

Abstract
A distinct advantage of nanosensor arrays is their ability to achieve ultra-low detection limits in solution by proximity placement to an analyte. Here, we demonstrate label-free detection of individual proteins from Escherichia coli (bacteria) and Pichia pastoris (yeast) immobilized in a microfluidic chamber, measuring protein efflux from single organisms in real time. The array is fabricated using non-covalent conjugation of an aptamer-anchor polynucleotide sequence to near-infrared emissive single-walled carbon nanotubes, using a variable chemical spacer shown to optimize sensor response. Unlabeled RAP1 GTPase and HIV integrase proteins were selectively detected from various cell lines, via large near-infrared fluorescent turn-on responses. We show the process of E. coli induction, protein synthesis, and protein export is highly stochastic, yielding variability in protein secretion, with E. coli cells undergoing division under starved conditions producing 66% fewer secreted protein products than their non-dividing counterparts. We further demonstrate the detection of a unique protein product resulting from T7 bacteriophage infection of E. coli, illustrating that nanosensor arrays can enable real-time, single-cell analysis of a broad range of protein products from various cell types.
INTRODUCTION
There is significant interest in the label-free optical detection of proteins from crude, unpurified biological samples, or directly from the protein production source. Protein expression and secretion guide a significant aspect of nearly every cellular metabolic or signaling pathway. In some systems, stochasticity in protein production is inherent. In other systems, aberrations in protein expression can be representative of disease states. In both cases, detection of protein from crude samples, or from the point-of-production, could significantly reduce the time necessary to detect variability or aberrations in protein production1. To detect and quantify protein expression and secretion, standard approaches rely primarily on immunological analytical methods including enzyme-linked immunosorbent assays (ELISA), western blotting, radial immunodiffusion, or mass spectrometry. Recent advances have explored electrochemical detection of proteins with antibodies and aptamers with great success for temporal quantification of protein from purified samples, several with picomolar detection capabilities 2, 3, 4, 5. Fluorescence labeling of target proteins has provided the additional dimension of spatial information for protein detection and studying protein-protein interactions, though most protein detection strategies are for intracellular protein detection 6, 7, 8. However, such methods rely on fluorescent modification of the target protein, and are restricted to use inside the cell. Moreover, these approaches for protein detection and quantification often require preliminary purification steps prior to analysis, further elongating the time between protein production and detection.
In light of the need for label-free spatio-temporal optical detection of proteins in complex biological environments, we have developed a sensitive and selective label-free protein detection platform imaged with a custom-built near-infrared microscope9,10. We base this platform on the coupling of aptamer-anchor polymers to semiconducting single-wall carbon nanotube (SWNT) near-infrared (nIR) emitters. This platform leverages the selectivity for specific protein targets via synthetic DNA aptamers adhered to SWNT. DNA aptamers for proteins are selected from reported systematic evolution of ligands by exponential enrichment polynucleotide segments with known affinities for protein targets. Protein binding to aptamer targets on SWNT subsequently relays an optical signal in the nIR optical window. Photon scattering in biological samples is low in the nIR emission window, enabling, optical detection of single proteins in complex biological media such as crude cell lysates and bacterial cultures. Furthermore, SWNT are the only fluorophores to-date that have essentially infinite lifetimes, and are not susceptible to on-off blinking as quantum dots are. Therefore, the signal produced by SWNT, combined with the selectivity provided by aptamers, provides much promise for long-term optical monitoring of specific protein targets from within crude biological samples over long timescales.
Herein, we: 1) develop a platform to optically detect specific proteins, 2) detect proteins of interest from unpurified crude cell lysates. We show the immediate utility of our platform by 3) detecting single secreted proteins from E. coli, HEK 293, and Pichia pastoris cells engineered to synthesize and secrete our protein target upon induction, and show that a single cell’s protein secretion “footprint” depends on whether it is undergoing cell division. Lastly, we 4) monitor the real-time lytic release of target protein from cells infected with a T7 bacteriophage engineered to transfect our target protein gene into its bacterial host, for various multiplicity of infection ratios. We henceforth confirm that the relationship between T7 infection-to-lysis time and viral load follows a power law relationship.
Aptamer-anchor design for selective protein recognition
We can engineer an optical response to a protein target via aptamer-anchoring to SWNT surfaces. Aptamers are nucleotide polymers with a high sequence-specific affinity for a particular target molecule, often a protein. The polymer for this platform is based upon an “anchor” domain that adheres the polymer to the SWNT surface 11, 12, and a molecular recognition “capture” domain that enables selective perturbation of the SWNT fluorescence13, 14, 15 by only the conjugate protein target. Here, the anchor segments are alternating AT nucleotide repeats that have been shown to adsorb strongly to the SWNT surface 16 and the molecular recognition is provided by a folded polynucleotide aptamer. In this manner, unlabeled proteins can be detected with SWNT via DNA heteropolymers with (AT)11 DNA “anchor” sequences and aptamer “binding domains” (Figure 1a).
Figure 1. Characterization of aptamer-anchor structure on nanotube.

