

Abstract
Fourier transform infrared (FTIR) spectra of biological cells can reveal clinically important information about cells’ composition, including their normal or cancerous status. The recently emerged diagnostic technique of spectral cytopathology (SCP) combines FTIR with multivariate statistical analysis to detect cell abnormalities, differentiate between cell types, and monitor desease progression. We demonstrate a new variant of SCP, a metasurface-enhaced infrared reflection spectroscopic cytopathology (MEIRSC), that utilises judiciously designed plasmonic metasurfaces to localize and enhance the evanescent field near the cell’s membrane, and to carry out spectroscopic interrogations of the cells attached to the metasurface using reflected infrared light. Our findings indicate that the MEIRSC approach enables us to differentiate between normal and cancerous human colon cells. The sensitivity of MEIRSC is such that a very small (about 50 nm deep) portion of the cell can yield valuable diagnostic information.
Graphical Abstract
Metasurface-enhanced infrared reflection spectroscopic cytopathology (MEIRSC) is used for label-free distinguishing between normal and cancerous colon cell lines.
Introduction
The ability to distinguish between different phenotypic states of a given cell, as well as between different types of cells, is crucial for a variety of fundamental and clinical life sciences applications. These include the monitoring of biochemical processes in a living cell [1] (including its response to therapeutics and other stimuli) and effective early cancer screening [2], just to name a few. The science of differentiating between tumorous and normal cells, commonly referred to as cytopathology (or sometimes simply cytology), is an important and established pre- and post-operative diagnostic tool. Cytology relies on the visual inspection of the morphology of stained cells by a pathologist, followed by an interpretation of their state (e.g., cancerous, pre-cancerous, normal, etc.). However, morphological features do not provide the needed diagnostic sensitivity, which is presently in the 30%-87% range [3]. For many diseases, such as cervical or lung cancer, both sensitivity and specificity of cytology are even lower [4], [5].
More specific approaches to differentiating between different cell types include immunological evaluation, i.e. they rely on antibodies attaching to specific antigens that are over-expressed by the cells. Immunologic and morphological evaluations can also be combined [6], [7] for better specificity. However, the specificity of immunologic approaches is also limited because different cell types may express the same antigens. For example, epithelial cell adhesion molecule (EpCAM) is a common antigen for a variety of tumor cells. Fluorescent staining for various positive markers can be used for cell type differentiation such as distinguishing between circulating tumor cells (CTCs) and leukocytes [8]; but, the same issue of limited specificity remains. Moreover, the viability of stained cells is not guaranteed.
Therefore, there is considerable interest in label-free approaches to cytology that rely entirely on the native properties of the cell. Infrared spectroscopic cytopathology (SCP) [9], [10] is one such promising technique. It relies on spectroscopic data obtained from coupling mid-infrared (MIR) light to the vibrational modes of the constituent molecules (e.g., proteins, lipids, phospholipids, etc.). The cell’s fingerprint associated with the MIR part of the electromagnetic spectrum which overlaps with molecular vibrations (wv = 900 – 1,800cm−1) provides a representation of the cellular structure/function based on chemical bonds’ vibrations. It provides an excellent (potentially clinical) tool for distinguishing between different cell types and populations [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], and can be applied to single cells or even to tissue sections [22].
MIR SCP is an inexpensive and automated tool that has already been used in a variety of rather subtle cell-characterization applications, including the investigations of the effects of anti-cancer drugs on tumour cells [23] because of its sensitivity to metabolic perturbations inside a cancer cell during the treatment. Other successful applications of MIR SCP include the characterization of blood [24], urine [25], and plasma [26] cells. Remarkably, for some of the cancer types (e.g., cervical cancer) SCP has already been shown [3] to be a more reliable diagnostic technique than conventional cytopathology. Guided by the key idea that biochemical information obtainable from the spectroscopic data can be at least as valuable for medical diagnoses as the morphological information [3], various spectroscopic modalities have been developed over the years [10] [27], [19].
Briefly, the most common vibrational spectroscopic methods for cellular analysis include Fourier Transform Infrared (FTIR) transmission spectroscopy [28], [29], [30], [31], [32], attenuated total reflection (ATR) FTIR spectroscopy [33], [22], [18], [34], [35], and transflection [36], [27] spectroscopy. With the exception of the ATR-FTIR spectroscopy, all these techniques involve the transmission of IR light through the entire cell. This can be a significant limitation, especially for performing the SCP of live cells in their natural aqueous environment. Because MIR radiation is absorbed by water, it cannot be sent through the water on its way towards the cells unless the water layer is narrower than roughly 10 μm [37]. However, the extremely shallow depth of such channels is not desirable because it can impose mechanical stress on the cells. These limitations of the transmission-based measurements explain why most of such measurements have been carried out with dried/fixed cells. ATR-FTIR measurements of live cells in the aqueous environment have been done in reflection because the technique does not involve MIR light propagation through water. However, the high-index prisms are too costly to be used as single-use optical devices.
The second limitation of the transmission-based SCP is more subtle, and is not related to the cells being alive or fixed. It has to do with the fact that the multi-organelle structure of a cell is rather complex, and the transmission spectra contain information about molecular composition of all organelles inside the cell. For many applications, it may be desirable to focus on a specific region of the cell (e.g., the cellular membrane), and the contributions from other, less relevant, regions of the cell obscure the target of interest. For example, it has been known for some time [38] that the cellular progression from normal to cancerous is frequently accompanied by significant molecular composition changes at the cell’s surface, such as significant changes in extracellular proteins [39] and downregulation of cell adhesion molecules [40]. Therefore, there is a well justified need to be able to “see” tens of nanometers deep into the cell, which can only be done using evanescent fields [41], [42].
Another application that could benefit from evanescent field spectroscopy is the identification of multi-drug resistant (MDR) cancer cells. MDR is a clinically important cellular-level phenomenon that is frequently caused by the drugs’ efflux from the cells. The latter is mediated by the activity of over-expressed trans-membrane transporter proteins [43], [44], [45], [46]. Therefore, if the primary goal of the spectroscopic interrogation is to distinguish between MDR and non-MDR cells, then it is clearly advantageous to collect the IR spectra from a small portion of the cell that includes the membrane region.
Quantitative interpretation of the observed physical differences between drug-resistant and drug-sensitive cells is another important application where it is advantageous to collect spectral signatures from the region around the cellular membrane. For example, bio-mechanical properties of the cellular surface, such as its stiffness [47], is a useful biomarker for drug resistance [48]. A cell’s biomechanics is largely determined by its near-surface biochemical properties, which include the membrane, the underlying cortical cytoskeleton, and the connecting proteins. All these sub-cellular structures are located within less than a hundred nanometres from the cellular surface [49], and could be potentially interrogated using a spectroscopic technique that emphasizes their contributions.
Thus, there is a clear incentive to develop a reflection-based spectroscopic technique that does not require passing MIR light through the entirety of a cell, and can collect vibrational signatures from a small region of a cell. An additional desirable feature of such technique would be enhanced optical field intensity at the cell that could potentially enhance the signal-to-noise ratio of the reflected signal [50]. Once the technique is developed, another important question can be experimentally addressed: is the spectral information from such a small region of a cell sufficient for distinguishing between different cell types (e.g., cancerous or non-cancerous, drug-resistant or drug-sensitive, pluripotent or differentiated)?
