

Abstract
The symptomologies of Alzheimer’s disease (AD) develop over decades suggesting modifiable life-style factors may contribute to disease pathogenesis. In humans, hyperinsulinemia associated with type 2 diabetes mellitus increases the risk for developing AD and both diseases share similar age-related etiologies including amyloidogenesis. Since we have demonstrated that soluble Aβ42 elicits glutamate release, we wanted to understand how diet-induced insulin resistance alters hippocampal glutamate dynamics, which are important for memory formation and consolidation. Eight to twelve week-old C57BL/6J and AβPP/PS1 mice were placed on either a low-fat diet (LFD) or high-fat diet (HFD) for eight months. A HFD led to significant weight increases as well as impaired insulin sensitivity, glucose tolerance, and learning in both C57BL/6J and AβPP/PS1 mice. AβPP/PS1 LFD mice had elevated hippocampal basal as well as stimulus-evoked glutamate release that was further increased with consumption of a HFD. Immunohistochemistry indicated an increase in vesicular glutamate transporter 1 and glial fibrillary acidic protein density in hippocampal subregions corresponding with this elevated extracellular glutamate. While no differences in hippocampal plaque load were observed, the elevated astrogliotic response surrounding the plaques in AβPP/PS1 HFD mice may have been a compensatory mechanism to control plaque accumulation. These data support that AβPP/PS1 mice have chronically elevated extracellular glutamate that is exacerbated by a HFD and that modifiable life-style factors such as obesity-induced insulin resistance can contribute to AD pathogenesis.
Keywords: Diabetes, Amyloid-beta, Cognition, Alzheimer’s disease, Astrogliosis, Excitotoxicity
Graphical Abstract
The aim of the present study was to address how obesity-induced insulin resistance alters hippocampal glutamate dynamics in both cognitively normal and AβPP/PS1 mice predisposed to Alzheimer’s disease (AD) pathology. AβPP/PS1 mice presented with insulin resistance, neuroinflammation, and elevated hippocampal glutamate dynamics compared to C57BL/6J mice. A high-fat diet exacerbated all of these phenotypes in C57BL/6J and AβPP/PS1 mice. The current study highlights elevated hippocampal glutamatergic neurotransmission associated with a diabetic phenotype. Additionally, our laboratory has observed a consistent theme of elevated hippocampal glutamate across ages of AβPP/PS1 mice, which may serve as an early therapeutic biomarker for AD pathogenesis.
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder characterized by a slow, but progressive, accumulation of extracellular aggregated beta-amyloid (Aβ) and intracellular hyperphosphorylated tau tangles (Jack et al. 2013). This accumulation leads to alterations in neurotransmitter dynamics, synapse loss, and cerebral atrophy that culminates in the eventual cognitive and functional decline associated with the disorder (Mota et al. 2014). To date, current therapeutics target cholinesterase inhibitors, to increase acetylcholine levels, or antagonism of the N-methyl-D-aspartate (NMDA) receptor, to prevent glutamate mediated excitotoxicity (Cummings et al. 2014; Godyń et al. 2016). However, these therapies have limited efficacy, only treat symptoms, and do not decelerate disease progression, possibly because they are administered at advanced AD stages. Without a well-established biomarker for AD, early diagnosis is difficult and underscores the lack of disease-modifying pharmacotherapy options. To further complicate diagnosis, evidence supports that AD symptomology develops over decades and modifiable lifestyle factors, such as obesity-induced type 2 diabetes mellitus (T2DM), may contribute to AD progression (Barnes and Yaffe 2011).
The peptide hormone, insulin, regulates glucose uptake and storage in the periphery and brain for use in energy production (De Felice 2013). However, excessive caloric consumption, particularly of hydrogenated or saturated fats, promotes a cascade of metabolic events starting with elevated circulating insulin concentrations that leads to insulin resistance and increases the risk factor for developing T2DM (Holland et al. 2007). In fact, T2DM and AD share several age-related etiologies including hyperinsulinemia, insulin resistance, hyperglycemia, amyloidogenesis and memory impairment (Zhao and Townsend 2009; Talbot et al. 2012; Moloney et al. 2010). These similar symptomologies suggest insulin resistance and the subsequent onset of T2DM is a risk for developing AD (Vandal et al. 2014). In support of this, the Mayo Clinic Alzheimer Disease Patient Registry has reported that 80% of their AD patients had either T2DM or impaired glucose tolerance (Janson et al. 2004) and T2DM in midlife increases the odds for developing mild cognitive impairment or AD later in life by 1.5- to 2-fold (Allen et al. 2004; Ott et al. 1999; Arvanitakis et al. 2004). While the mechanistic link between T2DM and AD is not fully elucidated, the metabolic hypothesis of AD supports altered insulin signaling promotes a cascade of neurological events that initiate the pathogenesis of AD (Hoyer 2002). For example, brain insulin degrading enzyme (IDE) regulates the metabolism of both insulin and Aβ, but at a lower affinity for the latter. As such, hyperinsulinemia prevents IDE from degradation of monomeric Aβ leading to its accumulation and aggregation (Farris et al. 2003). These small molecular weight isoforms of Aβ (monomers, dimers, and trimers) are hypothesized to be the bioactive component that causes synaptic dysfunction, neurotoxicity, and the eventual neurodegeneration associated with AD (Jin and Selkoe 2015; Yang et al. 2017).
Prior studies have demonstrated that soluble Aβ42 elicits glutamate release through the α7 nicotinic acetylcholine receptor (α7nAChR; Talantova et al., 2013; Hascup and Hascup, 2016). Because of glutamate’s role in learning and memory, it is hypothesized that persistent, excessive synaptic glutamate overstimulates the NMDA receptor thereby preventing detection of physiological signals leading to cognitive impairment (Parsons et al. 2007). In fact, our laboratory has demonstrated that double transgenic mice expressing a mutant amyloid precursor protein (Mo/HuAPP695swe) and Presenilin 1 (PS1-dE9) genes (AβPP/PS1) have elevated hippocampal glutamate as early as 2–4 months of age; prior to the onset of cognitive decline (Hascup and Hascup 2015). While previous studies have demonstrated that HFD exacerbates cognitive decline and disease neuropathology in animal models of AD (Vandal et al. 2014; Knight et al. 2014; Thériault et al. 2016; Julien et al. 2010), alterations to memory associated neurotransmitters have not been elucidated. The aim of the present study was to address how obesity-induced insulin resistance alters glutamate dynamics in both cognitively normal and AβPP/PS1 mice predisposed to AD pathology. Since previous studies have shown that HFD affects memory in cognitively normal rodents (Kanoski and Davidson 2011; Cordner and Tamashiro 2015), non-AD control mice help to understand changes associated with, or independent from, the metabolic hypothesis of AD pathogenesis.
Materials & Methods
Animals:
Protocols for animal use were approved by the Laboratory Animal Care and Use Committee at Southern Illinois University School of Medicine (Protocol #219–14-003) and the study was not preregistered. Eight to twelve week old male, C57BL/6J (RRID:IMSR_JAX:000664) and AβPP/PS1 (RRID:MMRRC_034832-JAX; Mo/HuAPP695swe / PS1-dE9), mice were obtained from Jackson Laboratory (Bar Harbor, ME), and group housed on a 12:12 hour light: dark cycle with food and water available ad libitum. All experiments were conducted during the light phase. Genotype was confirmed by TransnetYX®, Inc (Cordova, TN). All mice were ear tagged with unique numerical identifiers so as to blind researchers throughout the experimental paradigms. Pseudo randomization using the Microsoft Excel 2013 randomization function to generate random decimal numbers between 0 and 1 for each mouse and dietary treatment. These random numbers were then sorted into ascending order generating a list that categorized mice into the following groups C57BL/6J low fat diet (LFD), C57BL/6J HFD, AβPP/PS1 LFD, and AβPP/PS1 HFD. Similar methodology was used to determine the order of which animals were assessed. A n=15 for all treatment groups was allocated at study initiation; however, due to animal loss from normal aging and disease progression, the following indicates the remaining number of animals available at the end of the 8 month dietary treatment: C57BL/6J LFD (n=14), C57BL/6J HFD (n=11), AβPP/PS1 LFD (n=11), AβPP/PS1 HFD (n=12). All of these remaining mice underwent blood glucose monitoring, cognitive assessment, in vivo glutamate recordings, and immunohistochemical (IHC) analysis except for one AβPP/PS1 HFD mouse that died during in vivo glutamate recordings as outlined in Figure 1A. Following in vivo electrochemistry, all mice were euthanized by an overdose of isoflurane followed by rapid decapitation with sharp scissors.
Figure 1: Experimental Design, Mouse Weight, and Blood Glucose Measurements.