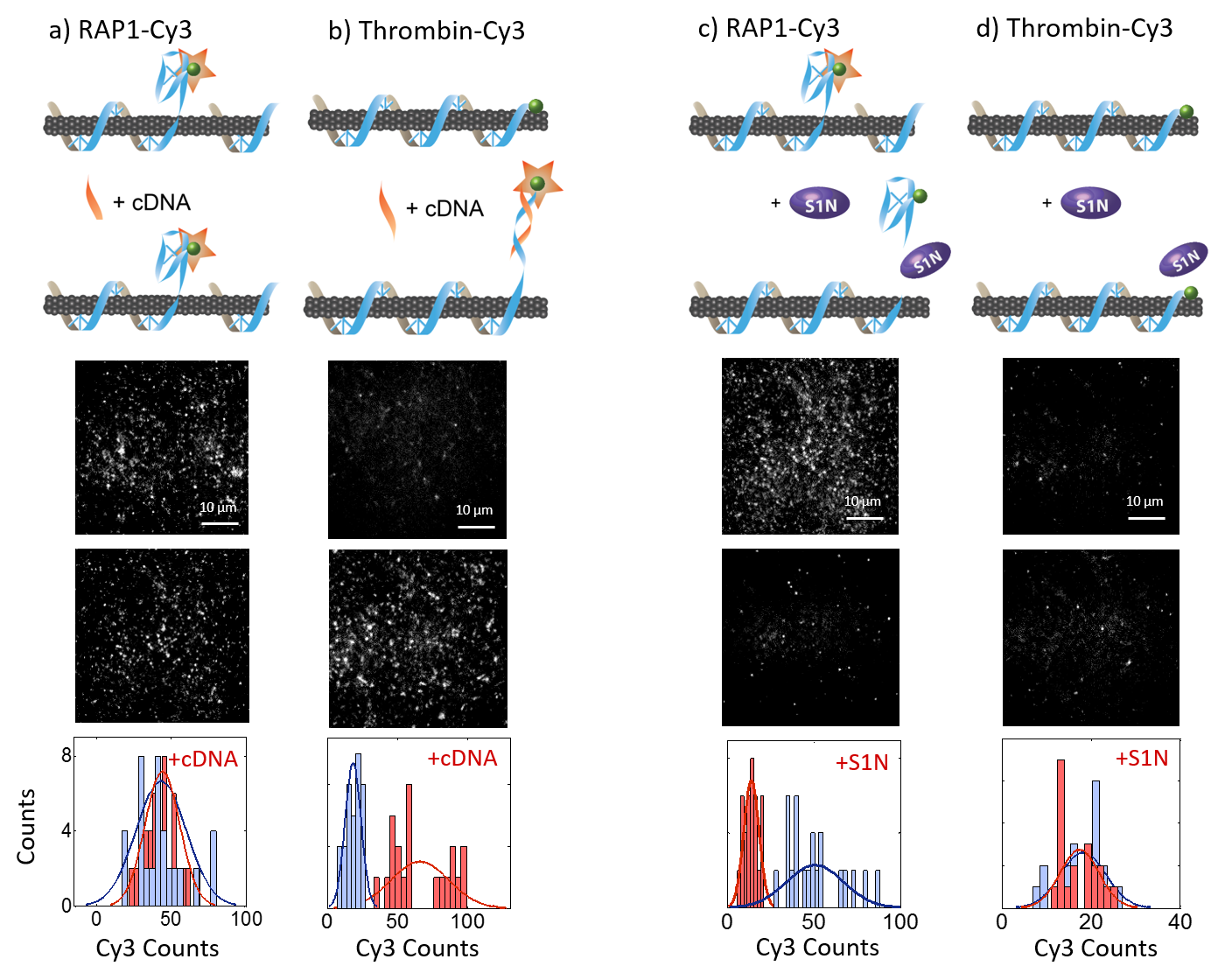
(a) 6,5 chirality RAP1 aptamer-SWNT response to addition of 3 μM RAP1 protein with schematic representation of aptamer-SWNT construct binding, with DNA anchor (blue), and DNA or RNA aptamer (purple). (b) Nine aptamer-SWNT screen (horizontal axis) against nine protein analytes (vertical axis). Red is sensor fluorescence turn-on, blue is sensor fluorescence turn-off, where off-diagonal elements represent SWNT fluorescence response to non-conjugate (nonspecific) protein-aptamer SWNT pairs, and the diagonal (highlighted by a dashed black line) represents fluorescence response to conjugate (specific) protein-aptamer SWNT pairs. We observe strong turn-on responses (red) for RAP1 protein and HIV1 integrase protein, with normalized fluorescence turn-on responses of (I-Io)/Io = 0.53 and 0.48, respectively. (c) RAP1 (top) aptamer-SWNT constructs with N = 1, 3, or 5 abasic spacers between anchor and aptamer detect RAP1 with a larger fluorescence turn-on response than constructs lacking a spacer. The response for thrombin (bottom), however, is unchanged regardless of spacer incorporation. Results suggest an aptamer equilibrium that fluctuates between a correctly folded aptamer (protein accessible) on the SWNT, and an incorrectly folded aptamer (protein inaccessible) on the SWNT surface Error bars are standard error. (d) Single-molecule TIRF visualization of aptamer-SWNT interaction for Cy3-labeled RAP1. Cy3 tag on Thrombin SWNT sensor is initially quenched (top panel, blue histogram) suggesting the thrombin aptamer is denatured on the SWNT. Addition of ssDNA complementary to the Thrombin aptamer, +cThrombin DNA, de-quenches the Cy3 tag and leads to an increase in visible Cy3 fluorophores (red histogram bars). Conversely, Cy3 tag on RAP1 SWNT sensor is initially de-quenched (bottom panel, blue histogram) suggesting the RAP1 aptamer is properly folded on the SWNT. Addition of ssDNA complementary to the RAP1 aptamer, +cRAP1 DNA, does not change the Cy3 count (red histogram bars). Results suggest a primarily SWNT surface-desorbed RAP1 aptamer, and primary SWNT surface-adsorbed Thrombin aptamer.
We tested this platform by constructing an aptamer-anchor SWNT sensor for RAP1 protein, a vital cytosolic protein for T-cell receptor signaling. Upon addition of 3 μM purified RAP1 protein to solution-phase sensor, RAP1 docks to the aptameric binding domain of our sensor, and we observe a 53% increase in the normalized SWNT intensity, ((I-Io)/Io) (Figure 1 a). The proximity of the protein to the SWNT surface produces a change in the local dielectric environment of the SWNT, which produces a nIR optical signal in the form of a SWNT fluorescence increase. We next probed the generalizability of our platform to a library of proteins and their aptameric targets conjugated to SWNTs. We constructed a library of 9 SWNT-aptamer sensors, and screened them against their conjugate proteins as well as the other non-target proteins in the library. Protein concentrations added varied from 110 nM to 6.6 μM, depending on the biologically relevant concentration of each protein (see Materials and Methods). Figure 1 b shows the fluorescence response heat map for each pair, as a function of the SWNT normalized intensity ((I-Io)/Io). Two aptamer-SWNT sensors in particular respond strongly and selectively to their protein targets: RAP1 and HIV1 integrase, which show a 53% and 48% fluorescence turn-on response in the presence of their protein targets, respectively. Off-diagonal nonspecific cross-responses were not observed. We subsequently performed a series of experiments to understand the response mechanism of our sensors.
