

Abstract
The use of metal complex immobilized/decorated porous materials as catalysts has found various applications. As such, finding a new and mild method for synthesis of metal complex immobilized over porous material is of great interest. Immobilized porous materials for styrene oxidation were reported in this work. Immobilized porous material of Cu‐Schiff base complex @MIL‐101 were described, in which immobilized Cu‐Schiff base complex within super cage of a metal‐organic framework (MOF)‐based porous material, chromium (III) terephthalate MIL‐101. They were systematically characterized by using elemental analysis, powder X‐ray diffraction, fourier transform infrared spectroscopy, N2 absorption‐desorption, and so on, also used as catalyst for the selective oxidation of styrene to benzaldehyde. Comparatively, the immobilized heterogeneous catalyst of Cu‐Schiff base complex@MIL‐101 acted as an efficient heterostructure catalyst in the oxidation of styrene to benzaldehyde up to six cycles, and showed superior activity for styrene oxidation over MIL‐101.
Keywords: immobilization, metal-organic frameworks, catalysis, MIL-101, styrene
1. Introduction
The Schiff bases containing heterocycles with donor atoms such as nitrogen, oxygen, sulfur, and so on,1 have a widely applications in analytical chemistry, catalysis, medicine, corrosion, biological activity, and photochromic discoloration.2 Schiff base metal complexes are very similar to the structure of metal porphyria complexes which have widely uses catalysis with its unique structural properties, so Schiff base metal complexes as catalysts also have a wide space for research. Their catalytic activity and biological activity have become the focus of research. In recent years, there have been many reports on the catalysis of Schiff base complexes, such as olefinic oxidation,3 asymmetric catalysis,4 oxidation of alcohol,5 suzuki coupling reactions6 The results show that the Schiff base complexes exhibit high catalytic performance. But the majority of Schiff base metal complexes are homogeneous, although homogeneous Schiff base metal complexes contain a variety of active sites and achieve high efficiency catalytic activity, however, small molecules of complexes are prone to react with each other to change the original structure and reduce or lose catalytic activity. Alao Schiff base metal complexes is generally dissolved in high‐temperature and is not easy to reuse, which will certainly limit their application in the industrial field. Therefore, the development of heterogeneous systems of inorganic or organic carriers has attracted extensive attention in recent years.
Metal organic frameworks (MOFs) or porous coordination poly‐mers(PCPs) are a class of low‐density and three‐dimensional (3D) crystalline materials consisting of metal ions and inorganic /organic ligands interconnected by coordination bonds. Their diversified and desgnable framework/pore structures, high porosity, high surface area, large frame‐work flexibility/dynamism, and so on have made them good candidates for a variety of potential application such as gas adsorption and separation,7 catalysis,8 drug delivery9 and other functions.10 In recent years, post synthetic modification11 is a powerful strategy for the introduction of functional groups in MOFs bases because it makes possible the late‐stage functionalization of MOFs. Here we report a heterogeneous catalysts based on Schiff base metal complexes and metal organic framework of MIL‐10112(chromium (III) terephthalate, Cr3F(H2O)2O[O2C‐C6H4‐(CO2)]3⋅nH2O, n≈25)with a mild method adopting post synthetic modification strategy. A series of heterogeneous catalysts of Cu‐Schiff base complex@MIL‐101 were prepared, in which Cu‐Schiff base complexes were immobilized in MIL‐101(denoted as Cu‐Schiff base complex@MIL‐101(x), Schiff Base=salicylaldehyde ethylenediamine Schiff base; x refers the molar ratio of Cu‐Schiff base complex/MIL‐101). Although impregnation approach has been demonstrated to be a successful method, but the maximum size of Cu‐ Schiff base complex molecule is 13 Å, so it is difficult to diffuse into the super cage of MIL‐101 (Figure 1A). To circumvent this problem, an in situ mild method was judiciously adopted to prepare Cu‐Schiff base complex@MIL‐101(x) heterogeneous catalysts. Namely, calculated amount of salicylaldehyde molecule of 6 Å in maximun size was first freely diffused into the super cage of MIL‐101 (Figure 1B), followed reacted with the added ethylenediamine to form bis(salicylaldiminato) Schiff base within the MIL‐101(Figure 1C). Bis(salicylaldiminato) Schiff base were crowded filled within the MIL‐101 and not allowed to escape out from the supercages of MIL‐101. Metal ion Cu was subsequently added to in‐situ react with the formed bis(salicylaldiminato) Schiff base to give the final Cu‐ Schiff base complex@MIL‐101(x) heterogeneous catalysts (Figure 1D).
