

Abstract
Epithelial cell polarity is linked to the control of tissue growth and tumorigenesis. The tumor suppressor and cell polarity protein lethal-2-giant larvae (Lgl) promotes Hippo signaling and inhibits Notch signaling to restrict tissue growth in Drosophila melanogaster. Notch signaling is greater in lgl mutant tissue than in wild-type tissue because of increased acidification of endosomal vesicles, which promotes the proteolytic processing and activation of Notch by γ-secretase. We showed that the increased Notch signaling and tissue growth defects of lgl mutant tissue depended on endosomal vesicle acidification mediated by the vacuolar adenosine triphosphatase (V-ATPase). Lgl promoted the activity of the V-ATPase by interacting with Vap33 (VAMP-associated protein of 33 kDa). Vap33 physically and genetically interacted with Lgl and V-ATPase subunits and repressed V-ATPase–mediated endosomal vesicle acidification and Notch signaling. Vap33 overexpression reduced the abundance of the V-ATPase component Vha44, whereas Lgl knockdown reduced the binding of Vap33 to the V-ATPase component Vha68–3. Our data indicate that Lgl promotes the binding of Vap33 to the V-ATPase, thus inhibiting V-ATPase–mediated endosomal vesicle acidification and thereby reducing γ-secretase activity, Notch signaling, and tissue growth. Our findings implicate the deregulation of Vap33 and V-ATPase activity in polarity-impaired epithelial cancers.
INTRODUCTION
Deregulation of cell polarity and the subsequent loss of tissue architecture is a hallmark of human epithelial cancers and contributes to the initiation and progression of tumorigenesis (1–3). Epithelial cell polarity (apico-basal polarity) is important for establishing and maintaining specific membrane domains along the apical-basal axis of epithelial cells and for positioning the adherens junctions that are required for cell-cell adhesion and communication (4, 5). Several key regulators of cell polarity were discovered in the vinegar fly, Drosophila melanogaster, as neoplastic tumor suppressors. Among these are lethal-2-giant larvae (Lgl), Scribbled (Scrib), and Discs large (Dlg), all of which promote apical-basal cell polarity through their interactions with other polarity regulators, including the Par complex component atypical protein kinase C (aPKC) and the transmembrane protein Crumbs (Crb). Lgl, Scrib, and Dlg also limit cell proliferation (2, 6–10) and localize to the basolateral (septate) junctions in Drosophila epithelial tissues (11–13). The Scribble module (Scrib, Dlg, and Lgl) is also involved in asymmetric cell division and the proliferation of neural stem cells (14–17). Because of their key roles in cell polarity and proliferation control, loss-of-function mutations in scrib, dlg, or lgl result in the formation of neoplastic epithelial and brain tumors in Drosophila (11, 17–26). The Scribble module proteins have a conserved tumor suppressor role in mammalian systems, as evidenced by the rescue of the corresponding Drosophila tumorous phenotypes by the expression of the mammalian orthologs (27–29) and their involvement in mammalian tumorigenesis (2, 9, 30, 31).
Apico-basal cell polarity regulators control tissue growth by modulating various signaling pathways (2, 7–9, 30–32). However, Drosophila Lgl has functions that are distinct from those of Dlg and Scrib in the control of signaling pathways and tissue growth (33). We have shown that Lgl’s role in tissue growth control is separable from its function in cell polarity, because clonal depletion of Lgl in the developing eye epithelium results in increased cell proliferation and tissue overgrowth effects without affecting apico-basal cell polarity (33, 34). Moreover, in the eye epithelium, clonal depletion of Lgl does not lead to activation of c-Jun N-terminal kinase (JNK) or apoptosis of the lgl mutant tissue (33–35), whereas the loss of lgl in the wing epithelium (36–39) or the loss of scrib or dlg in the eye epithelium (20, 40–42) does. Thus, the developing Drosophila eye represents an ideal system to observe the effect of Lgl depletion on cellular signaling without the confounding effects of apico-basal cell polarity disruption.
Lgl controls tissue growth independent of its apico-basal cell polarity role by regulating the Salvador-Warts-Hippo (Hippo) pathway (8, 33–35, 43, 44). The Hippo pathway negatively regulates tissue growth in response to cell-cell contact and tissue architectural cues (8, 45). We found that Lgl depletion inhibits the Hippo pathway by causing mislocalization of Hippo (Hpo) away from the apical cortex, where it is normally activated by apical cues (33, 43). We have also shown that Lgl inhibits signaling through the Notch pathway (46, 47). Notch activation depends on a complex sequence of processing and trafficking events. Upon binding to ligand, Notch is cleaved at the base of the transmembrane region by a metalloproteinase, thus releasing the extracellular domain and leaving behind a membrane-anchored cytoplasmic portion called the Notch extracellular truncation (NEXT). NEXT is further processed by γ-secretase to release the active form, the Notch intracellular domain (NICD), into the cytoplasm. NICD translocates into the nucleus to stimulate the transcription of target genes, such as those encoding the HES (Hairy/Enhancer of split) transcription factors in the Enhancer of split [E(spl)] complex (48). Our previous analysis revealed that increased Notch signaling contributes to the tissue growth effects of lgl mutant tissue by promoting the expression of the cell proliferation gene Cyclin A and the cell survival gene roughest (rst) (46, 47). Thus, increased Notch signaling, together with impaired Hippo pathway signaling, causes the overgrowth defects in lgl mutant tissue (33, 46, 47).
Endocytosis promotes Notch signaling (49), and endosomal vesicle acidification promotes the activity of γ-secretase, which cleaves and activates Notch (50–52). Consistent with the defects in Notch signaling in Lgl-depleted tissue, we found that lgl mutant tissue exhibited endocytic defects and increased endosomal vesicle acidification (46, 47). Moreover, reduction in intracellular acidification by treatment with the deacidifying drug chloroquine normalized Notch signaling in lgl mutant tissue and rescued the lgl mutant adult eye phenotype (46, 47). Thus, the higher Notch signaling in lgl mutant tissue is due to increased endosomal vesicle acidification and thereby increased γ-secretase activity and Notch cleavage–mediated activation. Whereas chloroquine has a documented role in blocking autophagy (53), lgl mutant eye disc epithelial clones do not show altered autophagy (47), and therefore, it is likely that chloroquine is rescuing the lgl mutant defects by normalizing the cell signaling defects. Together, our previous data suggest that Lgl regulates endosomal vesicle acidification and thereby Notch signaling; however, the mechanism of this regulation was unclear.
Here, we investigated the mechanism by which Lgl regulates endosomal vesicle acidification. The endosomal vesicle acidification and Notch signaling defects in lgl mutant tissue depended on the vacuolar adenosine triphosphatase (V-ATPase), a multisubunit proton pump that drives endosomal acidification (54). Through proteomics analysis of Lgl-interacting proteins, we identified Vap33 (VAMP-associated protein of 33 kDa, also known as VAPB) as a protein that links Lgl to the V-ATPase complex. Vap33 genetically and physically interacted with Lgl and V-ATPase components to inhibit Notch target gene expression and tissue growth. Moreover, Lgl depletion reduced the interaction of Vap33 with the V-ATPase component Vha68–3, consistent with a mechanism wherein Lgl promotes the ability of Vap33 to inhibit V-ATPase–mediated endosomal vesicle acidification. In addition, Vap33 reduced the abundance of the V-ATPase component Vha44. Thus, we have uncovered the molecular mechanism linking the tumor suppressor Lgl to the control of V-ATPase activity and Notch signaling, which may have important implications for the treatment of epithelial cancers in which cell polarity is impaired.
RESULTS
V-ATPase activity is required for increased Notch target gene expression in lgl mutant eye epithelial tissue
We have previously shown that lgl mutant clones in the third-instar Drosophila larval eye epithelium exhibit increased LysoTracker incorporation (46, 47), indicative of increased endosomal vesicle acidification, which is controlled by the V-ATPase (54). The V-ATPase has been previously shown to be important for Notch signaling because endosomal acidification promotes the activity of γ-secretase (50–52, 55). To confirm whether the increased Notch signaling in lgl mutant tissue depended on V-ATPase activity (Fig. 1, A to F), we examined whether a mutation in Vha68–2 (Vha68–2R6) (51), a V1 subunit component of the V-ATPase that has been shown to reduce V-ATPase activity (Fig. 1G) (51), would rescue the increased Notch target gene expression observed in lgl mutant clones. We monitored Notch activation using the Notch target reporter line E(spl)lacZm8−2.61(2) (56, 57) (denoted as E(spl)lacZ), which we have documented as a robust reporter of Notch activity in the posterior region of the developing eye epithelium (46, 47). LacZ expression in this line corresponds to high ligand-dependent Notch signaling within and posterior to the morphogenetic furrow of the Drosophila third-instar larval developing eye tissue, where the Notch ligand Delta is present and Delta-Notch signaling promotes G1-S cell cycle progression and the initiation of neural development (Fig. 1A) (58–61). lgl27S3 mutant clones, generated using ey-FLP–mediated recombination and marked by the absence of green fluorescent protein (GFP), showed increased E(spl)lacZ expression, indicating increased Notch signaling, in the posterior region of the eye epithelium relative to the surrounding normal tissue (Fig. 1C, quantified in Fig. 1H) and resulted in adults with distorted and disorganized eyes (Fig. 1D), as we have previously shown (46). In addition, as previously shown (51), Vha68–2R6 mutant clones showed reduced E(spl)lacZ expression (fig. S1A and Fig. 1H) and resulted in a slightly disorganized arrangement of ommatidia in the adult eye (fig. S1B). Consistent with V-ATPase activity contributing to the increased Notch signaling in lgl mutant tissue, lgl27S3 Vha68–2R6 double-mutant clones showed a partial restoration of the expression of E(spl)lacZ relative to the surrounding normal tissue (Fig. 1, E and H) and lgl27S3 mutant clones (Fig. 1B). Consistent with this, the Vha68–2R6 mutation partially rescued the lgl27S3 mutant mosaic adult eye phenotype (Fig. 1F). This rescue of the lgl27S3 mutant eye defect by Vha68–2R6 was not due to death of the lgl mutant tissue, because mutant clones (marked by the absence of GFP) were still observed in the pupal retinas (fig. S1D). Thus, these results show that genetically reducing V-ATPase activity normalizes Notch signaling in lgl mutant tissue.
Fig. 1. V-ATPase activity is required for the increase in Notch reporter expression in lgl mutant tissue in the larval eye epithelium.
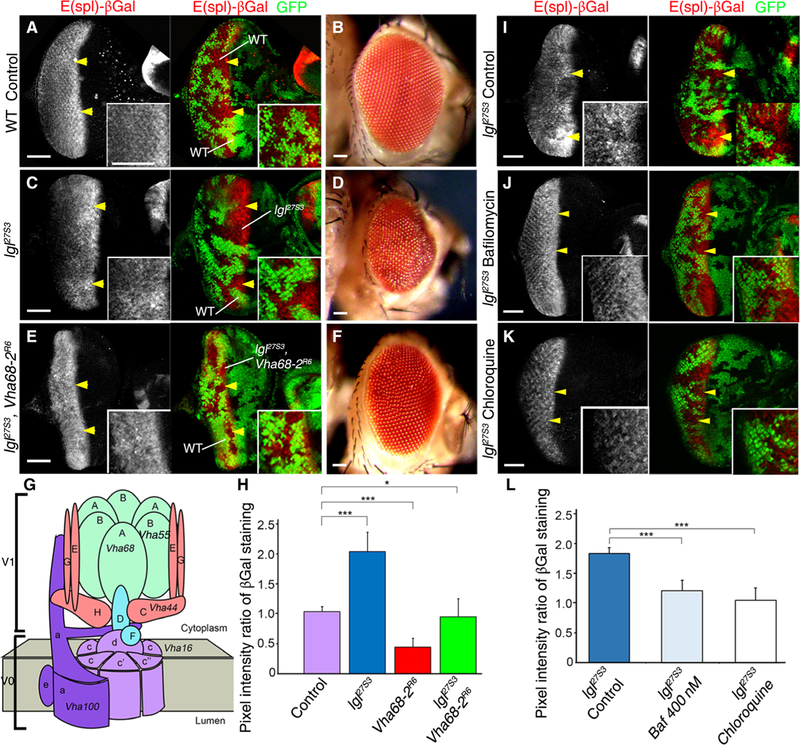
(A) Confocal planar section of a control (ey-FLP; FRT40A Ubi-GFP/FRT40A) mosaic eye-antennal disc from larvae expressing GFP in all tissues and harboring the Notch target reporter E(spl)lacZ stained for β-galactosidase (βGal; gray in single-channel images and red in merged images), showing expression of E(spl)lacZ within and posterior to the morphogenetic furrow. Ey-FLP mediated recombination generates GFP-positive and GFP-negative clones. Arrowheads indicate clones showing normal E(spl)lacZ expression. (B) WT mosaic adult female eye. (C) Mosaic disc containing lgl27S3 mutant clones (GFP-negative cells, arrowheads), stained for βGal (gray, red). (D) lgl27S3 mosaic adult female eye. (E) lgl27S3 Vha68–2R6 double-mutant mosaic disc stained for βGal (gray, red). Mutant tissue is GFP-negative. (F) lgl27S3 Vha68–2R6 mosaic adult female eye. (G) Schematic of the V-ATPase protein complex showing all the subunits and the corresponding names of the genes for those analyzed in this study. The V-ATPase complex consists of an integral membrane subunit (Vo) and a cytoplasmic subunit (V1). (H) Quantification of the βGal pixel intensity ratio between mutant clones relative to WT clones in experiments shown in (A), (C), and (E). n = 3 independent experiments, n = 3 samples analyzed for each genotype in each independent experiment. Error bars indicate SEM. ***P < 0.0001 (t tests with two-tailed distribution and unequal or equal variance) *P = 0.04 (Mann-Whitney U test). (I to K) Confocal planar images of lgl27S3 mosaic third-instar larval eye discs stained for βGal (gray). Mutant tissue is GFP-negative. Discs were from untreated flies (I) or flies that were exposed to bafilomycin A1 (J) or chloroquine (K). (L) Quantification of the βGal pixel intensity ratio between lgl27S3 and WT tissue, from experiments (I) to (K). n = 3 independent experiments, n = 3 or 4 samples analyzed for each genotype per experiment. Error bars indicate SEM. ***P < 0.0001 (t tests with two-tailed distribution and equal variance). Insets (A, C, E, and I to K) show high-magnification images. In all images, posterior is to the left. Scale bars, 50 µm.
To confirm the genetic interactions, we tested whether specific pharmacological inhibition of V-ATPase could rescue the increased Notch signaling in lgl mutant tissue (Fig. 1, I to L). Accordingly, we used bafilomycin A1, which binds to the V-ATPase Vo subunit c (62) and has been shown to inhibit V-ATPase–mediated, γ-secretase– dependent Notch activation (50). We orally administered bafilomycin A1 to developing Drosophila lgl27S3 mutant mosaic larvae and compared the effect of feeding the larvae with the vesicle de-acidifying compound chloroquine, which rescues the increased Notch signaling in lgl27S3 mutant tissue, as we have previously shown (46, 47). Bafilomycin A1 treatment nearly completely rescued the ectopic expression of the Notch reporter E(spl)lacZ that was observed in lgl27S3 mutant clones (Fig. 1J, quantified in Fig. 1L) relative to the vehicle (ethanol)–treated lgl27S3 mutant mosaic control (Fig. 1, I and L). Chloroquine treatment also restored the normal expression of the Notch reporter (Fig. 1, K and L). Thus, chemically inhibiting V-ATPase confirms our results from genetic studies, demonstrating that V-ATPase activity was required for the increased Notch signaling in lgl mutant tissue.
Vap33 links Lgl to the V-ATPase
To provide molecular insight into how Lgl affects the V-ATPase, we conducted an affinity purification–mass spectrometry (AP-MS) analysis of Lgl binding partners from Drosophila S2–cultured cells using Lgl tagged with streptavidin-binding peptide (SBP) at the C terminus (fig. S2A). We conducted two independent AP-MS analyses of Lgl-SBP–interacting proteins and ranked the results using the Significance Analysis of INTeractome (SAINT) statistical analysis software (data file S1) (63). Among the highly significant Lgl interactors (SAINT score = 1) were the expected cell polarity proteins, aPKC and Par6, both of which are known to interact with Lgl (7). We then assessed the other highly significant interactors (SAINT score >0.9) for those with links to V-ATPase subunits and other proteins involved in endocytosis. We identified one Lgl-interacting protein, Vap33 [VAP33A, VAP-33–1, VAMP (v-SNARE)–associated protein 33], that has also been shown to interact with endocytic regulators and the V-ATPase components, Vha68–2 and Vha100–4 (Fig. 1G), in a global Drosophila proteomics analysis, archived in the Drosophila protein interaction map (DPiM) database (Fig. 2A) (64). Vha68–2, Vha100–4, and other V-ATPase components were also identified among our Lgl interactors with SAINT scores <0.9, indicating interactors of lower confidence. Consistent with the interaction of Drosophila Vap33 with V-ATPase components from the DPiM database, we found that global proteomics analyses in human cells also identified that one of the human orthologs of Vap33, VAPA, interacts with several V-ATPase components with high confidence (BioGrid) (65–67): ATP6V1A, ATP6V1E1, ATP6V1B2, ATP6V1G1, and ATP6V0D1, which are orthologous to Drosophila Vha68–1 to Vha68–3, Vha26, Vha55, Vha13, and AC39–1 and AC39–2, respectively. In addition, Vap33’s human orthologs, VAPA and VAPB, physically interact with endocytic regulators (65, 67–70), and mutations in Drosophila Vap33 and human VAPB result in endocytic defects with the accumulation of the early endosome marker Rab5 (71), a phenotype we also observed in Drosophila lgl mutant tissue (46, 47). Thus, the interactions between Vap33 and V-ATPase components and the involvement of Vap33 in endocytosis appear to be evolutionarily conserved. However, physical or genetic interactions between human Lgl orthologs (LLGL1 and LLGL2) and VAPA or VAPB have not been documented, which, given our findings in Drosophila, warrants further analysis.
Fig. 2. Lgl interacts with Vap33 in vitro and in vivo.
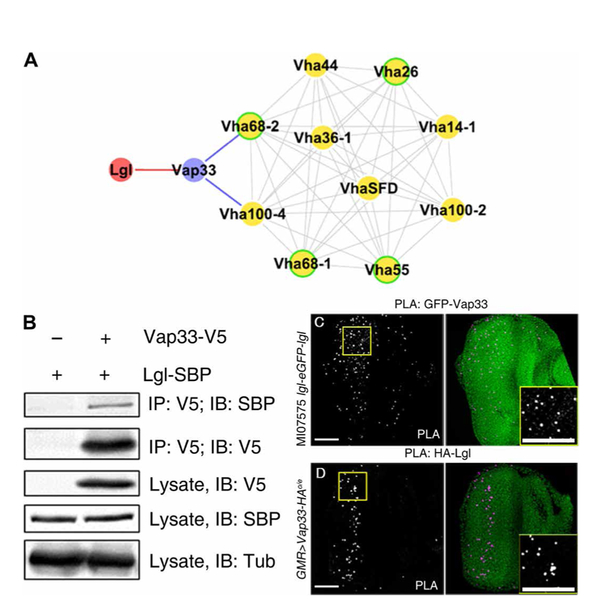
(A) Protein interaction map summarizing the results of our proteomic data linking Lgl to Vap33 (red) and data from the DPiM linking the indicated proteins to Vap33 (blue). Proteins shown in yellow are subunits of the V-ATPase, and their known interactions are indicated in light gray. Proteins for which the human homolog interacts with the human Vap33 ortholog VAPA are outlined in green. (B) Representative immunoblot showing coimmunoprecipitation of Lgl-SBP and Vap33-V5 from S2 cells. Vap33-V5 was immunoprecipitated (IP) with an antibody recognizing V5 and immunoblotted (IB) with an antibody recognizing SBP. The experiment was performed twice. (C and D) Confocal planar images showing in situ PLA in third-instar larval eye discs. The positive PLA signals are punctate and shown in gray and magenta. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI; green). Insets show high-magnification images of the PLA foci. (C) PLA on MI07575 lgl-eGFP-lgl eye discs using GFP and Vap33 antibodies. (D) PLA on GMR>Vap33-HA eye discs using HA and Lgl antibodies. The anterior region of the eye disc where the GMR>Vap33-HA is not expressed, and hence there is no HA epitope, acts as an internal negative control. n = 3 independent experiments, n = 3 or 4 samples per experiment. Scale bars, 50 µm.
To validate the interaction between Lgl and Vap33 detected by MS, we assessed whether the reverse interaction could be detected by coimmunoprecipitation. Lgl-SBP and simian virus 5 epitope (V5)–tagged Vap33 were expressed in S2 cells, and cell extracts were immunoprecipitated with an antibody recognizing V5. Immunoblotting of the eluted proteins with the SBP antibody revealed that Lgl-SBP coimmunoprecipitated with Vap33-V5 (Fig. 2B), thereby confirming their physical interaction in S2 cells. Using a similar approach, we also found that the V-ATPase component Vha44 (also called protein C) coimmunoprecipitated with SBP-tagged Lgl, although this interaction was not robust (fig. S2B).
We next tested whether Lgl and Vap33 interact in vivo. We determined whether Lgl and Vap33 were in close proximity in Drosophila epithelial cells using the in situ proximity ligation assay (PLA) (72), which detects interactions between proteins that are located within <40 nm of one another (73). We first confirmed that the PLA could detect the interaction between known binding partners, Crb and Patj, in the eye-antennal epithelium and that no PLA foci were detected using various negative controls (fig. S2, C to E). To determine whether interactions occurred between Lgl and Vap33, we made use of a MiMIC (Minos-mediated integration cassette) Lgl-GFP protein fusion line (MI07575, a homozygous viable line, indicating that a functional Lgl protein is produced) (74, 75) and examined interactions using antibodies recognizing GFP and Vap33. PLA foci were observed in the eye-antennal disc (Fig. 2C). We also overexpressed a hemagglutinin (HA)–tagged Vap33 (UAS-Vap33-HA) (76) using a GMR-GAL4 driver (77) that is expressed in the posterior region of the eye disc and used antibodies recognizing Lgl and HA to detect interactions between Lgl and Vap33 (Fig. 2D). We observed many intensely staining foci in the posterior region of the eye disc, indicating an interaction of Lgl and Vap33. In addition to the eye epithelia, we also detected PLA interactions between Lgl-GFP and Vap33 in epithelial cells of the wing and salivary gland (fig. S2, F and H) but not in the control without Vap33 antibody (fig. S2, G and L). In addition, using conventional immunofluorescence staining, Lgl-GFP colocalized with Vap33 at the cell cortex and in the cytoplasm in eye and salivary gland epithelial cells (fig. S2, J to M). Together, these data demonstrate that Lgl and Vap33 localized in close proximity to one another in epithelial cells at the cell cortex and in the cytoplasm.
Vap33 represses Notch signaling in the Drosophila eye epithelium
To test whether Vap33 represses Notch signaling, similarly to Lgl (46, 47), we analyzed the effect of Vap33 overexpression or knockdown on the expression of the Notch transcriptional target reporter E(spl)lacZ in the Drosophila third-instar larval eye epithelium and on the adult eye phenotype (Fig. 3, A to L). Relative to the GFP-negative wild-type control (Fig. 3, A and B) and lgl27S3 mutant clones (Fig. 3, C and D), overexpression of Vap33 (Vap33o/e) in lgl27S3 mutant clones [GFP-positive, achieved using the mosaic analysis using a repressible marker (MARCM) system and UAS-GFP to mark the lgl mutant tissue and express UAS-Vap33] in the Drosophila eye epithelium normalized Notch reporter expression (Fig. 3, E and F, quantified in Fig. 3M), whereas overexpression of Vap33 alone in wild-type, GFP-positive clones (MARCM system) did not significantly affect E(spl)lacZ expression (Fig. 3, G and H, quantified in Fig. 3M). These data indicate that Vap33 overexpression opposed the effect of Lgl depletion on Notch signaling. In addition, Vap33 overexpression in lgl27S3 mutant clones led to a strong suppression of the lgl mutant mosaic adult eye phenotype (Fig. 3F). To rule out the possibility that this suppression might simply be due to the death of the mutant tissue, we analyzed late-stage pupal retinas to determine whether clones were present after the developmental cell death phase that occurs during eye patterning. GFP-positive tissue, which marks the mutant tissue (using the MARCM system), was present in the Vap33-overexpressing lgl mutant mosaic pupal retinas (fig. S3, A and B), indicating that the lgl27S3 mutant adult eye tissue growth and morphology defects were rescued by Vap33 overexpression. Thus, Vap33 overexpression suppresses the increased Notch signaling and adult eye defects of lgl mutant eye tissue.
Fig. 3. Vap33 represses Notch reporter expression in the Drosophila eye epithelium.
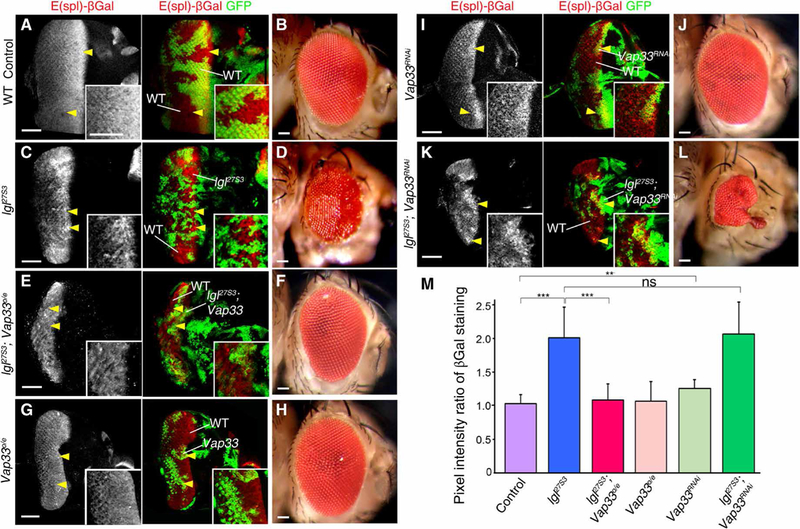
(A) Confocal planar sections of control mosaic third-instar larval eye-antennal discs (marked by GFP) from flies carrying the Notch reporter E(spl)lacZ stained for βGal (gray, red) showing expression of E(spl)lacZ in GFP-positive WT tissue and in GFP-negative clones. (B) WT mosaic adult female eye. (C) lgl27S3 mosaic eye-antennal disc stained for βGal. lgl27S3 mutant tissue is GFP-negative and marked by arrowheads. (D) lgl27S3 mosaic adult female eye. (E) lgl mutant and Vap33-overexpressing (lgl27S3 Vap33o/e) mosaic eye-antennal disc stained for βGal. Mutant tissue is GFP-positive and marked by arrowheads (and also in panels G, I, and K). (F) lgl27S3 Vap33o/e mosaic adult female eye. (G) Vap33o/e mosaic disc stained for βGal. (H) Vap33o/e mosaic adult female eye. (I) Vap33RNAi mosaic disc stained for βGal (P = 0.002). (J) Vap33RNAi mosaic adult female eye. (K) lgl27S3 Vap33RNAi mosaic eye-antennal disc stained for βGal. (L) lgl27S3 Vap33RNAi mosaic adult female eye. Scale bars, 50 µm. Insets show higher magnification. (M) Quantification of the pixel intensity ratio in control and mutant clones relative to WT clones. n = 3 independent experiments, n = 5 samples analyzed for each genotype per experiment. Error bars indicate SEM. ***P < 0.0001, *P = 0.002. ns, differences not significant (t tests with two-tailed distribution and unequal or equal variance).
We then tested whether knockdown of Vap33 in clones using UAS-Vap33RNAi, which effectively reduced Vap33 protein abundance (fig. S3C), modulated expression of the E(spl)lacZ Notch reporter in eye disc clones (Fig. 3, I and J). Vap33 knockdown clones (GFP-positive) led to a modest but significant increase (1.2-fold increase) in E(spl)lacZ expression relative to the surrounding wild-type cells (Fig. 3I, quantified in Fig. 3M), relative to the wild-type control eye discs (Fig. 3A), and similar to, albeit not as strong as, that observed with lgl27S3 mutant clones (GFP-negative) (Fig. 3C). However, stronger depletion of Vap33, using the Vap33∆20 null allele, resulted in a robust accumulation of intracellular Notch, as detected by an antibody specific to the NICD, N-intra (fig. S3, D to F), as occurs in lgl27S3 mutant clones and is associated with increased Notch reporter expression (46). In addition, Vap33∆20 clones (GFP-negative) led to a significant increase (1.35-fold increase) in E(spl)lacZ expression relative to the surrounding wild-type cells (fig. S3, H and I, quantified in fig. S3G). Together, these results provide evidence that Vap33 knockdown increases Notch signaling.
We then asked whether Vap33 knockdown could modulate the increased Notch signaling in lgl27S3 mutant clones (Fig. 3, K and L). In third-instar larval eye epithelia, the increased E(spl)lacZ expression observed in lgl27S3 mutant clones (GFP-positive) was not significantly further increased by Vap33 knockdown (Fig. 3, K and M). However, the adult eye phenotype of lgl27S3 Vap33RNAi double-mutant mosaic adult eyes was strongly affected (reduced in size with altered morphology; Fig. 3L) relative to lgl27S3 mosaic adult eyes (Fig. 3D) and Vap33RNAi mosaic adult eyes (Fig. 3J) compared to the wild-type control (Fig. 3B). The effect of Vap33RNAi on the lgl27S3 adult eye morphology, without apparent effects on Notch signaling in the third-instar larval eye epithelium, might be due to stronger Notch signaling activation occurring later in development, when it would be expected that the Lgl and Vap33 proteins would be further depleted. Alternatively, there might be additional effects of Vap33 knockdown on other cellular processes and on tissue architecture alterations during pupal development. Together, our results indicate that Vap33 functions to repress Notch target gene expression. Moreover, because overexpression of Vap33 rescues the increased Notch signaling defects of lgl mutant tissue and suppresses the lgl mutant mosaic adult eye phenotype, this suggests that Vap33 function might be defective in Lgl-depleted tissue.
Overexpression of Vap33 suppresses the tissue overgrowth phenotype caused by γ-secretase–mediated activation of Notch
We tested whether Vap33 could repress the γ-secretase–mediated activation of Notch, similarly to Lgl (47). To achieve this, we used NEXT, the truncated form of Notch resulting from metalloproteinase cleavage, which is the substrate for γ-secretase (51). We overexpressed a UAS-NEXT transgene (78) in the notum region of the developing wing epithelium, which generates the adult thorax, in wild-type and various transgenic and mutant flies using the Eq1-GAL4 (eq>) driver (79). The expression of red fluorescent protein (RFP) and lacZ (as neutral transgene controls) using the eq> driver did not affect the growth of the thorax (Fig. 4A). Knockdown of Vap33 (Vap33RNAi, Fig. 4B) or overexpression of Vap33 (Vap33o/e, Fig. 4C) using the eq> driver also did not affect thoracic growth, although overexpression of Vap33 resulted in a loss-of-bristle-and-socket phenotype in the thorax (fig. S4, A and B), consistent with decreased Notch signaling (80). Knockdown of the V-ATPase component Vha55 (Vha55RNAi, Fig. 4D) also had no effect on thorax growth, but overexpression of the V-ATPase component Vha44 (Vha44o/e, Fig. 4E) caused mild thoracic morphology defects. Expression of the NEXT transgene using the eq> driver resulted in overgrowth of the thorax (47) in flies expressing the neutral transgene control, lacZ (Fig. 4F). This overgrowth phenotype was not modified by expression of Vap33RNAi (Fig. 4G). In contrast, and consistent with previous studies showing that γ-secretase activity depends on endosomal vesicle acidity, overexpression of Vap33 (Fig. 4H) or knockdown of Vha55 (Fig. 4I) strongly suppressed the NEXT-driven thorax overgrowth phenotype. Overexpression of Vha44 in the eq>NEXT background resulted in lethality. Similar to our previous observations with Lgl (47), Vap33 overexpression did not suppress the overgrown thorax phenotype-induced overexpression of NICD (using a UAS-NICD transgene) (81), the active form of Notch that is generated by γ-secretase–mediated cleavage of NEXT (fig. S4, C and D). However, expression of a dominant-negative form of the Notch pathway transcriptional coactivator Mastermind (mamDN) (82) strongly suppressed the NICD overexpression phenotype (47). As expected, expression of mamDN suppressed the NEXT overexpression phenotype (Fig. 4J). The results of the overexpression or knockdown of Vap33 and V-ATPase components on thoracic size in the eq>RFP control and in the eq>NEXT flies were quantified and statistically analyzed (Fig. 4K), revealing the robustness of these genetic interactions.
Fig. 4. Overexpression of Vap33 or knockdown of the V-ATPase component Vha55 suppresses the NEXT overexpression phenotype.
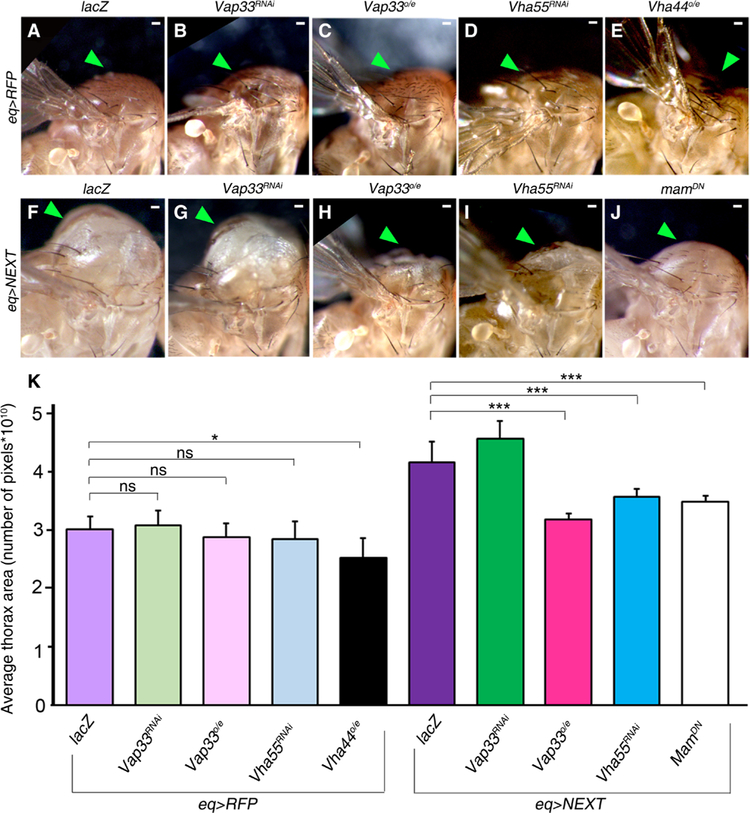
(A to E) Lateral views of the adult thorax of flies expressing the indicated transgenes in flies expressing RFP under the control of the eq> driver: (A) Control eq>RFP, lacZ; (B) eq>RFP, Vap33RNAi; (C) eq>RFP, Vap33o/e; (D) eq>RFP, Vha55RNAi; (E) eq>RFP, Vha44o/e. Arrowheads indicate the notum. (F to J) Lateral views of the adult thorax of flies expressing the indicated transgenes in the eq>NEXT background: (F) eq>NEXT, lacZ; (G) eq>NEXT, Vap33RNAi; (H) eq>NEXT, Vap33o/e; (I) eq>NEXT, Vha55RNAi; (J) eq>NEXT, mamDN. (K) Quantification of the thoracic area in all genotypes in (A) to (J). n = 2 independent experiments, n = 3 samples analyzed for each genotype per experiment. Error bars indicate SEM. ***P < 0.0001, *P = 0.01. ns, differences not significant (t tests with two-tailed distribution and equal variance or Mann-Whitney U test). Scale bars, 50 µm.
Thus, Vap33 modulates the NEXT overexpression phenotype, but not the NICD overexpression phenotype, consistent with Vap33 modulating endosomal vesicle acidification and γ-secretase–mediated cleavage and activation of Notch. Vap33 overexpression phenocopied the effect of Vha55 knockdown on the NEXT-induced thoracic phenotype, which is consistent with Vap33 inhibiting V-ATPase function and γ-secretase–mediated cleavage of NEXT. In addition, that overexpression of Vap33 and NEXT phenocopied the overexpression of Lgl with NEXT (47) indicates that Lgl and Vap33 function similarly in their regulation of Notch signaling.
Vap33 reduces endosomal vesicle acidification in the Drosophila eye epithelium
We tested whether Vap33 knockdown or overexpression affected V-ATPase activity in vivo, as assessed by LysoTracker staining, which indicates endosomal vesicle acidification. Relative to control third-instar larval eye discs (Fig. 5A), Vap33 null mutant (Vap33∆20) (83) clones in eye discs (GFP-negative) showed increased LysoTracker incorporation relative to the surrounding GFP-positive wild-type tissue (Fig. 5B), similar to lgl27S3 mutant tissue (Fig. 5C) (46, 47). In addition, we found that overexpression of Vap33 (Vap33o/e) strongly suppressed the increased LysoTracker staining in lgl27S3 mutant tissue (Fig. 5D), and clones of Vap33o/e alone also showed reduced LysoTracker staining (Fig. 5E). These results (quantified in Fig. 5F) are consistent with the notion that both Lgl and Vap33 negatively regulate V-ATPase activity and that Vap33 function is reduced in lgl mutant tissue.
Fig. 5. Vap33 reduces endosomal vesicle acidification in the Drosophila eye epithelium.
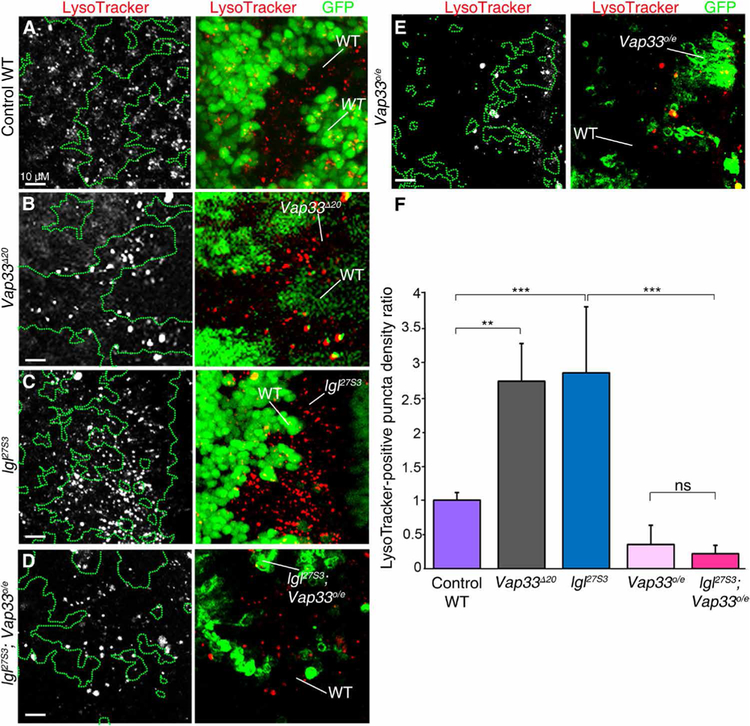
(A to E) Confocal images of mosaic third-instar larval eye discs stained with LysoTracker (gray in single-channel images and red in merged images) and showing GFP, which is absent in the mutant clones in B, C, and present in the mutant clones in D, E (MARCM system). (A) Control WT clones. (B) Vap33∆20 clones. (C) lgl27S3 clones. (D) lgl27S3 Vap33o/e clones. (E) Vap33o/e clones. Mutant and WT tissue is indicated in each panel. (F) Quantification of the number of LysoTracker+ puncta in the mutant clones relative to the same area of surrounding WT tissue. The graph shows the average mutant/WT ratio of the number of LysoTracker puncta per unit area for each genotype. n = 2 independent experiments, n = 3 samples analyzed per genotype per experiment. Error bars indicate SEM. ***P < 0.0001, **P = 0.002. ns, differences not significant (t tests with two-tailed distribution and unequal or equal variance). Scale bars, 10 µm.
Vap33 interacts with the V-ATPase component Vha44 and suppresses the Vha44 overexpression eye phenotype
Having shown that Vap33 represses V-ATPase–mediated endosomal vesicle acidification, we next investigated whether Vap33 interacts with V-ATPase components in vivo physically and genetically. First, we used the PLA to determine whether Vap33 interacts with the V-ATPase V1 subunit, Vha44, in situ in the third-instar larval eye-antennal epithelium (Fig. 6A and fig. S5A). Because we were unable to use the Vha44 antibody (which was raised in guinea pig, for which there is no established secondary antibody for use in the PLA), we used the MI11080 MiMIC line, which carries a transposon insertion that leads to a protein fusion of Vha44 with GFP (74), to mark Vha44 expression. Although MI11080 is homozygous lethal, the localization of the GFP fusion protein is similar to Vha44 antibody staining (fig. S5, B and C). Vha44-GFP and Vha44 proteins were present throughout the eye-antennal disc, concentrated around the cell cortex (fig. S5, B and C). We conducted the PLA using the MI11080 line and GFP and Vap33 antibodies, which revealed many foci throughout the eye-antennal disc (Fig. 6A) relative to the negative control (fig. S5A), indicating that Vha44 and Vap33 are in close proximity to one another. Consistent with this finding, conventional immunofluorescence staining showed Vap33 and Vha44-GFP to colocalize in many foci, although Vap33 staining showed a broader cellular distribution (fig. S5E) than Vha44. Together, these results show that Vap33 and Vha44 are in close proximity to one another in eye-antennal epithelial cells.
Fig. 6. Vap33 physically and genetically interacts with the V-ATPase component Vha44 and reduces Vha44 protein abundance.
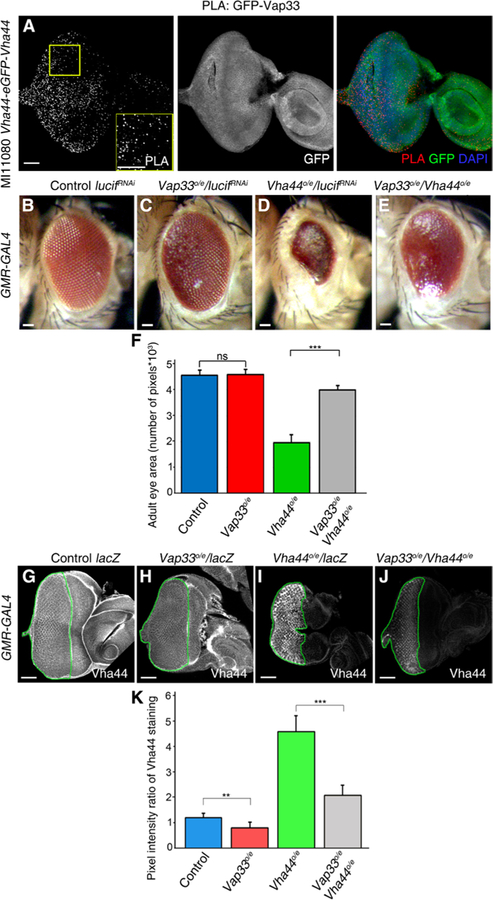
(A) Confocal planar images of the PLA in MI11080 Vha44-eGFP-Vha44 third-instar larval eye-antennal discs using GFP and Vap33 antibodies. The eye-antennal discs were also stained to show Vha44-GFP. Nuclei were stained with DAPI (blue). (B to E) Images of adult female eyes in which the indicated RNA interference (RNAi) or overexpression (o/e) constructs were expressed under the control of the eye-specific GMR-GAL4 driver. (B) GMR>luciferaseRNAi (control). (C) GMR>Vap33o/e and luciferaseRNAi. (D) GMR>Vha44 and luciferaseRNAi. (E) GMR>Vha44 and Vap33. (F) Quantification of eye size (pixels) from experiments illustrated in (B) to (E). n = 3 independent experiments, n = 3 samples per genotype per experiment. Error bars indicate SEM. ***P < 0.0001; ns, differences not significant (t tests with two-tailed distribution and unequal or equal variance). (G to J) Confocal planar images of third-instar larval eye-antennal discs from flies expressing the indicated transgenes under the control of the GMR-GAL4 driver and stained to show Vha44. (G) GMR>lacZ (control). (H) GMR>Vap33 and lacZ. (I) GMR>Vha44 and lacZ. (J) GMR>Vha44 and Vap33. Scale bars, 50 µm. (K) Quantification of the pixel intensity ratio of the mutant GMR posterior domain compared to the anterior WT domain from eye discs in (G) to (J). n = 3 independent experiments, n = 3 samples per genotype per experiment. Error bars indicate SEM. ***P < 0.0001, *P = 0.008. ns, differences not significant (t tests with two-tailed distribution and unequal or equal variance).
We next assessed whether Vap33 genetically interacts with Vha44 in the eye using the eye-specific driver GMR-GAL4 (Fig. 6, B to E) (77) to direct the expression of Vap33 and Vha44. As a control, we expressed a neutral transgene, UAS-luciferaseRNAi, which showed no effect on normal eye development (Fig. 6B). Vap33 overexpression caused minor defects in the arrangement of the ommatidia without affecting eye size (Fig. 6C), whereas overexpression of Vha44 (using a UAS-Vha44 transgene) (84) resulted in a small eye with a glassy appearance, indicating that there were defects in the differentiation of the cells that make up the ommatidia (facets) (Fig. 6D). Combined GMR-driven overexpression of both Vha44 and Vap33 resulted in a strong suppression of the reduced eye phenotype of Vha44 overexpression alone and a slight improvement of the differentiation defects, because some ommatidia could now be discerned (Fig. 6E). These results (quantified in Fig. 6F) show that Vap33 antagonizes the overexpression of Vha44 in the developing Drosophila eye epithelium.
We examined how overexpression of Vap33 affected the Vha44 overexpression phenotype by monitoring Vha44 protein abundance and localization. Vha44 protein, as detected by an antibody specific for Vha44, was present throughout the eye-antennal disc and concentrated around the borders of the ommatidia in the posterior region of the control (UAS-lacZ) eye disc (Fig. 6G). Expression of Vap33 (Vap33o/e) in the posterior region of the eye disc using GMR-GAL4 resulted in a reduction of Vha44 protein abundance (Fig. 6H). Expression of Vha44 (Vha44o/e) using the GMR driver resulted in increased abundance of Vha44 protein in the posterior region of the eye disc, as expected (Fig. 6I). Vap33o/e reduced Vha44 protein abundance in Vha44o/e tissue (Fig. 6J compared to Fig. 6I, quantified in Fig. 6K). In the overexpression experiment (Fig. 6J), because expression of Vap33 and Vha44 are under heterologous promoter control (GMR-UAS) and the endogenous abundance of Vha44 is low (Fig. 6G), the effect on Vha44 protein abundance is most likely at the level of protein stability, rather than reflecting the documented feedback loop of V-ATPase activity on Vha44 endogenous transcription (85). Together, these data reveal a role for Vap33 in regulating Vha44 protein abundance and therefore V-ATPase activity.
Lgl promotes the interaction of Vap33 with the V-ATPase component Vha68–3
Conceivably, the mechanism by which Lgl represses the activity of the V-ATPase might occur through Lgl modulating the abundance or localization of Vap33 protein, enabling Vap33 to interact with the V-ATPase. If so, then Vap33 abundance might be reduced or the Vap33 protein may be mislocalized in Lgl-depleted tissue. However, in lgl27S3 mutant larval eye epithelial tissue, Vap33 protein abundance was unaltered, and its localization was not obviously perturbed (fig. S5F). Furthermore, neither the abundance nor the localization of Vha44 was obviously perturbed in lgl27S3 mutant larval eye epithelial tissue (fig. S5D).
To further explore the mechanism by which Lgl and Vap33 regulate the V-ATPase, we examined the interaction of another V-ATPase subunit, Vha68–2 (protein A of the V-ATPase V1 subunit, Fig. 1G), and its paralog Vha68–3 with Vap33 in S2 cells by coimmunoprecipitation analysis. By expressing HA-tagged Vha68–3 and V5-tagged Vap33 and immunoprecipitating with the V5 antibody, we saw robust interaction of Vha68–3 with Vap33 (Fig. 7A), and a similar result was obtained using the opposite direction for the immunoprecipitation (fig. S6A). Likewise, HA-tagged Vha68–2 coimmunoprecipitated with V5-tagged Vap33 when the V5 antibody was used for the immunoprecipitation (fig. S6B). These results support the interaction of Vap33 with the V-ATPase complex as revealed from our MS analysis and data mining (Fig. 2A). Next, to determine the importance of Lgl for the interaction of Vap33 with Vha68–3, we knocked down Lgl using RNAi in S2 cells expressing Lgl-SBP, which resulted in robust depletion of the Lgl-SBP protein after 96 hours, relative to the β-lactamase (Bla) double-stranded RNA (dsRNA) control (Fig. 7B). We then assessed the interaction of Vap33-V5 with Vha68–3–HA in transfected S2 cells upon knockdown of Lgl or the Bla control (Fig. 7C). Lgl depletion resulted in a robust reduction in the amount of Vha68–3–HA that immunoprecipitated with Vap33 relative to the Bla-RNAi control, whereas Vha68–3–HA protein abundance in the lysate was not visibly altered (Fig. 7C). This result shows that Lgl is required for the strong association of Vap33 with the Vha68–3 component of the V-ATPase and acts to stabilize that interaction. A similar result was observed in an in vivo experiment using the PLA technique. Using the Vha44 MiMIC line, in which Vha44 is fused to GFP, we conducted the PLA using GFP and Vap33 antibodies in lgl27S3 mutant eye discs (Fig. 7D). The results revealed that there was a threefold reduction of the number of foci in lgl27S3 mutant clones (RFP-negative) relative to wild-type surrounding tissue (RFP-positive) in the eye epithelium (Fig. 7E). Overall, our results support a mechanism whereby Lgl binds to Vap33, which, in turn, promotes the interaction of Vap33 with the Vha68 component of the V-ATPase, thereby leading to the inhibition of V-ATPase activity, through destabilization of Vha44 and perhaps other V-ATPase proteins (Fig. 7F). Reduction of V-ATPase activity, in turn, reduces endosomal vesicle acidification, γ-secretase activity, Notch signaling, and tissue growth.
Fig. 7. Lgl facilitates Vap33 interaction with the V-ATPase component Vha68-3.
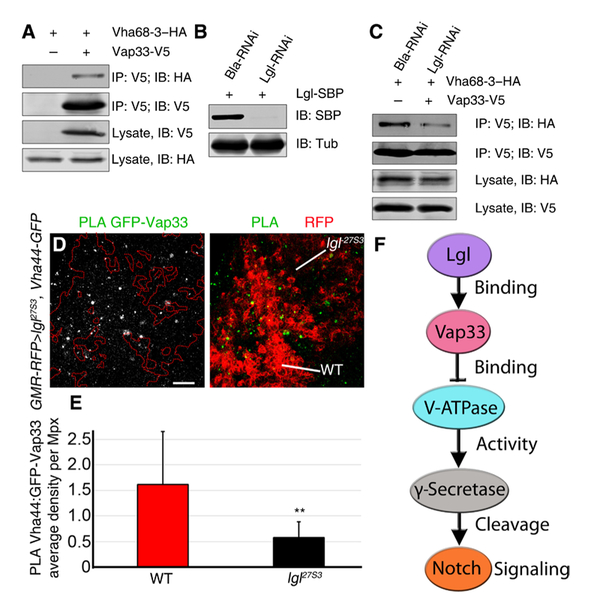
(A) Representative immunoblot showing coimmunoprecipitation of Vha68–3–HA and Vap33-V5 from S2 cells. Vap33-V5 was immunoprecipitated (IP) with an antibody specific to V5 and immunoblotted (IB) with an antibody specific for HA. (B) Representative immunoblot showing the abundance of Lgl-SBP in S2 cells in which Lgl or Bla (control) was knocked down by RNAi. (C) Representative immunoblot showing coimmunoprecipitation of Vha68–3–HA and Vap33-V5 from extracts of S2 cells in which Lgl was knocked down. Vap33-V5 was immunoprecipitated with an antibody specific for V5 and blotted with an antibody specific for HA. The experiments in (A) to (C) were conducted twice. (D) Confocal planar images showing the PLA for Vha44-GFP and Vap33 in the posterior region of third-instar larval lgl27S3 mosaic eye discs. Mutant tissue is marked by the absence of RFP in the posterior of the eye disc, using GMR-RFP as a clonal marker in eye discs expressing the Vha44 protein trap Vha44-GFP throughout the tissue. The positive PLA signal is punctate and shown in gray (left) or green (right). Scale bar, 10 µm. (E) Quantification of the PLA foci in lgl27S3 mutant tissue compared to WT tissue, plotted as average density per megapixel. n = 3 independent experiments, n = 4 samples analyzed per genotype per experiment. Error bars indicate SEM. **P = 0.005 (t tests with two-tailed distribution and unequal variance). (F) Model for the regulation of V-ATPase activity by Lgl through Vap33. Lgl binds to and activates or stabilizes Vap33, which, in turn, binds to V-ATPase subunits and inhibits V-ATPase activity, thereby reducing γ-secretase activity and Notch activation.
DISCUSSION
In addressing the mechanism by which Lgl controls vesicular acidification and Notch activity, we have shown that Lgl restricts endosomal V-ATPase activity by demonstrating that Lgl depletion increases vesicle acidification, as evidenced by LysoTracker staining, and that reducing V-ATPase function suppresses the increased Notch signaling and adult eye phenotype of lgl mutant mosaics. Using proteomic analysis of Lgl in S2 cells, we discovered Vap33 (an endosomal Vampinteracting protein and an ortholog of mammalian VAPA and VAPB) as an Lgl-interacting protein that mediates Lgl interactions with V-ATPase components. We show in S2 cells that Lgl binds to Vap33 and the V-ATPase component Vha44 and that Vap33 binds to the V-ATPase component Vha68–3. Consistent with this, in Drosophila tissues, Lgl is localized in close proximity to Vap33, and Vap33 to Vha44. We show that Vap33 genetically interacts with Lgl and V-ATPase components. Overexpression of Vap33 suppresses lgl mutant defects, consistent with Vap33 and Lgl working in a similar manner in the same genetic pathway. In regard to this, it should be noted that Vap33 overexpression probably does not bypass Lgl loss, because although lgl27S3 is a zygotic null mutant, Lgl is maternally supplied and maternal protein or preclonal protein function lasts to the third-instar larval stage (34, 36). Thus, in lgl mutant clonal tissue, overexpression of Vap33 might enable increased binding between the Vap33 and the remaining Lgl protein, thereby facilitating Vap33’s function in restricting V-ATPase activity. Furthermore, we also demonstrate that Vap33 represses V-ATPase–mediated endosomal vesicle acidification and Notch signaling. Mechanistically, we show that Lgl knockdown reduces the interaction of Vap33 with the V-ATPase component Vha68–3 and that Vap33 overexpression reduces the abundance of Vha44. Thus, our studies have revealed a role for Vap33 in linking Lgl to V-ATPase inhibition (Fig. 7F). Our results provide evidence that Lgl promotes the interaction of Vap33 with the V-ATPase component Vha68 and that Vap33 reduces Vha44 protein abundance. In turn, a reduction of Vha44 protein abundance would be expected to reduce V-ATPase–mediated endosomal vesicle acidification and thereby limit γ-secretase activity and Notch cleavage–mediated activation. Thus, upon Lgl depletion, Vap33-mediated repression of the V-ATPase is impaired, endosomal vesicle acidification is increased, and thereby γ-secretase–mediated cleavage and activation of Notch is increased.
The molecular mechanisms for how Lgl controls Vap33 association with Vha68 and how Vap33 affects Vha44 protein abundance and represses V-ATPase function remain to be resolved. We envisage three possible molecular mechanisms for Lgl-Vap33 regulation of V-ATPase. (i) Lgl and Vap33 may reduce V-ATPase subunit stability, as suggested by the reduction of Vha44 protein abundance by Vap33 overexpression (Fig. 6, G to K). (ii) Lgl and Vap33 may interfere with V-ATPase holoenzyme assembly, which occurs by association of the V1 cytoplasmic complex with the Vo integral membrane complex (Fig. 1G) (86). (iii) Lgl and Vap33 may inhibit the activity of the V-ATPase holoenzyme. However, these regulatory mechanisms may be connected, because studies in yeast have shown that deletion of any V-ATPase subunit, except H or c″, disrupts the formation of the V-ATPase holoenzyme (86); therefore, a reduction in Vha44 might result in a corresponding decrease in the V-ATPase holoenzyme assembly and thereby V-ATPase activity. Our results support the first of these potential mechanisms.
Comparison between Lgl and Vap33 functions
Our finding that Lgl binds to Vap33 and that Vap33 represses V-ATPase activity is a role not previously recognized for Vap33, although it has been implicated in many cellular processes (87). Vap33 (known as VAPA and VAPB in vertebrates) is evolutionarily conserved from yeast to humans (87, 88) and has been intensely studied in different organisms and different cell types. It contains at its N terminus a domain of ~120 amino acids that is homologous to the major sperm protein of Caenorhabditis elegans, and this motif interacts with proteins containing a Phe-Phe-Ala-Thr motif, such as the oxysterol-binding protein, which is involved in sterol lipid metabolism, and the phosphatidylinositol transfer proteins, which are involved in the transport of phospholipids (87, 89). At Vap33’s C terminus, there is a membrane-binding domain, tethering the protein to endosomal membranes (87). Mutations in human VAPB are causative for a form of the neurodegenerative disease amyotrophic lateral sclerosis type 8 (ALS8), and Drosophila models of the disease mutation (Vap33-P58S, which acts as a dominant-negative) show neurodegenerative phenotypes, which can be rescued by expression of the wild-type human VAPB (90–93). Tissues from ALS patients who did not harbor the C3orf72 expansion, which gives rise to another form of ALS, accumulate the early endosomal marker Rab5 (71). Rab5 accumulation is also observed in Drosophila tissues upon expression of Vap33-P58S (71). We have observed a similar phenotype in Drosophila lgl mutant tissue, wherein early, recycling, and early-multivesicular body endosomes accumulate (46, 47). Thus, lgl mutants show defects similar to Vap33 mutants, consistent with our data showing that Lgl and Vap33 physically and genetically interact. Whether the increased V-ATPase activity in lgl and Vap33 mutants is connected with the observed endocytosis defects remains to be determined. Although Lgl and Vap33 function similarly in V-ATPase regulation and endocytosis, they have distinct functions in other processes, because Vap33, in contrast to Lgl (2, 9), has not been implicated in cell polarity regulation or as tumor suppression (87).
The function of Lgl and Vap33 in Notch signaling pathway regulation
Our findings have revealed a regulatory role for Vap33 in inhibiting Notch signaling by inhibiting V-ATPase activity in Drosophila eye epithelial tissue. In comparison with the effect of Lgl loss on Notch reporter expression, Vap33 loss had much less of an effect (Fig. 3, C, I, and M, and fig. S3, G to I). Possible reasons for this might be Vap33 maternal protein perdurance or functional redundancy of Vap33 with its paralogs CG33523 and farinelli (CG7919), which are also expressed in Drosophila imaginal disc tissues. It is also possible that Lgl also inhibits Notch signaling through an additional mechanism that does not depend on Vap33 or Vap33 paralogs. Further studies are needed to elucidate these possibilities. Because we also identified CG33523 as an Lgl-binding protein in our AP-MS analysis (data file S1), its potential involvement in Lgl function is of particular interest.
Whether the Lgl-Vap33–V-ATPase regulatory mechanism we have uncovered in the Drosophila eye epithelial tissue occurs in other tissues remains to be determined. However, our analysis of the interaction of Lgl with Vap33 by PLA has revealed that Lgl and Vap33 interactions occur in the wing and salivary gland epithelial cells, and therefore, it is possible that Lgl also acts through Vap33 to inhibit the activity of the V-ATPase in these tissues. Our previous studies (47), as well as this study, have revealed that lgl or Vap33 overexpression or V-ATPase knockdown can repress the activation of NEXT in the developing wing notum, which becomes part of the adult thorax.
The relationship between Lgl regulation of Notch and Hippo signaling
Because we have previously shown that lgl mutant tissue has reduced Hippo signaling (33), and because altered Notch trafficking has been observed in Hippo pathway mutant tissues (94, 95), impaired Hippo signaling could potentially contribute to the increased Notch signaling in lgl mutant tissue. However, this is unlikely to be the case for the following reasons: (i) Maitra et al. (94) observed that Hippo pathway impairment affected the trafficking of the Notch receptor in a ligand-independent manner, whereas we found that lgl depletion increases ligand-dependent Notch signaling, and we saw no effect on the trafficking of full-length Notch (46, 47). (ii) The apical accumulation of Notch receptor in the Hippo pathway mutants is due to apical membrane expansion (95), and we do not observe an expansion of the apical membrane in third-instar larval lgl mutant clones, which only impairs the Hippo pathway mildly compared with core Hippo pathway mutants (33, 34). (iii) We have shown that lgl depletion results in impaired Hippo pathway signaling in an aPKC-dependent manner (33), whereas lgl mutants result in increased Notch signaling in an aPKC-independent manner (46). The precise manner by which Lgl and aPKC control Hippo signaling in Drosophila epithelial tissues remains to be determined; however, because inhibiting the V-ATPase rescues the adult eye defects of lgl mutant mosaics, which are due to both Hippo and Notch signaling perturbations [this study and (46, 47)], it is possible that Vap33-mediated regulation of the V-ATPase and vesicle acidification also affect the Hippo pathway.
Function of Lgl-Vap33 in V-ATPase regulation in tissue homeostasis and cancer
The inhibition of the V-ATPase by Lgl-Vap33 might be important physiologically in limiting Notch activation in developing epithelial tissues. Consistent with this, we found that the interaction of Lgl with Vap33 by PLA (Fig. 2C) appeared to be greater within and posterior to the morphogenetic furrow of the eye epithelium, corresponding to the region in which the Notch ligand Delta is present and stimulates Notch signaling. The regulation of V-ATPase activity by Lgl and Vap33 might also be important in response to epithelial wounding, where cell morphology and polarity changes would be expected to impair Lgl function (9), and the resulting increases in V-ATPase activity and endosomal vesicle acidification would then promote Notch signaling to induce cell proliferation and tissue repair. Moreover, because the human Vap33 ortholog VAPA also interacts with V-ATPase components (65–67), our findings suggest that the regulation of endosomal vesicle acidification by Lgl might also be evolutionarily conserved. Given the involvement of the human Lgl homologs Llgl1 (also known as Hugl1) and Llgl2 (also known as Hugl2) as tumor suppressors in mammalian cells and in human cancer (2, 9, 27, 31, 96, 97), our findings also suggest that Lgl-depleted and cell polarity–impaired epithelial cancers might have deregulated Vap33 and V-ATPase activity. However, Llgl1 or Llgl2 interaction with VAPA or VAPB has not been reported, and further studies are required to elucidate whether this mechanism is conserved in human cells. Although the mammalian Vap33 orthologs VAPA and VAPB have not been shown to be tumor suppressors (87), this might be due to genetic redundancy or pleiotropic functions of these genes. However, increased V-ATPase activity and endosomal vesicle acidification is prevalent in many human cancers and associated with deregulation of various signaling pathways including the Notch pathway (50, 55, 98–103). Because perturbations in cell polarity regulators are involved in the initiation of cancer and are connected to the deregulation of signaling pathways (2, 9, 30, 32, 104–110), our findings on the connection of Lgl and Vap33 to V-ATPase regulation might reveal new insights into, and avenues for the treatment of, cell polarity–impaired cancers.
MATERIALS AND METHODS
Mutants and transgenes
Fly stocks were generated in house previously or obtained from other laboratory or stock centers (as detailed in table S1). For the genotypes of samples analyzed in all figures, see table S2.
Clonal analysis
Negatively marked eye-antennal disc clones of lgl27S3 were generated using the ey-FLP/FRT system (111) using ey-FLP; FRT40, Ubi-GFP or ey-FLP; GMR-RFP, FRT40A. The FRT19A Vap33∆20 and FRT19A, Ubi-GFP; ey-FLP (generated in this study) were used for the Vap33∆20 clonal analysis. GFP positively marked mutant clones were generated using the mosaic analysis with a repressible cell marker [MARCM (GFP+)] system (112) using ey-FLP, UAS-GFP; Tub-GAL80, FRT40A; Tub-GAL4/TM6B (MARCM 2L), as previously described (33). Flies were raised on standard cornmeal-agar food at 25°C.
Immunofluorescence
Third-instar larval eye-antennal discs were dissected in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 30 min, washed in PBS + 0.1 or 0.3% Triton X-100 (PBT), and blocked in PBT + 1% bovine serum albumin (PBT/BSA). The tissues were incubated with the primary antibody in PBT/BSA overnight at 4°C. After washing off the primary antibodies, the tissues were incubated with the secondary antibodies in PBT for 1 hour at room temperature. After washing off the secondary antibodies, the samples were mounted in 80% glycerol or Vectashield (Vector Laboratories). For all eye imaginal disc experiments, we repeated the experiment at least twice with two independent biological samples from each genotype including the control genotype.
Antibodies used were as follows: mouse βGal (Sigma, 1:500), rabbit GFP (Invitrogen A11122, 1:500), mouse GFP (Invitrogen A11120, 1:500), rabbit Lgl (J. Knoblich, 1:500), mouse HA [Developmental Studies Hybridoma Bank (DSHB), 1:100], mouse N-intra (DSHB, 1:100), guinea pig Vha44 (M. Simmons, 1:100), rabbit Vap33 (H. Bellen, 1:1000), mouse Crb (DSHB, 1:50), and rabbit Patj (M. Bhat, 1:500). Secondary antibodies were as follows: anti-mouse Alexa 488, 568, 633, and 647; anti-rabbit Alexa 488, 568, 633, and 647; and anti– guinea pig Alexa 568. All secondary antibodies were used at a dilution of 1:500. DNA was stained with 1 µM DAPI.
Chloroquine and bafilomycin A1 treatment
Chloroquine (Sigma) diluted in PBS or PBS alone (vehicle control) was added to standard cornmeal-agar food for a final concentration of 1 mg/ml. Bafilomycin A1 dissolved in ethanol (Sigma) or ethanol along (vehicle control) was added to standard cornmeal-agar food to a final concentration of 400 nM. Crosses were set up in normal vials and transferred 24 hours later to chloroquine, bafilomycin A1, or the corresponding control vials. Third-instar larvae were collected, dissected, and processed for immunofluorescence.
LysoTracker assay
Third-instar larval eye-antennal discs were dissected in PBS, incubated with LysoTracker Red DND-99 (Invitrogen) for 5 min, and then fixed in 4% paraformaldehyde for 30 min, washed in PBS, and mounted in 80% glycerol or Vectashield (Vector Laboratories). LysoTracker analysis after fixation of Drosophila tissues is a documented procedure (113).
Proximity ligation assay
The interaction between Lgl and Vap33 or Vap33 and Vha44 in Drosophila larval tissues was detected in situ using the DUO92101 Duolink In Situ Red Starter Kit Mouse/Rabbit (Sigma) according to the instructions of the manufacturer. Briefly, primary antibody incubation was applied using the same conditions as immunohistofluorescence staining. Duolink secondary antibodies against the primary antibodies were then added. These secondary antibodies were provided as conjugates to oligonucleotides that were able to form a closed circle through base pairing and ligation using Duolink ligation solution when the antibodies were in close proximity (72) at a distance estimated to be <40 nm (73). The detection of the signals was conducted by rolling circle amplification using DNA polymerase incorporating fluorescently labeled nucleotides into the amplification products. The resulting positive signals were visualized as bright fluorescent dots, with each dot representing one interaction event. As a technical negative control, one of the primary antibodies was not added therefore, no positive signals were obtained from that assay. An additional negative control was performed in a tissue without one of the antigens (GFP), and the full protocol was performed in those tissues. As a positive control, antibodies against two well-known interactors in this tissue, Crb-Patj, were used. The tissues were visualized using a confocal microscope system (Leica TCS SP5, Zeiss Confocal LSM780 PicoQuant FLIM).
Primary antibody pairs used were mouse Crb with rabbit Patj (fig. S2D), mouse HA with rabbit Lgl (Fig. 2D), and mouse GFP with rabbit Vap33 (Figs. 2C, 6A, and 7D and fig. S2, F and H). A GFP-tagged version of Lgl was used to detect interactions with Vap33 because the Lgl and Vap33 antibodies were both raised in rabbit. GFP-tagged Vha44 was used because the available Vha44 antibody was raised in guinea pigs, and the PLA is not designed for use with antibodies raised in guinea pig.
Imaging
Fluorescently labeled samples were mounted in 80% glycerol and analyzed by confocal microscopy (Leica TCS SP5, Zeiss Confocal LSM780 PicoQuant FLIM). Images were processed using Leica LAS AF Lite and Fiji (ImageJ 1.50e). Images were assembled using Adobe Photoshop CS5.1 or CC. Adult eyes and thorax were imaged on a dissecting microscope using a Scitec Infinity1 camera.
Statistical analysis of signal intensity
Relative E(spl)m8-lacZ βGal staining within eye discs was determined from images taken at the same confocal settings. Average pixel intensity was measured using the measurement log tool from Adobe Photoshop 5.1 in an area between 400 and 2500 pixels depending on clone size. Clones were chosen just posterior to the morphogenetic furrow of each eye disc. Average pixel intensity was measured in mutant clones/domains and the wild-type adjacent tissue of the same areas (n = ~10 for each sample) and expressed as a ratio of mutant clone/domain pixel intensity/wild-type tissue pixel intensity.
The number of LysoTracker, PLA foci, or N-intra–positive puncta was measured in the mutant clones and in the adjacent wild-type tissue by using the Imaris 6.3.1 software. Quantification of Vha44 was determined from images taken at the same confocal settings. Vha44 staining of eye discs was determined by quantification of the average pixel intensity, by using measurement log tool from Adobe Photoshop 5.1, in the GMR domain relative to the same area of the adjacent wild-type tissue and expressed as a ratio. Data were plotted using Microsoft Excel 2013 and analyzed using t test or Mann-Whitney U test statistical analysis, and IBM SPSS Statistics 23.0 (SPSS Inc.) was used for statistical analyses. The α level for statistical significance was set at P = 0.05. Error bars represent SEM, and significance was **P ≤ 0.0001, **P ≤ 0.001, or *P ≤ 0.05 or 0.01, as indicated in the figure legends.
Cloning
The full length of Lgl open reading frame was amplified by polymerase chain reaction (PCR) with the addition of the Xho I and Pme I restriction sites at the N and C terminus, respectively. The primers used were as follows: Lgl–Xho I, 5′-ACGTCTCGAGCAACATGTTA-AAGTTTATC-3′; Lgl–Pme I, 5′-ACGTGTTTAAACGCAAATTG-GCTTTCTTC-3′. The PCR products were digested and inserted into the Xho I/Eco RV sites of the pMK33-C-SBP vector.
MS sample preparation
To identify Lgl binding proteins, we purified Lgl-containing protein complexes from cultured Drosophila S2 cells, using the single-step purification with the SBP tag (114–117), followed by nano–liquid chromatography–MS/MS analysis of the interactors. Drosophila S2 cells were transfected with 1.5 µg of pMK33-Lgl-SBP plasmid, and stable cell lines were selected for about 4 weeks in Schneider medium containing 0.5 mM hygromycin B (Sigma). Cells were amplified to 50 ml of dense culture. Proteins were then induced with 0.07 mM CuSO4 overnight and lysed in default lysis buffer [DLB; 50 mM tris (pH 7.5), 5% glycerol, 0.2% Igepal, 1.5 mM MgCl2, 125 mM NaCl, 25 mM NaF, 1 mM Na3VO4]. Lysates were centrifuged at 13,000g for 15 min and incubated with streptavidin beads (Pierce) for 2 hours at 4°C. Beads were washed with DLB three times, and protein complexes were eluted with 100 µl of 2 mM biotin (Sigma). The eluate was loaded on SDS–polyacrylamide gel and electrophoresed so that the dye front migrated ~1 cm in the separating gel. The gel was then stained with Coomassie blue, and the lane was cut into two 5 mm × 5 mm pieces and sent for MS analysis (Taplin Mass Spectrometry Facility, Harvard Medical School). The MS analysis was conducted on two biological replicates for our experimental samples (Lgl) and six biological replicates for controls (untransfected S2 cells).
Identification of proteins that interact with Lgl and Vap33
For each identified protein, total peptide numbers were analyzed by the SAINT program as a comparison between the two experimental (Lgl) samples and six controls. This analysis resulted in identification of Vap33 as a high-confidence Lgl interactor (AvgP SAINT value = 0.998; see data file S1). To construct a protein interaction map shown in Fig. 2A, we first downloaded interactions for the 10 core subunits of the V-ATPase complex from the STRING database (https://string-db.org/). These subunits are highlighted in yellow color and are connected by gray lines in Fig. 2A. We then searched the DPiM database (https://interfly.med.harvard.edu/) using Vap33 (FBgn0029687) as bait and identified an interaction with Vha68–2 with one peptide. By searching the DPiM database with Vha100–4 (FBgn0038613) as bait, we identified an interaction with Vap33 with five unique peptides. The Vap33/Vha100–4 and Vap33/Vha68–2 interactions from DPiM are shown as blue lines in Fig. 2A, and the Lgl/Vap33 interaction identified in our MS analysis is shown as a red line in Fig. 2A. Human orthologs of Vha68–1 and Vha68–2 (ATP6V1A), Vha55 (ATP6V1B2), and Vha26 (ATP6V1E1) are interactors of the human ortholog of Vap33 (VAPA), according to the BioGRID (https://thebiogrid.org/). These interactions are indicated as green circles around the corresponding Drosophila proteins in Fig. 2A. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (118) partner repository with the data set identifier PXD009568.
Coimmunoprecipitation
Full-length Vap33 and lgl open reading frames were amplified by PCR and inserted into pMT/V5-HisA vector (Life Technologies) and pMK33-C-SBP vector, respectively. Vha44-HA (FMO05545), Vha68–3-HA (FMO10703), and Vha68–2-HA (FMO11859) plasmids were purchased from the Drosophila Genomics Resource Center. Drosophila S2 cells were maintained in Schneider medium (Life Technologies), supplemented with 10% fetal bovine serum (Gibco), at 25°C. Transfections of S2 cells were performed using the Effectene transfection reagent (Qiagen). For immunoprecipitation, S2 cells were grown in 10-cm plates. Equal amounts of pMT-Vap33-V5 and pMK33-Lgl-SBP, pMT-Vha44-HA and pMK33-Lgl-SBP, or pMT-Vha68–2(3)-HA and pMT-Vap33-V5 plasmids were transfected (a total amount of 6 µg for each plate). For experiments involving RNAi (Fig. 7), the cells were treated as described in the section on RNAi below.
CuSO4 was added to culture media at a final concentration of 0.35 mM for inducing expression of the proteins. Cells were lysed by using DLB (see above) with 2× Complete Protease Inhibitor Tablets with EDTA (Roche). Lysates were centrifuged at 13,000g for 10 min, and the clear supernatants were incubated with anti-V5 beads or anti-HA beads (Sigma) for 2 hours at 4°C. Beads were washed three times with lysis buffer. The protein complexes were eluted with SDS buffer, transferred onto polyvinylidene difluoride membranes (Merck or Millipore), and probed with mouse anti-V5 (1:1000), rabbit antitubulin (1:1000), mouse anti-SBP (1:1000), and rabbit anti-HA antibodies (1:1000; Sigma). Secondary antibodies used were as follows: IRDye 800CW Donkey anti-Rabbit IgG and IRDye 680CW Donkey anti-Mouse IgG (LI-COR). Most coimmunoprecipitation experiments were conducted at least twice.
RNAi
UAS-Vap33-RNAi was obtained from the Vienna Drosophila Resource Center [VDRC ID: 30404; library: GD Stocks (GD) (119); construct ID: 3990; CG number: CG5014; hairpin length: 310; inserted chromosome: 3]. Left primer, CGCGAATTCCCCTTCACCCGACCCGTTGT; right primer, CGCTCTAGACGCACTTCAGTTTGGCGTCCAT.
UAS-Vha55-RNAi was obtained from the VDRC [VDRC ID: 46553; library: GD Stocks (GD) (119); construct ID: 9363; CG number: CG17369; hairpin length: 373; inserted chromosome: 3]. Left primer, CGCGAATTCCCACACCGACAACTTCGCCATC; right primer, CGCTCTAGAGTCCGTTGCGTCCCTCCACAC.
For knocking down Lgl in S2 cells, dsRNA (lgl-RNAi) was prepared (using the primers listed below), using the RiboMAX Kit (Promega). As a negative control, β-lactamase dsRNA (Bla-RNAi) was also generated, as we have previously described (120). Cells were treated with 15 µg of dsRNA specific for lgl or the Bla negative control, for 96 hours in six-well culture plates. After 96 hours, RNAi-treated cells were seeded into 10-cm tissue culture dishes and transfected with the indicated plasmids.
Primers, used to generate the lgl dsRNA, corresponding to the lgl-RNAi construct v51247 (GD stocks, from VDRC) (119), which generates a 334-nucleotide dsRNA targeting the third exon region, were as follows: Lgl-T7-forward, 5′-TAATACGACTCACTATAGGGCGTT-GCTGCCAATCAAAACACTACC-3′; Lgl-T7-reverse, 5′-TAATA-CGACTCACTATAGGGGGGGCTATGTATGCTCGCTGGAC-3′.
Supplementary Material
www.sciencesignaling.org/cgi/content/full/11/533/eaar1976/DC1
Fig. S1. Vha68–2 mutant clones show reduced Notch reporter expression, and lgl Vha68–2 mutant tissue is retained in pupal retinas.
Fig. S2. Lgl physically interacts with Vap33 and the V-ATPase component Vha44.
Fig. S3. Vap33-overexpressing tissue is retained in the pupal retina, and Vap33 mutant tissue accumulates N-intra and increases expression of the Notch reporter.
Fig. S4. Overexpression of Vap33 results in reduced bristle formation but does not suppress the NICD-induced thoracic overgrowth phenotype.
Fig. S5. Vap33 colocalizes with Vha44-GFP, and Vap33 protein abundance and localization are unaffected in lgl mutant tissue.
Fig. S6. Vap33 physically interacts with Vha68–3 and Vha68–2.
Table S1. Drosophila stocks and references.
Table S2. List of genotypes of the samples used in each figure.
Data file S1. Lgl-interacting proteins.
Acknowledgments:
We thank J. Manent and H. Bellen for helpful discussions, A. Ferrús and S. Casas-Tinto for supporting M.P. in this project, P. Burke for maintaining the fly stocks, and past and present members of our laboratories for their inputs in this project. We are grateful to J. Knoblich, H. Bellen, T. Vaccari, M. Simons, M. Bhat, H. Sun, A. Bergmann, G. Baeg, the Vienna Drosophila Resource Centre, the Bloomington Drosophila Stock Centre, and the DSHB for supplying fly stocks and antibodies, and FlyBase for its wealth of information. We acknowledge the support of the Bioimaging Facility at the La Trobe Institute for Molecular Science and at the Microscope Core at the Peter MacCallum Cancer Centre for help with this project.
Funding: The project and M.P. were supported by Cancer Council Victoria grant APP1041817 (to H.E.R. and A.V.), a Juan de la Cierva-Incorporacion postdoctoral fellowship (IJCI-2014–19272) from the Spanish Ministerio de Ciencia e Innovacion (to M.P.), Contributing to Australian Scholarship and Science Foundation grant SM/14/5398 (to L.M.P.), and NIH grants HD085870 and GM123136 (to A.V.). H.E.R. was supported by a National Health and Medical Research Council Fellowship level B (APP1020056), a Bridging Fellowship from La Trobe University, and funds from the La Trobe Institute of Molecular Science.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The MS data have been deposited in the ProteomeXchange Consortium via the PRIDE Partner repository database (accession number PXD009568). All other data needed to evaluate the conclusions in the paper are present in the paper.
REFERENCES AND NOTES
- 1.Hanahan D, Weinberg RA, Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Elsum I, Yates L, Humbert PO, Richardson HE, The Scribble–Dlg–Lgl polarity module in development and cancer: From flies to man. Essays Biochem 53, 141–168 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Wodarz A, Näthke I, Cell polarity in development and cancer. Nat. Cell Biol 9, 1016–1024 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Tepass U, Tanentzapf G, Ward R, Fehon R, Epithelial cell polarity and cell junctions in Drosophila. Annu. Rev. Genet 35, 747–784 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Ohno S, Intercellular junctions and cellular polarity: The PAR–aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr. Opin. Cell Biol 13, 641–648 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Assémat E, Bazellières E, Pallesi-Pocachard E, Le Bivic A, Massey-Harroche D, Polarity complex proteins. Biochim. Biophys. Acta 1778, 614–630 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Tepass U, The apical polarity protein network in Drosophila epithelial cells: Regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu. Rev. Cell Dev. Biol 28, 655–685 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Richardson HE, Portela M, Tissue growth and tumorigenesis in Drosophila: Cell polarity and the Hippo pathway. Curr. Opin. Cell Biol 48, 1–9 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Humbert PO, Russell SM, Smith L, Richardson HE, The Scribble–Dlg–Lgl module in cell polarity regulation, in Cell Polarity 1, Ebnet K, Ed. (Springer International Publishing Switzerland, 2015), chap. 4, pp. 65–111. [Google Scholar]
- 10.Cheng L, Parsons LM, Richardson HE, Modelling cancer in Drosophila genome, in Encyclopedia Life Sciences (eLS Wiley, 2013). [Google Scholar]
- 11.Bilder D, Li M, Perrimon N, Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289, 113–116 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Bilder D, Schober M, Perrimon N, Integrated activity of PDZ protein complexes regulates epithelial polarity. Nat. Cell Biol 5, 53–58 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Tanentzapf G, Tepass U, Interactions between the crumbs, lethal giant larvae and bazooka pathways in epithelial polarization. Nat. Cell Biol 5, 46–52 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Peng C-Y, Manning L, Albertson R, Doe CQ, The tumour-suppressor genes lgl and dlg regulate basal protein targeting in Drosophila neuroblasts. Nature 408, 596–600 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Albertson R, Doe CQ, Dlg, Scrib and Lgl regulate neuroblast cell size and mitotic spindle asymmetry. Nat. Cell Biol 5, 166–170 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Betschinger J, Mechtler K, Knoblich JA, The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature 422, 326–330 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Lee C-Y, Robinson KJ, Doe CQ, Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 439, 594–598 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Woodhouse E, Hersperger E, Shearn A, Growth, metastasis, and invasiveness of Drosophila tumors caused by mutations in specific tumor suppressor genes. Dev. Genes Evol 207, 542–550 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Beaucher M, Goodliffe J, Hersperger E, Trunova S, Frydman H, Shearn A, Drosophila brain tumor metastases express both neuronal and glial cell type markers. Dev. Biol. 301, 287–297 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brumby AM, Richardson HE, scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J 22, 5769–5779 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gateff E, Malignant neoplasms of genetic origin in Drosophila melanogaster. Science 200, 1448–1459 (1978). [DOI] [PubMed] [Google Scholar]
- 22.Bryant PJ, Levinson P, Intrinsic growth control in the imaginal primordia of Drosophila, and the autonomous action of a lethal mutation causing overgrowth. Dev. Biol 107, 355–363 (1985). [DOI] [PubMed] [Google Scholar]
- 23.Woods DF, Bryant PJ, Molecular cloning of the lethal(1)discs large-1 oncogene of Drosophila. Dev. Biol 134, 222–235 (1989). [DOI] [PubMed] [Google Scholar]
- 24.Mechler BM, McGinnis W, Gehring WJ, Molecular cloning of lethal(2)giant larvae, a recessive oncogene of Drosophila melanogaster. EMBO J 4, 1551–1557 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merz R, Schmidt M, Torok I, Protin U, Schuler G, Walther HP, Krieg F, Gross M, Strand D, Mechler BM, Molecular action of the l(2)gl tumor suppressor gene of Drosophila melanogaster. Environ. Health Perspect 88, 163–167 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal N, Kango M, Mishra A, Sinha P, Neoplastic transformation and aberrant cell-cell interactions in genetic mosaics of lethal(2)giant larvae (lgl), a tumor suppressor gene of Drosophila. Dev. Biol 172, 218–229 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Grifoni D, Garoia F, Schimanski CC, Schmitz G, Laurenti E, Galle PR, Pession A, Cavicchi S, Strand D, The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 23, 8688–8694 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Dow LE, Brumby AM, Muratore R, Coombe ML, Sedelies KA, Trapani JA, Russell SM, Richardson HE, Humbert PO, hScrib is a functional homologue of the Drosophila tumour suppressor Scribble. Oncogene 22, 9225–9230 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Thomas U, Phannavong B, Muller B, Garner CC, Gundelfinger ED, Functional expression of rat synapse-associated proteins SAP97 and SAP102 in Drosophila dlg-1 mutants: Effects on tumor suppression and synaptic bouton structure. Mech. Dev 62, 161–174 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Halaoui R, McCaffrey L, Rewiring cell polarity signaling in cancer. Oncogene 34, 939–950 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Stephens R, Lim K, Portela M, Kvansakul M, Humbert PO, Richardson HE, The scribble cell polarity module in the regulation of cell signaling in tissue development and tumorigenesis. J. Mol. Biol S0022–2836(0018)30037–30038 (2018). [DOI] [PubMed]
- 32.McCaffrey LM, Macara IG, Signaling pathways in cell polarity. Cold Spring Harb. Perspect. Biol 4, a009654 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE, Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr. Biol 20, 573–581 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Grzeschik NA, Amin N, Secombe J, Brumby AM, Richardson HE, Abnormalities in cell proliferation and apico-basal cell polarity are separable in Drosophila lgl mutant clones in the developing eye. Dev. Biol 311, 106–123 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grzeschik NA, Parsons LM, Richardson HE, Lgl, the SWH pathway and tumorigenesis: It’s a matter of context and competition! Cell Cycle 9, 3202–3212 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Froldi F, Ziosi M, Garoia F, Pession A, Grzeschik NA, Bellosta P, Strand D, Richardson HE, Grifoni D, The lethal giant larvae tumour suppressor mutation requires dMyc oncoprotein to promote clonal malignancy. BMC Biol 8, 33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun G, Irvine KD, Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev. Biol 350, 139–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu M, Xin T, Weng S, Gao Y, Zhang Y, Li Q, Li M, Activation of JNK signaling links lgl mutations to disruption of the cell polarity and epithelial organization in Drosophila imaginal discs. Cell Res 20, 242–245 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Calleja M, Morata G, Casanova J, Tumorigenic properties of Drosophila epithelial cells mutant for lethal giant larvae. Dev. Dyn 245, 834–843 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Igaki T, Pagliarini RA, Xu T, Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol 16, 1139–1146 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Uhlirova M, Bohmann D, JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J 25, 5294–5304 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leong GR, Goulding KR, Amin N, Richardson HE, Brumby AM, Scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol 7, 62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsons LM, Grzeschik NA, Richardson HE, lgl regulates the Hippo pathway independently of Fat/Dachs, Kibra/Expanded/Merlin and dRASSF/dSTRIPAK. Cancers 6, 879–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parsons LM, Grzeschik NA, Allott ML, Richardson HE, Lgl/aPKC and Crb regulate the Salvador/Warts/Hippo pathway. Fly 4, 288–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grusche FA, Richardson HE, Harvey KF, Upstream regulation of the hippo size control pathway. Curr. Biol 20, R574–R582 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Parsons LM, Portela M, Grzeschik NA, Richardson HE, Lgl regulates Notch signaling via endocytosis, independently of the apical aPKC-Par6-Baz polarity complex. Curr. Biol 24, 2073–2084 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Portela M, Parsons LM, Grzeschik NA, Richardson HE, Regulation of Notch signaling and endocytosis by the Lgl neoplastic tumor suppressor. Cell Cycle 14, 1496–1506 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andersson ER, Sandberg R, Lendahl U, Notch signaling: Simplicity in design, versatility in function. Development 138, 3593–3612 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Fortini ME, Bilder D, Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev 19, 323–328 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobia F, Duchi S, Deflorian G, Vaccari T, Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol. Oncol 8, 207–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaccari T, Duchi S, Cortese K, Tacchetti C, Bilder D, The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 137, 1825–1832 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan Y, Denef N, Schüpbach T, The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev. Cell 17, 387–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasquier B, Autophagy inhibitors. Cell. Mol. Life Sci 73, 985–1001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cotter K, Stransky L, McGuire C, Forgac M, Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci 40, 611–622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun-Wada GH, Wada Y, Role of vacuolar-type proton ATPase in signal transduction. Biochim. Biophys. Acta 1847, 1166–1172 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Kramatschek B, Campos-Ortega JA, Neuroecotdermal transcription of the Drosophila neurogenic genes E(spl) and HLH-m5 is regulated by proneural genes. Development 120, 815–826 (1994). [DOI] [PubMed] [Google Scholar]
- 57.Christiansen AE, Ding T, Fan Y, Graves HK, Herz H-M, Lindblad JL, Bergmann A, Non-cell autonomous control of apoptosis by ligand-independent Hedgehog signaling in Drosophila. Cell Death Differ 20, 302–311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baonza A, Freeman M, Notch signalling and the initiation of neural development in the Drosophila eye. Development 128, 3889–3898 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Baonza A, Freeman M, Control of cell proliferation in the Drosophila eye by Notch signaling. Dev. Cell 8, 529–539 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Firth LC, Baker NE, Extracellular signals responsible for spatially regulated proliferation in the differentiating Drosophila eye. Dev. Cell 8, 541–551 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Mollereau B, Domingos PM, Photoreceptor differentiation in Drosophila: From immature neurons to functional photoreceptors. Dev. Dyn 232, 585–592 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Bowman EJ, Bowman BJ, V-ATPases as drug targets. J. Bioenerg. Biomembr 37, 431–435 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Choi H, Larsen B, Lin Z-Y, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras A-C, Nesvizhskii AI, SAINT: Probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods 8, 70–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, McKillip E, Shah S, Stapleton M, Wan KH, Yu C, Parsa B, Carlson JW, Chen X, Kapadia B, VijayRaghavan K, Gygi SP, Celniker SE, Obar RA, Artavanis-Tsakonas S, A protein complex network of Drosophila melanogaster. Cell 147, 690–703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M, A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, Babu M, Craig SA, Hu P, Wan C, Vlasblom J, Dar VU, Bezginov A, Clark GW, Wu GC, Wodak SJ, Tillier ER, Paccanaro A, Marcotte EM, Emili A, A census of human soluble protein complexes. Cell 150, 1068–1081 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, Chessman K, Pal S, Cromar G, Papoulas O, Ni Z, Boutz DR, Stoilova S, Havugimana PC, Guo X, Malty RH, Sarov M, Greenblatt J, Babu M, Derry WB, Tillier ER, Wallingford JB, Parkinson J, Marcotte EM, Emili A, Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baron Y, Pedrioli PG, Tyagi K, Johnson C, Wood NT, Fountaine D, Wightman M, Alexandru G, VAPB/ALS8 interacts with FFAT-like proteins including the p97 cofactor FAF1 and the ASNA1 ATPase. BMC Biol 12, 39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP, The BioPlex network: A systematic exploration of the human interactome. Cell 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, Szpyt J, Tam S, Zarraga G, Pontano-Vaites L, Swarup S, White AE, Schweppe DK, Rad R, Erickson BK, Obar RA, Guruharsha KG, Li K, Artavanis-Tsakonas S, Gygi SP, Harper JW, Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanhueza M, Chai A, Smith C, McCray BA, Simpson TI, Taylor JP, Pennetta G, Network analyses reveal novel aspects of ALS pathogenesis. PLOS Genet 11, e1005107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L-G, Landegren U, Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Koos B, Andersson L, Clausson C-M, Grannas K, Klaesson A, Cane G, Söderberg O, Analysis of protein interactions in situ by proximity ligation assays. Curr. Top. Microbiol. Immunol 377, 111–126 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Nagarkar-Jaiswal S, Lee P-T, Campbell ME, Chen K, Anguiano-Zarate S, Gutierrez MC, Busby T, Lin WW, He Y, Schulze KL, Booth BW, Evans-Holm M, Venken KJ, Levis RW, Spradling AC, Hoskins RA, Bellen HJ, A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. eLife 4, e05338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, Bellen HJ, MiMIC: A highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods 8, 737–743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsuda H, Han SM, Yang Y, Tong C, Lin YQ, Mohan K, Haueter C, Zoghbi A, Harati Y, Kwan J, Miller MA, Bellen HJ, The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133, 963–977 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hay BA, Maile R, Rubin GM, P element insertion-dependent gene activation in the Drosophila eye. Proc. Natl. Acad. Sci. U.S.A 94, 5195–5200 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vaccari T, Lu H, Kanwar R, Fortini ME, Bilder D, Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J. Cell Biol 180, 755–762 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang CY, Sun YH, Use of mini-white as a reporter gene to screen for GAL4 insertions with spatially restricted expression pattern in the developing eye in Drosophila. Genesis 34, 39–45 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Hartenstein V, Posakony JW, A dual function of the Notch gene in Drosophila sensillum development. Dev. Biol 142, 13–30 (1990). [DOI] [PubMed] [Google Scholar]
- 81.Kidd S, Lieber T, Young MW, Ligand-induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes Dev 12, 3728–3740 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Helms W, Lee H, Ammerman M, Parks AL, Muskavitch MA, Yedvobnick B, Engineered truncations in the Drosophila mastermind protein disrupt Notch pathway function. Dev. Biol 215, 358–374 (1999). [DOI] [PubMed] [Google Scholar]
- 83.Pennetta G, Hiesinger PR, Fabian-Fine R, Meinertzhagen IA, Bellen HJ, Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron 35, 291–306 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Petzoldt AG, Gleixner EM, Fumagalli A, Vaccari T, Simons M, Elevated expression of the V-ATPase C subunit triggers JNK-dependent cell invasion and overgrowth in a Drosophila epithelium. Dis. Model. Mech 6, 689–700 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tognon E, Kobia F, Busi I, Fumagalli A, De Masi F, Vaccari T, Control of lysosomal biogenesis and Notch-dependent tissue patterning by components of the TFEB-V-ATPase axis in Drosophila melanogaster. Autophagy 12, 1–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forgac M, Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol 8, 917–929 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Lev S, Ben Halevy D, Peretti D, Dahan N, The VAP protein family: From cellular functions to motor neuron disease. Trends Cell Biol 18, 282–290 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Nishimura Y, Hayashi M, Inada H, Tanaka T, Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem. Biophys. Res. Commun 254, 21–26 (1999). [DOI] [PubMed] [Google Scholar]
- 89.Loewen CJ, Levine TP, A highly conserved binding site in vesicle-associated membrane protein-associated protein (VAP) for the FFAT motif of lipid-binding proteins. J. Biol. Chem 280, 14097–14104 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Chai A, Pennetta G, Insights into ALS pathomechanisms: From flies to humans. Fly 9, 91–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nishimura AL, Mitne-Neto M, Silva HCA, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M, A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet 75, 822–831 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ratnaparkhi A, Lawless GM, Schweizer FE, Golshani P, Jackson GR, A Drosophila model of ALS: Human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLOS ONE 3, e2334 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chai A, Withers J, Koh YH, Parry K, Bao H, Zhang B, Budnik V, Pennetta G, hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet 17, 266–280 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maitra S, Kulikauskas RM, Gavilan H, Fehon RG, The tumor suppressors Merlin and Expanded function cooperatively to modulate receptor endocytosis and signaling. Curr. Biol 16, 702–709 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Genevet A, Polesello C, Blight K, Robertson F, Collinson LM, Pichaud F, Tapon N, The Hippo pathway regulates apical-domain size independently of its growth-control function. J. Cell Sci 122, 2360–2370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grifoni D, Garoia F, Bellosta P, Parisi F, De Biase D, Collina G, Strand D, Cavicchi S, Pession A, aPKCζ cortical loading is associated with Lgl cytoplasmic release and tumor growth in Drosophila and human epithelia. Oncogene 26, 5960–5965 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Russ A, Louderbough JM, Zarnescu D, Schroeder JA, Hugl1 and Hugl2 in mammary epithelial cells: Polarity, proliferation, and differentiation. PLOS ONE 7, e47734 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Di Cristofori A, Ferrero S, Bertolini I, Gaudioso G, Russo MV, Berno V, Vanini M, Locatelli M, Zavanone M, Rampini P, Vaccari T, Caroli M, Vaira V, The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget 6, 17514–17531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hernandez A, Serrano-Bueno G, Perez-Castineira JR, Serrano A, Intracellular proton pumps as targets in chemotherapy: V-ATPases and cancer. Curr. Pharm. Des 18, 1383–1394 (2012). [DOI] [PubMed] [Google Scholar]
- 100.Kartner N, Manolson MF, V-ATPase subunit interactions: The long road to therapeutic targeting. Curr. Protein Pept. Sci 13, 164–179 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Sennoune SR, Martinez-Zaguilan R, Vacuolar H+-ATPase signaling pathway in cancer. Curr. Protein Pept. Sci 13, 152–163 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Stransky L, Cotter K, Forgac M, The function of V-ATPases in cancer. Physiol. Rev 96, 1071–1091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McConnell M, Feng S, Chen W, Zhu G, Shen D, Ponnazhagan S, Deng L, Li YP, Osteoclast proton pump regulator Atp6v1c1 enhances breast cancer growth by activating the mTORC1 pathway and bone metastasis by increasing V-ATPase activity. Oncotarget 8, 47675–47690 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klezovitch O, Fernandez TE, Tapscott SJ, Vasioukhin V, Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev 18, 559–571 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Elsum IA, Yates LL, Pearson HB, Phesse TJ, Long F, O’Donoghue R, Ernst M, Cullinane C, Humbert PO, Scrib heterozygosity predisposes to lung cancer and cooperates with KRas hyperactivation to accelerate lung cancer progression in vivo. Oncogene 33, 5523–5533 (2013). [DOI] [PubMed] [Google Scholar]
- 106.Pearson HB, Perez-Mancera PA, Dow LE, Ryan A, Tennstedt P, Bogani D, Elsum I, Greenfield A, Tuveson DA, Simon R, Humbert PO, SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J. Clin. Invest 121, 4257–4267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pearson HB, McGlinn E, Phesse TJ, Schlüter H, Srikumar A, Gödde NJ, Woelwer CB, Ryan A, Phillips WA, Ernst M, Kaur P, Humbert P, The polarity protein Scrib mediates epidermal development and exerts a tumor suppressive function during skin carcinogenesis. Mol. Cancer 14, 169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Godde NJ, Sheridan JM, Smith LK, Pearson HB, Britt KL, Galea RC, Yates LL, Visvader JE, Humbert PO, Scribble modulates the MAPK/Fra1 pathway to disrupt luminal and ductal integrity and suppress tumour formation in the mammary gland. PLOS Genet 10, e1004323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Archibald A, Al-Masri M, Liew-Spilger A, McCaffrey L, Atypical protein kinase C induces cell transformation by disrupting Hippo/Yap signaling. Mol. Biol. Cell 26, 3578–3595 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCaffrey LM, Montalbano J, Mihai C, Macara IG, Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 22, 601–614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu T, Rubin GM, Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237 (1993). [DOI] [PubMed] [Google Scholar]
- 112.Lee T, Luo L, Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 24, 251–254 (2001). [DOI] [PubMed] [Google Scholar]
- 113.DeVorkin L, Gorski SM, LysoTracker staining to aid in monitoring autophagy in Drosophila. Cold Spring Harb. Protoc 2014, 951–958 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Kyriakakis P, Tipping M, Abed L, Veraksa A, Tandem affinity purification in Drosophila: The advantages of the GS-TAP system. Fly 2, 229–235 (2008). [DOI] [PubMed] [Google Scholar]
- 115.Veraksa A, Bauer A, Artavanis-Tsakonas S, Analyzing protein complexes in Drosophila with tandem affinity purification-mass spectrometry. Dev. Dyn 232, 827–834 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Gilbert MM, Tipping M, Veraksa A, Moberg KH, A screen for conditional growth suppressor genes identifies the Drosophila homolog of HD-PTP as a regulator of the oncoprotein Yorkie. Dev. Cell 20, 700–712 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang L, Veraksa A, Single-step affinity purification of ERK signaling complexes using the streptavidin-binding peptide (SBP) tag. Methods Mol. Biol 1487, 113–126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H, 2016 update of the PRIDE database and related tools. Nucleic Acids Res 44, D447–D456 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dietzl G, Chen D, Schnorrer F, Su K-C, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ, A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156 (2007). [DOI] [PubMed] [Google Scholar]
- 120.Anjum SG, Xu W, Nikkholgh N, Basu S, Nie Y, Thomas M, Satyamurti M, Budnik BA, Ip YT, Veraksa A, Regulation of Toll signaling and inflammation by β-arrestin and the SUMO protease Ulp1. Genetics 195, 1307–1317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencesignaling.org/cgi/content/full/11/533/eaar1976/DC1
Fig. S1. Vha68–2 mutant clones show reduced Notch reporter expression, and lgl Vha68–2 mutant tissue is retained in pupal retinas.
Fig. S2. Lgl physically interacts with Vap33 and the V-ATPase component Vha44.
Fig. S3. Vap33-overexpressing tissue is retained in the pupal retina, and Vap33 mutant tissue accumulates N-intra and increases expression of the Notch reporter.
Fig. S4. Overexpression of Vap33 results in reduced bristle formation but does not suppress the NICD-induced thoracic overgrowth phenotype.
Fig. S5. Vap33 colocalizes with Vha44-GFP, and Vap33 protein abundance and localization are unaffected in lgl mutant tissue.
Fig. S6. Vap33 physically interacts with Vha68–3 and Vha68–2.
Table S1. Drosophila stocks and references.
Table S2. List of genotypes of the samples used in each figure.
Data file S1. Lgl-interacting proteins.