To better understand which aspects of our sensors yield strong and selective fluorescence signals to their conjugate proteins, we constructed three chemical variants of each anchor-aptamer polymer by incorporating either one, three, or five consecutive 18-atom hexa-ethyleneglycol spacers between the (AT)11 anchor sequence, and the aptamer sequence. The spacers are abasic and do not adsorb onto the SWNT surface, creating a physical separation between the SWNT surface anchor and aptamer sequence, where the physical length of a single spacer is approximately 1.3 nm.
Our results show that the incorporation of 1 or 3 abasic spacers between the anchor and RAP1 aptamer domains drastically improve our sensor response. Alternatively, removal of the (AT)11 anchor sequence abolishes the sensor response and produces unstable DNA-SWNT suspensions (Supplemental Figure S1). The response of each of the 1 and 3 abasic spacer SWNT sensors increases nearly 4-fold compared to a sensor without a spacer. These results suggest that 1 or 3 spacers distance the aptamer to disfavor the aptamer adhesion to the SWNT surface, while keeping the aptamer within the dielectric environment of the SWNT surface that enables perturbation of the fluorescence emission. In contrast, when incorporating 1, 3, or 5 abasic spacers between the anchor and aptamer sequence of the thrombin polymer, no response is observed, similar to the construct without a spacer (Figure 1 c). This could be a result of a strongly adherent thrombin aptameric sequence that will not desorb from the SWNT surface. It is also possible that the binding of thrombin to the aptamer near the surface of the SWNT does not sufficiently perturb the SWNT dielectric environment to produce a change in the SWNT fluorescence.
To probe the structure of the aptamer in the case of the responsive RAP1 sensor compared to the non-responsive thrombin sensor, we designed a series of single-molecule experiments. We tagged each our (AT)11-RAP1 and (AT)15-thrombin sequences with a 3’ terminal Cy3 dye, which is known to quench as a function of proximity to the surface of SWNT 17,18. Therefore, we tested the predominant structure of the aptamer by surface-immobilizing our SWNT sensors on the surface of a microfluidic chamber (see Materials and Methods) and observing the number of fluorescent spots observed upon excitation of the Cy3 dye. For Cy3-tagged RAP1 sensors, addition of the complementary sequence resulted in no significant net increase in Cy3 emitters (from 43.2 ± 3.8 pre-(AC)6 to 44.0 ± 2.6 post-(AC)6 (mean ± SE)). In contrast, for Cy3-tagged thrombin sensors, addition of the complementary sequence resulted in a significant net increase in Cy3 emitters (from 18.3 ± 1.3 pre-(AC)6 to 66.0 ± 4.6 post-(AC)6 (mean ± SE); Figure 1 d). These results, along with both positive and negative controls (see Supplemental Information & Figure S2) suggest that the RAP1 aptamer is primarily found in its correctly folded G-quadruplex structure, whereas the thrombin aptamer is primarily stacked onto the SWNT surface.
Nanosensors enable detection of unlabeled proteins in vitro and from crude cell lysates
We quantified the sensitivity of our RAP1 aptamer sensor by surface-immobilizing our sensors and flowing in varying concentrations of recombinant human RAP1 protein (Abcam, ab162732). Upon flowing in RAP1, we observe an immediate increase in fluorescence intensity, ((I-Io)/Io) for all sensors in our field of view, where ((I-Io)/Io) ranged from 0.73 ± 0.06 to 1.82 ± 0.08 (mean ± SE) for 0.25 μg/mL and 25 μg/mL of RAP1 protein, respectively (Figure 2 a). We observed this turn-on signal simultaneously from all sensors in our field of view at all concentrations (Figure 2 b), except for sensors exposed to 0.25 μg/mL of RAP1 protein (Figure 2 c). At 0.25 μg/mL, the response observed was primarily that of individual aptamer-SWNT sensors turning on independently and often reversibly (Figure 2 d). Given the molecular mass of our RAP1 protein (~30 kDa), this corresponds to a protein concentration of 3.5 nM, well within a single-molecule concentration regime. We developed a model based on intensity response for the bulk-response regime, and the first passage time response for the single-sensor regime, to explain the response profile of our sensor as a function of RAP1 concentration (Supplemental Information for model) and fit it to our experimental concentration curves.
Figure 2. Calibration of nanosensor response to recombinant protein.

(a) Concentration-response curve for RAP1 protein shows two regimes of response. Above 0.15 μg/mL RAP1, a bulk sensor response starts being observed for the integrated sensor response for all sensors in the field of view (red), whereas below 0.15 μg/mL RAP1, individual sensor responses are observed (blue). (b) Example nIR intensity response upon addition of RAP1, and corresponding integrated intensity curves for RAP1 concentrations from 0 to 25 μg/mL. (c) First passage time until first sensor turn-on response at various RAP1 concentrations within the single-senor response regime. First passage times show a decaying exponential, as expected by protein diffusion-limited kinetics, with semilog(y) plot inset. (d) Example single-sensor nIR intensity response upon addition of RAP1 protein (red box). Sample traces are shown to demonstrate variance in first passage times for two sample RAP1 concentrations, 0.25 μg/mL (blue) and 0.06 μg/mL (green). All error bars are standard error. Sensor sensitivity can measure individual binding events in the single-sensor regime.