Figure 1.

Schematic illustration of the encapsulation of Cu‐Schiff base complex within the MIL‐101 cage: A) scheme representation of the mesoporous cage of MIL‐101; A1) perspective view of the mesoporous cage of =MIL‐101 with hexagonal window; B) diffusion of salicylaldehyde into the MIL‐101; C) immobilization of bis salicylaldehyde Ethylenediamine Schiff Base in MIL‐101; D) Cu‐Schiff complex@MIL‐101(x) heterogeneous catalysts via in‐situ reaction.
We successfully synthesized immobilized heterogeneous catalysts base on Cu‐Schiff Base Complex and Metal‐Organic Framework MIL‐101, and also have compared catalytic activity of pure metal organic frameworks and immobilized catalysts on the oxidation of styrene.
2. Results and Discussion
Preparation of samples. Porous host MIL‐101 was prepared as described previously12 .XRD analysis indicates the formation of pure MIL‐101 (see in Figure 2). For the preparation of Cu‐Schiff base complex@MIL‐101(x) heterogeneous catalysts, when y mmol low molecular weight salicylaldehyde (y=0.4, 0.8, 1.6) was firstly diffused into the supercages of MIL‐101 (0.2 mmol), the molar ratio of Cu‐Schiff base complex toMIL‐101 is x. A a Series of immobilzed heterogeneous catalysts of Cu‐Schiff base complex@MIL‐101(x=2,4,6) were synthesized. Figure 2 presents the X‐ray diffraction patterns of the parent materials and the composites. Cu‐Schiff base complex exhibits a single peak at the black inverted triangle in Figure 2, about 6.5°, 10.3°, 14.2°, 14.5°. It has to be mentioned that a peak below 6° on the Cu‐Schiff base complex XRD pattern has its origin in the sample holder. Due to the mesoporous character of MIL‐101, most peaks for the MIL sample are observed in a small angle region and are characteristic of the structure of that particular MOF. For Cu‐Schiff Base complex@MIL‐101(x) material, most peaks from MIL are preserved except those of low intensity (2 Theta∼4.2 and 4.9°), suggesting that the MOF structure is mainly preserved and the framework from Cu‐Schiff complex@MIL‐101(x) only induced slight distortion. XRD results also indicated that with the increasing of the loading amount the intensity of characteristic peaks of MIL‐101 was gradually reduced with the increase of Cu‐Schiff complex loading in MIL‐101, but the MIL‐101 structure was still remained. It concluded that Cu‐Schiff Base complex were successfully immobilized within the cavities of MIL‐101.
Figure 2.

XRD patterns the Cu‐Schiff complex@MIL‐101(x) catalysts: a) the simulated XRD powder patterns of MIL‐101 in reference 12; b) the synthesized MIL‐101; c) x=2 : 1; d) x=4 : 1; e) x=8 : 1; f) Cu‐Schiff base complex.
Catalytic measurements. Catalytic oxidation of styrene(Scheme 1) was carried out in a 250 ml round bottom flask using metal Schiff base complexes incorporated MIL‐101 as catalysts. In a typical catalytic reaction, 200 ml of Tris‐HCl buffers solution pH 7.6 and H2O2 (3.4×10−3 mol/L) were mixed and heated in a water bath with continuous stirring at 30 °C. Then the catalytic oxidation reaction was initiated with the addition of appropriate amount of catalyst (0.0010 g) and styrene (1×10−4 mol/L, 0.0022 g) to this solution. The progress of oxidation styrene was monitored by analyzing the maximum adsorbance intensity of styrene at 246 nm using a Shimadzu UV‐3600 double‐beam scanning spectrophotometer via withdrawing small aliquots of the reaction mixture at a specific interval of time. The reactions followed apparent first order rate kinetics for at least three half‐lives, because the concentration of hydrogen peroxide in the catalytic oxidation reaction is far greater than the concentration of styrene, therefore the concentration of hydrogen peroxide can be regarded as a constant, the catalytic oxidation of styrene follows apparent pseudo‐first‐order reaction kinetics. the reaction rate equation can be expressed as: r=kobsd [S]t, kobsd is first order reaction rate constant, [S] is the total concentration of styrene. For such a pseudo‐first‐order reaction, one reactant is greatly excessive, which satisfies the principle of the isolation method, so polynomial fitting method can be used, the concentration data of reactant S and product y at different times were measured. The experimental data [y] ∼ t were processed by nonlinear fitting method, and then the nonlinear fitting was conducted by excel‘s programming solution in office software. So in the paper kobsd was determined from the plots of At (absorbance at time t) versus t using the non‐linear fitting method. Figure 3A shows a representative UV‐vis spectrum for the oxidation of Styrene catalyzed by Cu‐Schiff complex@MIL‐101(2). Successive decrease in the absorption maximum of Styrene at 246 nm were observed with increasing reaction time. The reaction appears to be first order with respect to Styrene concentration (Figure 3B). Using nonlinear fitting of the experimental data, correlation coefficient is 0.876, the kobsd is 0.0278 and conversion rate is 93.54 %,
Scheme 1.