Plasmonic metasurfaces (see Fig. 1 for a specific metasurface design used in this work) are periodic arrays of engineered metallic antennas with tailored electromagnetic response over a broad range of frequencies [51], [52], [53], [54], [55], [56], [57], [58], [59]. Especially important for the SCP applications are infrared metasurfaces because biomolecules constituting live cells possess mid-infrared vibrational fingerprints that can be used for their identification, thereby improving the specificity of optical biosensing. Metasurfaces enable strong concentration and enhancement of optical energy in their immediate proximity [59], [60], [61]. The most straightforward qualitative reason for the optical field enhancement at the metasurface is the “lightning rod” effect: the electric field of the incident light wave is enhanced because individual elements of the metasurface act as highly-polarizable optical antennas. For the more complex multi-antenna designs of the unit cell, even stronger enhancement can be achieved as described below. Not surprisingly, MIR metasurfaces have already been utilized to detect and quantify the presence and spatial orientation properties of protein monolayers [59], [50].
Fig. 1.

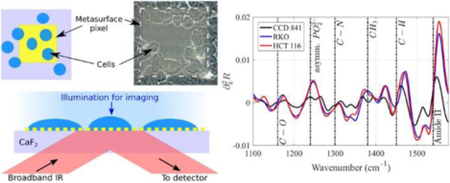
Schematic of the device for metasurface-enhanced infrared reflection spectral cytopathology (MEIRSC). (a) Plasmonic metasurface pixels composed of two rows (Pixel 1 row and Pixel 2 row) of pixels. Each pixel is a square array of unit cells. Each unit is comprised of two unequal gold antennas (straight and bent) deposited on IR-transparent substrate (CaF2). The units comprising Pixels 1 and 2 are scaled versions of each other. (b) Left: sketch of a metasurface pixel (yellow shaded region) with deposited cells (blue ellipses) on top. Cells do not cover the entire metasurface. Right: image acquired from the experimental sample clearly depicting the metamaterial pixel (light square) and cells (darker circular patches). (c) Cells are deposited onto the metasurface sensor with the aid of a PDMS chamber. PDMS and CaF2 are held together with acrylic clamps while the cell solution is injected into the chamber. (d) The cells are simultaneously imaged (visible light) in transmission and spectroscopically interrogated (MIR light) in reflection through the substrate and metasurface. Visible light illumination: from the top, MIR illumination: from the bottom. Both IR and visible collection optics: on the bottom.
Similarly to other internal reflection elements (IREs) [62], such as high-index ATR prisms, plasmonic metasurfaces produce a locally enhanced optical field that exponentially decreases away from the surface. However, unlike the ATR prisms that produce spatial localization of the evanescent fields on a scale of a micron, plasmonic metasurfaces enable field localization over a distance of < 100nm. Moreover, the field enhancement produced by the metasurfaces can be considerably higher than that produced by the high-index prisms [50]. High field enhancement is highly beneficial for spectroscopic applications because stronger fields enhance the light-matter interaction, thereby increasing the transduced signal (e.g., the spectral features in the reflected signal corresponding to molecular vibrations) from the biological material (cells, tissues, protein monolayers) placed on top of the IRE. Moreover, strong field localization enables us to probe the outer region of the cell that is in more intimate contact with the metasurface than the rest of the cell [63]. The localization depth of the field at the metasurface is comparable to the thickness of the cellular membrane (several nanometers, including the extracellular molecules), but much smaller than the total thickness of a cell (several microns).
Motivated by the above properties of MIR plasmonic metasurfaces for SCP applications, we describe a novel spectroscopic technique, Metasurface-Enhanced Infrared Reflection Spectroscopic Cytopathology (MEIRSC), and apply it to several types of human cells (normal and cancerous). While metasurfaces have already been used for enhanced spectroscopy and identification of protein monolayers [59], so far there has not been an experimental study of metasurface-enhanced spectroscopy of the whole cells. It is not a priori obvious that metasurface-enhanced spectroscopy is a good match for studying adherent cells.
For example, the very small localization length of the evanescent fields presents not only an opportunity to study cellular membranes, but also a challenge because a cell needs to be in intimate contact with the metasurface in order for this technique to work. It is reasonable to expect that the MEIRSC should work because the separation between cells and the sensing substrate is estimated [64] to be from 10–15 nm (for focal contacts) to 30–50 nm (for close contacts). However, this question can only be resolved experimentally, as we do in this study using fixed cells from three cell lines. We note that, while MEIRSC of live cells in their natural aqueous environment is beyond the scope of this study, there are many applications that require fixed cells [65]. For example, it may be desirable to transfer the samples to other laboratories, or to pathologists for further examination. Our study confirms that spectral information collected from a small region of human cells located in close vicinity of their membrane is sufficient for differentiating between cell lines of normal and cancerous colon. Multivariate analysis of the spectra [66], [22], [67], [68] enabled us to clearly differentiate between human colon normal (CCD 841) cell line and two colon carcinoma (RKO and HCT 116) cell lines. However, it was not possible to differentiate between the above colon carcinoma cell lines.
Experimental Procedure
Sensor Fabrication
The plasmonic metasurface sensors were fabricated with standard electron beam lithography followed by metal deposition (70 nm of Au on top of 10 nm of Cr adhesion layer) and liftoff process. Infrared-transparent CaF2 windows (12.5 mm × 12.5 mm, 0.5 mm thickness, Laser Optex Inc.) were used as substrates for the sensors. The periodic unit cell of the designed structure is shown in Fig. 1a. The units are arranged as two dimensional (2D) arrays with unequal periods Px and Py. Further details about the unit cell of the metasurface, including individual antenna sizes and periodicities, are given in the Supplementary Information (SI) section, Fig. S1†. A 2D array of these unit cells forms one individual sensor ‘pixel’. Each pixel is a 120 × 120 μm2 square. Two pixel types with different spectral responses (due to their different geometric sizes) were used in this study. The fabricated sensor array had 5 separate copies of each pixel type (Pixel 1 and Pixel 2 on Fig. 1a) on each CaF2 substrate. Tuning of the spectral response for the different pixels was achieved by scaling all the in-plane dimensions, including the periods Px and Py of the unit cell. Pixels’ geometry definitions, corresponding scaling factors, and the two dominant resonance frequencies (dipole resonance and Fano) are given in SI section, Table S1†.
Cells Preparation
The three human colon cell lines used in the experiments are listed in Table 1. All the cells were cultured in cell culture medium (Thermo Fisher Scientific MEM solution) supplemented with 10% fetal bovine serum (FBS, Corning) at 37°C and 5% CO2. Prior to the experiments, the adherent cells were detached using TrypLE Express (obtained from Gibco). The cell suspension was then centrifuged at 1200 rpm for 2 min to pellet the cells. The supernatant was discarded and the cell pellet was re-suspended in fresh cell culture medium (without FBS). The number of cells in a pellet ranges from 1–5 million, which corresponds to roughly ¼ to ½ of cells in a culture flask at moderate to high coverage (on the culture surface of the flask). This procedure removes cellular debris as well as dead cells prior to incubation with the metasurface samples.