Our aptamer- protein screen in Figure 1 b shows promise of selectivity for use of aptamer-SWNT sensors to detect proteins from complex molecular environments. We next tested the ability of our RAP1 aptamer sensor to function amid other biomolecules. We first performed a screen for RAP1 aptamer-SWNT sensor response to two proteins that are known to bind to exposed surfaces of DNA-SWNT constructs, bovine serum albumin and neutravidin. Neither protein produced a significant change in the fluorescence of the RAP1 aptamer-SWNT (Figure S3). Next, we tested the RAP1 aptamer-SWNT sensor for its ability to detect RAP1 from crude E. coli cell lysates. The RAP1 gene was transfected into E. coli BL21 cells, and cells were IPTG induced to produce RAP1 protein (Figure 3 a). A control batch of cells lacking the RAP1 gene insertion was also prepared. Each sample was lysed in a sonicator, and the crude lysate was isolated via centrifugation (see Materials and Methods). We next immobilized our RAP1 aptamer-SWNT sensor on the surface of a microfluidic chamber and monitored the nIR fluorescence response of our surface-immobilized sensors upon addition of a 1:100 dilution of RAP1-transfected E. coli cell lysate, and the RAP1-free control cell lysate. Immediately upon addition of the crude RAP1 cell lysate supernatant dilution, we observe an increase in sensor intensity ((I-Io)/Io) = 1.7 within the 0.5 second frame acquisition rate of our microscope (Figure 3 b). The increase in fluorescence is notably absent in the crude cell lysate lacking RAP1 expressed protein ((I-Io)/Io) = - 0.1. Because our platform has single-protein sensitivity, if a generating source, instead of a pool, of individual proteins is to be placed in the proximity of the sensor array, a singular protein could be detected within the 200 μL sample volume of our chamber. Because 1 protein = 1.66e−24 moles, within our 200 μL sample volume, a source generating single proteins can be used to achieve effectively zeptomolar (1.66e−24 moles/200 μL) protein detection, although the analyte arrival time is a better metric of sensitivity in this stochastic limit. We explore this in subsequent measurements of proteins from individual live cells.
Figure 3. Detection of protein from crude cell lysate and from E. coli engineered to secrete target protein.

(a) Schematic of RAP1 transformation and E. coliRAP1 growth. (b) Surface-immobilized RAP1 aptamer-SWNT sensors (schematic) show an instantaneous turn-on response upon addition of crude cell lysate from cells grown to produce RAP1. The fluorescence of crude cell lysate from cells lacking the RAP1 gene remains largely invariant. (c) Concentration-response curves for 0 (cyan), 1e8 (orange), 4e8 (blue), 7e8 (red), 1e9 (green), and 1.6e9 (black) cfu/mL E. coli engineered to secrete RAP1, and corresponding sensor nIR responses prior to, and post-induction with 50 nM aTc. (d) Two representative single-sensor fluorescence intensity traces, plotted in red and blue, for an agarose gel-immobilized E. coli bacterium, circled yellow in brightfield image, engineered to secrete RAP1. Corresponding nIR intensity images of surface-immobilized sensors (boxed in red and blue) pre- and post-induction with aTc.
Real-time detection of protein secreted from Escherichia coli
We engineered E. coli to express and secrete RAP1 peptide upon induction with anhydrotetracycline (aTc), as described previously 19, 20. We tested the response of our protein sensor platform by introducing a titration of RAP1-secreting E. coli concentrations into our microfluidic chambers. For E. coli concentrations of 1e8 cfu/mL through 1.6e9 cfu/mL, we observe a bulk sensor response across the entire sensor surface field of view, as expected by the dense packing of E. coli cells observed in the corresponding brightfield view of our microfluidic chamber (Figure 3 c). For E. coli concentrations of approximately 1.5e7 cfu/mL, the brightfield view of our chamber shows sparse dispersions of E. coli cells, approximately 1 cell found per ~ 60 × 80 micron field of view. At this low cell concentration, we limited the mobility of individual E. coli cells within the microfluidic chamber to allow for an hour-long observation of the same cell, and induced this cell with aTc (see Materials and Methods). After a time-lag attributed to protein transcription and translation, we observed individual RAP1 aptamer-SWNT sensor responses upon RAP1 protein efflux from individual E. coli cells. From our sensor response calibration curve and the corresponding model (Figure 2), we can attribute the single-sensor responses to the binding of a RAP1 protein to an aptameric docking site near the SWNT. Figure 3 c shows a representative bulk turn-on response of our surface-immobilized sensors when E. coli concentrations of 1e8 cfu/mL through 1.6e9 cfu/mL are introduced into the microfluidic chamber. Conversely, Figure 3 d shows representative single-sensor turn-on responses as the result of protein secretion from a single immobilized E. coli cell. The E. coli cell in Figure 3 d behaves as a protein generation source within our microfluidic chamber. We note that the ability of the array to monitor singular protein efflux from the E. coli source within the 200 μL chamber effectively demonstrates a 8.3 zeptomolar (1.66e−24 moles/200 μL) protein detection, although in reality, the concentration no longer limits detection in this stochastic limit, but rather the analyte arrival time as in Fig 2c.