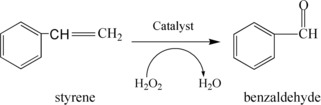
Reaction Scheme for Oxidation of Styrene to Benzaldehyde
Figure 3.

(A) Decrease in the absorption maximum of benzoquinone at 246 nm catalyzed by Cu‐ Schiff complex@MIL‐101(2) with increasing reaction time. [Styrene]=1×10−4 mol L−1, [Cu‐Schiff base complex@MIL‐101(2)]/[ Styrene]=0.04, pH=7.6 (Tris−HCl buffer), T=30 °C. (B) Typical plot of At versus time for oxidation of styrene catalyzed by Cu‐Schiff base complex@MIL‐101(2)@MIL‐101(2) using experimental data in Figure 3A, and nonlinear fitting of the experimental data: dots represent the experimental data, and the line shows the fitting results
Preparation and characterization of samples.The successful incorporation of the Cu‐Schiff base complex within the MIL‐101 super cage was confirmed by X‐ray powder diffraction, infrared spectroscopy, N2 adsorption measurement, and elemental analysis. X‐ray diffraction confirmed that the framework of MIL‐101 was kept intact upon the inclusion of Cu‐Schiff base complex. However, noticeable changes of relative intensities were observed with the inclusion of Cu‐Schiff base complex (Figure 1). It is also worthy to note that the diffraction peaks of Cu‐Schiff base complex gradually became evident with the increase of x values, as shown by Figure 1. FT‐IR spectra of the Cu‐Schiff base complex@MIL‐101(x) verify the presence of Cu‐Schiff base complexes in the MIL‐101 matrix (Figure 4). For comparison, the IR spectra of bare MIL‐101 and Cu‐Schiff base complexe were also presented. The characteristic bands of the Cu‐Schiff base in 906–1341 cm−1, and 443–523 cm−1 region were observed within the Cu‐Schiff base comple@MIL‐101(x) solids, indicating the inclusion of Cu‐Schiff base in the cages of MIL‐101. The new bands in the low‐frequency region of 443–523 cm−1 are assigned to strethcing mode of v(M−O) and v(M−N) bonds, indicating the coordinaiton between Cu ions and oxygen and nitrigen atoms. As the feed ratio of Cu‐Schiff base to MIL‐101 was increased from 2 : 1 to 8 : 1, the intensity of these characteristic peaks becomes more obvious, indicating an increased Cu‐Schiff base content within the MIL‐101 cage. These results indicate the presence of Cu‐Schiff base complex within the MIL‐101 solids. The presence of Cu‐Schiff base complex within the super cage of MIL‐101 was further visualized by N2 adsorption isotherms for MIL‐101 and Cu‐Schiff base complex@MIL‐101(x). Compared with the bare MIL‐101, Cu‐Schiff base complex@MIL‐101(x) exhibit a significant decrease of the N2 amount adsorbed at P/P 0>0.2. The higher the molar ratio of Cu‐schiff base to MIL‐101 is, the lower the adsorbed N2 amount is (see Figure 5). The BET surface area decreases from 2646 m2 g−1 of bare MIL‐101 to 2425 m2 g−1 of Cu‐Schiff base complex@MIL‐101(2) (see Table 1). An increased Cu‐Schiff base/MIL‐101 ratio leads to an evident decrease of surface area, also as increasing Cu‐Schiff base/MIL‐101 ratio, pore size diameter decreased from 2 nm to 1.72 nm, as show in Figure S1, it further demonstrating the incorporation of Cu‐Schiff base within the MIL‐101 super cage. The presence of Cu‐Schiff base in the super cage of MIL‐101 was further confirmed by the decreased pore volume and the increased Cu content in Cu‐MIL‐101(x) determined by AES‐ICP analysis, as summarized in Table 1. The scanning electron microscope(SEM) observation of Cu‐Schiff base complex@MIL‐101with different Cu‐Schiff base complex dosage ( Figure 6) shown that the loading of the Cu‐Schiff base complex does not affect the MOF morphology and the MOF caystals remain intact in the composite. So we can conclude that the Cu‐Schiff base complex complex was successfully immobilized within the cavities of MIL‐101 based on the above mentioned analysis results.