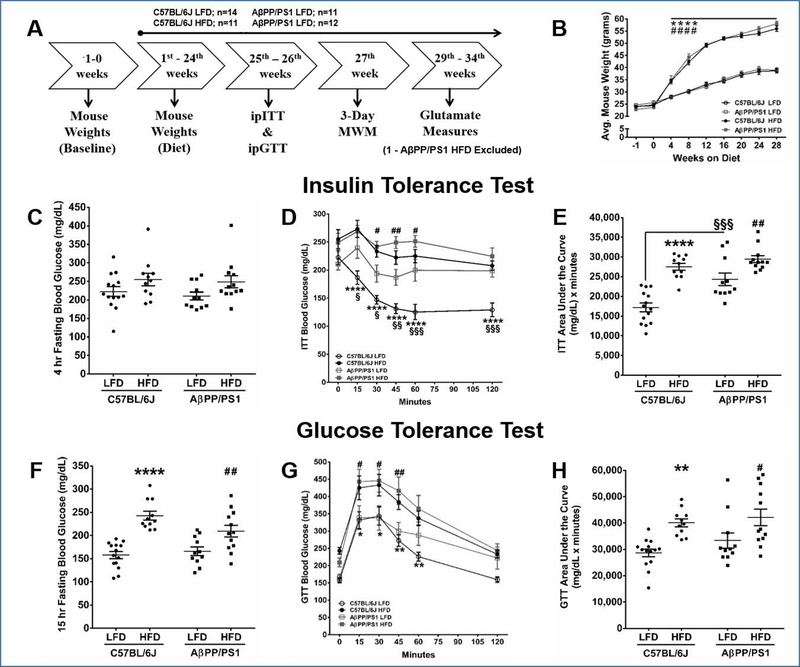
A) An outline of the experimental design. Abbreviations: intraperitoneal insulin tolerance test (ipITT), intraperitoneal glucose tolerance test (ipGTT), Morris water maze (MWM). B) Analysis of mouse weight gain through 28 weeks on either LFD or HFD. Results from the four hour fasting ipITT and 15 hour fasting ipGTT. Mouse genotypes and diet group are indicated on each graph. C) Four hour fasting blood glucose prior to ip injection of 1 IU/kg b.w. of insulin, D) blood glucose during the 120 minute ipITT, E) area under the curve of the 120 minute ipITT, F) Fifteen hour fasting blood glucose prior to ip injection of 2 g/kg b.w. of glucose, G) blood glucose during the 120 minute ipGTT, H) area under the curve of the 120 minute ipGTT. *p<0.05, **p<0.01, ****p<0.0001 C57BL/6J LFD (n=14) vs C57BL/6J HFD (n=11); #p<0.05, ##p<0.01, ####p<0.0001 AβPP/PS1 LFD (n=11) vs AβPP/PS1 HFD (n=12); §p<0.05, §§p<0.01, §§§p<0.001 C57BL/6J LFD vs AβPP/PS1 LFD; where n refers to the number of animals.
Chemicals:
All chemicals were prepared and stored according to manufacturer recommendations unless otherwise noted. L-glutamate oxidase (EC 1.4.3.11) was obtained from Cosmo Bio USA Co. (Carlsbad, CA; Cat: YMS-80049) and reconstituted in distilled, deionized water to make a 1U/µl stock solution and stored at 4°C. Sodium phosphate monobasic monohydrate (Cat: BP330–500), sodium phosphate dibasic anhydrous (Cat: S375–500), 1,3-phenylenediamine dihydrochloride (mPD; Cat: P017225G), sodium chloride (Cat: S271–3), calcium chloride dehydrate (Cat: BP510–100), dextrose monohydrate (Cat: D15–500), and hydrogen peroxide (H2O2; Cat: H325–100) were obtained from Thermo Fisher Scientific (Waltham, MA). L-glutamic acid sodium salt (Cat: G1626), potassium chloride (Cat: P9333), bovine serum albumin (BSA; Cat: A3059), glutaraldehyde (Cat: G5882), dopamine hydrochloride (DA; Cat: H8502), L-ascorbic acid (AA; Cat: A7056), and dibutyl phthalate and xylene (DPX; Cat: 06522) were obtained from Sigma-Aldrich Co. (St. Louis, MO). Rabbit polyclonal glial fibrillary acidic protein (GFAP) antibody was obtained from Dako (Carpinteria, CA; RRID:AB_10013382). Guinea pig polyclonal vesicular glutamate transporter 1 (VGLUT1) antibody was obtained from Millipore (Burlington, MA; RRID:AB_2301751). Biotinylated goat anti-rabbit serum (RRID:AB_2313606), biotinylated goat anti-guinea pig serum (RRID:AB_2336132), avidin-biotin complex (ABC) kit (RRID:AB_236818), and VIP peroxidase substrate kit (RRID:AB_2336819) were obtained from Vector Laboratories (Burlingame, CA). Amylo-Glo® RTD™ with ethidium bromide (EtBr) was obtained from Biosensis (Temecula, CA; Cat: TR-400-AG).
Low-fat and high-fat diet:
All mice were switched from standard rodent chow (13% kcal fat, 57% kcal carbohydrate, 30% kcal protein, 4% sucrose, 4.09 kcal/gm; LabDiet, St. Louis, MO; Cat: 5001) to either a LFD (10% kcal fat, 70% kcal carbohydrate, 20% kcal protein, 7% sucrose, 3.85 kcal/gm; Cat: D12450J) or HFD (60% kcal fat, 20% kcal carbohydrate, 20% kcal protein, 7% sucrose, 5.24 kcal/gm; Cat: D12492) obtained from Research Diets Inc. (New Brunswick, NJ). A LFD diet was used as the control diet so as to match protein and sucrose content with the HFD. Mouse weight was monitored throughout the study (Fig 1B).
Intraperitoneal (ip) Insulin tolerance test (ITT) and Glucose Tolerance Test (GTT):
To determine insulin sensitivity, an initial blood glucose measurement (time = 0) was taken from the tail vein of four hour fasted mice and measured using a Presto® glucometer (AgaMatrix, Salem, NH) followed by ip injection of 1 IU / kg body weight (b.w.) Humulin® R (Henry Schein, Melville, NY: Cat: 1238578). To determine glucose tolerance, an initial blood glucose measurement was taken (time = 0) from fifteen hour fasted mice followed by an ip injection of 2 g of glucose / kg b.w. Following either injection, blood glucose levels were measured sequentially at 15, 30, 45, 60, and 120 min (Fang et al. 2017).
Morris water maze:
The MWM paradigm consisted of 2 consecutive training days where the mouse learned to remain on the platform for 60 seconds before rescue. For the first training day, a visible platform protruded 1 cm out of the opaque pool of water to aid in platform location. Mice underwent three consecutive 60 second maximum trials with a 15 minute inter-trial interval. On the second training day, the visible platform was removed and mice underwent 3 training blocks (30 minute inter-block interval) of 3, sixty second maximum trials (15 minute inter-trial interval) to learn the location of the submerged platform (1 cm below the surface). Starting quadrant was varied for each trial. The probe challenge consisted of a single 60 s trial. The ANY-maze video tracking system (Stoelting Co., Wood Dale, IL; RRID:SCR_014289) records and analyzes maze navigation. The three training sessions for Day 1 and for each training block in Day 2 were averaged.
Enzyme-based microelectrode arrays:
Enzyme-based MEAs with platinum (Pt) recording surfaces were fabricated, assembled, coated, and calibrated for in vivo mouse glutamate measurements as previously described (Burmeister et al., 2000; Hascup et al., 2006; Hascup et al., 2013). One of the MEA (Quanteon LLC; Cat: R2) Pt sites was coated with an L-glutamate oxidase, BSA, glutaraldehyde solution. BSA and glutaraldehyde increase the adhesion and crosslink L-glutamate oxidase to the MEA surface, while L-glutamate oxidase enzymatically degrades glutamate to α-ketoglutarate and H2O2, the electroactive reporter molecule. The second Pt recording site (self-referencing or sentinel site) was coated with a BSA and glutaraldehyde solution that is unable to enzymatically generate H2O2 from L-glutamate. A potential of +0.7V vs a Ag/AgCl reference electrode was applied to the Pt recording surfaces, resulting in a two electron oxidation of H2O2 and the subsequent current was amplified and digitized by the Fast Analytical Sensing Technology (FAST) 16mkIII (Quanteon, LLC; Nicholasville, KY) electrochemistry instrument.
mPD Electropolymerization:
Pt recording surfaces were electroplated with 5 mM mPD in 0.05 M phosphate buffered saline (PBS) for 20 min to restrict the passage of AA, DA, uric acid, and 3,4-dihydroxyphenylacetic acid (Hascup et al. 2016).
Calibration:
MEAs were calibrated in 0.05 M PBS (37°C) to create a standard curve for the conversion of current to glutamate concentration. Final beaker concentrations of 250 µM AA, 20, 40, and 60 µM L-glutamate, 2 µM DA, and 8.8 µM H2O2 were used to assess MEA performance. A total of 49 MEAs were used in the present study. The average ± standard error of the mean (SEM) for glutamate sensitivity was 5.7 ± 0.3 pA/μM (R2 = 0.998 ± 0.001), selectivity ratio of 367 ± 48 to 1, and limit of detection of 0.20 ± 0.03 μM based on a signal-to-noise ratio of 3.
In Vivo Anesthetized Recordings:
A glass micropipette (World Precision Instruments, Inc.; Cat: 1B100–6) was used for local application studies. The tip of the micropipette (12–15 µm internal diameter) was positioned between the pair of recording sites and mounted ~100 µm above the MEA surface. Mice were anesthetized using 1.5% isoflurane (Henry Schein; Cat: 029405) in a calibrated vaporizer (Parkland Scientific; V3000) and placed in a stereotaxic frame with a mouse anesthesia mask (David Kopf Instruments; Cat: 900 / 907) with a mouse anesthesia mask. Body temperature was maintained at 37°C. The MEA / micropipette assembly was lowered into the dentate gyrus (DG; AP: −2.0, ML: ± 1.0, DV: −2.2 mm), CA3 (AP: −2.0, ML: ± 2.0, DV: −2.2 mm) and CA1 (AP: −2.0, ML: ± 1.0, DV: −1.7 mm) from Bregma (Paxinos and Franklin 2004). A Ag/AgCl reference wire was positioned beneath the skull and rostral to the right hemisphere craniotomy. Constant voltage amperometry (4Hz) was performed using the FAST16mkIII. Calibration data, in conjunction with a MATLAB (MathWorks, Natick, MA; RRID:SCR_014289) graphic user interface program (Version 6.1) was used to calculate extracellular glutamate. The sentinel site current (pA) was subtracted from the glutamate recording site current (pA) and divided by the slope (pA/µM) obtained during the calibration (Burmeister and Gerhardt 2001; Burmeister et al. 2002; Hascup et al. 2010; Hascup et al. 2011).