Net protein secretion measured from single E. coli, HEK 293, and Pichia pastoris cells
We studied N = 22 individual E. coli cells from induction through the course of an hour. We did so as described above and as depicted in Figure 4 a, by casting a low concentration of E. coli in 0.2% agarose minimal media above a dense array of our RAP1 protein nanosensors. We subsequently model our system, as depicted in Figure 4 a, for each of four discrete stages of RAP1 model protein detection from our E. coli cells. Our model begins with the introduction of inducer aTc into our single-cell containing matrix at t = 0, through observation of secreted proteins at our sensor array: I – introduction of aTc into the cell matrix, and diffusion into the cell, II – expression (transcription of gene and translation of product), III – secretion of protein, and IV – diffusion of protein to sensor array. With the exception of protein secretion (step III), a process that has not yet been studied in the literature at the single-cell and single-protein level, each of these mechanisms can be modeled with diffusion kinetics (steps I & IV), or derived kinetically from the literature (step II): Step I, the diffusion of aTc to the E. coli cell, is described by the Brownian diffusion of the aTc inducer through the agarose matrix to the cell, and is calculated to be near-instantaneous at 0.4 seconds as described by the sharp peak in Figure 4 c. Step II, the transcription, translation, and intra-cellular diffusion of RAP1 has been detailed in the literature. We combine the literature values of the transcription and translation rates for E. coli to model the timing of RAP1 production, also considering the depletion of mRNA and protein that is likely to occur in a carbon-source depleted environment such as the minimal media in which our experiments are conducted (Figure 4 c, dotted lines). Step III, the secretion of RAP1, is our experimentally-determined parameter. As we show in Figure 4 c secretion time histograms, the time lag between the predicted RAP1 protein synthesis and observed secretion represents the time required for protein secretion from our E. coli cell. Lastly, step IV represents the diffusion of RAP1 from its secretion point along the cell membrane to the nanosensors array. RAP1 diffusion from point source (the E. coli cell) to sensor (the nanosensor array) contributes minimal time to the secretion process (~12 seconds) and is expected to produce a spatial distribution along the nanosensor array as modeled by the Brownian diffusion of RAP1 protein from the cell to the surface (Figure 4 b). The kinetics described here, and the spatial distribution model are described in Supplemental Information, equations S1-S5).
Figure 4. Imaging the secretion of single proteins from individual microorganisms.

(a) Schematic of single immobilized E. coli above a RAP1-specific nanosensor array surface. Zoom-in of cell shows four stages of protein production from I) induction with aTc, II) transcription and translation, III) diffusion to secretion site and secretion, and IV) diffusion from secretion site to nanosensor array. (b) Model of expected RAP1 protein secretion “footprint” from E. coli cell above nanosensor array surface, with a red-blue heatmap corresponding to predicted areas of high-low relative protein detection from the E. coli source above. (c) Typical protein footprint 1-hour post-induction with aTc of RAP1 secretion for Histogram (counts) of first-response times for each cell for N =22 individual protein secretion events for dividing (green) or non-dividing (magenta) cells, showing quicker first-protein-secretion events for non-dividing cells. Kinetic model for protein secretion pathway for RAP1 secretion: aTc diffusion to the cell (red spike), protein transcription (mRNA concentration; dashed black), and protein translation (protein concentration; dashed blue). Our assay has the unique capability to discern the kinetic variation. Black dotted line models expected protein content in cell supernatant if measured experimentally using an SDS-PAGE gel to monitor for protein band intensity. Typical nIR protein footprint 1-hour post-induction with aTc of RAP1 secretion for (d) a dividing E. coli cell (brightfield inset) and, (e) a non-dividing cell (brightfield inset). Dividing cells show a noticeably smaller protein secretion footprint than non-dividing cells (154.0 ± 69.2 vs. 486.2 ± 113.0 protein-responding sensors, respectively). Our device can also be functionalized with sensors that specifically detect HIV-1 integrase proteins. We utilize a microfluidic device for HIV1 integrase detection to monitor HIV1 integrase protein secreted from (f) HEK cells, (g) bulk yeast cells, and (h) a single yeast cell, with brightfield insets for each. Sensor device can measure protein secretion from a variety of different cell types, and measure differences in protein secretion patterns and kinetics.
Our ability to observe protein secretion from an individual E. coli cell enables us to quantify the protein secreted by individual E. coli cells by counting the net number of responsive sensors underneath each cell: the E. coli ‘protein footprint’. We studied the protein footprint of N = 22 cells, where each footprint was formed by the intensity response of each sensor for 1 hour from induction with aTc in starved conditions with minimal media. None of the cells studied underwent cellular division, and most did not significantly elongate during the experiment. However, 6 of the 22 cells showed noticeable formation of an invaginated septal wall throughout the course of the experiment (Figure 4 d). Cells showing no signs of cell division were found to have larger protein secretion footprints in the nanosensors array than cells undergoing cell division, as observed by the protein footprint underneath each cell type (Figure 4 e). Non-dividing cells produced an average of 486.2 ± 113.0 protein-responsive sensors, as compared to the 154.0 ± 69.2 response from sensors that responded underneath dividing cells.
We next monitored the secretion of a second unlabeled protein product, HIV1 integrase, from both HEK 293 and P. pastoris cells. HEK cells were seeded into our microfluidic device in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. HEK cells were monitored for 75 minutes and the protein footprint underneath a collection of HEK cells was quantified. HEK cells constitutively secreted HIV1 integrase, which we monitored for the duration of the image acquisition. In addition, we monitored HIV1 integrase secretion from P. pastoris cells by transferring cells into induction medium BMMY to initiate HIV1 integrase protein production. P. pastoris cells were monitored for 75 minutes both at bulk-cell and single-cell levels, and the secreted HIV1 protein footprint was quantified throughout the course of the image acquisition. For bulk-cell levels (Figure 4 f), HIV1 integrase constitutively secreted from HEK cells 24 hours post-trypsinization was monitored. Trypsinized HEK cells were seeded into the device for a duration of 75 minutes of constitutive secretion. In these trials, HIV1 secreting HEK cells produced an average intensity turn-on response of 76.0 ± 27% from the HIV1 integrase-specific protein sensor array. Also for bulk-cell levels (Figure 4 g), HIV1 integrase secreted from P. pastoris cells was monitored from the time that P. pastoris cells were seeded into the device with induction medium BMMY to initiate HIV1 integrase protein production, for a duration of 75 minutes post-induction. In these trials, HIV1-secreting P. pastoris cells produced an average intensity turn-on response of 170.3 ± 32%, from the HIV1 integrase-specific protein sensor array. Single-cell levels of protein secretion were not detected from a single HEK cell seeded into a microfluidic HIV1 integrase sensor device within a 75 minute time window. However, single-HIV1 integrase proteins were successfully detected for single P. pastoris cells within a 75 minute time window (Figure 4 h). Further time-lapse analysis shows that HEK cells showed greatest protein efflux 24 hours post-trypsinization, whereas P. pastoris cells began to secrete protein on average 30 minutes post-induction.
Real-time detection of virally-induced cell lysis
We engineered a T7 bacteriophage to express RAP1 (T7RAP1). Due to the lytic cycle of the T7, we expect a sudden release of RAP1 protein from infected E. coli cells undergoing lysis. We used our sensor platform to study the release of RAP1 from the resulting lysis of E. coli infected with T7RAP1, for varying concentrations of engineered phage. The ratio between the virus particles and bacteria is defined as the multiplicity of infection (MOI), which dictates the infection rate for phage infections:
| Equation 1. |
We can statistically calculate the number of E. coli cells that will be infected by a phage for a given MOI with a Poisson distribution21 where P(n) is the probability that a single E. coli will be infected by n phages for a given MOI, abbreviated M:
| Equation 2. |
A MOI of 1 defines a closely synchronized infection, and theoretically, all the host cells get infected at the same time and multiply, where each infection produces about 200 new viruses 22. Therefore, we chose to perform a series of experiments where the MOI is varied from 10 to 1e−5 and studied the real-time release of RAP1 resulting from the E. coli lysis.
The fluorescence of surface-immobilized sensors was monitored for up to 1 hour, where experiments with lower viral loads showed a longer lysis-time. Figure 5 a shows a schematic of the T7 bacteriophage infection pathway, culminating with cell lysis and RAP1 release. Figure 5 b shows the brightfield images, and the nIR response of our RAP1 sensors prior to, and after E. coli infection by T7RAP1. Post-infection, we observe mostly lysed cells and a strong nIR fluorescence increase across the field of view, suggesting RAP1 release. Similarly, Figure 5 c shows the brightfield images, and the nIR response of our RAP1 sensors prior to, and after E. coli infection by phage lacking the RAP1 gene, T7WT. Post-infection, we observe mostly lysed cells in our brightfield channel, but lack a nIR sensor response. These results show the advantage of a protein sensor platform in its ability to detect the specific protein product of a phage infection, instead of only reporting cell lysis.
Figure 5. Real-time monitoring of bacteriophage T7RAP1 infection of E. coli cells, and resulting cell lysis.

(a) Schematic of T7RAP1 infection and lytic cycle of E. coli, culminating with cell lysis and release of RAP1 protein. (b) Brightfield (left) and nIR images (right) of 5e10 cfu/mL E. coli cells prior to (‘before infection’) and post (‘after infection’)- addition of 5e9 pfu/mL T7RAP1. (c) Corresponding brightfield and nIR images of 5e10 cfu/mL E. coli cells prior to (‘before infection’) and post (‘after infection’)- addition of 5e9 pfu/mL T7WT. While brightfield can detect lytic infection via E. coli lysis, nIR protein sensors detect the RAP1 protein product from phage genome infection. (d) Concentration-response curve for 5e10 cfu/mL E. coli and 5e6 pfu/mL T7RAP1 (orange), fit to a sigmoid (red), and the corresponding first time-derivative of the sigmoid (green dotted line). (e) Peak of first-derivatives of 5e10 cfu/mL E. coli and 5e4 (grey), 5e5 (magenta), 5e6 (orange), 5e7 (green), 5e8 (blue), and 5e9 (red) 5e10 (black) pfu/mL T7RAP1, error bars are standard error. Sensors can distinguish between infections that produce target RAP1 proteins, and infections that do not.
We performed an analysis of each E. coli – T7RAP1 ratio, and the normalized sensor surface intensity response to each condition as a function of time (Figure 5 d). We notice that the sensor response as a function of post-infection time is sigmoidal, with a clear intensity inflection shortly after addition of T7RAP1 for high phage concentrations (5e9 pfu/mL). Similarly, the sensor response is also sigmoidal after addition of less T7RAP1 (5e3 pfu/mL), however the inflection is slower and therefore less marked at lower phage concentrations. Therefore, to describe both the T7RAP1 infection peak and breadth, sensor intensity response curves were analyzed by fitting the curve to a sigmoid, where the point of inflection was determined by taking the first time-derivative of the sigmoidal fit.
| Equation 3. |
The resulting derivative function can be fit with a Gaussian, where the peak represents the greatest rate of RAP1 protein release from cells lysed due to viral infection. The peak of each Gaussian represents the time-point of most E. coli lysis and RAP1 release in our microfluidic chamber. We show a representative response trace with sigmoidal fit and derivative Gaussian in Figure 5 d. As expected, we show that the first-derivative of our sensor intensity response curves are sharply peaked Gaussians at short time-scales for our highest bacteriophage dilution, 5e9 pfu/mL (Figure 5 e). As the viral load decreases in order-of-magnitude increments, the first-derivative peaks broaden and peak at longer times, suggesting that the E. coli infection process becomes increasingly stochastic and lengthy. We find a primarily linear relationship between peak response time and log-scale phage concentration, suggesting an exponential relationship between viral load and E. coli cells for cell lysis.
CONCLUSIONS
The design of SWNT-based nanosensors for protein detection from crude and live cell samples requires a robust approach that will produce stable sensors under a variety of conditions optimized for cell viability. To expedite protein detection and push protein detection limits, efforts have turned primarily to electrochemical 23, 24, immunosorbent (ELISA) 25, mass spectrometric 26, plasmonic 27, 28, 29, 30, fluorescence labeling 7, 31, and raman 32,33 protein and analyte detection techniques. A common limitation faced by these techniques is an inability to simultaneously optimize spatial resolution, temporal resolution, or low (single)-protein detection limits, due to non-optical signal transduction. Our results provide an orthogonal approach to discover optical sensors for proteins.