Table 1:
List of the cell types used and their origins.
| Cell Type | Origin |
|---|---|
| CCD 841 | Human colon normal (ATCC CRL-1790) |
| RKO | Human colon carcinoma (ATCC CRL-2577) |
| HCT 116 | Human colon carcinoma (ATCC CCL-247) |
Cell Deposition
Because colorectal cancer cells are known to over-express the epidermal growth factor receptor (EGFR) proteins [69], [70], we facilitated their attachment to the metasurface by functionalizing it with anti-EGFR antibodies (E2760, Sigma). To facilitate antibodies’ attachment to gold metasurfaces, thiol linkers (dithiolaromatic PEG6-NHNH2, SPT-0014B, SensoPath Technologies) were attached to the antibodies using a method described elsewhere [71]. The sensor surface was submerged in antibody solution and placed on a rocking shaker (Fisher Scientific Open Air Rocker) for 4 hours. Afterwards, it was gently washed three times with milli-Q water to remove excess unbound antibodies. Consequently, the plasmonic metasurface was functionalized with antibodies to capture EGFR-expressing cells.
A PDMS fluidic chamber was attached to the metasurface with acrylic clamps, and a cell solution (~106 cells/mL, 90 μL total volume) was injected into the chamber with the aid of a syringe and tubing (Fig. 1c). The cells were allowed to settle onto the metasurface for about 10 minutes (Fig. 1 and Fig. 2). The sample was then incubated at 37°C and 5% CO2 for four hours. We found that incubation enhances the spectroscopic signal originating from molecular vibrational lines. The incubation time can be shortened to two hours and still observe significant increase of spectral lines’ strength compared to samples that were not incubated after cell deposition. Cell coverage was roughly 20–30 cells per pixel. After allowing the cells to spread on the metasurface in an incubator, the cells were fixed by incubating them in a 4% paraformaldehyde (PFA) solution for 20 minutes. Subsequently, the PDMS chamber was removed and the sample with cells was washed with milli-Q water three times to remove any residual PFA. After the fixation, the samples were allowed to dry for a day at ambient conditions, followed by the measurements of the IR spectra in reflection.
Fig. 2.

Device preparation steps. (1)-(2) Metasurface pixels are fabricated onto IR transparent CaF2 substrate. (3) A protein (antibody) layer is deposited onto the metasurface to form an artificial surface for the anchoring of the cells. (4) Live cells are allowed to get immobilized on the antibody-presenting metasurface, and (5) adhere to the surface during a 4 hour incubation at 37°C. (6) Cells are fixed with 4% paraformaldehyde (PFA) solution and left to dry for one day before collecting IR spectra.
Data Collection
Reflection spectra from the metasurface pixels covered with cells were measured through the substrate as illustrated in Fig. 1d. The spectra were collected with a Thermo Scientific Continuum microscope coupled to a Nicolet 6700 FTIR spectrometer equipped with XT-KBr beamsplitter and liquid nitrogen cooled MCT detector. The y –polarized (see Fig. S1a) incident light was used for all measurements. Data was recorded at 4°cm−1 resolution in 650–4000 cm−1 range. The measurement area under the microscope was limited to a 100 μm × 100 μm square. The collected spectra were then analyzed using internally developed MATLAB scripts. Two independent experiments were run for each cell line.
Data Analysis
Principal component analysis (PCA) was employed to analyze the collected reflection spectra. MATLAB software was used for all data analysis. Measured spectra were smoothed using moving average filter to lower noise. Due to slight variations in experiment conditions, the absolute values of reflectance can fluctuate from measurement to measurement. Therefore, all reflectance spectra were normalized to their peak reflectance.
Second derivatives of the normalized reflection spectra were calculated and used as input data for PCA because it has been shown [27], [65] to emphasize the vibrational lines. In order to give larger weight to native molecular vibrations, the spectral regions that are close to plasmonic resonances of the metasurface were discarded in the PCA. Full spectra were truncated to include the data in the 1100–1580 cm−1 spectral range for Pixel 1, and 1100–1770 cm−1 for Pixel 2. Additionally, all data in the 1300–1640 cm−1 range was discarded for Pixel 2 because of the strong plasmonic resonance in that range.
PCA of all second derivative spectra was conducted using MATLAB’s built-in functions with default parameters (singular value decomposition (SVD) algorithm and data centering before computing SVD). The two pixel types used in this study were analyzed separately.
Experimental Measurement of the Evanescent Field Penetration Distance from the Metasurface
We experimentally characterized the penetration depth of the enhanced optical field produced in the proximity of the two plasmonic metasurfaces (Pixels 1 and 2) used in this study. The field penetration depth of the plasmonic metasurface was quantified by using different thicknesses of polymethyl methacrylate (PMMA) that was spin coated onto the metasurface. The addition of PMMA changes the refractive index surrounding the metasurface from that of the air, and causes the spectral shift of the reflectivity curves. The magnitude of the spectral shift is dependent on the PMMA layer thickness t.
PMMA was spin coated onto the metasurfaces at four different thicknesses: t = 44 nm, 57 nm, 67 nm, and 136 nm. The reflection spectra from the coated metasurfaces were measured using the same methods as described in the Data Collection section above. Spectral shift of the broad (“dipole” [59]) peak in the reflection spectra was used to quantify the field penetration depth. Experimental data of dipole peak shift (Δλ) versus PMMA thickness (t) was fitted with a function in the form of where a and tsat (the sought after penetration depth) are free parameters. The experimental data presented in Fig. 4 indicates that for both pixels (1 and 2) the saturation thickness is tsat~45 nm. These observations are in good agreement with the results obtained from numerical simulations (Fig. 3b).
Fig. 4.

Measurements of the penetration depth tsat of the metasurface evanescent field using variable-thickness PMMA coverage. The wavelength shift of the reflectivity maximum for (a) Pixel 1, and (b) Pixel 2 depends on the PMMA thicknesses t. At t = tsat. the shift reaches 63% of its asymptotic value.
Fig. 3.

The principles and advantages of MEIRSC: evanescent field enhancement and localization, high SNR. (a) Enhancement of evanescent optical fields by the plasmonic metasurface. Field enhancement has been averaged over all the surfaces of gold, except for the gold-substrate interface. Spectrally flat “shoulders” enable broadband spectroscopy. Gray shaded area at the Fano resonance position includes the peak of field enhancement (~220), but has been omitted from the figure because this spectral region is not used in analysis of cell spectra. The inset shows the optical field distribution. (b) Field intensity averaged over a plane positioned at z = ℎ above the metasurface. (c)-(d) Optical microscopic images of CCD 841 cells on the metasurface and on the bare CaF2, respectively. White dotted line in (c): outline of the sensor pixel. (e) Comparison of signal strengths (second derivative of reflectivity) from the cell-covered areas with (red) and without (black) metasurface. The curve without metasurface is flipped with respect to the bare substrate because features are π out of phase for the two surfaces. The metasurface used in this case is Pixel 2 and the cells are CCD 841. It is clear that the metasurface significantly enhances the vibrational lines from the cells. The signal from the bare substrate (black line) is difficult to discern from the noise (gray region), except for the strongest vibrational lines (Amide I and II).
Results and discussion
Plasmonic Metasurfaces as IR-Reflective and Optically Transparent Substrates for Vibrational Spectroscopy
One of the most desirable features of the plasmonic metasurfaces is a large light intensity enhancement factor equal to the near-field electric field intensity at the metasurface (|E2|) divided by the electric field intensity in the incident pulse . The field enhancement, calculated directly on the metasurface and plotted in Fig. 3a,b, has two important properties: (a) a sharp and narrow field enhancement resonance related to the so-called Fano resonance [59], [72] of the metasurface, and (b) a spectrally broad “shoulder” in the field enhancement curve that enables broadband sensing using a single pixel. While in this study we are not taking advantage of property (a), property (b) enables us to include spectrally distant molecular vibrations into the PCA. This is particularly important for a pilot study, like the one presented here, because it is not a priori clear which molecular vibrations provide the clearest differentiation between different cell types.