Immunohistochemistry & Semi-Quantification:
Following in vivo electrochemistry, the brains were removed and post-fixed in 4% paraformaldehyde for 48 hours and then transferred into 30% sucrose in 0.1M PB for 24 hours prior to sectioning. Forty-five micron sections of the hippocampus were obtained using a Microm cryostat (Zeiss; Cat: HM 500). Serial sections (every 6th) of the hippocampus were processed for free-floating immunohistochemistry (IHC) using rabbit polyclonal GFAP (1:2,000) or guinea pig polyclonal VGLUT1 antibody (1:1,000) (Farrand et al. 2017; Hascup et al. 2016). Endogenous peroxidase activity was quenched by treating sections with 10% H2O2 in 20% methanol for 10 minutes. Sections for primary antibodies were permeabilized in Tris-buffered saline with 0.25% TritonX-100 following treatment for 20 minutes with sodium metaperiodate. Nonspecific binding was controlled by 1 hour incubation in 10% normal goat serum. Sections were incubated overnight in the primary antibody at room temperature. The next day, sections were incubated for 1 hour with the secondary antibody (1:200; biotinylated goat anti-rabbit serum or biotinylated goat anti-guinea pig serum) and 1 hour with the Vectastain ABC kit (Vector). The reaction was developed using the VIP peroxidase substrate kit (Vector) to enhance the reaction and produce a color stain. This reaction was stopped using 0.1 M PB, and the sections were mounted on glass slides, dehydrated, and cover-slipped with DPX. To control for staining intensity, staining of all sections for each antibody was conducted on the same day and developed with VIP for the same amount of time (GFAP: 3 minutes, VGLUT1: 2 minutes). For plaque staining, slides containing serial sections (every 6th) of the hippocampus were incubated for 10 min in freshly prepared Amylo-Glo® RTD™ solution followed by a 5 min rinse in 0.9% saline without shaking, then 1 minute incubation with EtBr based on product protocol (1:100). Staining intensities of GFAP, VGLUT1, and plaque formation in the hippocampus were determined using National Institutes of Health Image J Software 1.48 (RRID:SCR_003070) to measure a gray scale value within the range of 0–256, where 0 represents white and 256 represents black (Farrand et al. 2017). A template for the DG, CA3, and CA1 hippocampal subregions for VGLUT1 and GFAP while a template for the whole hippocampus was created for plaque formation. Templates were used on all brains similarly, and images were captured with a Nikon Eclipse E-600 microscope equipped with an Olympus-750 video camera system, and a Dell Pentium III computer. Measurements were performed blinded, and approximately six sections were averaged to obtain one value per subject. If six sections per stain were not obtained, the subject was excluded from data analysis. Staining density was obtained when background staining was subtracted from mean staining intensities on every sixth section through the hippocampus.
Aβ42 ELISA:
A separate cohort of mice were used for insoluble Aβ42 determination. Mice were euthanized as described above and the hippocampus was dissected and stored at −80°C until tissue processing. Protein concentrations were determined using the BCA method and assessment of the insoluble fractions of Aβ42 were performed using the Human / Rat β amyloid (42) ELISA kit (WAKO Chemicals; Cat: 292–64501).
Data Analysis:
Sample size was determined based on previous MWM, electrochemical, and IHC data using C57BL/6J and AβPP/PS1 mice. A power calculation indicated a minimum of 10 mice per group for MWM and electrochemical recordings and 5 mice per group for IHC analysis (Boger et al. 2007, Hascup & Hascup, 2015) to detect differences with 95% confidence (α=0.05) and 0.8 power. Prism (GraphPad Software, Inc., La Jolla, CA; RRID:SCR_002798) software was used for all statistical analyses including D’Agostino-Pearson omnibus normality tests. A one-way Analysis of Variance (ANOVA) was used for MWM and for electrochemical stimulus volume comparisons, while a two-way ANOVA (diet vs genotype) was used for all other analyses. When the ANOVA indicated a statistically significant main effect, a Holm-Sidak’s multiple comparisons post-hoc test was used. Outliers were determined with a single Grubb’s test (α=0.05). Data are represented as mean ± SEM and significance was defined as p<0.05. The unit of analysis “n” for each data set refers to the number of mice and data is available upon request.
Results
Changes in weight gain induced by a HFD:
An outline of the experimental design is presented in Figure 1A. All mice were given a two week acclimation period when they arrived at our animal facility, placed on standard chow for 2 weeks, and weighed weekly until study completion (Figure 1B). Following the two week acclimation period, pseudo randomization was used to assign mice to either the C57BL/6J LFD (n=14), C57BL/6J HFD (n=11), AβPP/PS1 LFD (n=11), or AβPP/PS1 HFD (n=12) groups. All mice remained on their respective diets until study completion. A diet effect was observed by four weeks (F[3,44] = 81.29; P<0.0001). As expected, both C57BL/6J and AβPP/PS1 mice on HFD gained more weight compared to genotype matched LFD groups No differences in weight gain were observed between genotypes within each diet group.
HFD impairs peripheral insulin sensitivity:
After 24 weeks on their respective diets, the effects of a HFD on the sensitivity of blood glucose levels to the action of insulin were tested with an ipITT. No difference in blood glucose levels were observed during a four hour fast (Figure 1C). A HFD significantly impaired peripheral insulin sensitivity compared to genotype matched LFD mice when examining the 120 minute blood glucose response to the insulin challenge (F[3,44] = 23.58; P<0.0001; Figure 1D) as well as the subsequent area under the curve analysis (F[1,44] = 45.41; P<0.0001; Figure 1E) indicating an obesity-induced T2DM phenotype. Additionally, an effect of genotype (F[1,44] = 15.61; P=0.0003) was observed supporting that AβPP/PS1 mice have impaired insulin sensitivity that is independent of diet (Figure 1D-E).
Glucose metabolism is impaired in mice fed a HFD:
The effects of a HFD on glucose metabolism was tested using the ipGTT. Mice fed a HFD had significantly (F[1,44] = 42.28; P<0.0001; Figure 1F) elevated 15 hour fasting blood glucose compared to genotype matched LFD mice as a result of the obesity-induced insulin resistance. Mice fed a HFD metabolized glucose slower during the 120 minute glucose challenge (F[3,44] = 8.019; P=0.0002) and subsequent area under the curve analysis (F[1,44] = 18.32; P<0.0001) compared to genotype matched LFD mice (Figure 1G-H).
Mice on a HFD have impaired spatial learning and memory:
One week following ipGTT and 27 weeks into dietary feeding, mice underwent a 3 day MWM. The MWM tests spatial learning and memory recall by requiring the mouse to utilize visual cues to repeatedly swim to a static, submerged platform, regardless of the starting quadrant. During the MWM, mice were trained to locate a visible escape platform on Day 1 and a hidden platform on Day 2. The visible platform verifies visual acuity while simultaneously habituating the mice to the novel environment thereby reducing stress while encouraging a motivation to escape the pool (Gulinello et al. 2009). The three training sessions for Day 1 and for each training block in Day 2 were averaged for individual mice for analysis. Latency to reach to the platform as well as cumulative distance from the platform were assessed. Cumulative distance is a proximity measure designed to reflect search error through summation of the distance from the platform calculated at one second intervals while accounting for trial variations in starting quadrant and swimming speed (Gallagher et al. 2015). As shown in Figure 2A, AβPP/PS1 HFD took longer to locate the hidden escape platform during the first training block of Day 2 compared to their Day 1 performance (F[3,44) = 4.118; P=0.0117). In Figure 2B, C57BL/6J LFD mice were the only group that did not travel further from the hidden escape platform on the first training block of Day 2 and continued to significantly decrease over successive training blocks (F[3,52] = 3.449; P=0.0231) compared to their Day 1 performance. On the contrary, the other three groups of mice traveled further from the hidden escape platform on the first training block of Day 2, which was significant in AβPP/PS1 HFD mice (F[3,44] = 5.178; P=0.0038) compared to their Day 1 performance. While C57BL/6J HFD and both AβPP/PS1 diet groups decreased the cumulative distance from the hidden escape platform over successive training blocks, a significant improvement over Day 1 performance was not observed supporting decreased learning in these three groups of mice. During the MWM probe challenge for memory recall, no differences were observed in the number of annulus 40 crossings (Figure 2C).
Figure 2: MWM Training and Probe Challenge.