The platform we present herein provides label-free protein detection in a wavelength range that enables selective protein recognition in crude samples, from living cells, and from bacteriophage infected cells. Our experimental design accomplishes two key design features for fluorescence-based protein recognition: 1) providing a molecular recognition element for reversible detection of a protein target, and 2) maintaining a stable linkage between the SWNT and the molecular recognition element to ensure reproducibility and reversibility of protein detection based on our previous work of polynucleotide corona phase stabilities 34, 35, 36. The selectivity of our protein + aptamer-SWNT screen for RAP1 and HIV1 integrase suggests that, in these two cases, the aptamer serves the purpose of selective protein docking and subsequent perturbation of the SWNT fluorescent corona. We confirm, through the introduction of abasic spacers between the anchor and aptamer sequence, and single-molecule studies, that aptamer denaturation on the SWNT surface is the primary contributor to non-responsive aptamer-SWNT constructs.
We confirm the immediate utility of our platform by addressing the challenge of protein detection from living E. coli cells engineered to secrete our target protein. We further extend our platform to detect HIV1 integrase from HEK 293 and P. pastoris. E. coli is a heavily-studied organism for understanding transcription and translation, and for genetic engineering. HEK 293 (Human Embryonic Kidney 293) and P. pastoris are widely used eukaryotic protein expression hosts in both academic labs and the pharmaceutical industry. Although intensive efforts have been devoted to improve protein expression through vector design, host cell engineering and upstream process development, there is limited knowledge and experience regarding single-cell analysis of protein secretion. Our approach offers a new way to study eukaryotic protein secretion at the single-cell level, and provides insights of protein secretion processes in industrial protein expression systems. Herein, we show successful engineering of the RAP1 gene into E. coli, and the HIV1 integrase gene into HEK 293 and P. pastoris. Subsequently, we monitor the productivity of RAP1 and HIV1 integrase protein export by individual engineered E. coli and P. pastoris cells in real-time. For E. coli, we show that the process of induction, protein synthesis, and protein export is a highly stochastic process yielding variability in protein secretion on a single-cell level, with cells undergoing cell division under starved conditions producing fewer secreted protein products than their non-dividing counterparts. The production of protein products in E. coli has been an active area of investigation in recent years, considering both intrinsic and extrinsic sources of protein production fluctuation 37. Previous studies have indirectly hypothesized that production of recombinant protein in E. coli may be ‘diluted’ during and immediately following cell division, based on mRNA counts 32. Others have suggested that cell division could affect transcription rates, and gene-regulatory polymerases and ribosomes 38. Our platform enables direct visualization of net protein production from a single E. coli over the course of an hour, confirming both the stochasticity of the process, and the effect of cell cycle on the production and export of a recombinant protein product. We further show that our platform can be extended to the detection of protein from E. coli infected with T7 bacteriophages carrying the RAP1 gene. Our platform shows promise of extension for the real-time and single-cell optical detection of a broad range of metabolic products.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported by a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), the Simons Foundation, a BBRF young investigator award, and a Beckman Foundation Young Investigator Award (M.P.L.). M.S.S acknowledges a grant from the National Science Foundation for the support of this work. H.A. is supported by fellowships from the Japan Society for the Promotion of Science, and the Naito Foundation. D.Y. acknowledges support from an NSF GRFP fellowship. And L.C. acknowledges support from a LAM research fellowship. A.Y.C. acknowledges graduate research support from the Hertz Foundation, the Department of Defense, and NIH Medical Scientist Training Program grant T32GM007753. This work was also supported by the National Institutes of Health (DP2 OD008435, P50 GM098792), the Office of Naval Research (N00014–13-1–0424), and the National Science Foundation (MCB-1350625). Authors thank Pablo Perez-Pinera (University of Illinois Urbana-Champaign) for providing the parental Pichia cells.
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT
No conflicts of interest
REFERENCES
- 1.Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab Eng 2012, 14(3): 189–195. [DOI] [PubMed] [Google Scholar]
- 2.Kingsmore SF. Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nat Rev Drug Discov 2006, 5(4): 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chikkaveeraiah BV, Bhirde AA, Morgan NY, Eden HS, Chen X. Electrochemical immunosensors for detection of cancer protein biomarkers. Acs Nano 2012, 6(8): 6546–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurita R, Arai K, Nakamoto K, Kato D, Niwa O. Development of electrogenerated chemiluminescence-based enzyme linked immunosorbent assay for sub-pM detection. Anal Chem 2010, 82(5): 1692–1697. [DOI] [PubMed] [Google Scholar]
- 5.Hammock ML, Knopfmacher O, Naab BD, Tok JBH, Bao ZA. Investigation of Protein Detection Parameters Using Nanofunctionalized Organic Field-Effect Transistors. Acs Nano 2013, 7(5): 3970–3980. [DOI] [PubMed] [Google Scholar]
- 6.Jain A, Liu R, Xiang YK, Ha T. Single-molecule pull-down for studying protein interactions. Nat Protoc 2012, 7(3): 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain A, Liu RJ, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, et al. Probing cellular protein complexes using single-molecule pull-down. Nature 2011, 473(7348): 484–U322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mogalisetti P, Walt DR. Stoichiometry of the alpha-Complementation Reaction of Escherichia coil beta-Galactosidase As Revealed through Single-Molecule Studies. Biochemistry-Us 2015, 54(8): 1583–1588. [DOI] [PubMed] [Google Scholar]
