

Abstract
The interpretation of high-dimensional structure–activity data sets in drug discovery to predict ligand–protein interaction landscapes is a challenging task. Here we present Drug Discovery Maps (DDM), a machine learning model that maps the activity profile of compounds across an entire protein family, as illustrated here for the kinase family. DDM is based on the t-distributed stochastic neighbor embedding (t-SNE) algorithm to generate a visualization of molecular and biological similarity. DDM maps chemical and target space and predicts the activities of novel kinase inhibitors across the kinome. The model was validated using independent data sets and in a prospective experimental setting, where DDM predicted new inhibitors for FMS-like tyrosine kinase 3 (FLT3), a therapeutic target for the treatment of acute myeloid leukemia. Compounds were resynthesized, yielding highly potent, cellularly active FLT3 inhibitors. Biochemical assays confirmed most of the predicted off-targets. DDM is further unique in that it is completely open-source and available as a ready-to-use executable to facilitate broad and easy adoption.
Introduction
Chemical space is vast and can only be explored to a small extent with experimental methods to find suitable hits for drug discovery programs.1,2 The search for new chemical starting points to modulate therapeutic targets is essential for the development of novel drugs. It has been postulated that the best way to find a new drug is to start with an old drug.3 This is in line with the central paradigm in medicinal chemistry that similar structures exert similar biological activities.4 Protein kinases are an important class of drug targets because of their key role in intracellular signal transduction processes involved in cancer, autoimmune diseases, and (neuro)inflammation.5,6 The therapeutic value of the protein kinase family is demonstrated by the 38 kinase inhibitors (KIs) currently approved by the FDA and a plethora of molecules being tested in clinical trials for this enzyme family.7 It is anticipated that these clinically approved KIs may serve as starting points to identify novel drug candidates for other kinases.
Most KIs interact with a structurally and functionally conserved ATP-binding site that is present in all 518 human protein kinases. It is well established that KIs bind multiple members of the kinase family and that this may affect their efficacy and toxicity.8 Detailed investigation of the target interaction landscape of KIs is therefore important to understand their molecular mode of action and offers the opportunity to identify new starting points for other therapeutically interesting kinases. Many complex, high-dimensional data sets with structure–activity relationships (SARs) of KIs over a broad selection of kinases have become available (Table S1).9−14 These empirical data sets may serve as guides to explore chemical space around this drug target family and predict (off-)target activity using advanced computational chemistry methods, such as quantitative SAR (QSAR) models, the similarity ensemble approach (SEA), support vector machines, k-nearest neighbor, random forest, naïve Bayes, (deep learning) neural networks (NNs), and principal component analysis (PCA).15−18
Advanced machine learning models promise to revolutionize the field of drug discovery. Employing high-dimensional data sets, these models are used to predict a wider range of biological activities for a compound compared with traditional drug design methods (e.g., molecular modeling, docking, and early QSAR models such as Hansch and Free–Wilson analyses19). However, advanced machine learning models are hampered in their applicability by a lack of clear interpretation and a tendency to overfit high-dimensional data. Many of the best-performing machine learning models are black boxes in which it is unclear how the data are used to generate novel hypotheses. They also require in-depth knowledge of advanced cheminformatics and highly specialized or purpose-built software. These technical requirements slow the implementation of the tools in the daily practice of drug discovery and consequently prevent the research community from taking full advantage of the wealth of data becoming available. Therefore, there is a clear need for better tools to interpret and visualize complex, high-dimensional SAR data sets in an easy and intuitive manner and to predict the biological activity profiles of novel hits for drug discovery programs. Here we present Drug Discovery Maps (DDM), a machine learning tool that allows the visualization and prediction of target–ligand interaction landscapes.
Results
t-SNE Maps the Molecular Similarity of Experimental Drugs in Chemical Space
On the basis of the principle that the chemical structure of a compound determines its biological and chemical properties, a machine learning algorithm that predicts target–ligand interaction landscapes should be able to recognize molecular similarity between different molecules. Traditionally, chemical similarity is measured by the Tanimoto coefficient (Tc).20 A molecular fingerprint, which is a high-dimensional bit vector that captures the presence or absence of chemical groups in a molecule, is used by the Tc to calculate the similarity between compounds. As a similarity metric the Tc has its limitations, predominantly because it averages differences over all bits, thereby losing information.21 Thus, we envisioned that the data contained in the molecular fingerprint could be used more efficiently by a machine learning algorithm to determine molecular similarity.
In recent years, the t-distributed stochastic neighbor embedding (t-SNE) algorithm has been shown to be a powerful tool to visualize complex high-dimensional data sets in diverse experimental settings.22−26 This state-of-the-art unsupervised machine learning technique is especially powerful in preserving local data structures in high-dimensional data. It can be readily applied to bit strings of any length and as such is easily applicable to chemical structures represented by molecular fingerprints. We aimed to use t-SNE at the core of our prediction model, where the algorithm is used to find and cluster the most similar molecules in a large data set and visualize that similarity clustering in two-dimensional space.