Figure 4.

Infrared spectra of the Cu‐Schiff base complex@MIL‐101(x) catalysts: a) bare MIL‐101; b) x=2 : 1; c) x=4 : 1; d) x=8 : 1; e) Cu‐Schiff base complex.
Figure 5.

Nitrogen adsorption isotherms 77 K of Cu‐Schiff base complex@MIL‐101(x) catalysts; a) bare MIL‐101, b) x=2 : 1, c) x=4 : 1, d) x=8 : 1.
Table 1.
| Catalyst | BET [m2g−1][b] | Pore‐Volume [m3g−1][b] | Cu content [wt %][c] | K min−1 | Conversion R |
|---|---|---|---|---|---|
| MIL‐101 | 2646 | 1.37 | — | 0.187 | 48.46 % |
| Cu‐Schiff base complex@MIL‐101(2) | 2325 | 1.07 | 2.16 | 0.223 | 87.30 % |
| Cu‐Schiff base complex@MIL‐101(4) | 1274 | 0.58 | 6.17 | 0.378 | 93.54 % |
| Cu‐Schiff base complex@MIL‐101(8) | 947 | 0.42 | 8.77 | 0.310 | 84.40 % |
[a] Reaction was carried out using 0.0010 g Cu‐Schiff base complex@MIL‐101(x) catalysts, 0.2 μM tyrene in 200 ml Tris‐HCl buffers solution of pH 7.6 containing H2O2 (3.4×10−3 mol L−1) as oxidant at 30 °C. [b] BET surface areas obtained from N2 adsorption isotherms. [c] Determined by AES‐ICP analysis.
Figure 6.

SEM images for: a) MIL‐101, b) Cu‐Schiff base complex@MIL‐101(2), c) Cu‐Schiff base complex@MIL‐101(4), d) Cu‐Schiff base complex@MIL‐101(8).
Influences of Different Loading Capacity
The catalytic performance of Cu‐Schiff base complex@MIL‐101(x) was evaluated using the styrene oxidation as a model reaction. H2O2 was used as oxidant. Table 2 lists the catalytic results of the Styrene oxidation over Cu‐Schiff base complex@MIL‐101(x) catalysts. Free Cu‐Schiff base complex in Tris‐HCl buffer solution demonstrate a fast reaction rate of 0.41 min−1, (Table 1), Without Cu‐Schiff base complex, the reaction for Bare MIL‐101 is going very slowly with a rate 0.087 min−1, in comparison, Cu‐Schiff base complex@MIL‐101(x) reacts more than four times faster with the rate of 0.378 min−1, (Table 1)Notably, the reaction rate constant k was evidently affected by the content of Cu‐Schiff base complex complex within the MIL‐101. The rate constant k increases to 0.223 min−1 and 0.378 min−1 at x=2 : 1 and x=4 : 1, respectively. Further increase in x value leads to an evident decrease of k of 0.31 min−1. This phenomenon could be attributed to the blocking effect. Large amount of immobilized Cu‐Schiff base complex complex in the pore of MIL‐101 could block the easily accessible Cu‐Schiff base complex functional groups, thus resulting in a decreased catalytic activity.
Table 2.