- 9.Beyene AG, Demirer GD, Landry MP. Nanoparticle-Templated Molecular Recognition Platforms for Detection of Biological Analytes. Current Protocols in Chemical Biology 2016, 8: 197–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salem DP, Landry MP, Bisker G, Ahn J, Kruss S, Strano MSS. Chirality dependent corona phase molecular recognition of DNA-wrapped carbon nanotubes. Carbon 2016, 97: 147–153. [Google Scholar]
- 11.Roxbury D, Mittal J, Jagota A. Molecular-Basis of Single-Walled Carbon Nanotube Recognition by Single-Stranded DNA. Nano Lett 2012, 12(3): 1464–1469. [DOI] [PubMed] [Google Scholar]
- 12.Tu XM, Manohar S, Jagota A, Zheng M. DNA sequence motifs for structure-specific recognition and separation of carbon nanotubes. Nature 2009, 460(7252): 250–253. [DOI] [PubMed] [Google Scholar]
- 13.Bisker G, Dong J, Park HD, Iverson NM, Ahn J, Nelson JT, et al. Protein-targeted corona phase molecular recognition. Nat Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson JT, Kim S, Reuel NF, Salem DP, Bisker G, Landry MP, et al. Mechanism of Immobilized Protein A Binding to Immunoglobulin G on Nanosensor Array Surfaces. Anal Chem 2015, 87(16): 8186–8193. [DOI] [PubMed] [Google Scholar]
- 15.Oliveira SF, Bisker G, Bakh NA, Gibbs SL, Landry MP, Strano MS. Protein functionalized carbon nanomaterials for biomedical applications. Carbon 2015, 95: 767–779. [Google Scholar]
- 16.Zhang JQ, Landry MP, Barone PW, Kim JH, Lin SC, Ulissi ZW, et al. Molecular recognition using corona phase complexes made of synthetic polymers adsorbed on carbon nanotubes. Nat Nanotechnol 2013, 8(12): 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang R, Jin J, Chen Y, Shao N, Kang H, Xiao Z, et al. Carbon nanotube-quenched fluorescent oligonucleotides: probes that fluoresce upon hybridization. J Am Chem Soc 2008, 130(26): 8351–8358. [DOI] [PubMed] [Google Scholar]
- 18.Kruss S, Landry MP, Vander Ende E, Lima BMA, Reuel NF, Zhang JQ, et al. Neurotransmitter Detection Using Corona Phase Molecular Recognition on Fluorescent Single-Walled Carbon Nanotube Sensors. J Am Chem Soc 2014, 136(2): 713–724. [DOI] [PubMed] [Google Scholar]
- 19.Callura JM, Cantor CR, Collins JJ. Genetic switchboard for synthetic biology applications. Proc Natl Acad Sci U S A 2012, 109(15): 5850–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen AY, Deng Z, Billings AN, Seker UO, Lu MY, Citorik RJ, et al. Synthesis and patterning of tunable multiscale materials with engineered cells. Nat Mater 2014, 13(5): 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellis EL, Delbruck M. The growth of bacteriophage. J Gen Physiol 1939, 22(3): 365–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen HM, Kang C. Lysis delay and burst shrinkage of coliphage T7 by deletion of terminator Tphi reversed by deletion of early genes. J Virol 2014, 88(4): 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taleat Z, Cristea C, Marrazza G, Mazloum-Ardakani M, Sandulescu R. Electrochemical immunoassay based on aptamer-protein interaction and functionalized polymer for cancer biomarker detection. J Electroanal Chem 2014, 717: 119–124. [Google Scholar]
- 24.Jacobs CB, Peairs MJ, Venton BJ. Review: Carbon nanotube based electrochemical sensors for biomolecules. Analytica Chimica Acta 2010, 662(2): 105–127. [DOI] [PubMed] [Google Scholar]
- 25.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol 2010, 28(6): 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc Natl Acad Sci U S A 2012, 109(38): 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zijlstra P, Paulo PMR, Orrit M. Optical detection of single non-absorbing molecules using the surface plasmon resonance of a gold nanorod. Nat Nanotechnol 2012, 7(6): 379–382. [DOI] [PubMed] [Google Scholar]
- 28.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Biosensing with plasmonic nanosensors. Nat Mater 2008, 7(6): 442–453. [DOI] [PubMed] [Google Scholar]
- 29.Ament I, Prasad J, Henkel A, Schmachtel S, Sonnichsen C. Single unlabeled protein detection on individual plasmonic nanoparticles. Nano Lett 2012, 12(2): 1092–1095. [DOI] [PubMed] [Google Scholar]
- 30.Qian XM, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, et al. In vivo tumor targeting and spectroscopic detection with surface-enhanced Raman nanoparticle tags. Nat Biotechnol 2008, 26(1): 83–90. [DOI] [PubMed] [Google Scholar]
- 31.Sakabe M, Asanuma D, Kamiya M, Iwatate RJ, Hanaoka K, Terai T, et al. Rational Design of Highly Sensitive Fluorescence Probes for Protease and Glycosidase Based on Precisely Controlled Spirocyclization. J Am Chem Soc 2013, 135(1): 409–414. [DOI] [PubMed] [Google Scholar]
- 32.Paulus GLC, Nelson JT, Lee KY, Wang QH, Reuel NF, Grassbaugh BR, et al. A graphene-based physiometer array for the analysis of single biological cells. Scientific Reports 2014, 4 (6865): 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Gu C, Qian F, Li Y, Zhang J. Highly Sensitive Detection of Proteins and Bacteria in Aqueous Solution Using Surface-Enhanced Raman Scattering and Optical Fibers. Anal Chem 2011, 83(15): 5888–5894. [DOI] [PubMed] [Google Scholar]
- 34.Landry MP, Vukovic L, Kruss S, Bisker G, Landry AM, Islam S, et al. Comparative Dynamics and Sequence Dependence of DNA and RNA Binding to Single Walled Carbon Nanotubes. J Phys Chem C Nanomater Interfaces 2015, 119(18): 10048–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giraldo JP, Landry MP, Kwak SY, Jain RM, Wong MH, Iverson NM, et al. A Ratiometric Sensor Using Single Chirality Near-Infrared Fluorescent Carbon Nanotubes: Application to In Vivo Monitoring. Small 2015. [DOI] [PubMed] [Google Scholar]
- 36.Roxbury D, Mittal J, Jagota A. Molecular-basis of single-walled carbon nanotube recognition by single-stranded DNA. Nano Lett 2012, 12(3): 1464–1469. [DOI] [PubMed] [Google Scholar]
- 37.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science 2002, 297(5584): 1183–1186. [DOI] [PubMed] [Google Scholar]
- 38.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet 2004, 36(2): 147–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.