We decided to apply the t-SNE algorithm to visualize the molecular similarity of molecules from the Drug Repurposing Hub, an online repository containing compounds that have been clinically tested in humans.27 We selected only the launched drugs (2274) and manually classified them into 27 chemotypes. Morgan fingerprints (RDKit, 4096 bits, radius = 2) were generated for each of these 2274 clinical compounds using KNIME, an open-source software package.28,29 The fingerprints were fed into the Python implementation of the Barnes–Hut t-SNE algorithm to generate a map of the drug-like chemical space.30 The resulting map (Figure 1A) shows remarkable colocalization of most of the chemotypes. As an example, the family of penicillin-like structures at the far right of the plot (cyan) is completely separated from all other chemical matter. Some unannotated molecules (in gray) are visible in the cluster, but upon detailed inspection they all constitute β-lactams in which the sulfur is either substituted or omitted. In addition, many other highly dense clusters are visible at the boundaries of the map, corresponding to highly defined chemotypes such as the rapamycin, conazole, and oxytocin analogues. It is noteworthy that even in the apparently less defined center of the map, clear colocalization of similar molecules can be observed, for example, a cluster of aspirin-like molecules (orange, near the origin). Thus, t-SNE is able to map the chemical space of approved drugs following a chemist’s intuition and recognizes molecular similarity in a broad set of diverse drug-like molecules.
Figure 1.

t-SNE visualization of chemical space. (a) t-SNE embedding of the “launched” drugs in the Drug Repurposing Hub. Embedding is based on the 4096-bit Morgan fingerprint. t-SNE settings: perplexity = 25, learning rate = 50, iterations = 10 000. Markers are colored according to 27 manually attributed chemotypes. An animation of the process of embedding is included in the supporting video. (b) t-SNE embedding of the Published Kinase Inhibitor Set. Embedding is based on the 4096-bit Morgan fingerprint. t-SNE settings: perplexity = 50, learning rate = 50, iterations = 10 000. Markers are colored according to 31 manually attributed chemotypes.
Next, we wanted to test whether t-SNE is still able to recognize molecular similarity within a smaller set of drug-like molecules that is more homogeneous and has higher molecular similarity. To this end, we performed t-SNE-mediated clustering of the molecules from the Published Kinase Inhibitor Set (PKIS).31 The PKIS is a 364-member library of molecules assembled by GSK that are all classified as inhibitors of protein kinases. The PKIS represents 31 chemotypes, and their activities have been measured on 200 kinases.13 The resulting map of chemical space representing the KIs (Figure 1B) again shows clear colocalization of specific chemotypes. A more in-depth analysis (see the Supporting Information and Figure S1) confirms the initial visual inspection and shows high statistical correlation between the autonomously derived clustering and the human annotation. Of the 31 chemotypes annotated, 23 were fully collected in one computationally assigned cluster. For example, the orange and gold clusters on the left of the map are completely isolated and comprise all of the compounds of those chemotypes (Figure S1). This illustrates how t-SNE is capable of recognizing and clustering molecular entities in a highly specific manner and allows the visual inspection of high-dimensional chemical structural data, or chemical space, in an easy and intuitive way.
t-SNE Map of the Target Space of Kinases Recapitulates Phylogenetic Information
On the basis of the observation that binding sites in closely related proteins bind similar endogenous molecules and (experimental) drugs, we wanted to determine whether the t-SNE algorithm is capable of clustering proteins on the basis of the chemical similarity of their amino acids in the binding pocket. Conceptually, this approach is analogous to proteochemometric modeling.32 To this end, we chose the protein kinase family as the drug target class because this is a large family of over 500 members that all use ATP in their active site and often show cross-reactivity toward (experimental) drugs. To quantify the similarity of kinases, we aligned the amino acid sequences of the whole kinase domains containing the ATP-binding pocket and used a fingerprint based on physicochemical properties of the amino acids.33 The fingerprints were used to create a two-dimensional map of the target space by the t-SNE algorithm. The resulting map (Figure 2A) is striking, as it almost seamlessly recreates the phylogenetic tree published by Manning et al. in 2002.34 To assign the kinases to clusters, the coordinates of the t-SNE embedding were fed into the unsupervised clustering algorithm DBSCAN (see Supporting Information for details).35 All 10 assigned clusters were significantly (P < 0.0001, hypergeometric test) enriched for a specific kinase group as assigned by Manning et al. (Figure 2A). Closer inspection of some of the kinases unassigned by DBSCAN reveals that they belong to distinct branches of the phylogenetic tree, corresponding to their separation from the main clusters. As an example, the four TK kinases at the far right of the embedding (burgundy) all belong to the JAK family (JAK1, -2, and -3 and Tyk2) but only represent their second kinase domain. The first kinase domain is more closely associated with the rest of the TK group and lies just outside the DBSCAN-assigned cluster. The close association of the second kinase domains with the RGC cluster (colored brown) is especially striking, as these domains, just like the RGC kinases, are considered to be pseudokinases. The same holds true for MLKL, IRAK2, and IRAK3. Intriguingly, the IRAK family of TKL kinases has four members, of which IRAK1 and IRAK4 are catalytically active whereas IRAK2 and IRAK3 are not.36 In the t-SNE embedding, the former are located in the major TKL cluster (orange), whereas the latter are actually assigned to the RGC-dominated cluster. MLKL has also been shown to lack catalytic activity in at least one report.37
Figure 2.

t-SNE visualization of kinase domains reveals phylogenetic information. (a) t-SNE embedding of physicochemical fingerprints of the kinase domains of 535 human kinase domains. t-SNE settings: perplexity = 50, learning rate = 50, iterations = 25 000. Arbitrary t-SNE coordinates are rotated to match the dendrogram orientation of Manning et al.34 Markers are colored according to the 12 groups defined by Manning et al., and the background is colored on the basis of the DBSCAN-generated clustering, colored by the dominant kinase group in that cluster (blanks are unclustered kinases). (b) Manning et al. manually curated kinome dendrogram overlaid with circles colored according to the background coloring from the t-SNE map in (A) based on the unsupervised DBSCAN clustering.39
Another interesting feature is the separation of a group (left of the plot) of TKL kinases from the major cluster. This subset features all but one of the STKR family of cell-surface-bound receptor kinases. Upon closer inspection, even the subfamilies of STRK1 and -2 are discernible. Strikingly, the MISR2 (AMHR2) kinase receptor is located with kinases categorized as “Other”. This receptor kinase has an atypical DFG motif (DLG) and as such can indeed be classified as a pseudokinase, although phosphorylation activity has experimentally been shown.38 The other members of the STKR family do all share the conserved DFG motif. Finally, on the lower side of the t-SNE plot, several AGC-colored kinases have been clustered with the CAMK kinases. These actually represent the second kinase domains of the RSK family, which were also attributed to the CAMK group by Manning et al.34
In summary, this analysis of target space of the binding site of protein kinase domains ensured us that this embedding is able to recognize overall similarity but also detect subtle differences between the different binding domains of most kinase inhibitors.
DDM Can Predict Target–Ligand Interaction Landscapes
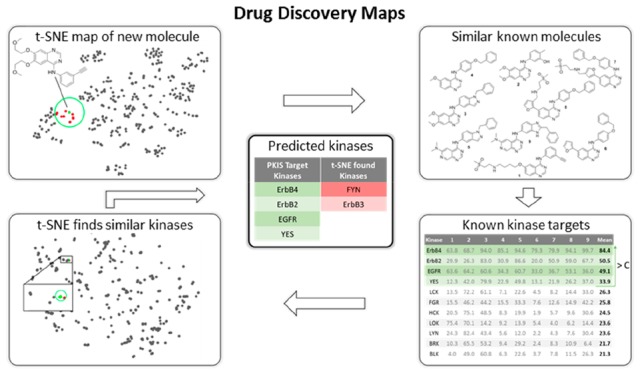
On the basis of chemical and target space maps of kinases and their inhibitors, we envisioned that these could provide a workflow to predict the activity of novel compounds for the entire kinome. We dubbed this approach Drug Discovery Maps (DDM). The bioactivity data measured by Elkins et al.13 for the PKIS were used as the training set, as the PKIS contains the most unique interactions of all open data sets (Table S1). The optimization of the workflow with all of the parameters is described in more detail in the Supporting Information. The final architecture of the algorithm is depicted in Figure 3 and illustrated for the EGFR inhibitor erlotinib. At first, a t-SNE embedding is generated in which erlotinib is mapped onto the chemical space of the PKIS (top left). This information is used to find the nine most similar molecules (top right). Of these, the inhibition data measured by Elkins et al. are averaged, and all of the kinases above a threshold value C are considered targets (bottom right). A view the inhibition profiles for this process is included in Figure S5. These kinases are then looked up in the target space map (Figure 2), and the most similar kinases are appended (bottom left) to yield the final prediction (center). As the molecular t-SNE embedding is slightly stochastic, the described process is repeated several times (R), and the number of times a kinase is predicted is tracked. Our DDM model was validated using an independent data set generated by Karaman et al.9 The resulting prediction statistics for each of the 38 compounds in this test set are summarized in Table S2. The average positive prediction value (PPV) was 40% with a Matthews correlation coefficient (MCC) of 0.21. We compared these statistics with previously published methods and found that DDM was better than QSAR models and equal in performance to random-forest-based proteochemometric models (Figure S2). A receiver operating characteristic (ROC) analysis of the performance of DDM on this test set showed an area under the curve (AUC) of 0.76 (Figure S3). Taken all together, these result show that we have developed and validated a novel machine learning model to predict kinome inhibitor landscapes.
Figure 3.

Schematic overview of the DDM workflow. In this example, the targets of erlotinib are predicted. On the basis of a t-SNE embedding (top left), the PKIS inhibitors nearest to erlotinib are found (top right). For these, the inhibition data as measured by Elkins et al.13 are averaged and used as an initial prediction (bottom right). These targeted kinases are then looked up in the t-SNE embedding (bottom left), where the most similar kinases are added to yield the final prediction (center).
Discovery of Novel FLT3 Inhibitors Using DDM
To investigate the utility of the model in early drug development, it was applied for the identification of new inhibitors for FMS-like tyrosine kinase 3 (FLT3). FLT3 is implicated in advanced myeloid leukemia, where approximately 30% of patients carry an internal tandem duplication (ITD) in their FLT3 gene that activates the kinase and acts as a driver mutation.40 Recently, midostaurin has been approved by the FDA for the treatment of acute myeloid leukemia (AML) patients, and several other inhibitors are currently being tested in clinical trials. However, fast adaptive mutations in the FLT3 gene quickly result in drug-induced resistance of the AML, warranting the search for novel chemotypes to inhibit this kinase. To this end, the DDM model was used to predict the kinome–ligand interaction landscape of a small kinase-focused library of 1152 molecules. They were analyzed using various values for the activity cutoff C and were ultimately filtered with C = 40% and a prediction count of at least nine out of 10 runs in order to have a balanced number of molecules to be tested. These stringent cutoffs yielded a set of 44 compounds predicted to be active at FLT3.
To validate our virtual DDM screen, we performed a time-resolved fluorescence resonance energy transfer (FRET)-based biochemical assay with all 1152 compounds against FLT3 at an initial concentration of 10 μM. This screen yielded 184 actives with >50% loss of activity (16% of all compounds). Of these compounds, the pEC50 values were measured, resulting in 135 compounds with pEC50 > 5, with a mean of 6.7 ± 0.9. Eighteen of the 184 compounds were also identified by our DDM screen, which results in a PPV (or hit rate) of 41% (Figure 4A, P < 0.0001 (hypergeometric test)), which is almost 3-fold higher than the hit rate of the biochemical assay. Interestingly, 15 of the predicted compounds demonstrated EC50 values of <2 μM (34%, P < 0.0001 (hypergeometric test)) with an average pEC50 of 7.3 ± 1.1; this group included the most active compound found in the screen, crenolanib (pEC50 = 9.0). The hit rate was nearly identical to the validation statistics for the test set (Figure S2), where an overall PPV of 40% was achieved. The same holds for the negative predictive value (89%) and the sensitivity (11%). The successful application of our model for the FLT3 screen may partially be attributed to the high coverage for the TK family of kinases. It should be noted that the relatively low sensitivity (11%) is a balanced choice between minimizing the number of compounds to screen and finding more actual hits. This can easily be tuned by varying the cutoff parameter.
Figure 4.

Discovery of novel FLT3 inhibitors using DDM. (a) Scatter plot of all compounds and their inhibitory effects at 10 μM as measured in the high-throughput screen. DDM-predicted molecules are marked red. (b) Structures and syntheses of the two compounds resynthesized and tested in situ against MV4:11 cells. Reagents and conditions: (i) cyanamide, nitric acid, ethanol, 78 °C, 76%; (ii) dimethylformamide diethyl acetal, toluene, 80 °C, 80%; (iii) K2CO3, ethanol, 78 °C, 31%; (iv) 4-aminophenol, NaOH, DMSO, 100 °C, 65%; (v) triphosgene, DCM, 40 °C; (vi) 1,4-dioxane, 110 °C, 44% over two steps. (c) Dose–response curves for compounds 1 and 2 against recombinant FLT3 in a FRET-based activity assay. Markers denote mean ± SD (N = 4). Dotted lines denote the 95% confidence intervals of the EC50 fits. (d) Dose–response curves of compounds 1 and 2 against MV4:11 leukemia cells. Markers denote mean ± SD (N = 3). Dotted lines denote the 95% confidence intervals of the EC50 fits. (e) Docking poses of 1 and 2 in the 3D models of FLT3 and the corresponding 2D interaction plots.
Two of the predicted compounds, 1 and 2 (Figure 4B), were selected on the basis of their chemical properties, novelty regarding FLT3 inhibition, and predicted interaction profiles (vide infra). These compounds were resynthesized using established methods (see Figure 4B and the Supporting Information). The activity of the compounds was confirmed in a FRET assay using recombinant human FLT3 (Figure 4C). Compounds 1 and 2 showed a concentration-dependent activity with pEC50 values of 7.3 ± 0.1 and 8.8 ± 0.1, respectively. To determine the cellular activities of these two compounds, a cell proliferation assay using the FLT3-dependent AML cell line MV4:11 was performed. Both 1 and 2 showed clear cellular activity with pEC50 values of 6.3 ± 0.1 and 8.5 ± 0.1, respectively (Figure 4D). In summary, the experimental validation of the hits illustrates the power of our DDM workflow for compound selection in the lab.
Finally, to explain the potential binding mode of compounds 1 and 2, these compounds were docked using a DFG-in model for 1 and a DFG-out structure (PDB entry 4RT7) for 2 (Figure 4E). Compound 1 binds to the hinge region with the aminopyrimidine moiety in a fashion typical for type 1 kinase inhibitors. Compound 2 binds in the DFG-out conformation much like RIPK2 (PDB entry 5AR7) by forming hydrogen bonds to the DFG motif using the urea functionality and to the hinge region using the pyridine nitrogen.41
Kinome Activity Spectrum Prediction Using DDM
To reduce potential toxic side effects, kinase cross-reactivity is ideally minimized. DDM enables rapid assessment of the predicted cross-reactivity because by default DDM predicts the interactions with the entire kinome. Thus far, however, only the FLT3 prediction has been taken into account. As final validation, we tested the activities of the two inhibitors on the predicted off-targets in biochemical assays. In addition to FLT3, compounds 1 and 2 were predicted to be active against 35 and 33 kinases, respectively (C = 40%, R > 0.5). The off-targets were validated using KinaseProfiler by Eurofins at 10 μM. The inhibition data per compound are shown in Table S3. For compound 1 the predictions were 69% accurate (24 of the 35 off-targets confirmed (<50% remaining activity) with two additional off-targets in the low 50% residual activity range). For compound 2 the prediction was exceedingly accurate, as 26 of the 33 targets (79%) were indeed inhibited >50%. To conclude, DDM was able to predict the kinome–inhibitor interaction landscape with a relatively high accuracy.
Discussion
Drug discovery is still largely an empirical process that is challenging, time-consuming and hard.42 Multiparameter optimization of chemical structures, which is needed to balance the activity and selectivity of a drug candidate, requires the understanding of high-dimensional data sets. Machine learning algorithms have been employed to analyze and predict compound activity using large data sets with varying success.15−17 Some of the major drawbacks of most computational models are the complexity of the algorithm and the “black box” nature of the systems. Implementation and interpretation of such systems is not trivial, and consequently, they have not been widely adopted by the drug discovery community.
Here we present DDM, which is an intuitive, data-driven (bio)molecule similarity clustering procedure using state-of-the-art machine learning techniques. The model is based on the t-distributed stochastic neighbor embedding (t-SNE) algorithm to generate a visualization of molecular similarity in two dimensions.43,44 Color is used as a third dimension to interactively visualize the biological activity or compound class (chemotype). DDM combines two different maps. The first map depicts the chemical space, in which compounds are clustered on the basis of their molecular similarity, whereas in the second map protein targets are clustered on the basis of the chemical similarity of the amino acids making up the kinase domain. By combining the two maps, DDM is able to predict bioactivities of small molecules across a protein family. We applied DDM to visualize the chemical space of currently available drugs, the published kinase inhibitor set (PKIS) and the target space of the protein kinase family (kinome). DDM was able to predict the kinome activity profile of another independent set of kinase inhibitors with comparable or better scores than the currently available machine learning techniques. We applied DDM to identify new hits for the oncogene FMS-like tyrosine kinase 3 (FLT3), a validated therapeutic target for the treatment of acute myeloid leukemia.45 The hits were resynthesized, and their biological activities were validated in biochemical and cellular assays. Finally, the off-target profiles of the hits as predicted by DDM were validated in a panel of kinase assays.
Although our model performs equally well or better than the current computational drug discovery tools, it is envisioned that our model can be further improved when more comprehensive data sets become available in the public domain. In the PKIS training set, 364 inhibitors were tested at only two concentrations on approximately 200 unique wild-type kinases. A more expansive data set of a broader set of more diverse compounds tested on a larger number of kinases in a concentration–response fashion would inherently improve the predictions generated over the entire kinome.
The added value of direct knowledge of the off-targets of these compounds enables prioritization in medicinal chemistry efforts, as demonstrated by the KinaseProfiler screen of predicted off-targets. This allows medicinal chemists to rank scaffolds on the basis of acceptable off-targets, which in turn depends on biological questions or medical indications. The information obtained from the docking poses of these molecules can also be used for structure-based design, directly incorporating the knowledge derived from the clinically relevant mutations into the hit-optimization project.
The DDM concept presented here can easily be adapted to work with any data set available. Because all data, algorithms, and data processing tools used are in the public domain or open-source, it is highly adaptable and extensible. Concrete examples include different druggable protein classes, such as G-protein-coupled receptors, ion channels, or nuclear hormones, or the ability to be trained on a different molecular set altogether, e.g., solubility, membrane permeability, metabolic stability, pharmacokinetics, or toxicological data.
To aid in the implementation of our tool as it is presented here, a Python-based executable including a graphical user interface (Figure 5) has been made available online via Github.46 The unpackaged Python script with a list of dependencies is also available. Also included is a fully annotated KNIME workflow to allow step-by-step execution and analysis. This set of tools should enable the integration of this data-driven approach into any project without any need of investments a priori.
Figure 5.

Graphical user interface (left) and generated output (right) of the Python implementation of the DDM algorithm presented here. Only a SMILES string is required as input, and the output is provided as depicted on the right. The packaged executable as well as the original Python script have been made available online.46
To conclude, the machine learning algorithm Barnes–Hut t-SNE was successfully implemented in a drug discovery setting to predict ligand–protein interaction landscapes. The concept of DDM is applicable to a multitude of drug discovery challenges, which, given the proper data set, can be used to design a small molecule with a balanced set of physicochemical and biological properties as required for drug candidates. It is envisioned that DDM may make the drug discovery process more efficient.
Acknowledgments
The Open Source community is gratefully acknowledged for their invaluable contribution to this work. J. van Groningen and S. van Helden are kindly acknowledged for their assistance with the high-throughput screening assay. We thank Prof. B. Lelieveldt for critical reading of the manuscript. The Agentschap Innoveren en Ondernemen (AIO) is acknowledged for financial support (AIO Project 155028) to E.B.L. NWO/ECHO is acknowledged for financial support to M.v.d.S. and A.P.A.J. The Cancer Drug Discovery Initiative is acknowledged for financial support to S.H.G.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.8b00640.
Author Contributions
A.P.A.J., S.H.G., R.H.M.W., E.B.L., C.A.A.v.B., and G.P.J.W. performed the experiments and analyzed the results. A.P.A.J., C.A.A.v.B., G.P.J.W., J.N., and M.v.d.S. designed the experiments. A.P.A.J., G.P.J.W., and M.v.d.S. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Reymond J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick P.; Ellis C. Chemical Space. Nature 2004, 432, 823–823. 10.1038/432823a. [DOI] [Google Scholar]
- Black J.The Practice of Medicinal Chemistry. In The Practice of Medicinal Chemistry; Wermuth C. G., Ed.; Elsevier, 2011; p 54. [Google Scholar]
- Bender A.; Glen R. C. Molecular Similarity: A Key Technique in Molecular Informatics. Org. Biomol. Chem. 2004, 2, 3204. 10.1039/b409813g. [DOI] [PubMed] [Google Scholar]
- Johnson L. N.; Lewis R. J. Structural Basis for Control by Phosphorylation. Chem. Rev. 2001, 101, 2209–2242. 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Adams J. A. Kinetic and Catalytic Mechanisms of Protein Kinases. Chem. Rev. 2001, 101, 2271–2290. 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- Klaeger S.; Heinzlmeir S.; Wilhelm M.; Polzer H.; Vick B.; Koenig P.-A.; Reinecke M.; Ruprecht B.; Petzoldt S.; Meng C.; Zecha J.; Reiter K.; Qiao H.; Helm D.; Koch H.; Schoof M.; Canevari G.; Casale E.; Depaolini S. R.; Feuchtinger A.; Wu Z.; Schmidt T.; Rueckert L.; Becker W.; Huenges J.; Garz A.-K.; Gohlke B.-O.; Zolg D. P.; Kayser G.; Vooder T.; Preissner R.; Hahne H.; Tõnisson N.; Kramer K.; Götze K.; Bassermann F.; Schlegl J.; Ehrlich H.-C.; Aiche S.; Walch A.; Greif P. A.; Schneider S.; Felder E. R.; Ruland J.; Médard G.; Jeremias I.; Spiekermann K.; Kuster B. The Target Landscape of Clinical Kinase Drugs. Science 2017, 358, eaan4368. 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S.; Chaikuad A.; Gray N. S.; Knapp S. The Ins and Outs of Selective Kinase Inhibitor Development. Nat. Chem. Biol. 2015, 11, 818–821. 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Metz J. T.; Johnson E. F.; Soni N. B.; Merta P. J.; Kifle L.; Hajduk P. J. Navigating the Kinome. Nat. Chem. Biol. 2011, 7, 200–202. 10.1038/nchembio.530. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins J. M.; Fedele V.; Szklarz M.; Abdul Azeez K. R.; Salah E.; Mikolajczyk J.; Romanov S.; Sepetov N.; Huang X.-P.; Roth B. L.; Al Haj Zen A.; Fourches D.; Muratov E.; Tropsha A.; Morris J.; Teicher B. A.; Kunkel M.; Polley E.; Lackey K. E.; Atkinson F. L.; Overington J. P.; Bamborough P.; Müller S.; Price D. J.; Willson T. M.; Drewry D. H.; Knapp S.; Zuercher W. J. Comprehensive Characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- Miduturu C. V.; Deng X.; Kwiatkowski N.; Yang W.; Brault L.; Filippakopoulos P.; Chung E.; Yang Q.; Schwaller J.; Knapp S.; King R. W.; Lee J.-D.; Herrgard S.; Zarrinkar P.; Gray N. S. High-Throughput Kinase Profiling: A More Efficient Approach toward the Discovery of New Kinase Inhibitors. Chem. Biol. 2011, 18, 868–879. 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann-Franck S.; van Westen G. J. P.; Papadatos G.; Beltran Escudie F.; Roberts A.; Overington J. P.; Domine D. Unprecedently Large-Scale Kinase Inhibitor Set Enabling the Accurate Prediction of Compound–Kinase Activities: A Way toward Selective Promiscuity by Design?. J. Chem. Inf. Model. 2016, 56, 1654–1675. 10.1021/acs.jcim.6b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgenfrei F. A.; Fulle S.; Merget B. Kinome-Wide Profiling Prediction of Small Molecules. ChemMedChem 2018, 13, 495–499. 10.1002/cmdc.201700180. [DOI] [PubMed] [Google Scholar]
- Merget B.; Turk S.; Eid S.; Rippmann F.; Fulle S. Profiling Prediction of Kinase Inhibitors: Toward the Virtual Assay. J. Med. Chem. 2017, 60, 474–485. 10.1021/acs.jmedchem.6b01611. [DOI] [PubMed] [Google Scholar]
- Cichonska A.; Ravikumar B.; Parri E.; Timonen S.; Pahikkala T.; Airola A.; Wennerberg K.; Rousu J.; Aittokallio T. Computational-Experimental Approach to Drug-Target Interaction Mapping: A Case Study on Kinase Inhibitors. PLoS Comput. Biol. 2017, 13, e1005678. 10.1371/journal.pcbi.1005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C.; Fujita T. ρ-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. 10.1021/ja01062a035. [DOI] [Google Scholar]
- Willett P.; Winterman V. A Comparison of Some Measures for the Determination of Inter-Molecular Structural Similarity Measures of Inter-Molecular Structural Similarity. Quant. Struct.-Act. Relat. 1986, 5, 18–25. 10.1002/qsar.19860050105. [DOI] [Google Scholar]
- Baldi P.; Nasr R. When Is Chemical Similarity Significant? The Statistical Distribution of Chemical Similarity Scores and Its Extreme Values. J. Chem. Inf. Model. 2010, 50, 1205–1222. 10.1021/ci100010v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahfouz A.; van de Giessen M.; van der Maaten L.; Huisman S.; Reinders M.; Hawrylycz M. J.; Lelieveldt B. P. F. Visualizing the Spatial Gene Expression Organization in the Brain through Non-Linear Similarity Embeddings. Methods 2015, 73, 79–89. 10.1016/j.ymeth.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Amir E. D.; Davis K. L.; Tadmor M. D.; Simonds E. F.; Levine J. H.; Bendall S. C.; Shenfeld D. K.; Krishnaswamy S.; Nolan G. P.; Pe’er D. ViSNE Enables Visualization of High Dimensional Single-Cell Data and Reveals Phenotypic Heterogeneity of Leukemia. Nat. Biotechnol. 2013, 31, 545–552. 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmoula W. M.; Balluff B.; Englert S.; Dijkstra J.; Reinders M. J. T.; Walch A.; McDonnell L. A.; Lelieveldt B. P. F. Data-Driven Identification of Prognostic Tumor Subpopulations Using Spatially Mapped t-SNE of Mass Spectrometry Imaging Data. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12244–12249. 10.1073/pnas.1510227113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N.; Smith J.; Briscoe J.; Watkins C. An Intuitive Graphical Visualization Technique for the Interrogation of Transcriptome Data. Nucleic Acids Res. 2011, 39, 7380–7389. 10.1093/nar/gkr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskesen E.; Reinders M. J. T. 2D Representation of Transcriptomes by T-SNE Exposes Relatedness between Human Tissues. PLoS One 2016, 11, e0149853. 10.1371/journal.pone.0149853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello S. M.; Bittker J. A.; Liu Z.; Gould J.; McCarren P.; Hirschman J. E.; Johnston S. E.; Vrcic A.; Wong B.; Khan M.; Asiedu J.; Narayan R.; Mader C. C.; Subramanian A.; Golub T. R. The Drug Repurposing Hub: A Next-Generation Drug Library and Information Resource. Nat. Med. 2017, 23, 405–408. 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold M. R.; Cebron N.; Dill F.; Gabriel T. R.; Kötter T.; Meinl T.; Ohl P.; Sieb C.; Thiel K.; Wiswedel B.. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Preisach C., Burkhardt H., Schmidt-Thieme L., Decker R., Eds.; Springer, Berlin, 2008; pp 319–326. [Google Scholar]
- Landrum G. RDKit: Open-Source Cheminformatics; http://www.rdkit.org (accessed Oct 24, 2018).
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay É. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Drewry D. H.; Willson T. M.; Zuercher W. J. Seeding Collaborations to Advance Kinase Science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top. Med. Chem. 2014, 14, 340–342. 10.2174/1568026613666131127160819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Westen G. J. P.; Wegner J. K.; IJzerman A. P.; van Vlijmen H. W. T.; Bender A. Proteochemometric Modeling as a Tool to Design Selective Compounds and for Extrapolating to Novel Targets. MedChemComm 2011, 2, 16–30. 10.1039/C0MD00165A. [DOI] [Google Scholar]
- Heil B.; Ludwig J.; Lichtenberg-Frate H.; Lengauer T. Computational Recognition of Potassium Channel Sequences. Bioinformatics 2006, 22, 1562–1568. 10.1093/bioinformatics/btl132. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Ester M.; Kriegel H.-P.; Sander J.; Xu X.. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In KDD-96 Proceedings; AAAI, 1996; pp 226–231. [Google Scholar]
- Murphy J. M.; Zhang Q.; Young S. N.; Reese M. L.; Bailey F. P.; Eyers P. A.; Ungureanu D.; Hammaren H.; Silvennoinen O.; Varghese L. N.; Chen K.; Tripaydonis A.; Jura N.; Fukuda K.; Qin J.; Nimchuk Z.; Mudgett M. B.; Elowe S.; Gee C. L.; Liu L.; Daly R. J.; Manning G.; Babon J. J.; Lucet I. S. A Robust Methodology to Subclassify Pseudokinases Based on Their Nucleotide-Binding Properties. Biochem. J. 2014, 457, 323–334. 10.1042/BJ20131174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. M.; Czabotar P. E.; Hildebrand J. M.; Lucet I. S.; Zhang J.-G.; Alvarez-Diaz S.; Lewis R.; Lalaoui N.; Metcalf D.; Webb A. I.; Young S. N.; Varghese L. N.; Tannahill G. M.; Hatchell E. C.; Majewski I. J.; Okamoto T.; Dobson R. C. J.; Hilton D. J.; Babon J. J.; Nicola N. A.; Strasser A.; Silke J.; Alexander W. S. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity 2013, 39, 443–453. 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Josso N.; Clemente N. di. Transduction Pathway of Anti-Müllerian Hormone, a Sex-Specific Member of the TGF-β Family. Trends Endocrinol. Metab. 2003, 14, 91–97. 10.1016/S1043-2760(03)00005-5. [DOI] [PubMed] [Google Scholar]
- Eid S.; Turk S.; Volkamer A.; Rippmann F.; Fulle S. KinMap: A Web-Based Tool for Interactive Navigation through Human Kinome Data. BMC Bioinf. 2017, 18, 16. 10.1186/s12859-016-1433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrosa-Garcia M.; Baer M. R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. 10.1158/1535-7163.MCT-16-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnley A. K.; Convery M. A.; Lakdawala Shah A.; Jones E.; Hardwicke P.; Bridges A.; Ouellette M.; Totoritis R.; Schwartz B.; King B. W.; Wisnoski D. D.; Kang J.; Eidam P. M.; Votta B. J.; Gough P. J.; Marquis R. W.; Bertin J.; Casillas L. Crystal Structures of Human RIP2 Kinase Catalytic Domain Complexed with ATP-Competitive Inhibitors: Foundations for Understanding Inhibitor Selectivity. Bioorg. Med. Chem. 2015, 23, 7000–7006. 10.1016/j.bmc.2015.09.038. [DOI] [PubMed] [Google Scholar]
- Scannell J. W.; Bosley J. When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis. PLoS One 2016, 11, e0147215. 10.1371/journal.pone.0147215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Maaten L.; Hinton G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Reutlinger M.; Schneider G. Nonlinear Dimensionality Reduction and Mapping of Compound Libraries for Drug Discovery. J. Mol. Graphics Modell. 2012, 34, 108–117. 10.1016/j.jmgm.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Zarrinkar P. P.; Gunawardane R. N.; Cramer M. D.; Gardner M. F.; Brigham D.; Belli B.; Karaman M. W.; Pratz K. W.; Pallares G.; Chao Q.; Sprankle K. G.; Patel H. K.; Levis M.; Armstrong R. C.; James J.; Bhagwat S. S. AC220 Is a Uniquely Potent and Selective Inhibitor of FLT3 for the Treatment of Acute Myeloid Leukemia (AML). Blood 2009, 114, 2984–2992. 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A. P. A. Drug Discovery Maps. https://github.com/APAJanssen/DrugDiscoveryMaps/ (accessed Oct 24, 2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.