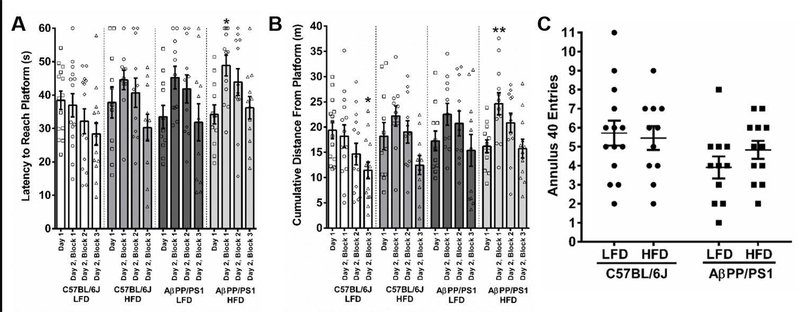
The 2-day MWM training session consisted of a visible platform on Day 1 that was changed to a hidden platform on Day 2 and the probe challenge. A) Latency to reach the escape platform for each training session. B) The cumulative distance each mouse spent away from the platform for the training sessions. A & B) *p<0.05, **p<0.01, hidden platform vs Day 1 visible platform. C) The number of annulus 40 crossings. C57BL/6J LFD, n=14; C57BL/6J HFD, n=11; AβPP/PS1 LFD, n=11; AβPP/PS1 HFD, n=12; where n refers to the number of animals.
HFD elevates hippocampal basal glutamate:
A minimum of 2 weeks post MWM (29 weeks into dietary feeding) an enzyme based MEA was used to measure glutamate dynamics in the DG, CA3, and CA1. Representative glutamate traces showing basal and stimulus-evoked glutamate release are presented in Figure 3. Basal glutamate was calculated by taking a 10 s baseline average prior to start of pressure ejection in the DG, CA3, and CA1. When examining basal glutamate (Figures 4A-C), a genotype effect was only observed in the CA1 (F[1,43] = 12.23; P=0.0011), indicating increased tonic glutamate in AβPP/PS1 mice. A diet effect was observed in the DG (F[1,43] = 6.232; P=0.0165), CA3 (F[1,43] = 12.90; P=0.0008), and CA1 (F[1,43] = 12.78; P=0.0009), supporting that a HFD elevates basal glutamate with synergistic effects observed in AβPP/PS1 mice.
Figure 3: Stimulus-Evoked Glutamate Release Traces.
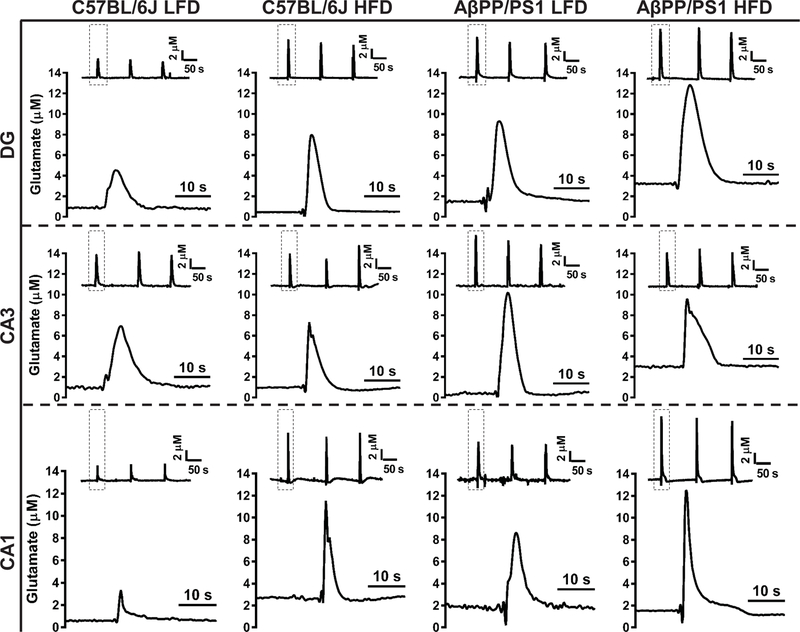
Representative traces of glutamate release from 70 mM KCl stimulation. Columns indicate genotype and diet while rows indicate hippocampal subfield. Within each panel, the inset trace depicts the reproducibility of the glutamate signals with the dashed box indicating a single response magnified beneath for a clearer presentation of glutamate dynamics. Concentration and time axes are consistent in all panels for comparative interpretation.
Figure 4: Hippocampal Glutamate Measures.
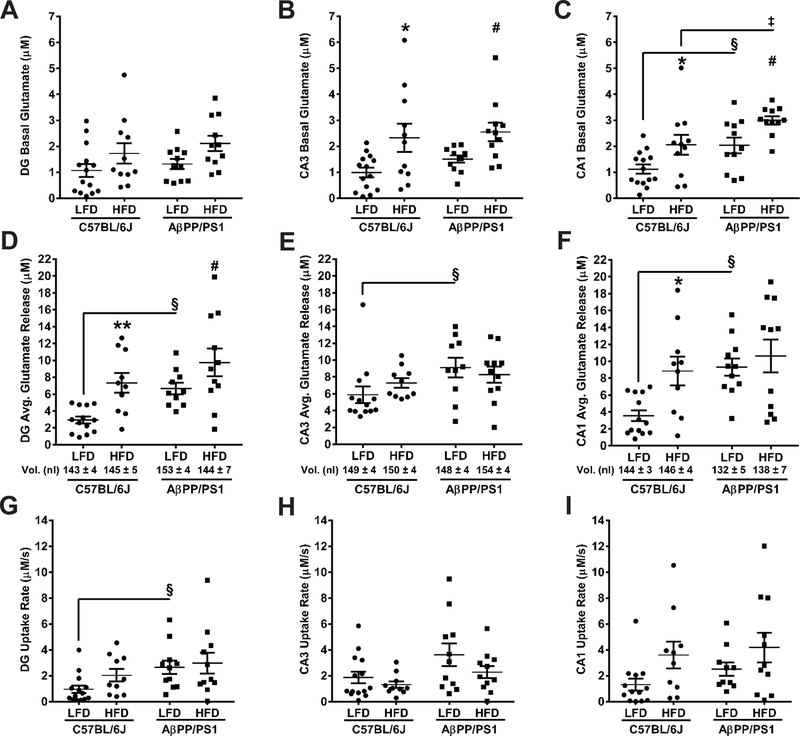
Basal glutamate, stimulus-evoked glutamate release, and evoked glutamate uptake rate in the DG (A, D, G), CA3 (B, E, H), and CA1 (C, F, I). Basal glutamate was measured prior to local application of stimulus. Stimulus volumes (mean ± SEM) are shown beneath the bar graphs in D-F. *p<0.05, **p<0.01 C57BL/6J LFD (n=13–14; 1 subject excluded) vs C57BL/6J HFD (n=10–11; 1 subject excluded); #p<0.05 AβPP/PS1 LFD (n=10–11; 1 subject excluded) vs AβPP/PS1 HFD (n=11; 1 subject died); §p<0.05 C57BL/6J LFD (n=13–14) vs AβPP/PS1 LFD (n=10–11); ‡p<0.05 C57BL/6J HFD (n=10) vs AβPP/PS1 HFD (n=11); where n refers to the number of animals and at most a single subject per group was excluded by Grubb’s test.
HFD alters hippocampal glutamate dynamics:
A glass micropipette attached to the enzyme-based MEA was used to locally apply sterile filtered (0.20 µm) 70 mM KCl (70 mM KCl, 79 mM NaCl and 2.5 mM CaCl2, pH 7.4) by pressure ejection (5–15 psi, 1–2 s pulses) using a Picospritzer III (Parker-Hannafin Corp.). Ejection volumes were maintained between 100–200 nl in each hippocampal subfield and monitored using a stereomicroscope (Luxo Corp. Cat:, Elmsford, NY) fitted with a calibrated reticule (Hascup and Hascup 2016). Similar volumes of stimulus were locally applied in the DG (F[3,40] = 0.7720; P=0.5165), CA3 (F[3,40] = 0.3397; P=0.7967), and CA1 (F[3,41] = 1.655; P=0.1915) of all mouse groups to elicit glutamate release (Figures 4D-F). A genotype effect was observed in the DG (F[1,40] = 8.429; P=0.0060), CA3 (F[1,40] = 4.720; P=0.0358), and CA1 (F[1,41] = 7.559; P=0.0088), whereby AβPP/PS1 mice release more glutamate upon depolarization. A diet effect was observed in the DG (F[1,40] = 12.57 P = 0.0010) and CA1 (F[1,41] = 5.772; P=0.0209), but not the CA3 (F[1,40] = 0.0870; P=0.7695), indicating a HFD increased stimulus-evoked glutamate release. The clearance of glutamate is predominantly mediated by uptake in high-efficiency excitatory amino acid transporters (EAAT) located on glia (Zhou and Danbolt 2013). A significant effect of genotype was observed for glutamate uptake rate in the DG (F[1,42] = 6.050; P=0.0181) and CA3 (F[1,42] = 5.793; P = 0.0206), but not the CA1 (Figures 4G-I). A significant effect from diet led to increased glutamate uptake rate only in the CA1 (F[1,40] = 5.721; P=0.0216).
Increased expression of VGLUT1 in HFD mice:
IHC was used to determine changes in VGLUT1 expression in the DG, CA3, and CA1. Representative images of VGLUT1 from the DG are shown in Figures 5A-D (40x magnification) and average mean density for each hippocampal subfield are presented in Figures 5E-G. A significant effect of genotype on VGLUT1 expression was observed in the DG (F[1,34] = 12.40; P=0.0012), CA3 (F[1,33] = 12.59; P=0.0012) and CA1 (F[1,36] = 32.74; P<0.0001), supporting that the elevated hippocampal basal and stimulus-evoked glutamate release in APP/PS1 mice is a result of increased glutamatergic vesicles. Likewise, a significant effect of diet existed on VGLUT1 expression in the DG (F[1,34] = 17.67; P=0.0002), CA3 (F[1,33] = 27.63; P<0.0001) and CA1 (F[1,36] = 17.54; P=0.0002), indicating that a HFD increased glutamatergic vesicles corresponding with the elevated hippocampal glutamate.