Summary of catalysis results of oxidizing Styrene in the presence of H2O2.
| Free Cu‐Schiff base complex[a] | MIL‐101 | Cu‐Schiff base complex@MIL‐101(2) | Cu‐Schiff base complex@MIL‐101(4) | Cu‐Schiff base complex@MIL‐101(8) | |
|---|---|---|---|---|---|
| Rate (min−1) | 0.41 | 0.087 | 0.223 | 0.378 | 0.31 |
| Conversion(%) | 85.42 % | 48.46 % | 87.3 % | 93.54 % | 84.4 % |
[a] pH=7.6 (Tris–HCl buffer), Rate calculated from the first 40 min. Final conversion after 24 h.
Recyclability of catalysts. We evaluated the recyclability of by checking its catalytic activities at different cycles. As shown in Figure 7, the reaction rate of Cu‐Schiff base complex@MIL‐101(4) fluctuates from 0.36 to 0.22 min−1 in the first six cycles; it decreases to 0.18 min−1 at seventh cycle. Representing about 50 % activity drop compared to that of the first cycle. In comparison, the activity of MIL‐101 decreases abruptly with more than 47 % activity lost after the first cycle, and only 28 % activity remains at the fourth cycle(table S1). the fast decay of Cu‐Schiff base complex@MIL‐101(4) originates from the leaching was observed for Cu‐Schiff base complex@MIL‐101(4) over seven cycles, and the MOF host could still maintain its framework intergrity after catalytic cycles as evidenced by powder X‐ray diffraction studytic (Figure 8). We concluded that the capability of Cu‐Schiff base complex@MIL‐101(4) for at least six cycles could be attributed to the strong hydrophobic interaction between the MIL‐101 and Cu‐Schiff base complex molecules trapped in hydrophobic nanoscopic cages, preventing their escape from the MOF host matrix, which was detected Cu‐Schiff base complex@MIL‐101(4) after reaction by ICP, the content of Cu only 1.4 %, So The catalytic activity decreased rapidly.
Figure 7.

reaction rate of MIL‐101 and Cu‐Schiff base complex@MIL‐101(4) at different cycles
Figure 8.

PXRD of patterns for Cu‐Schiff complex)@MIL‐101(4) after catalytic cycles.
Mechanisms for Catalytic Oxidation of Styrene
We attempted to elucidate possible mechanisms based on our experimental results and previously published literature13. It is speculated that the unique structural characteristics of the Cu‐Schiff base complex@MIL‐101 composites are responsible for their superior activity. First, the abundant interconnected nanoporosity accessible through open pore of MOF offers numerous opportunities for fast diffusion and capture of guest molecules. In turn, the capture and enrichment of styrene within the cavities will result in an improved local concentration effect around the encapsulated Cu‐Schiff base complex, which is beneficial to prolong their stay time on the active sites, enhancing catalytic activity. Second, the group in the MIL101 organic linkers could serve as antenna to enhancing absorption and catalytic active sites, Subsequently, the generated intermediates could be quickly transferred, due to the strong internal interactions between Cu‐Schiff base complex and MIL101, Finally, the encapsulated structure of Cu‐Schiff base complex guarantees ready accessibility of the styrene and generated intermediates, immediately oxidized captured substrates on the catalyst surface, thus maximizing the catalytic activity.
Conclusions
In summary, a series of Cu‐Schiff base complex were successfully immobilized in the cage of MIL‐101 via a mild method, which exhibited superior catalysis performances compared to pure metal organic framework MIL‐101 and Cu‐Schiff base complex. More over, the activity of the catalyst changed little after 6 times of reuse. The results show that Cu‐Schiff base complex did not break away from metal organic framework during the reaction process, indicating that the immobilized method can effectively improve the stability of the Cu‐Schiff base complex catalyst, considering the richness of metal organic frameworks structures, the present studies open a new avenue for Schiff‐ base complex immobilization as heterogeneous catalysis.
Experimental Section
Reagents and instrument. Styrene was purchased from Aldrich. 30 % hydrogen peroxide, Hydrochloric Acid, ethylenediamine, salicylaldehyde, Cobalt(II) nitrate 6‐hydrate, nickel(II) sulfate 6‐hydrate, copper(II) chloride(III) dihydrate, chromic nitrate 9‐hydrate, Styrene, hydrochloric acid, and tri‐(hydroxymethyl)aminomethane (Tris) were all of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All chemicals and solvents were used as received without further purification. Water used for all kinetic measurel was redistilled water. Kinetic investigations were carried out by using UV‐vis spectrophotometer on a Shimadzu UV‐3600 double‐beam scanning spectrophotometer.he surface areas of the samples were obtained by theBET method using N2 adsorption data at 77 K. Powder XRD patterns of the prepared catalysts were measured with a Philips X'Pert MPD X‐ray powder diffractometer with monochromatic CuKα1 radiation (40KV, 40 mA). IR spectra were recorded on a Perkin‐Elmer Spectrum One Fouriertransform infrared spectrometer by using the conventional KBr pellet method (3 wt.%). element analysis were detected by M6.