When a cell is near such a metasurface, the field penetrates a small distance (tsat <100 nm) into the cell, probing only a peripheral region of the cell. In addition, probing the cells in reflection from the metasurface makes it greatly insensitive to the thickness of the cell layer. The metasurface also significantly increases the reflectivity, thereby improving the signal-to-noise (SNR) ratio and enabling shorter spectrum acquisition times. Although not essential for this study, the fact that the reflected signal is collected from the substrate side is beneficial for conducting SCP studies in aqueous environment, where water absorption is an important factor.
To demonstrate the effectiveness of the MEIRSC approach, we have compared the reflection spectra for the CCD 841 cells that were deposited either on the metasurface (see the microscopy image in Fig. 3c) or on the bare CaF2 substrate (see the microscopy image in Fig. 3d). Reflectivity R(k) from the cells on the bare substrate is very small: R(k) ~ 1 ÷ 2% across the entire spectral range of interest, 650 cm−1 < k < 4000 cm−1, where k (measured in inverse centimeters) is the wavenumber which is related to the wavelength λ (measured in microns) according to. k = 104/λ. On the contrary, the reflectivity from the cells placed on the metasurface is more than an order of magnitude larger.
More significantly, the reflected spectrum from the cells on a bare substrate does not have a sufficient SNR to identify the molecular vibrational lines with certainty. To demonstrate that, we have compared the second derivatives of the reflectivity, , for the metasurface-enhanced (red line in Fig. 3e) and bare-substrate (black line in Fig. 3e) data. Clearly, the signal from the cells on the bare substrate is at the noise level (gray zone in Fig. 3e), making it impossible to identify the vibrational features. On the contrary, the metasurface-enhanced vibrational features are much stronger than the noise and can be easily identified (see Table 2 for a list of identified vibrational lines and their origin).
Table 2:
Table of molecular vibrational lines of interest. These lines correspond to the peaks in vs k plot. The listed modes have been extracted from references [27] [36] [79] [80] [81] [82] [83].
| ω (cm−1) | Vibrational mode | Notes |
|---|---|---|
| 970 | C-O-P stretch, phosphodiester | Phospholipids; generally weak |
| 1080 | Symmetric stretch of phosphate group | Phospholipids; carbohydrate vibrations nearby: ν(C-C), ν(C-O), ν(C-O-H), ν(-CO-O-C) |
| 1150–1170 | C−O stretching | Carbohydrates |
| 1230–1245 | Asymmetric stretch of a phosphate group | Phospholipids, phosphorylated protein |
| 1300 | Amide III, C-H stretch, N-H bend | Proteins |
| 1380 | δ(CH3) symmetric bending mode | Glycolipids, proteins |
| 1450 | C−H vibration: δas(CH3), δ(CH2) |
Lipids, proteins |
| ≈1550 | Amide II: δ(NH) + v(CN) |
Proteins |
| ≈ 1660 | Amide I: v(C = O) + δ(NH) |
Proteins; merged with H2O absorption |
| 1740 | v(C = O) vibration | Lipids |
To put MEIRSC in a proper perspective vis a vis other evanescent field techniques that enable sub-micron penetration into a cell, we note that the penetration depth of IR field for the metasurfaces used in this work, tsat ~ 45nm, is comparable to the excitation depth (~60 ÷ 100 nm) in the Total Internal Reflection Fluorescence (TIRF) microscopy. TIRF is a widely used method for investigating processes in and around a cellular membrane [73], [74]. While TIRF imaging provides excellent spatial and temporal resolution, it relies on the use of fluorescent molecules. In contrast, metasurface-enhanced spectroscopy requires no labelling of cells or molecules in them because IR spectra of the molecules in and around the membrane are the natural “fingerprints” of a cell.
Analysis of Cell Spectra
We used several human cell lines (see Table 1) to investigate spectroscopic differences between different cell types. All of these cell lines are derived from the same organ — colon. The CCD 841 cells are normal, while RKO and HCT 116 cells are cancerous. This allows us to look at the differences between normal and cancerous cells originating from the same organ (albeit derived from different patients).
We have used a monolayer of anti-EGFR antibodies to immobilize the cells on the metasurface pixels (Fig. 2). Note that this antibody is not very specific because all cell types analyzed here express at least some EGFR on their surface. Therefore, the antibody used here is employed for better attachment, not for better specificity. Other moderately-specific surface engineering strategies for immobilizing cells on metasurfaces can be utilized as well, e.g., using surfaces presenting adhesion proteins or peptides [75], [76]. However, the main specificity in SCP comes from the infrared spectra, not from the cell binding ligands. Even though the antibodies alone also contribute to the spectrum because they constitute a protein monolayer, we have experimentally found that their spectrum contains much weaker spectral features than those from the cellular spectra. The reason for that is the small thickness (under 10nm) of the protein monolayer.
All the cells were fixed prior to the collection of the IR spectra. Fixing a cell causes the cell components to be immobilized in their final state. This allows for a more controlled experiment as compared to live cells’ experiments, where the cells’ conditions change with time. Recent studies [65] indicate that spectral changes introduced by fixation are negligible in comparison to changes induced by disease. In our case, fixation had another benefit—it enhanced signals from vibrational lines. Additionally, the presence of water dampens the signal from the vibrational lines of interest. However, fixing the cells allows us to dry the cells, thus making these vibrational fingerprints become more prominent. There are nevertheless some water molecules left even in dry fixed cells, however, as evidenced by their (albeit diminished) spectral signature at 1670 cm−1. These are presumably water molecules that are hydrogen bonded with other molecules in the cell.
Two representative IR reflection spectra ((k), see Fig. 5a) and their second derivatives (, see Fig. 5d) for the RKO cell type are shown on Fig. 5. In the as-measured IR reflection spectra, the vibrational lines except for Amide I and Amide II are difficult to see with the bare eye (see Fig. 5a). Note that the reflection spectra in Fig. 5a do not look like the standard absorbance spectra in transmission FTIR spectroscopy. Similar phenomena have been observed in a different context of measuring reflection and transflection spectra of single cells [36], [77]. The unusual spectral shapes were explained as a convolution of broadband Mie resonances of a cell with vibrational modes of the individual constituent biomolecules. In the case of metasurface-enhanced spectroscopy, the Mie resonance of a cell is replaced by the plasmonic (dipole) resonance of the metasurface. While it is possible, in principle, to follow the same strategy as Refs. [36], [77] to obtain the effective absorbance, we did not find that to be necessary. Note that similar effects of spectral lines’ modification by plasmonic resonances were observed for protein monolayers deposited on top of plasmonic metasurfaces [59] and antenna arrays [50].
Fig. 5.

(a) Two representative normalized reflectivity spectra (k) as a function of the wavenumber k for the cell-covered metsurface Pixels 1 and 2. Only a few molecular vibrational lines are clearly discernible from the overall shape of (k). (b)-(c) Optical microscope images of the cells on top of the Pixels 1 and 2 (white dotted outlines). (d) Second derivatives of the spectra shown in (a). The molecular vibrational lines from the deposited cells are much clearer in the second derivative spectra. In (a) and (d) Pixels 1 and 2 are represented by the blue and red lines, respectively. Pixel 1 is a scaled-down version of Pixel 2, with the in-plane scaling ratio of 0.86. The fixed/dried cells are from the RKO (human colon carcinoma) cell line.
In the second derivative spectra , approximately 10 vibrational lines are much more clearly discernible (see Fig. 5d). The most prominent vibrational features identified from our measurements in the 900 cm−1 < k < 1800 cm−1 spectral range are as follows: kC−O−P ≈ 970cm−1, , kC−O ≈ 1150cm−1, , kA−III ≈ 1300cm−1, , , kA−II ≈ 1540cm−1, kA−I ≈ 1660cm−1, and . Here the A−I, A−II, and A−III labels refer to Amide I-III vibrational modes of proteins, respectively [68], [78].
Separate Amide III features at 1300 cm−1 (combined C-H stretch and N-H bend) and at 1240 cm−1 (combined C-N stretch and N-H bend) have also been identified in the literature [78]. However, distinguishing the latter from the exceeds the accuracy of our technique. The original assignment of the particular vibrations reflects the small penetration depth of the MEIRSC technique compared with more conventional transmission-based or ATR-based approaches to SCP. For example, the phosphate vibrations at and wavenumbers are frequently assigned to either nucleic acids (DNA/RNA), or to phospholipids [22]. However, DNA/RNA are primarily localized inside the nucleus which is too far away from the cellular membrane to be probed by MEIRSC. Therefore, we conclude that these spectral lines, which are visible in Fig. 5d, originate from phospholipids in the cellular membrane.
Therefore, based on our assumption that only a small portion of the cell next to the membrane is sampled by MEIRSC, the modes listed in Table 2 were identified as vibrational fingerprints of proteins, lipids, and carbohydrates using references [27] [36] [79] [80] [81] [82] [83]. There can be slight shifts between table values of molecular resonance positions and reflection dips derived from metasurface spectra, since in the latter case the exact dip position is dictated by the interaction between IR resonances of the molecular vibrations and the much broader resonances of the metasurface itself.
Note that the Amide I line overlaps with the water absorption line at k = 1670 cm−1, and as a result is overshadowed by the absorption of water. De-convoluting these two contributions requires precise control over water content. In our experiments, this is achieved by drying the fixed cells for a day. The Amide II line, however, is clearly visible in the spectrum, and is not distorted by water absorption.
In the absence of the cells, the reflection spectrum exhibits a very strong and sharp (see Fig. S1b,c) Fano feature [59], [72] that manifests itself in ultra-high field enhancement of the evanescent fields of the metasurface (see Fig. 3a, shaded zone). However, after the cells are deposited onto the sample and the reflection spectrum is measured, the Fano feature diminishes for those pixels with low cell coverage and disappears completely in case of high cell coverage (see Fig. S2). This is due to the loss of homogeneity because of the addition of cells. Those units of a metasurface that have a cell attached to them experience a significantly higher refractive index (and as a result experience a redshift) compared to those not covered by cells. This disparate behavior results in the disappearance of the sharp Fano feature (which is a collective phenomenon that depends on the existence of a large array of identical strongly interacting unit cells), while preserving the broader dipole feature. This phenomenon is discussed in more detail in supplementary information†. We note that the situation would be different if the cells were embedded in an aqueous environment because the refractive index of a cell is not very different from that of water. This is not the case for the dry cells SCP presented here; therefore, in this work we do not rely on the extremely high evanescent field enhancement near Fano resonances. Instead, we employ moderately high (just over an order of magnitude, see Fig. 3a) field enhancement outside of the Fano resonance region.
Quantitative Analysis of Cell spectra
Principal component analysis (PCA) was utilized to analyze how the spectral signatures of different cell lines vary quantitatively. We note that the reflection spectra and their second derivatives plotted in Fig. 5 are only representative of a single experiment on a single pixel. There is a considerable variation between the spectra obtained from two nominally identical pixels with similar coverage and distribution of the cells on the metasurface. The key question to be answered by our study is whether the difference between normal and cancerous (or cancerous, but originating from different cell lines) cells is larger than the variation produced by the variation of the state of the cells that belong to the same type. This question is answered by carrying out multivariate (PCA) analysis that enables us to group the cells based on their spectra, and to quantify the difference within the same cell type and between different cell types.
Second derivatives of cell spectra were used as input data for PCA because they are more sensitive to small spectral differences between fixed cells [65]. The resulting 1st and 2nd principal component scores, for both pixel sizes, are plotted on Fig. 6a and 7a, for the Pixel 1 and Pixel 2, respectively. In both cases, the first two principal components capture more than 85% of the data variance. Different symbols (ten for each cell) correspond to the three different cell lines as explained in the captions to Fig. 6 and 7. Each cell is represented by ten spectra: two separate experiments, with five duplicates for each experiment. Contrasting the CCD 841 cell line with RKO and HCT 116 cell lines is especially interesting because they are all human epithelial cells found in colon, with one (CCD 841) being normal (i.e. non-cancerous), and the others (RKO, HCT 116) being cancerous.
Fig. 6.
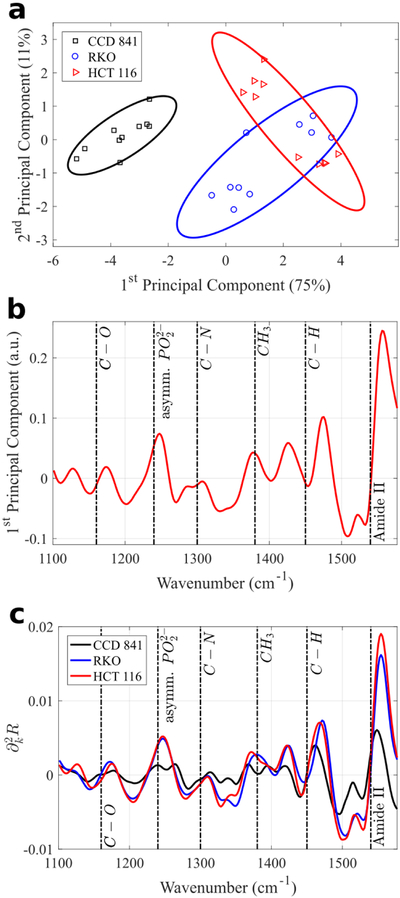
Principal Components Analysis (PCA) of the MEIRSC data collected from the three human colon cell types deposited on Pixel 1. (a) Scatter plot of the 1st and 2nd principal component scores. Normal and cancerous cell types occupy different parts of the plot, despite considerable variation within each cell type. Points corresponding to the CCD 841 cells (normal cells) clearly reside within an area on the left side of the plot, whereas the RKO and HCT 116 cells (cancerous cells) occupy the right side of the plot. Solid lines in (a) represent 80% confidence ellipses. Percentages in parentheses show the variance captured by the respective principal component. The first two principal components explain over 85% of variance. (b) 1st principal component spectrum (loading vector) reveals multiple identifiable molecular vibrational lines. (c) Average second derivative spectra for each cell type across all experimental measurements. While the differences between the two cancer cell lines (RKO and HCT 116) are small, the difference between those and the normal cell line (CCD 841) is significant.
Fig. 7.
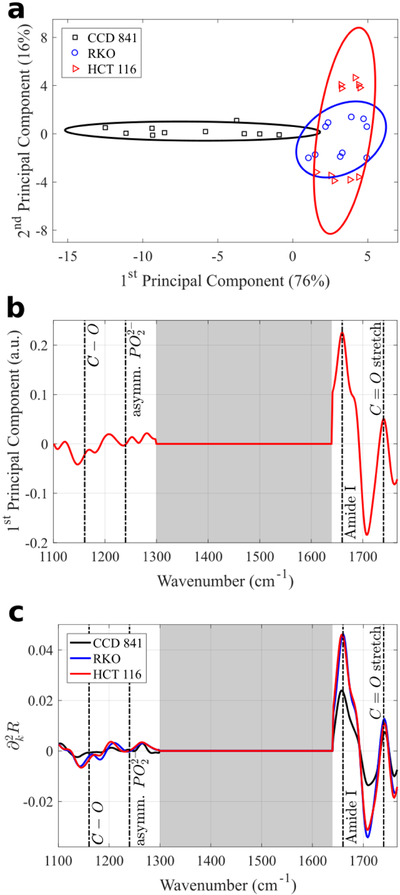
PCA of the MEIRSC data collected from the three human colon cell types deposited on Pixel 2. (a) Scatter plot of the 1st and 2nd principal component scores. Normal and cancerous cell types occupy different parts of the plot, despite considerable variation within each cell type. The separation of CCD 841 cells (normal cells) from the RKO and HCT 116 cells (cancerous cells) is less dramatic than for Pixel 1. Solid lines in (a) represent 80% confidence ellipses. Percentages in parentheses show the variance captured by the respective principal component. The first two principal components explain over 90% of variance. (b) 1st principal component spectrum (loading vector) reveals multiple identifiable molecular vibrational lines. Only low and high wavenumber features are represented due to discarding of the Fano resonance region in the spectra. Grey shaded areas in (b) and (c) mark the regions excluded from PCA. (c) Average second derivative spectra for each cell type.
The two different types of metasurface pixels are tuned to different resonant frequencies and hence provide the possibility of probing wider spectral regions. Moreover, comparing the results for Pixels 1 and 2 enables us to choose the pixel that is most suitable for differentiating between human colon cells. For example, for both pixels it can be observed that cancerous and normal cells occupy separate regions on the 2-dimensional principal component scatter plots (Fig. 6a and 7a). However, the separation between the normal and cancerous cells is clearer for Pixel 1. Therefore, we can conclude that Pixel 1 would be preferable for a blind study that requires assigning an unknown cell to a cancer/normal category.
The reason for that is that we had excluded the Fano resonance region from the analyzed spectral data sets for two reasons. First, the close presence of the Fano feature in the second derivative spectra distorts accurate identification of molecular vibrational lines due to the strong convolution of the Fano and vibrational features. Second, differences in cell coverage on each individual sensor pixel can significantly change the magnitude of Fano feature (as seen on Fig. S2), further complicating interpretation of PCA results. In practice, the excluded spectral ranges containing Fano resonances were selected from the steepest slope regions (large |∂kR|) for the bare (Fig. S1b,c) metasurfaces. Even though these spectral regions coincide with the desirable highest field enhancement of the metasurface (grey area on Fig. 3a for Pixel 2), they had to be discarded to avoid the extreme sensitivity of the spectra to the cell coverage. Nevertheless, more than an order of magnitude field enhancement outside of the Fano regions was sufficient to develop a highly sensitive SCP tool.
Now we can interpret why the performance of Pixel 1 was superior to that of Pixel 2. The difference between Pixels 1 and 2 lies in the spectral location of the Fano feature. Because all dimensions of Pixel 1 are smaller than that of Pixel 2, its Fano feature is blue-shifted with respect to that of Pixel 2 (see Fig. S1b,c). Specifically, the Fano resonance range lies entirely above the Amide II frequency. Therefore, the analyzed dataset for Pixel 1 does not include features with wavenumbers above Amide II, but does include most of the major mid-IR molecular vibrational lines identified in Table 2 (Fig. 6b,c). On the other hand, the dataset for Pixel 2 has a large portion of the spectrum cut out from the analysis: approximately 1300–1640 cm−1 (gray shaded area on Fig. 7b,c) corresponding to the Fano spectral region. Therefore, a smaller analyzed data set results in less differentiation between the normal (CCD 431) and cancerous (RKO and HCT 116) cells, as can be observed by comparing Fig. 7a with Fig. 6a. Remarkably, even for Pixel 2, it is still possible to distinguish cancerous and normal cells on the principal component scatter plot despite the reduced dataset.
We further observe that the 1st principal component spectra (the loading vectors) shown in Fig. 6b and 7b reveal that most of the spectral features differentiating between the cells correspond to the vibrational frequencies identified earlier and listed in Table 2. For example, Fig. 6b reveals that most of the variance (the first principal component vector) between the normal and cancerous cells is contained in the strengths of the Amide II line (protein), C-H vibration (from lipids and proteins), as well as the Amide III/asymmetric phosphate vibration (phospholipids and proteins).
The averaged (over ten data sets) spectra from Pixel 1 plotted in Fig. 6c reveal that the cancerous cells (RKO and HCT 116) have almost identical average spectra in the 1100 cm−1 to 1600 cm−1 spectral range; whereas the average spectra of the non-cancerous (CCD 841) cells are quite different in the same spectral range. The datasets collected from Pixel 2 metasurface provide complementary information about the difference between normal and cancerous cells: Fig. 7b,c indicate that there is considerable difference in the strength of the Amide I (proteins) spectral line. Remarkably, only the first two principal components are needed to clearly distinguish a normal cell from a cancerous one. Therefore, the combined SCP results from different pixels can be used to further increase the accuracy of the, potentially clinical, cancer diagnosis. Because different pixel types produce coverage sensitivity in different spectral ranges, earlier attempts to combine the spectra from multiple pixel types into a single PCA analysis produced less clear results [63], highlighting the importance to analyze data from different pixel types (Pixel 1 and Pixel 2 in this work) separately with PCA.