Figure 5: VGLUT1 Immunohistochemistry.
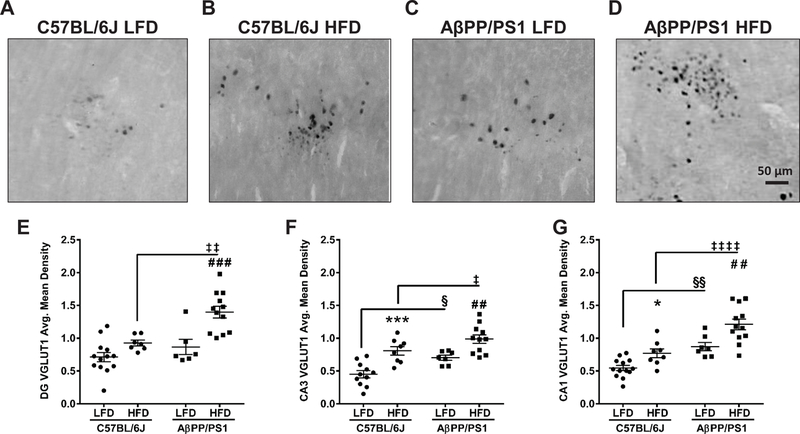
Representative images of VGLUT1 staining in the DG at 40x magnification from C57BL/6J LFD (A), C57BL/6J HFD (B), AβPP/PS1 LFD (C), and AβPP/PS1 HFD (D) mice. Scale bar = 50 µm. Average mean density of VGLUT1 staining in the DG (E), CA3 (F), and CA1 (G). *p<0.05, ***p<0.001 C57BL/6J LFD (n=11–13; 1–3 subjects excluded) vs C57BL/6J HFD (n=7–8; 3–4 subjects excluded); ##p<0.01, ###p<0.001 AβPP/PS1 LFD (n=6–7; 4–5 subjects excluded) vs AβPP/PS1 HFD (n=11–12; 1 subject excluded); §p<0.05, §§p<0.01 AβPP/PS1 LFD (n=6–7) vs C57BL/6J LFD (n=11–13); ‡p<0.05, ‡‡p<0.01, ‡‡‡‡p<0.0001 AβPP/PS1 HFD (n=11–12) vs C57BL/6J HFD (n=7–8); where n refers to the number of animals.
GFAP expression is increased by HFD:
IHC was used to determine changes in GFAP expression in the DG, CA3, and CA1. Whole hippocampal representative images (10x magnification) of GFAP expression are shown in Figures 6A-D and average mean density for each hippocampal subfield are presented in Figures 6E-G. A significant effect of genotype on GFAP expression was observed in the DG (F[1,36] = 14.83; P = 0.0005), CA3 (F[1,37] = 23.07; P<0.0001), and CA1 (F[1,39] = 100.60; P<0.0001), as indicated by greater hippocampal GFAP expression in APP/PS1 mice. In addition, a significant effect of diet exists on GFAP expression in the C57BL/6J DG (F[1,36] = 9.006; P=0.0049) and AβPP/PS1 CA3 (F[1,37] = 20.13; P<0.0001) and CA1 (F[1,39] = 15.98; P=0.0003), indicating that a HFD results in greater astrogliosis.
Figure 6: GFAP Immunohistochemistry.
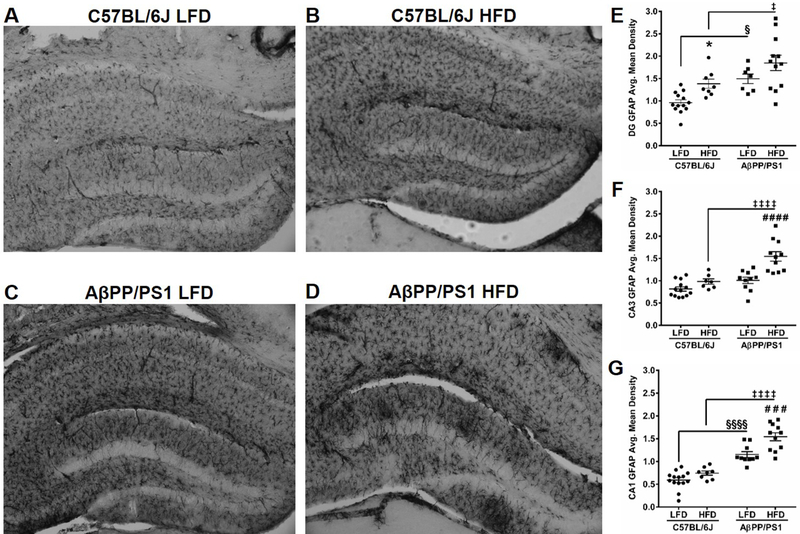
Whole hippocampal representative images of GFAP staining in C57BL/6J LFD (A), C57BL/6J HFD (B), AβPP/PS1 LFD (C), and AβPP/PS1 HFD (D) mice at 10x magnification. Average mean density of GFAP staining in the DG (E), CA3 (F), and CA1 (G). *p<0.05 C57BL/6J LFD (n=13–14; 1 subject excluded) vs C57BL/6J HFD (n=7–8; 3–4 subjects excluded); ###p<0.001, ###p<0.0001 AβPP/PS1 LFD (n=7–10, 1–4 subjects excluded) vs AβPP/PS1 HFD (n=11–12; 1 subject excluded); §p<0.05, §§§§p<0.0001 AβPP/PS1 LFD (n=7–10) vs C57BL/6J LFD (n=13–14); ‡p<0.05, ‡‡‡‡p<0.0001 AβPP/PS1 HFD (n=11–12) vs C57BL/6J HFD (n=7–8); where n refers to the number of animals.
A HFD does not alter plaque formation:
Hippocampal plaque formation was determined by staining with Amylo-Glo® RTD™. Whole hippocampal representative images (10x magnification) of plaque accumulation (blue) and EtBr counter stain (red) are shown in Figures 7A-D and average mean density of whole hippocampal plaque accumulation is presented in Figure 7E. As indicated by the arrows in Figures 7C and 7D, a genotype effect caused increased plaque accumulation (F[1,16] = 1410.0; P<0.0001). However, a diet effect was not observed (F[1,16] = 0.1096; P=0.7449), indicating that a HFD does not increase plaque deposition in AβPP/PS1 mice. This was further supported by ELISA determination in a separate cohort of mice showing a genotype effect (F[1,24] = 49.95; P<0.0001), but not a diet effect (F[1,24] = 0.79; P=0.3828).
Figure 7: Amyloid Plaque Formation.
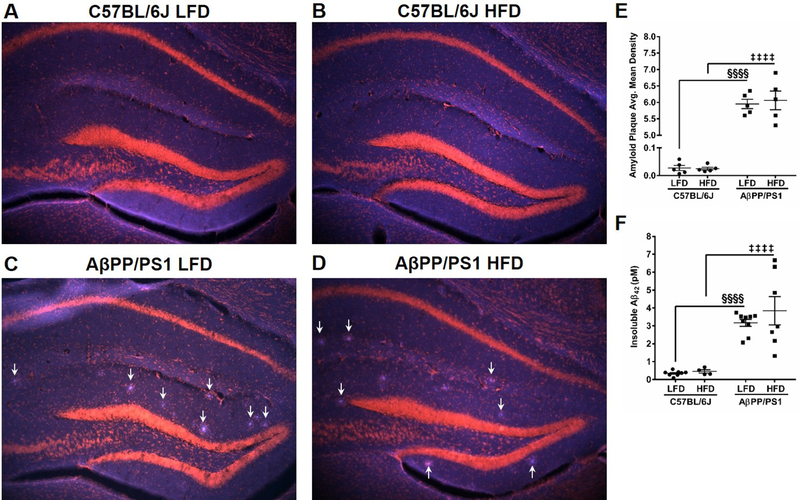
Whole hippocampal representative images of amyloid plaque formation (blue) and nuclei staining (red) by EtBr in C57BL/6J LFD (A), C57BL/6J HFD (B), AβPP/PS1 LFD (C), and AβPP/PS1 HFD (D) mice at 10x magnification. Arrows indicate plaque formations in C and D. Average mean density of plaque formation in the hippocampus (E) and insoluble Aβ42 ELISA determination (F). §§§§p<0.0001 AβPP/PS1 LFD (n=5,9) vs C57BL/6J LFD (n=5,8); ‡‡‡‡p<0.0001 AβPP/PS1 HFD (n=5,7) vs C57BL/6J HFD (n=5,4); where n refers to the number of animals and 7 subjects per group were excluded from IHC plaque analysis.
Discussion
Half of AD cases are attributable to modifiable lifestyle factors (Barnes and Yaffe 2011) including obesity-induced T2DM that has been suggested to increase the risk for developing AD 1.5- to 2-fold (Allen et al. 2004; Ott et al. 1999; Arvanitakis et al. 2004). While the exact molecular events linking T2DM to AD have not been fully elucidated, the metabolic hypothesis of AD (Hoyer 2002) supports impaired insulin signaling initiates a series of events including, Aβ accumulation (Farris et al. 2003), neuroinflammation (Granic et al. 2009), oxidative stress (De Felice and Ferreira 2014) and calcium dyshomeostasis (Zhang et al. 2017) leading to AD pathogenesis. The results of the present study support addition of elevated hippocampal glutamatergic signaling to this growing body of molecular parallels.
In the present study, starting at 3 months of age C57BL/6J and AβPP/PS1 mice were placed on either LFD (10% kcal from fat) or HFD (60% kcal from fat) with matching protein and sucrose content. Mice fed a HFD developed an obese phenotype starting one month after diet initiation and continued until study completion. Since insulin sensitivity is positively correlated to increased longevity and healthspan in vertebrates (Arum et al. 2014) we choose to examine peripheral blood glucose clearance in these mice. The obese phenotype resulted in impaired peripheral insulin signaling and glucose tolerance as observed in the ipITT and ipGTT. The insulin resistance observed in the HFD mouse groups explains the elevated 15 hr fasting blood glucose levels compared to genotype matched LFD groups. Interestingly, AβPP/PS1 mice fed a LFD had a similar metabolic profile to HFD mice supporting a naturally occurring insulin resistance in these mice, which has been reported elsewhere (Pedrós et al. 2014; Macklin et al. 2017). Despite the innate insulin resistance observed in AβPP/PS1 mice, HFD led to a further disruption of their metabolic profile.
A HFD has been shown to negatively affect learning and memory in cognitively normal rodents (Kanoski and Davidson 2011; Cordner and Tamashiro 2015) as well as exacerbate cognitive decline and AD-related neuropathology in animal models (Vandal et al. 2014; Knight et al. 2014; Thériault et al. 2016; Julien et al. 2010). For this study, diet treatments began prior to the onset of typically reported AD-related pathology and continued through an age when pathology and plaque burden are well developed in the AβPP/PS1 mouse model. During the visible portion (Day 1) of the MWM behavioral task, similar performances in the latency to the platform and cumulative distance traveled from the platform were observed in all groups of mice, indicating comparable visual acuity and physical activity despite weight differences. Learning impairments in AβPP/PS1 mice were discerned during the second training day with the hidden escape platform. Throughout the training blocks, we observed a higher percentage of C57BL/6J LFD and HFD mice successfully navigate the MWM while traveling less distance from the platform. To the contrary, AβPP/PS1 mice presented with learning impairments that were worsened when fed a HFD as supported by 1) a slower escape latency and 2) the cumulative distance from the submerged escape platform during the first two training blocks. By the third training block, AβPP/PS1 performance was similar to that observed during the Day 1 visible platform, but no significant improvements were observed. However, during the MWM probe challenge on the third day, no differences in the number of annulus 40 crossing was observed. The MWM paradigm employed in this study, while atypical from previously published reports from our laboratory (Hascup and Hascup 2015), was designed to reduce stress and anxiety in the mouse. The visible platform on Day 1 helped to habituate mice to the novel pool environment while testing for visual acuity since some inbred mouse strains develop retinal degeneration (Chang et al. 2002). Furthermore, the shorter training duration avoided learning limitations from multiple practice sessions, prevented fatigue, and increased throughput (Alamed et al. 2006). These data support that a HFD can negatively affect learning in both C57BL/6J and AβPP/PS1 mice.
Glutamate, the predominant excitatory neurotransmitter in the mammalian CNS, plays an essential role in learning and memory (Riedel et al. 2003) and has been implicated in several neurodegenerative disorders including Huntington’s, Parkinson’s, and Alzheimer’s diseases. To measure glutamate, we used an enzyme-based MEA with high spatial resolution (50 × 100 µm recording sites) that allowed for independent measures from the DG, CA3, and CA1 dorsal hippocampus, a region that is important for consolidation and retrieval of spatial memory during the MWM task (Cimadevilla et al. 2005). In the present study, AβPP/PS1 LFD mice exhibited elevated basal glutamate (CA1) and stimulus-evoked glutamate release (DG, CA3, and CA1) compared to C57BL/6J LFD mice. The elevated basal glutamate may result from a combination of mechanisms affecting soluble Aβ42 levels that are known to elicit glutamate release (Hascup and Hascup 2016; Talantova et al. 2013). First, the transgene expressions in AβPP/PS1 result in progressive Aβ42 accumulation (Alley et al. 2010), and second, the insulin resistance observed in these mice may prevent IDE from degrading monomeric Aβ42 leading to further accumulation and overactivation of α7nAChR on presynaptic glutamatergic terminals. Since hippocampal tissue were used for IHC, none was available for biochemical analysis. Further studies examining soluble Aβ42 are needed in order to validate this hypothesis.
VGLUT1, the predominant subtype of vesicles that store hippocampal glutamate (Liguz-Lecznar and Skangiel-Kramska, 2007), was increased in the CA3 and CA1 of AβPP/PS1 LFD compared to C57BL/6J LFD mice. Increased expression of VGLUT1 has been demonstrated to cause excess glutamate release (Daniels et al. 2011), however, homeostatic mechanisms exist to limit aberrant synaptic firing that may arise from environmental or genetic variations. But, during the early stages of AD, it is hypothesized that these negative feedback mechanisms begin to destabilize in cortical and hippocampal regions (Frere and Slutsky 2018). For example, epileptiform spikes have been observed in both AβPP/PS1 mice (Minkeviciene et al. 2009) and amnestic mild cognitively impaired patients (Vossel et al. 2013) as well as hyperexcitability of CA1 pyramidal neurons in AβPP/PS1 mice (Šišková et al. 2014). This hyperexcitability coupled with the increased VGLUT1 expression may explain the increased stimulus-evoked glutamate release observed throughout the hippocampus of the present study.
Both C57BL/6J and AβPP/PS1 mice fed a HFD presented with elevated basal (CA3 and CA1) and stimulus-evoked glutamate release (DG and CA1) compared to genotype-matched LFD mice. As described above, the increased extracellular glutamate observed in AβPP/PS1 HFD mice may be explained by the accumulation of Aβ42 stimulating glutamate release, but this would not be the case in C57BL/6J HFD mice. However, a HFD would initiate a cascade of separate events in both genotypes leading to the increased basal and stimulus-evoked glutamate release. The insulin resistance and subsequent increase in circulating blood glucose levels observed in both HFD mice would lead to an increase in neuronal glucose accumulation. Since neuronal glutamate synthesis can be derived from glucose (Sonnewald 2014) the higher blood glucose levels would increase the neurotransmitter pool of glutamate, which is supported by the elevated VGLUT1 density observed in the DG, CA3, and CA1 of HFD mice.
A HFD can upregulate glial glutamate transporter expression while increasing the maximal velocity of clearance (Valladolid-Acebes et al. 2012). While the present study did not examine EAAT density, increased expression of GFAP is indicative of astrogliosis (Brahmachari et al. 2006) that is associated with an increase in glial glutamate transporters in response to chronic cerebral injuries and neurodegenerative disorders (Haroon et al. 2017). At first, this appears counterintuitive. More transporters supports faster glutamate clearance that would decrease basal and evoked glutamate concentrations. But, elevated glutamate release causes increased EAAT density as a mechanism to prevent chronic accumulation of extracellular glutamate and potential excitotoxicity (Munir et al. 2000). Although not significant in all hippocampal subregions, stimulus-evoked glutamate uptake was increased in AβPP/PS1 LFD vs diet-matched C57BL/6J mice and a HFD further increased these rates in the DG and CA3. In other words, glutamate uptake rate generally increased in response to increases in basal and stimulus-evoked glutamate release.
As expected, hippocampal plaque pathology was only observed in AβPP/PS1 mice, however, a HFD did not increase plaque density which is similar to previous reports in these mice (Thériault et al. 2016). This may be due to the astrogliotic response to control plaque deposition in the pathogenesis of AD (Kraft et al. 2013). As such, we observed increased hippocampal GFAP density in AβPP/PS1 LFD and HFD compared to diet-matched C57BL/6J control mice that did not present with plaque pathology. GFAP density throughout the hippocampus was further elevated in AβPP/PS1 HFD compared to LFD mice, but this was only mildly observed in the DG of C57BL/6J HFD mice. Since astroglia play a role in Aβ clearance (Ries and Sastre 2016), the elevated astrogliotic inflammatory response, particularly in AβPP/PS1 HFD mice may have prevented additional plaque accumulation.
The elevated extracellular hippocampal glutamate levels observed in C57BL/6J HFD as well as AβPP/PS1 LFD and HFD mice would contribute to their decreased performance on the MWM task reported here and elsewhere (Thériault et al. 2016). The NMDA receptor is important for spatial learning and memory tasks (Morris et al. 1986), but mild, chronic overactivation would be detrimental to synaptic plasticity. This argument is based on the signal-to-noise hypothesis of NMDA receptor activation. Elevated tonic glutamate levels (as observed in this study) prevents detection of phasic signals thereby blocking formation of new learning (Parsons et al. 2007). This process could occur over an extended period of time before calcium overload, excitotoxicity, and eventual neurodegeneration as observed in AD. As such, this mechanism helps to explain the cognitive-improving effects of memantine, an NMDA receptor antagonist, in some AD patients (Parsons et al. 2007). In support of this, AβPP/PS1 mice fed a HFD followed by treatment with memantine saw significant reductions in insulin resistance, neuroinflammation, and cognitive deficits (Ettcheto et al. 2018). Alternatively, a HFD has been shown to decrease NMDA receptor subunit GluN2B leading to desensitization that may account for cognitive deficits (Valladolid-Acebes et al. 2012).
The concentration of extracellular basal glutamate is debated throughout the scientific literature with reports ranging from nanomolar to micromolar concentrations (Herman and Jahr 2007; Burmeister et al. 2013; Messam et al. 1995). These discrepancies are frequently attributed to methodological considerations that often times yield similar results when additional factors are taken into consideration. The size of our MEA recording sites limits our recording capabilities to the extracellular matrix (ECM) where we are measuring glutamate release and clearance from multiple synapses. As such, a summation of multiple extrasynaptic spillover events may cause elevated levels compared to those reported using patch clamp techniques in slice preparations. Additionally, the diffusion capabilities of neurotransmitters and other membrane impermeable molecules in the ECM are subject to both volume fraction (α = 0.2) and tortuosity (λ = 1.6) (Syková and Nicholson 2008). The α effectively amplifies the concentration of extrasynaptic spillover of glutamate in the ECM while the λ simultaneously slows down its diffusion and uptake into high-affinity transporters. Furthermore, the depolarizing stimulus used in the present study creates a positive net charge on the ECM resulting in a drag effect on the negatively charged glutamate molecules (Gundelfinger et al. 2010), and changes the membrane potential which EAATs rely upon for efficient uptake of glutamate (Takahashi et al. 1997). The net effect is a slower clearance of stimulus-evoked glutamate release when compared to other methods. Of course, tissue damage is always a concern with any invasive technique including, but not limited to, slice preparations, microdialysis, and MEA recordings. However, the ceramic substrate (Al2O3) on the MEAs used in this study have good biocompatibility helping to limit CNS damage allowing for single-unit neuronal activity measurements for at least 6 months post-implantation (Hascup et al. 2009). The MEAs employed in this study have routinely demonstrated that basal extracellular glutamate are sensitive to Na+-channel (tetrodotoxin), Ca2+-channel (ω-conotoxin) and EAAT (DL-threo-β-Benzyloxyaspartic acid) blockade lending credence to a healthy parenchyma surrounding the implanted MEA (Hascup et al. 2010; Hascup and Hascup 2016; Hascup et al. 2007). Furthermore, the extracellular glutamate concentrations reported in this manuscript fall below the Km (~20 µM) for EAATs (Zhou and Danbolt 2013). And basal hippocampal glutamate concentrations for C57BL/6J LFD mice (~1 µM) are below the reported EC50 (3.7 µM) for the NR1/NR2B NMDA receptor (Banke and Traynelis 2003), further strengthening our premise that chronic overactivation of NMDA receptors, as observed in the HFD and AβPP/PS1 groups would be detrimental to synaptic plasticity, cognition, and may lead to eventual neurodegeneration. Regardless, the current study was not designed to be a definitive assessment of basal glutamate concentrations. In fact, the level of basal glutamate is dependent upon a number of criteria including transporter density (Herman and Jahr 2007), ECM developmental stage (Gundelfinger et al. 2010) and the glia-neuron ratio (Azevedo et al. 2009) resulting in marked variation between brain regions, maturation, and species (Burmeister et al. 2013).
Conclusion
Excitotoxicity is a proposed mechanism underlying the neurodegeneration associated with AD. However, the basal and stimulus-evoked glutamate release values reported here are not considered neurotoxic for an intact nervous system. Rather, the current study (when combined with previous research from our laboratory) highlights a consistent theme of elevated hippocampal glutamate starting as early as 2–4 months in AβPP/PS1 mice (Hascup and Hascup 2015) that is potentially mediated by soluble Aβ42 (Hascup and Hascup 2016). Furthermore, obesity-induced insulin resistance caused cognitive impairments and increased extracellular glutamate in AβPP/PS1 mice. The progressive deposition of Aβ42 with AD progression may chronically elevate glutamate leading to the cognitive and function decline observed in AD, which can be exacerbated by modifiable life-style factors such as obesity-induced insulin resistance. While additional studies are ongoing to elucidate mechanisms associated with dietary influences on glutamate dynamics in AβPP/PS1 mice, hippocampal glutamate levels may serve as a viable early therapeutic biomarker for AD pathogenesis.
Acknowledgments
Funding
This work was supported by NIH R01 AG057767, NIH R01 AG061937, Center for Alzheimer’s Disease and Related Disorders at Southern Illinois University School of Medicine, the Kenneth Stark Endowment, and the Fraternal Order of Eagles (KNH, SOB, ERH), NIA AG051869 (YF, AB) and by the MUSC Barmore Foundation (MKR, HAB)
Abbreviations:
- mPD
1,3 phenylenediamine dihydrochloride
- α7nAChR
α7 nicotinic acetylcholine receptor
- AD
Alzheimer’s disease
- AA
ascorbic acid
- ANOVA
analysis of variance
- ABC
avidin-biotin complex
- Aβ
beta-amyloid
- b.w.
body weight
- BSA
bovine serum albumin
- DG
dentate gyrus
- DPX
dibutyl phthalate and xylene
- DA
dopamine hydrochloride
- EAAT
excitatory amino acid transporter
- FAST
Fast Analytical Sensing Technology
- GFAP
glial fibrillary acidic protein
- GTT
glucose tolerance test
- HFD
high-fat diet
- IDE
insulin degrading enzyme
- ITT
insulin tolerance test
- ip
intraperitoneal
- IHC
immunohistochemistry
- LFD
low-fat diet
- MEA
microelectrode array
- MWM
Morris water maze
- NMDA
N-methyl-D-aspartate
- PBS
phosphate buffered saline
- RRID
research resource identifiers
- SEM
standard error of the mean
- T2DM
Type 2 diabetes mellitus
- VGLUT1
vesicular glutamate transporter 1
Footnotes
Conflicts of Interest
The authors declare no competing financial interests.
Open Science Badges
This article has received a badge for *Open Materials* and for *Open Data* because it made the data publicly available. The data can be accessed at DOI: 10.13140/RG.2.2.11180.10888 and https://osf.io/5whvu (figures for data) and https://osf.io/gd5vf (materials and methods) . The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
References
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc 1, 1671–9. [DOI] [PubMed] [Google Scholar]
- Allen KV, Frier BM, Strachan MWJ (2004) The relationship between type 2 diabetes and cognitive dysfunction: longitudinal studies and their methodological limitations. Eur. J. Pharmacol 490, 169–75. [DOI] [PubMed] [Google Scholar]
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, Banerjee PK, Lahiri DK (2010) Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J. Neurosci. Res 88, 143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arum O, Boparai RK, Saleh JK, Wang F, Dirks AL, Turner JG, Kopchick JJ, Liu J-L, Khardori RK, Bartke A (2014) Specific suppression of insulin sensitivity in growth hormone receptor gene-disrupted (GHR-KO) mice attenuates phenotypic features of slow aging. Aging Cell 13, 981–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol 61, 661–6. [DOI] [PubMed] [Google Scholar]
- Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, Jacob Filho W, Lent R, Herculano-Houzel S (2009) Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol 513, 532–41. [DOI] [PubMed] [Google Scholar]
- Banke TG, Traynelis SF (2003) Activation of NR1/NR2B NMDA receptors. Nat. Neurosci 6, 144–152. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Yaffe K (2011) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet. Neurol 10, 819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Patrick KS, Ramamoorthy S, Denehy ED, Zhu H, Pacchioni AM, Granholm A-C, McGinty JF (2007) Long-term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line-derived neurotrophic factor heterozygous mice. J. Neurosci 27, 8816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari S, Fung YK, Pahan K (2006) Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci 26, 4930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Davis VA, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA (2013) Glutaraldehyde cross-linked glutamate oxidase coated microelectrode arrays: selectivity and resting levels of glutamate in the CNS. ACS Chem. Neurosci 4, 721–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA (2001) Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal. Chem 73, 1037–42. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Moxon K, Gerhardt GA (2000) Ceramic-based multisite microelectrodes for electrochemical recordings. Anal. Chem 72, 187–92. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA (2002) Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J. Neurosci. Methods 119, 163–71. [DOI] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR (2002) Retinal degeneration mutants in the mouse. Vision Res 42, 517–525. [DOI] [PubMed] [Google Scholar]
- Cimadevilla JM, Miranda R, López L, Arias JL (2005) Partial unilateral inactivation of the dorsal hippocampus impairs spatial memory in the MWM. Brain Res. Cogn. Brain Res 25, 741–6. [DOI] [PubMed] [Google Scholar]
- Cordner ZA, Tamashiro KLK (2015) Effects of high-fat diet exposure on learning & memory. Physiol. Behav 152, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers. Res. Ther 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Miller BR, DiAntonio A (2011) Increased vesicular glutamate transporter expression causes excitotoxic neurodegeneration. Neurobiol. Dis 41, 415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettcheto M, Sánchez-López E, Gómez-Mínguez Y, Cabrera H, Busquets O, Beas-Zarate C, García ML, et al. (2018) Peripheral and Central Effects of Memantine in a Mixed Preclinical Mice Model of Obesity and Familial Alzheimer’s Disease. Mol. Neurobiol 55, 7327–7339. [DOI] [PubMed] [Google Scholar]
- Fang Y, McFadden S, Darcy J, Hill CM, Huber JA, Verhulst S, Kopchick JJ, Miller RA, Sun LY, Bartke A (2017) Differential effects of early-life nutrient restriction in long-lived GHR-KO and normal mice. GeroScience 39, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrand AQ, Helke KL, Gregory RA, Gooz M, Hinson VK, Boger HA (2017) Vagus nerve stimulation improves locomotion and neuronal populations in a model of Parkinson’s disease. Brain Stimul 10, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Gue S (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta amyloid precursor protein intracellular domain in vivo 100, 4162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice F. G. (2013) Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J. Clin. Invest 123, 531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice F. G., Ferreira ST. (2014) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63, 2262–72. [DOI] [PubMed] [Google Scholar]