Synthesis of metal organic framework MIL101. Typically, a mixture of Cr(NO3)3⋅H2O (400 mg at 1 mmol), terephthalic acid (166 mg at 2 mmol), and HF (0.2 ml at 1 mmol) in 4.8 ml H2O was heated at 220 °C for 8 h in a Telfon‐lined stainless steel bomb. The resulting green products were further purified in refluxing ethanol for several times to remove the unreacted carboxylic acid.
Synthesis of Cu‐Schiff complex@MIL‐101(x). For the preparation of Cu‐Schiff complex@MIL‐101(x) heterogeneous catalysts, y mmol low molecular weight salicylaldehyde (y=0.4, 0.8, 1.6) was firstly diffused into the supercages of MIL‐101 (0.2 mmol) in a round bottom flask containing ethanol (4 mL) by heating to reflux. After 30 min, y/2 mmol ethylenediamine was drop‐wise added to give bis(salicylaldiminato) Schiff base incorporated MIL‐101. After a careful purification of the obtained product by repeated washing with a large mount of water, stoichiometric amount of CuCl2 was consequently added to give the final Cu‐ complex @MIL‐101(x) heterogeneous catalysts.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by Excellent Young Talents Fund Program of Higher Educaton Institutions of Anhui Province (CN)(grant GXYQ2018072), the National Ntural Science Foundation of China (NSFC, Grants 51505121 and 51375139), the Natural Sciece Foundation of hefei university (Grants 18ZR17ZDA).
Y. Wu, W. Wang, L. Liu, S. Zhu, X. Wang, E. Hu, K. Hu, ChemistryOpen 2019, 8, 333.
References
- 1.
- 1a. Schiff H., Liebigs Ann. Chem. 1897, 99, 236–226; [Google Scholar]
- 1b. Schiff H., Annalen 1864, 131, 118–119. [Google Scholar]
- 2.
- 2a. Das S., Bhunia S., Maity T., J. Mol. Catal. A 2014, 394, 188–197; [Google Scholar]
- 2b. Liu D., Zhang X., Zhu L., Catal. Sci. Technol. 2015, 5, 562–571; [Google Scholar]
- 2c. Zhou W. J., Albela B., He M. Y., Polyhedron 2013, 64, 371–376; [Google Scholar]
- 2d. Rao G. K., Kumar A., Singh M. P., J. Organomet. Chem. 2014, 753, 42–47; [Google Scholar]
- 2e. Xiao Y., Bi C., Fan Y., J. Coord. Chem. 2009, 62, 3029–3039; [Google Scholar]
- 2f. Saini R. P., Rumar V., Gupta A. K. ,Med. Chem. Res. 2014, 23, 690–698. [Google Scholar]
- 3.
- 3a. Lou L. L., Yu K., Ding F., J. Catal. 2007, 249, 102–110; [Google Scholar]
- 3b. Kureshy R. I., Ahmad I., Noor-ul H. K., J. Catal. 2005, 235, 28–34; [Google Scholar]
- 3c. Silva A. R., Wilson K., Clar J. H., Microporous Mater. 2006, 91, 128–138; [Google Scholar]
- 3d. Hu J., Li K., Li W., Appl. Catal. A 2009, 364, 211–220. [Google Scholar]
- 3e. Belokon Y. N., Maleev V. I., North M., ACS Catal. 2013, 3, 1951–1955. [Google Scholar]
- 3f. Gong L., Chen L. A., Meggers E., Angew. Chem. Int. Ed. 2014, 53, 10868–10874; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11046–11053. [Google Scholar]
- 4.
- 4a. Baleizao C., Gigante B., Garcia H., J. Catal. 2003, 215, 199–207; [Google Scholar]
- 4b. Thakur S. S., Chen S. W., Li W., J. Organomet. Chem. 2006, 691, 1862–1872; [Google Scholar]
- 4c. Kim J. H., Kim G. J., Catal. Lett. 2004, 92, 123–130. [Google Scholar]
- 5.
