK and Palakurthi S, J. Ocul. Pharmacol. Ther, 2016, 32, 67–82. [DOI] [PubMed] [Google Scholar]
- 15.Dubald M, Bourgeois S, Andrieu V and Fessi H, Pharmaceutics, 2018, 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macoul KL and Pavan-Langston D, Arch. Ophthalmol, 1975, 93, 587–590. [DOI] [PubMed] [Google Scholar]
- 17.Strohmaier K, Snyder E and Adamsons I, J. Am. Optom. Assoc, 1998, 69, 441–51. [PubMed] [Google Scholar]
- 18.Cheng YH, Tsai TH, Jhan YY, Chiu AWH, Tsai KL, Chien CS, Chiou SH and Liu CJL, Carbohydr. Polym, 2016, 144, 390–399. [DOI] [PubMed] [Google Scholar]
- 19.Iohara D, Okubo M, Anraku M, Uramatsu S, Shimamoto T, Uekama K and Hirayama F, Mol. Pharm, 2017, 14, 2740–2748. [DOI] [PubMed] [Google Scholar]
- 20.Song J, Bi H, Xie X, Guo J, Wang X and Liu D, Int. Immunopharmacol, 2013, 17, 99–107. [DOI] [PubMed] [Google Scholar]
- 21.Mammen A, Romanowski EG, Fedorchak MV, Dhaliwal DK, Shanks RM and Kowalski RP, Transl. Vis. Sci. Technol, 2016, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iqbal M, Zafar N, Fessi H and Elaissari A, Int. J. Pharm, 2015, 496, 173–190. [DOI] [PubMed] [Google Scholar]
- 23.Ramazani F, Chen W, Van Nostrum CF, Storm G, Kiessling F, Lammers T, Hennink WE and Kok RJ, Int. J. Pharm, 2016, 499, 358–367. [DOI] [PubMed] [Google Scholar]
- 24.Fedorchak MV, Conner IP, Medina CA, Wingard JB, Schuman JS and Little SR, Exp. Eye Res, 2014, 125, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang TI, Huang YC, Yang SC, Yeh JM and Peng YY, Mater. Chem. Phys, 2015, 152, 158–166. [Google Scholar]
- 26.Park H, Guo X, Temenoff JS, Tabata Y, Caplan AI, Kasper FK and Mikos AG, Biomacromolecules, 2009, 10, 541–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rwei SP, Tuan HNA, Chiang WY and Way TF, Materials (Basel)., 2018, 11, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schild HG, Muthukumar M and Tirrell DA, Macromolecules, 1991, 24, 948–952. [Google Scholar]
- 29.Lanzalaco S and Armelin E, Gels, 2017, 3, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang G and Wu C, J. Am. Chem. Soc, 2001, 123, 1376–1380. [Google Scholar]
- 31.Van Durme K, Van Assche G and Van Mele B, Macromolecules, 2004, 37, 9596–9605. [Google Scholar]
- 32.Ding Y and Zhang G, J. Phys. Chem. C, 2007, 111, 5309–5312. [Google Scholar]
- 33.Lee WF and Lin YH, J. Mater. Sci, 2006, 41, 7333–7340. [Google Scholar]
- 34.Gurdag G and Mutlu Oz G, Polym. Adv. Technol, 2009, 20, 216–224. [Google Scholar]
- 35.Pollock JF and Healy KE, Acta Biomater, 2010, 6, 1307–1318. [DOI] [PubMed] [Google Scholar]
- 36.Almeida H, Amaral MH, Lobão P and Lobo JMS, Drug Discov. Today, 2014, 19, 400–412. [DOI] [PubMed] [Google Scholar]
- 37.Wilson SL, Ahearne M and Hopkinson A, Toxicology, 2015, 327, 32–46. [DOI] [PubMed] [Google Scholar]
- 38.D’souza AA and Shegokar R, Expert Opin. Drug Deliv, 2016, 13, 1257–1275. [DOI] [PubMed] [Google Scholar]
- 39.Acheampong AA, Small D, Baumgarten V, Welty D and Tang-Liu D, J. Ocul. Pharmacol. Ther, 2002, 18, 325–37. [DOI] [PubMed] [Google Scholar]