- Frere S, Slutsky I (2018) Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 97, 32–58. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Burwell R, Burchinal M (2015) Severity of spatial learning impairment in aging: Development of a learning index for performance in the Morris water maze. Behav. Neurosci 129, 540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godyń J, Jończyk J, Panek D, Barbara M (2016) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Reports 68, 127–138. [DOI] [PubMed] [Google Scholar]
- Granic I, Dolga AM, Nijholt IM, Dijk G. van, Eisel ULM (2009) Inflammation and NF-κB in Alzheimer’s Disease and Diabetes. J. Alzheimer’s Dis 16, 809–821. [DOI] [PubMed] [Google Scholar]
- Gulinello M, Gertner M, Mendoza G, Schoenfeld BP, Oddo S, LaFerla F, Choi CH, McBride SMJ, Faber DS (2009) Validation of a 2-day water maze protocol in mice. Behav. Brain Res 196, 220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundelfinger ED, Frischknecht R, Choquet D, Heine M (2010) Converting juvenile into adult plasticity: a role for the brain’s extracellular matrix. Eur. J. Neurosci 31, 2156–2165. [DOI] [PubMed] [Google Scholar]
- Haroon E, Miller AH, Sanacora G (2017) Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 42, 193–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Littrell OM, Hinzmann JM, Werner CE, Davis VA, Burmeister JJ, Pomerleau F, Quintero J, Huettl P, Gerhardt GA (2013) Microelectrode Array Fabrication and Optimization for Selective Neurochemical Detection, in Microelectrode Biosens, (Marinesco S, Dale N, eds), Vol. 80, pp. 27–54. Humana Press, Totowa, NJ. [Google Scholar]
- Hascup ER, af Bjerkén S, Hascup KN, Pomerleau F, Huettl P, Strömberg I, Gerhardt GA (2009) Histological studies of the effects of chronic implantation of ceramic-based microelectrode arrays and microdialysis probes in rat prefrontal cortex. Brain Res 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup ER, Hascup KN, Stephens M, Pomerleau F, Huettl P, Gratton A, Gerhardt GA (2010) Rapid microelectrode measurements and the origin and regulation of extracellular glutamate in rat prefrontal cortex. J. Neurochem 115, 1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER (2015) Altered neurotransmission prior to cognitive decline in AβPP/PS1 mice, a model of Alzheimer’s disease. J. Alzheimer’s Dis 44, 771–776. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER (2016) Soluble Amyloid-β42 Stimulates Glutamate Release through Activation of the α7 Nicotinic Acetylcholine Receptor. J. Alzheimer’s Dis 53, 337–347. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Pomerleau F, Huettl P, Gerhardt GA (2007) Second-by-Second Measures of L-Glutamate in the Prefrontal Cortex and Striatum of Freely Moving Mice. J. Pharmacol. Exp. Ther 324, 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Stephens ML, Glaser PEA, Yoshitake T, Mathé AA, Gerhardt GA, Kehr J (2011) Resting glutamate levels and rapid glutamate transients in the prefrontal cortex of the Flinders Sensitive Line rat: a genetic rodent model of depression. Neuropsychopharmacology 36, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, Lynn MK, Fitzgerald PJ, Randall S, Kopchick JJ, Boger HA, Bartke A, Hascup ER (2016) Enhanced Cognition and Hypoglutamatergic Signaling in a Growth Hormone Receptor Knockout Mouse Model of Successful Aging. J. Gerontol. A. Biol. Sci. Med. Sci 72, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, Rutherford EC, Quintero JE, Day BK, Nickell JR, Pomerleau F, Huettl P, Burmeister JJ, Gerhardt GA (2006) Second-by-Second Measures of L-Glutamate and Other Neurotransmitters Using Enzyme-Based Microelectrode Arrays - Electrochemical Methods for Neuroscience - NCBI Bookshelf, in Electrochem. Methods Neurosci, (Borland AC, Michael LM, eds), pp. 407–450. CRC Press. [PubMed] [Google Scholar]
- Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J. Neurosci 27, 9736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Brozinick JT, Wang L-P, Hawkins ED, Sargent KM, Liu Y, Narra K, et al. (2007) Inhibition of Ceramide Synthesis Ameliorates Glucocorticoid-, Saturated-Fat-, and Obesity-Induced Insulin Resistance. Cell Metab 5, 167–179. [DOI] [PubMed] [Google Scholar]
- Hoyer S (2002) The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J. Neural Transm 109, 341–360. [DOI] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, et al. (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet. Neurol 12, 207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–81. [DOI] [PubMed] [Google Scholar]
- Jin M, Selkoe DJ (2015) Systematic analysis of time-dependent neural effects of soluble amyloid β oligomers in culture and in vivo: Prevention by scyllo-inositol. Neurobiol. Dis 82, 152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Émond V, Julien P, Calon F (2010) High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 31, 1516–1531. [DOI] [PubMed] [Google Scholar]
- Kanoski SE, Davidson TL (2011) Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol. Behav 103, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight EM, Martins IVA, Gümüsgöz S, Allan SM, Lawrence CB (2014) High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol. Aging 35, 1821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, et al. (2013) Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J 27, 187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguz-lecznar M, Skangiel-kramska J (2007) Vesicular glutamate transporters ( VGLUTs ): The three musketeers of glutamatergic system [DOI] [PubMed] [Google Scholar]
- Macklin L, Griffith CM, Cai Y, Rose GM, Yan X-X, Patrylo PR (2017) Glucose tolerance and insulin sensitivity are impaired in APP/PS1 transgenic mice prior to amyloid plaque pathogenesis and cognitive decline. Exp. Gerontol 88, 9–18. [DOI] [PubMed] [Google Scholar]
- Messam CA, Greene JG, Greenamyre JT, Robinson MB (1995) Intrastriatal injections of the succinate dehydrogenase inhibitor, malonate, cause a rise in extracellular amino acids that is blocked by MK-801. Brain Res 684, 221–4. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, Penke B, et al. (2009) Amyloid -Induced Neuronal Hyperexcitability Triggers Progressive Epilepsy. J. Neurosci 29, 3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C (2010) Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–43. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M (1986) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319, 774–6. [DOI] [PubMed] [Google Scholar]
- Mota SI, Ferreira IL, Rego AC (2014) Dysfunctional synapse in Alzheimer’s disease – A focus on NMDA receptors. Neuropharmacology 76, 16–26. [DOI] [PubMed] [Google Scholar]
- Munir M, Correale DM, Robinson MB (2000) Substrate-induced up-regulation of Na(+)-dependent glutamate transport activity. Neurochem. Int 37, 147–62. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Harskamp F. van, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53, 1937–42. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Stöffler A, Danysz W (2007) Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology 53, 699–723. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2004) The Mouse Brain in Stereotaxic Coordinates Gulf Professional Publishing. [Google Scholar]
- Pedrós I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, Auladell C, et al. (2014) Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1556–66. [DOI] [PubMed] [Google Scholar]
- Riedel G, Platt B, Micheau J (2003) Glutamate receptor function in learning and memory. Behav. brain Res. brain Res 140, 1–47. [DOI] [PubMed] [Google Scholar]
- Ries M, Sastre M (2016) Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, et al. (2014) Dendritic Structural Degeneration Is Functionally Linked to Cellular Hyperexcitability in a Mouse Model of Alzheimer’s Disease. Neuron 84, 1023–1033. [DOI] [PubMed] [Google Scholar]
- Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation - where do all the carbons go? J. Neurochem 131, 399–406. [DOI] [PubMed] [Google Scholar]
- Syková E, Nicholson C (2008) Diffusion in brain extracellular space. Physiol. Rev 88, 1277–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Billups B, Rossi D, Sarantis M, Hamann M, Attwell D (1997) The role of glutamate transporters in glutamate homeostasis in the brain. J. Exp. Biol 200, 401–409. [DOI] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, et al. (2013) Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U. S. A 110, E2518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Wang H-Y, Kazi H, Han L-Y, Bakshi KP, Stucky A, Fuino RL, et al. (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest 122, 1316–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thériault P, ElAli A, Rivest S (2016) High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 7, 67808–67827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valladolid-Acebes I, Merino B, Principato A, Fole A, Barbas C, Lorenzo MP, García A, et al. (2012) High-fat diets induce changes in hippocampal glutamate metabolism and neurotransmission. Am. J. Physiol. Endocrinol. Metab 302, E396–402. [DOI] [PubMed] [Google Scholar]
- Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, Lefrançois D, et al. (2014) Insulin reverses the high-fat diet-induced increase in brain aβ and improves memory in an animal model of Alzheimer disease. Diabetes 63, 4291–301. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, et al. (2013) Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70, 1158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, Selkoe DJ (2017) Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci 37, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chai R, Yang Y-Y, Guo S-Q, Wang S, Guo T, Xu S-F, Zhang Y-H, Wang Z-Y, Guo C (2017) Chronic diabetic states worsen Alzheimer neuropathology and cognitive deficits accompanying disruption of calcium signaling in leptin-deficient APP/PS1 mice. Oncotarget 8, 43617–43634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W-Q, Townsend M (2009) Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta 1792, 482–96. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Danbolt NC (2013) GABA and Glutamate Transporters in Brain. Front. Endocrinol. (Lausanne) 4, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]