- 5a. Rueping M., Vila C., Szadkowska A., ACS Catal.; [Google Scholar]
- 5b. Dayan S., Kalaycioglu N. O., Appl. Organomet. Chem. 2013, 27, 52–58; [Google Scholar]
- 5c. Wong W. L., Ho K. P., Lee L. Y. S., Appl. Catal. A 2013, 453, 244–249. [Google Scholar]
- 6.
- 6a. Bhunia S., Sen R., Koner S., Inorg. Chim. Acta. 2010, 363, 3993–3999; [Google Scholar]
- 6b. Dhara K., Sarkar K., Srimani D., Dalton Trans. 2010, 39,6395-6402. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Rosi N. L., Eckert J., Eddaoudi M., Vodak D. T., Sci. 2003, 300, 1127–1129; [DOI] [PubMed] [Google Scholar]
- 7b. Murray L. J., Dinca M., Long J. R., Chem. Soc. Rev. 2009, 38, 1294–1314; [DOI] [PubMed] [Google Scholar]
- 7c. Dtiren T., Sarkisov L., Yaghi O. M., Langmuir, 2004, 20, 2683–2689; [DOI] [PubMed] [Google Scholar]
- 7d. Ma S., Zhou H. C., Chem. Commun. 2010, 44–53; [DOI] [PubMed] [Google Scholar]
- 7e. Fedrey G., Chem. Soc. Rev. 2008, 37, 191–214; [DOI] [PubMed] [Google Scholar]
- 7f. Millward A. R., Yaghi O. M., J. Am. Chem. Soc. 2005, 127, 17998–17999; [DOI] [PubMed] [Google Scholar]
- 7g. Llewellyn P. L., Bourrelly S., Serre C., Langmuir, 2008, 24, 7245–7250; [DOI] [PubMed] [Google Scholar]
- 7h. Kitagawa S., Uemura K., Chem. Soc. Rev. 2005, 34, 109–119; [DOI] [PubMed] [Google Scholar]
- 7i. eeflFe M. O., Yaghi O. M., Chem. Rev. 2012, 112, 675–702; [DOI] [PubMed] [Google Scholar]
- 7j. Yang Q., Li L., Tan W., Chem. Commun. 2017, 53, 9797–9800; [DOI] [PubMed] [Google Scholar]
- 7k. Zhang G., Wei G., Liu Z., Chem. Mater. 2016, 28, 6276–6281; [Google Scholar]
- 7l. Cadiau A., Adil K., Bhatt P. M., Science 2016, 353, 137–140. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Yoon M., Srirambalaji R., Kim K., Chem. Rev. 2012, 112, 1196–1231; [DOI] [PubMed] [Google Scholar]
- 8b. Dybtsev D. N., Nuzhdin A. L., Chun H., Angew. Chem. Int. Ed. 2006, 45, 916–920; [DOI] [PubMed] [Google Scholar]
- 8c. Banejee M., Das S., Yoon M., J. Am. Chem. Soc. 2009, 131, 7524–7525. [DOI] [PubMed] [Google Scholar]
- 8d. Zhang G., Wei G., Liu Z., Chem. Mater. 2016, 28, 6276–6281. [Google Scholar]
- 8e. Xia Q., Li Z., Tan C., J. Am. Chem. Soc. 2017, 139, 8259–8266. [DOI] [PubMed] [Google Scholar]
- 8f. Zhu C., Mao Q., Li D., Catal. Commun. 2018, 104, 123–127. [Google Scholar]
- 9.