Conclusions
A novel characterization technique of biological cells – metasurface-enhance infrared reflection spectroscopic cytopathology (MEIRSC) – has been introduced and tested for its ability to differentiate between cancerous and normal human colon cells. We have measured MIR spectra of three immortalized cell lines (CCD 841, RKO, HCT 116) using plasmonic metasurfaces. These structures have spatially localized (~100 nm) high field enhancement (>20) over a broad spectral range. These properties make metasurfaces ideal for probing membrane and near-membrane regions of the cells, where significant differences between the molecular composition of normal and cancerous cells could be expected. Spectral measurements of fixed cells were carried out in air on an area of 100 μm × 100 μm, requiring only a few tens of cells. We have identified several characteristic molecular vibrations from the measured IR spectra, corresponding to the bonds present in the cells’ constituent molecules. Principal component analysis of second derivatives of the reflection spectra revealed spectral differences of cell types used. We were able to distinguish between normal cells (CCD 841) and cancerous cells (RKO, HCT 116) from the same organ – colon – demonstrating the power of this method. An important finding is that even a very small region of the cell (within tens of nanometers from the membrane) contains sufficient biomarkers to enable cell differentiation. Future work will investigate the possibility of applying MEIRSC to live cells in their natural aqueous environment.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R21CA173663. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was performed in part at the Cornell NanoScale Facility, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542081). The support of R.D. by Cornell’s Engineering Learning Initiatives (ELI) grant is also acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Grenci G, Birarda G, Mitri E, Businaro L, Pacor S, Vaccari L and Tormen M, Microelectronic Engineering, 2012, 98, 698–702. [Google Scholar]
- 2.Seddon AB, Phys. Status Solidi B, 2013, 250, 1020–1027. [Google Scholar]
- 3.Gajjar K, Ahmadzai AA, Valasoulis G, Trevisan J, Founta C, Nasioutziki M, Loufopoulos A, Kyrgiou M, Stasinou SM, Karakitsos P, Paraskevaidis E, Da Gama-Rose B, Martin-Hirsch PL and Martin FL, PLoS One, 2014, 9, e82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoler M and Schiffman M, JAMA, 2001, 285, 1500. [DOI] [PubMed] [Google Scholar]
- 5.Raghu G, Mageto YN, Lockhart D, Schmidt RA, Wood DE and Godwin JD, Chest, 1999, 116, 1168. [DOI] [PubMed] [Google Scholar]
- 6.Martin S, Zhang H-Z, Magyarosy E, Jaffe E, Hsu S-M and Chu E, AJCP, 1984, 82, 666. [DOI] [PubMed] [Google Scholar]
- 7.Char D, Ljung B-M, Deschenes J and Miller T, Brit. J. Ophthalmol, 1988, 72, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pantel K and Alix-Panabieres C, Cancer Res., 2013, 73, 6384–6388. [DOI] [PubMed] [Google Scholar]
- 9.Papamarkakis K, Bird B, Schubert JM, Miljkovic M, Wein R, Bedrossian K, Laver N and Diem M, Lab. Invest, 2010, 90, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diem M, Miljković M, Bird B, Chernenko T, Schubert J, Marcsisin E, Mazur A, Kingston E, Zuser E, Papamarkakis K and Laver N, Spectrosc.-Int. J, 2012, 27, 463–496. [Google Scholar]
- 11.Wong P, Wong RK, Caputo TA, Godwin TA and Rigas B, Proc. Natl. Acad. Sci. U.S.A, 1991, 88, 10988–10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohenford MA and Rigas B, Proc. Natl. Acad. Sci. U.S.A, 1998, 95, 15327–15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Argov S, Ramesh J, Salman A, Sinelnikov I, Goldstein J, Guterman H and Mordechai S, J. Biomed. Opt, 2002, 7, 248–254. [DOI] [PubMed] [Google Scholar]
- 14.Tfayli A, Piot O, Durlach A, Bernard P and Manfait M, Biochim. Biophys. Acta Gen. Subj, 2005, 1724, 262–269. [DOI] [PubMed] [Google Scholar]
- 15.Mordechai S, Sahu R, Hammody Z, Mark S, Kantarovich K, Guterman H, Podshyvalov A, Goldstein J and Argov S, J. Microsc, 2004, 215, 86–91. [DOI] [PubMed] [Google Scholar]
- 16.Petibois C and Deleris G, Trends Biotechnol., 2006, 24, 455–462. [DOI] [PubMed] [Google Scholar]
- 17.Tobin MJ, Chesters MA, Chalmers JM, Rutten FJ, Fisher SE, Symonds IM, Hitchcock A, Allibone R and Dias-Gunasekara S, Faraday Discuss., 2004, 126, 27–39. [DOI] [PubMed] [Google Scholar]
- 18.Walsh MJ, German MJ, Singh M, Pollock HM, Hammiche A, Kyrgiou M, Stringfellow HF, Paraskevaidis E, Martin-Hirsch PL and Martin FL, Cancer Lett., 2007, 246, 1–11. [DOI] [PubMed] [Google Scholar]
- 19.Martin FL, Kelly J, Llabjani V, Martin-Hirsch PL, Patel I, Trevisan J, Fullwood N and Walsh M, Nat. Protoc, 2010, 5, 1748–1760. [DOI] [PubMed] [Google Scholar]
- 20.Mostaco-Guidolin LB and Bachmann L, Appl. Spectrosc. Rev, 2011, 46, 388–404. [Google Scholar]
- 21.Sahu R, Argov S, Salman A, Zelig U, Huleihel M, Grossman N, Gopas J, Kapelushnik J and Mordechai S, J. Biomed. Opt, 2005, 10, 054017. [DOI] [PubMed] [Google Scholar]
- 22.German MJ, Hammiche A, Ragavan N, Tobin MJ, Cooper LJ, Matanhelia SS, Hindley AC, Nicholson CM, Fullwood NJ and Pollock HM, Biophys. J, 2006, 90, 3783–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derenne A, Gasper R and Goormaghtigh E, Analyst, 2011, 136, 1134–1141. [DOI] [PubMed] [Google Scholar]
- 24.Budinova G, Salva J and Volka K, Appl. Spectrosc, 1997, 51, 631–635. [Google Scholar]
- 25.Shaw RA, Kotowich S, Mantsch HH and Leroux M, Clin. Biochem, 1996, 29, 11–19. [DOI] [PubMed] [Google Scholar]
- 26.Heise H and Bittner A, J. Mol. Struct, 1995, 348, 127–130. [Google Scholar]
- 27.Baker MJ, Trevisan J, Bassan P, Bhargava R, Butler HJ, Dorling KM, Fielden PR, Fogarty SW, Fullwood NJ, Heys KA, Hughes C, Lasch P, Martin-Hirsch PL, Obinaju B, Sockalingum GD, Sulé-Suso J, Strong RJ, Walsh MJ, Wood BR, Gardner P and Martin FL, Nat. Protocols, 2014, 9, 1771–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hammody Z, Argov S, Sahu RK, Cagnano E, Moreha R and Mordechai S, Analyst, 2008, 133, 372–378. [DOI] [PubMed] [Google Scholar]
- 29.Ly E, Piot O, Durlach A, Bernard P and Manfait M, Analyst, 2009, 134, 1208. [DOI] [PubMed] [Google Scholar]
- 30.Kastl L, Kemper B, Lloyd GR, Nallala J, Stone N, Naranjo V, Penaranda F and Schnekenburger J, Proc. of SPIE, 2016, 9703, 970307. [DOI] [PubMed] [Google Scholar]
- 31.Seddon AB, Phys. Status Solidi B Basic Solid State Phys, 2013, 250, 1020. [Google Scholar]
- 32.Parnell H, Butterworth JH, Sakr H, Tang Z, Furniss D and Benson TM, Proc. of SPIE, 2016, 9703, 970309. [Google Scholar]
- 33.Chan KLA and Kazarian SG, Appl. Spect, 2003, 57, 381. [DOI] [PubMed] [Google Scholar]
- 34.Kuimova MK, Chan KLA and Kazarian SG, Appl. Spectr, 2009, 63, 164. [DOI] [PubMed] [Google Scholar]
- 35.Chan KLA and Kazarian SG, Chem. Soc. Rev, 2016, 45, 1850. [DOI] [PubMed] [Google Scholar]
- 36.Bassan P, Byrne HJ, Lee J, Bonnier F, Clarke C, Dumas P, Gazi E, Brown MD, Clarke NW and Gardner P, Analyst, 2009, 134, 1171–1175. [DOI] [PubMed] [Google Scholar]
- 37.Grenci G, Birarda G, Mitri E, Businaro L, Pacor S, Vaccari L and Tormen M, Microelectron. Eng, 2012, 98, 698. [Google Scholar]
- 38.Boveri T, Zur Frage der Entstehung maligner Tumoren, Gustav Fischer, Jena, 1914. [Google Scholar]
- 39.Hollingsworth M and Swanson B, Nat. Rev. Cancer, 2004, 4, 45–60. [DOI] [PubMed] [Google Scholar]
- 40.Cavallaro U and Christofori G, Nat. Rev. Cancer, 2004, 4, 118. [DOI] [PubMed] [Google Scholar]
- 41.Jiang W, Saxena A, Song B, Ward BB, Beveridge TJ and Myneni SCB, Langmuir, 2004, 20, 11433. [DOI] [PubMed] [Google Scholar]
- 42.Steyer J and Almers W, Nat. Rev. Mol. Cell Biol, 2001, 2, 268. [DOI] [PubMed] [Google Scholar]
- 43.Gottesman MM, Fojo T and Bates SE, Nat. Rev. Cancer, 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- 44.Bush JA and Li G, Int. J. Cancer, 2002, 98, 323–330. [DOI] [PubMed] [Google Scholar]
- 45.Stavrovskaya AA, Biochemistry, 2000, 65, 95–106. [PubMed] [Google Scholar]
- 46.Borst P, Evers R, Kool M and Wijnholds J, Biochim. Biophys. Acta Biomembr, 1999, 1461, 347. [DOI] [PubMed] [Google Scholar]
- 47.Diz-Muñoz A, Weiner OD and Fletcher DA, Nat. Phys, 2018, 14, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Islam M, Mezencev R, Brink BMH, Campbell B, Tasadduq B, Waller EK, Lam W, Alexeev A and Sulchek T, Cell Death Dis., 2018, 9, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clausen MP, Colin-York H, Schneider F, Eggeling C and Fritzsche M, J. Phys. D, 2017, 50, 064002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adato R and Altug H, Nat. Commun, 2013, 4, 2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holloway CL, Kuester EF, Gordon JA, O'Hara J, Booth J and Smith DR, IEEE Antennas Propag. Mag, 2012, 54, 10–35. [Google Scholar]
- 52.Kildishev AV, Boltasseva A and Shalaev VM, Science, 2013, 339, 1232009. [DOI] [PubMed] [Google Scholar]
- 53.Yu N, Genevet P, Kats M, Aieta F, Tetienne J-P, Capasso F and Gaburro Z, Science, 2011, 334, 333–337. [DOI] [PubMed] [Google Scholar]
- 54.Fedotov VA, Mladyonov PL, Prosvirnin SL, Rogacheva AV, Chen Y and Zheludev NI, Phys. Rev. Lett, 2006, 97, 167401. [DOI] [PubMed] [Google Scholar]
- 55.Fedotov VA, Schwanecke AS, Zheludev NI, Khardikov VV and Prosvirnin SL, Nano Lett., 2007, 7, 1996–1999. [DOI] [PubMed] [Google Scholar]
- 56.Hao F, Sonnefraud Y, Dorpe PV, Maier SA, Halas NJ and Nordlander P, Nano Lett., 2008, 8, 3983–3988. [DOI] [PubMed] [Google Scholar]
- 57.Liu N, Tang ML, Hentschel M, Giessen H and Alivisatos AP, Nat. Mater, 2011, 10, 631–636. [DOI] [PubMed] [Google Scholar]
- 58.Zheludev NI, Prosvirin SL, Papasimakis N and Fedotov VA, Nat. Photonics, 2008, 2, 351–354. [Google Scholar]
- 59.Wu C, Khanikaev AB, Adato R, Arju N, Yanik AA, Altug H and Shvets G, Nat. Mater, 2012, 11, 69–75. [DOI] [PubMed] [Google Scholar]
- 60.Arju N, Ma T, Khanikaev A, Purtseladze D and Shvets G, Phys. Rev. Lett, 2015, 114, 237403. [DOI] [PubMed] [Google Scholar]
- 61.Aksu S, Yanik AA, Adato R, Artar A, Huang M and Altug H, Nano Lett., 2010, 10, 2511–2518. [DOI] [PubMed] [Google Scholar]
- 62.Kazarian SG and Chan KLA, Appl. Spectr, 2010, 64, 135A. [DOI] [PubMed] [Google Scholar]
- 63.Arju N, Gupta SD, Kelp G, Cook J, Emelianov S and Shvets G, Advanced Photonics 2016 (IPR, NOMA, Sensors, Networks, SPPCom, SOF), 2016, SeW3E.1. [Google Scholar]
- 64.Izzard CS and Lochner LR, J. Cell. Sc, 1976, 21, 129. [DOI] [PubMed] [Google Scholar]
- 65.Mazur AI, Marcsisin EJ, Bird B, Miljkovic M and Diem M, Anal. Chem, 2012, 84, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams MJ, Chemometrics in Analytical Spectroscopy, 2nd ed., Royal Society of Chemistry, Cambridge, 2004. [Google Scholar]
- 67.Romeo M, Jennings BMM and Diem M, Biochim. Biophys. Acta Biomembr, 2006, 1758, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jimenez-Hernandez M, Hughes C, Bassan P, Ball F, Brown MD, Clark NW and Gardner P, Analyst, 2013, 138, 3957. [DOI] [PubMed] [Google Scholar]
- 69.Salomon D, Brandt R, Ciardiello F and Normanno N, Crit. Rev. Oncol. Hematol, 1995, 19, 183. [DOI] [PubMed] [Google Scholar]
- 70.Schaeybroeck S, Karaiskou-McCaul A, Kelly D, Longley D, Galligan L, Cutsem EV and Johnston P, Clin. Cancer. Res, 2005, 11, 7480. [DOI] [PubMed] [Google Scholar]
- 71.Kumar S, Aaron J and Sokolov K, Nat. Protoc, 2008, 3, 314–320. [DOI] [PubMed] [Google Scholar]
- 72.Khanikaev AB, Wu C and Shvets G, Nanophotonics, 2013, 2, 247–264. [Google Scholar]
- 73.Mattheyses AL, Simon SM and Rappoport JZ, J. Cell Sci, 2010, 123, 3621–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steyer JA and Almers W, Nat. Rev. Mol. Cell Biol, 2001, 2, 268. [DOI] [PubMed] [Google Scholar]
- 75.Peelen D, Kodoyianni V, Lee J, Zheng T, Shortreed MR and Smith LM, J. Proteome Res, 2006, 5, 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robertus J, Browne WR and Feringa BL, Chem. Soc. Rev, 2010, 39, 354. [DOI] [PubMed] [Google Scholar]
- 77.Bassan P, Byrne HJ, Bonnier F, Lee J, Dumas P and Gardner P, Analyst, 2009, 134, 1586–1593. [DOI] [PubMed] [Google Scholar]
- 78.Katukuri VK, Hargrove J, Miller SJ, Rahal K, Kao JY, Wolters R, Zimmermann EM and Wang TD, Biomed. Opt. Exp, 2010, 1, 1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yashunsky V, Lirtsman V, Golosovsky M, Davidov D and Aroeti B, Biophys. J, 2010, 99, 4028–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teh SK, Zheng W, Ho KY, Teh M, Yeoh KG and Huang Z, Int. J. Cancer, 2010, 126, 1920–1927. [DOI] [PubMed] [Google Scholar]
- 81.Lee SY, Yoon K-A, Jang SH, Ganbold EO, Uuriintuya D, Shin S-M, Ryu PD and Joo S-W, J. Vet. Sci, 2009, 10, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bellisola G and Sorio C, Am. J. Cancer Res, 2012, 2, 1–21. [PMC free article] [PubMed] [Google Scholar]
- 83.Richter T, Steiner G, Abu-Id MH, Salzer R, Bergmann R, Rodig H and Johannsen B, Vib. Spectrosc, 2002, 28, 103–110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.