- 9a. Rieter W. J., Pott K. M., Taylor K. M. L., J. Am. Chem. Soc. 2008, 11584–1 1585; [DOI] [PubMed] [Google Scholar]
- 9b. Imaz I., Martinez M. R., Fernandez L. G., Chem. Commun. 2010, 46, 4737–4739; [DOI] [PubMed] [Google Scholar]
- 9c. Horcajada P., Chalati T., Serre C., Nat. Mater. 2010, 9, 172–178. [DOI] [PubMed] [Google Scholar]
- 9d. Horcajada P., Serre C., Vallet-Regi M., Angew. Chem. 2006, 118, 6120–6124. [DOI] [PubMed] [Google Scholar]
- 9e. Ji R., Manna K., Lin Z., J. Am. Chem. Soc. 2017, 139, 7004–7011. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Wang Z., Zhang B., Fujiwara H., Chem. Commun. 2004, 416–4 17. [DOI] [PubMed] [Google Scholar]
- 10b. Jayaramulu K., Kanoo P., George S. J., Chem. Commun. 2010, 46, 7906–7908. [DOI] [PubMed] [Google Scholar]
- 10c. An J. Y., Shade C. M, Chengelis-Czegan D. A., J. Am. Chem. Soc. 2011, 133, 1220–1223. [DOI] [PubMed] [Google Scholar]
- 10d. Pramanik S., Zheng C., Zhang X., J. Am. Chem. Soc. 2011, 133, 4153–4155. [DOI] [PubMed] [Google Scholar]
- 10e. Akashima Y., Martinez V. M., Fumkawa S., Nat. Commun. 2011, 2, 168–171.21266971 [Google Scholar]
- 10f. Dong M. J., Zhao M., Ou S., Angew. Chem. Int. Ed. 2014, 53, 1575–1579; [DOI] [PubMed] [Google Scholar]
- 10g. Ke F., Li Y. Z., Zhang C. Y., J. Colloid Interface Sci. 2018, 522, 283–290; [DOI] [PubMed] [Google Scholar]
- 10h. Ke F., Peng C. Y., Zhang T., Sci. Rep. 2018. , 8, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Cohen S. M., Chem. Rev. 2011, 112, 970–1000; [DOI] [PubMed] [Google Scholar]
- 11b. Wang Z., Cohen S. M., Angew. Chem. Int. Ed. 2008, 47, 4699–2707; [DOI] [PubMed] [Google Scholar]
- 11c. Wang Z., Tanabe K. K., Cohen S. M., Chem. Eur. J. 2010, 16, 212–217; [DOI] [PubMed] [Google Scholar]
- 11d. Garibay S. J., Wang Z., Cohen S. M., Inorg. Chem. 2010, 49, 8086–8091; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Savonnet M., Bazer-Bachi D., Bats N, J. Am. Chem. Soc. 2010, 132, 4518–4519; [DOI] [PubMed] [Google Scholar]
- 11f. Falcaro P., Ricco R., Doherty C. M., Chem. Soc. Rev. 2014, 43, 5513–5560; [DOI] [PubMed] [Google Scholar]
- 11g. Lustig W. P., Mukherjee S., Rudd N. D., Chem. Soc. Rev. 2017, 46, 3242–3285; [DOI] [PubMed] [Google Scholar]
- 11h. Li Y. A., Zha C. W., Zhu N. X., Chem. Commun. 2015, 51, 17672–17675; [DOI] [PubMed] [Google Scholar]
- 11i. Zhao D., Cui Y., Yang Y., CrystEngComm 2016, 18, 3746–3759; [Google Scholar]
- 11j. Zbang X., Hu Q., Xia T., ACS Appl. Mater. Interfaces 2016, 8, 32259–32265; [DOI] [PubMed] [Google Scholar]
- 11k. Tanabe K. K., Wang Z. Q., Cohen S. M., J. Am. Chem. Soc. 2008, 130, 8508–8517; [DOI] [PubMed] [Google Scholar]
- 11l. Kitaura R., Onoyama G., Sakamoto H., Angew. Chem. Int. Ed. 2004, 43, 2684–2687; [DOI] [PubMed] [Google Scholar]
- 11m. Cohen S. M., Chem. Rev. 2012, 112, 970–1000; [DOI] [PubMed] [Google Scholar]
- 11n. Radshaw D., Garal A., Huo J., Chem. Soc. Rev. 2012, 41, 2344–2381; [DOI] [PubMed] [Google Scholar]
- 11o. Ke F., Jiang J., Li Y. Z., Appl. Surf. Sci. 2017, 413, 266–274. [Google Scholar]
- 12. Ferey G., Draznieks C. M., Serre C., Science, 2005, 309, 2040–2042. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Yao P. Z., Liu H. L., Wang D. T., J. Colloid Interface Sci. 2018, 522, 174–182; [DOI] [PubMed] [Google Scholar]
- 13b. Zeng L., Guo X., He C., ACS Catal. 2016, 6, 7935–7947; [Google Scholar]
- 13c. Dhakshinamoorthy A., Asiri A. M., García H., Angew. Chem. Int. Edit. 2016, 55, 5414–5445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary