

Abstract
Tailorable sorption properties at the molecular level are key for efficient carbon capture and storage and a hallmark of covalent organic frameworks (COFs). Although amine functional groups are known to facilitate CO2 uptake, atomistic insights into CO2 sorption by COFs modified with amine-bearing functional groups are scarce. Herein, we present a detailed study of the interactions of carbon dioxide and water with two isostructural hydrazone-linked COFs with different polarities based on the 2,5-diethoxyterephthalohydrazide linker. Varying amounts of tertiary amines were introduced in the COF backbones by means of a copolymerization approach using 2,5-bis(2-(dimethylamino)ethoxy)terephthalohydrazide in different amounts ranging from 25 to 100% substitution of the original DETH linker. The interactions of the frameworks with CO2 and H2O were comprehensively studied by means of sorption analysis, solid-state NMR spectroscopy, and quantum-chemical calculations. We show that the addition of the tertiary amine linker increases the overall CO2 sorption capacity normalized by the surface area and of the heat of adsorption, whereas surface areas and pore size diameters decrease. The formation of ammonium bicarbonate species in the COF pores is shown to occur, revealing the contributing role of water for CO2 uptake by amine-modified porous frameworks.
Introduction
Covalent organic frameworks (COFs) are a recently developed class of porous polymers with high chemical and thermal stability and well-defined crystal structures. COFs are promising for a range of applications, for example, in gas storage and separation,1−3 optoelectronics,4,5 and energy conversion.6,7 COFs are formed by condensation reactions of organic linkers that are covalently bound under reversible conditions, which provides a mechanism of error correction.8 All but a few COFs reported to date have two-dimensional (2D) network topologies, where the COF sheets are held together in the third dimension by noncovalent van der Waals interactions. The structure of COFs and their versatility allows for engineering these systems and their properties in a targeted manner.9−11 One way to do so is by pore-surface engineering, where the surface of preformed pores carrying specific functional sites can be further transformed postsynthetically, if desired.12 Another possibility is to modify the organic linkers presynthetically according to the targeted properties. A linker that is suitable for diverse transformations and has been successfully used in several COF syntheses is 2,5-diethoxyterephthalohydrazide (DETH).7,13,14 An example is the DETH-based COF-JLU4 which is synthesized by condensation with triformylphloroglucinol (TFG) and has been used in fluorescent pH sensing systems for aqueous solutions.15 Another chemically strongly related COF containing methoxy instead of ethoxy groups in the hydrazide linker, NUS-3, has been reported for the use in mixed-matrix membranes with high H2/CO2 permselectivity.16
Utilization of functional porous solids with custom-made pores has seen a burst of activity over the past decades, specifically in the context of carbon capture and storage (CCS). Carbon dioxide emissions are known to be the major source of global warming, and in order to reduce this effect, technically viable solutions for the capture and long-term storage of the greenhouse gas CO2 are needed and actively sought. The chemisorption of CO2 into aqueous alkanolamine solutions - known as amine scrubbing - is widely practiced in the downstream processing of flue gases produced at the mega ton scale by coal-fired fire plants.17 Primary or secondary amines form carbamates with CO2, whereas tertiary or sterically hindered amines act as bases accepting a proton from carbonic acid formed by dissolution of CO2 in water.18−20 It is worth noting that unhindered alkanolamines absorb only half a mole of CO2 per mole of amine by a zwitterion mechanism, whereas tertiary amines undergo base-catalyzed hydration of CO2 to form bicarbonate ions which increases the theoretical capacity to 1 mol of CO2 per mole of amine.18 The amine solutions that are used decompose over time, and their CO2 capturing ability decreases significantly.21 Additional problems are the corrosivity and toxicity of these amine solutions. Key goals in CCS are high adsorption capacity and cycle stability as well as full reversibility and adequate heats of adsorption. Materials such as activated carbons,22,23 metal–organic frameworks (MOFs),24,25 or COFs26,27 are, in contrast to the standard method, easy to regenerate at moderate temperatures and allow a great variety of functional designs. In this context, heterogeneous adsorbents such as COFs with precisely tunable pores decorated with functional groups are attracting increasing interest in the field. Another challenge in this context is the presence of small amounts of water, which can be competitively adsorbed by hydrophilic adsorbents, thus reducing the overall CO2 sorption capacity.28−30 Although mesoporous materials such as zeolites and activated carbons were already tested in pilot plants,31,32 the potential of COFs as alternative sorbents in the CCS technology has not been explored. However, to fully develop the potential of heterogeneous sorbents in CCS, understanding the interactions that account for CO2 adsorption at the molecular level is key.33−35
Here, we address this challenge by studying CO2 sorption in tertiary amine-functionalized COFs by a combination of adsorption isotherm measurements and solid-state nuclear magnetic resonance (NMR) spectroscopy, complemented by quantum chemical calculations, obtained on B97-2/pcS-2/PBE0-D3/def2-TZVP level of theory36−41 using the Turbomole42,43 program package for geometries and the FermiONs++44,45 program package for the calculation of NMR chemical shifts. DETH linker molecules were modified by insertion of a terminal tertiary amine group and integrated into two different hydrazone-linked COF systems. To adjust the linker functionalization level and study the influence of linker modification on the structural and sorption properties of the COF, a three-linker approach was developed inspired by classical copolymerization. We show that CO2 sorption capacities as well as heats of adsorptions can be increased by this strategy. To the best of our knowledge, this is the first time the molecular interaction of CO2 with a COF material was studied. CO2 was found to adsorb at tertiary amine sites through water-mediated formation of a bicarbonate species.
Results and Discussion
Synthesis and Structural Characterization
Two COF systems with different amounts of the amine-functionalized linker of 2,5-bis(2-(dimethylamino)ethoxy)terephthalohydrazide (DtATH, see Figure 1) were synthesized by a copolymerization approach using two generic COF systems.
Figure 1.

Synthesis of COF-42 (coCOF–H) from DETH and TFB (left) and HTFG–COF (coCOF–OH) from DETH and 2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde (TFG, right). Structure of DtATH, center.
The first system, named amine–coCOF–OH, is based on the hydrazone-linked HTFG–COF (coCOF–OH) that is synthesized by solvothermal condensation of DETH (see Figure 1) and 2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde (TFG, see Figure 1). The second system, COF-42 (coCOF–H), was synthesized similarly by condensation of DETH and 1,3,5-triformylbenzene (TFB, see Figure 1) and is referred to as amine–coCOF–H in the following.
For the synthesis of DtATH-containing samples, various amounts of DETH (25, 50, 75, and 100% substitution of the original DETH linker) were substituted by the respective amount of DtATH and the mixed linkers exposed to the initial COF synthesis protocol of the underlying coCOF systems coCOF–OH and coCOF–H. The solvent compositions were optimized to obtain high surface area and crystallinity in the modified samples.
COFs were characterized by Fourier transform infrared (FT-IR) spectroscopy, sorption analysis, powder X-ray diffraction (PXRD), and solid-state NMR. As seen in Figure S1, the FT-IR spectrum of coCOF–OH shows the characteristic C=O stretching vibrations of the β-ketoenamine carbonyl group at 1680 cm–1. No residual aldehyde stretches are visible, indicating the complete transformation of the starting material. Comparison with the monomers corroborates the formation of the hydrazone bond. The same was found in coCOF–H as well as in the amine-containing samples. Addition of the tertiary amine linker further leads to color deepening in the samples from light yellow to orange in coCOF–OH and from orange to reddish-brown in coCOF–H as is also visible in the solid-state UV/vis absorption spectra (see Figure S3).
Solid-state 1D 13C{1H} CP-MAS NMR further supports the bond formation and linker integration in both systems. The 13C NMR signals are assigned to the different carbon atoms shown schematically in Figure 2a,e as indicated by the labels in Figure 2b,f. The signals assigned to the ethoxy group were observed at 66 and 15 ppm. Amine-containing samples show additional peaks at 45 and 57 ppm (Figure 2c,d,g,h) that can be attributed to the aminoethoxy and dimethylamine groups, respectively, as corroborated by quantum chemical calculations for a model compound (see Table S3). In the molecular linker, the respective carbon center shows a 13C chemical shift of 45.1 ppm (see the Supporting Information). The relative intensity of the 13C NMR signal at 45 ppm increases with higher amount of amine in the synthesis mixture, consistent with greater incorporation of the amine linker into the COF framework.
Figure 2.
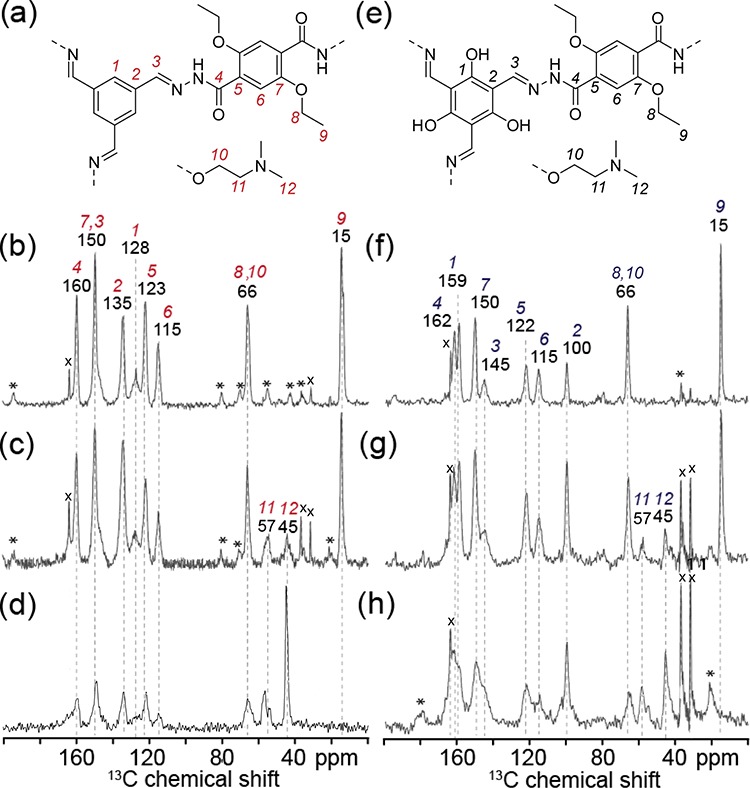
Schematic structural diagrams showing subsections of the (a) coCOF–H framework, (e) coCOF–OH framework, and the tertiary amine linker DtATH. Solid-state one-dimensional (1D) 13C{1H} CP-MAS NMR spectra of (b–d) coCOF–H and (f–h) coCOF–OH with (b,f) 0%, (c,g) 50%, and (d,h) 100% of DtATH substitution of the original DETH linker. The spectra in (b–d) and (f–h) were acquired at 11.7 T, 10 kHz MAS, 298 K, using cross-polarization contact times of 5 ms. The NMR spectrum (d) was acquired at 11.7 T, 12 kHz MAS, 298 K, and using cross-polarized contact times of 5 ms. Spinning sidebands are marked with asterisks. Distinct carbon atoms in the schematic structures in (a–e) are numbered and their associated 13C NMR signals labeled accordingly in (b–d) and (f–h). The narrow signals labeled with crosses at 164, 37, and 32 ppm correspond to residual dimethylformamide and at 25 ppm to residual tetrahydrofuran.46
PXRD confirms the formation of crystalline COF networks with unit cell dimensions being consistent with the structural models shown in Figure 3c. For coCOF–OH, a strong reflection at 3.4° and weaker ones at 5.5, 7.0, and 26.2° are assigned to the 100, 110, 200, and 001 Miller indices, respectively (see Figure 3a). The PXRD data match well with an AA eclipsed stacking structure with an interlayer distance of 3.48 Å because of π–π-stacking interactions (see Figure 3c). It should be noted that a lateral offset of 1.7–1.8 Å is expected but cannot be distinguished from the AA eclipsed stacking structure because of broadening of the reflections.47,48 Further investigations were carried out with the AA eclipsed model. Pawley refinement on the simulated structure suggests a P6/m space group with a = b = 29.6 Å and α = β = 90°, y = 120°.
Figure 3.
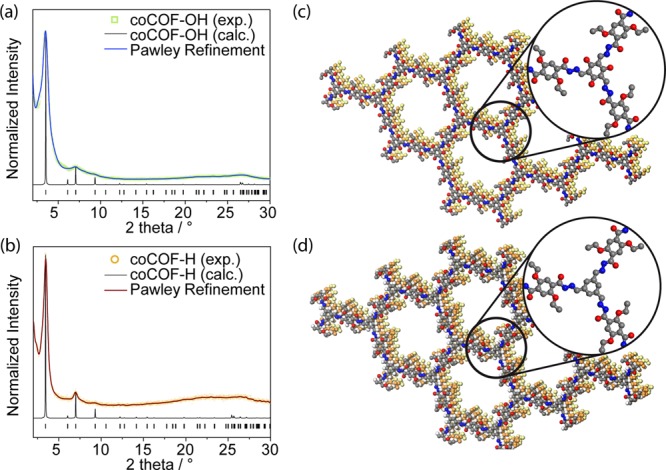
(a) PXRD pattern of coCOF–OH (open green squares), Pawley refined profile (blue line), and calculated XRD pattern for the idealized eclipsed (AA) stacking (black line). (b) PXRD pattern of coCOF–H (open orange circles), Pawley refined profile (red line), and calculated XRD pattern for the idealized eclipsed (AA) stacking (black line). (c) and (d) Eclipsed stacking model for coCOF–OH and coCOF–H, respectively. C, N, and O atoms are represented in gray, blue, and red, respectively. H atoms are omitted. The second and third layers are represented in orange and yellow for clarity, respectively.
In general, coCOF–H appears more crystalline than coCOF–OH. Introducing the modified linker leads to a further loss in crystallinity, whereas the architecture and dimensions of the unit cell are maintained. The 100, 110, 200, and 001 reflections of 100%-amine–coCOF–OH are found at 3.4, 5.7, 6.9, and 26.3°, respectively, which suggests retention of the stacking structure discussed above. The crystallinity decreases with higher amine content (see Figure S2). The loss of crystallinity is more distinct in amine–coCOF–OH which we attribute to a loss of reversibility in the bond formation and the coexistence of different tautomeric forms which is known for COFs based on the TFG linker.49
Adsorption Performance
According to argon sorption measurements at 87 K, both systems show characteristic type IV isotherms that are typical for mesoporous materials (see Figure 4). Brunauer–Emmett–Teller (BET) surface areas were calculated to be 998 m2 g–1 for coCOF–OH and 2336 m2 g–1 for coCOF–H, which surpasses the published values for both COFs (757 and 710 m2 g–1 for coCOF–OH and coCOF–H, respectively46). Pore size distributions (PSD) were derived from experimental data using nonlocal density functional theory and quenched solid-state functional theory calculations.50 In coCOF–H, the experimental pore size of 2.4 nm is in agreement with the theoretical value based on the structural model. Additionally, micropores of 0.92 and 0.61 nm are observed which points to structural effects such as mismatch stacking, leading to reduced pore sizes or pore blocking. The mesopores with 2.4 nm diameter account for 65% of the pore volume, whereas the smaller micropores represent 7.4% (for 0.92 nm) and 8.3% (for 0.61 nm) of the total pore volume. A similar trend is observed in coCOF–OH with a broader distribution of mesopores at 2.3 nm (55% pore volume) caused by the loss of long-range order because of tautomerism (see Figure S5). The smaller pores are found at 0.91 nm (26% pore volume) and 0.61 nm (16% pore volume).
Figure 4.

(a) Argon adsorption isotherms of coCOF–H (red) and coCOF–OH (blue). Water sorption at 273 K of (b) amine–coCOF–H and (c) amine–coCOF–OH with 0, 50, and 100% DtATH substitution of the original DETH linker. Adsorption is represented by filled symbols, desorption by open symbols. (d) Relative CO2 adsorption capacities at 273 K and BET surface areas of amine–coCOF–OH (blue and purple) and amine–coCOF–H (red and orange). BET surface area is indicated by triangles.
For amine-containing samples, BET surface areas decrease linearly with an increasing amount of tertiary amine. Respective values for all samples are listed in Table 1. We derived PSDs from Ar isotherms for the samples containing 50 and 100% modified linker for both systems (see Figures S4 and S5). Although the mean pore size of the pristine COFs is around 2.4 nm, additional smaller pores in the range of 1.4–1.8 nm are found for the amine-containing samples. This is in agreement with theoretical values for amine-modified pores, which vary between 1.6 and 2.2 nm depending on the amine conformation. A stochastic distribution of the different linkers in the systems along with stacking faults will lead to pores with different amounts of amine and therefore to different pore sizes, especially in the mixed systems. A broader distribution of pore sizes with more regular distribution of pore volumes is found in the 50% amine samples (see Table S2). In the 100% amine samples, two distinct pore sizes of 2.2 and 1.6 nm are found which are attributed to different pore surface architectures with amines either at the pore wall or protruding into the pore (see Figure S6).
Table 1. BET Surface Areas, CO2 Uptake at 273 K, Relative CO2 Adsorption at 273 K, and Heats of CO2 Adsorption of the Presented COFs.
| COF system | amount of DtATH (%) | BET SAa [m2 g–1] | CO2 uptake at 273 K [mmol g–1] | relative CO2 adsorption [μmol m–2] | Qstb [kJ mol–1] |
|---|---|---|---|---|---|
| amine–coCOF–H | 0 | 2336 | 2.66 | 1.14 | 24.0 |
| 25 | 1705 | 2.12 | 1.24 | 37.0 | |
| 50 | 811 | 1.60 | 1.97 | 40.4 | |
| 75 | 573 | 1.11 | 1.93 | 54.0 | |
| 100 | 514 | 1.14 | 2.22 | 72.4 | |
| amine–coCOF–OH | 0 | 998 | 1.74 | 1.75 | 36.7 |
| 25 | 822 | 1.60 | 1.95 | 47.9 | |
| 50 | 675 | 1.42 | 2.10 | 49.6 | |
| 75 | 581 | 1.27 | 2.19 | 66.0 | |
| 100 | 412 | 1.04 | 2.52 | 48.5 |
From Ar sorption measurements.
At zero coverage.
With respect to the CO2 sorption isotherms of both systems, a linear decrease was observed for the uptake capacity ranging from 2.66 to 1.14 mmol g–1 for coCOF–H (0–100% modification) and from 1.74 to 1.04 mmol g–1 for coCOF–OH (0–100% modification; see Table 1). Interestingly, the loss in uptake capacity with increasing amine functionalization is significantly less than the decrease of surface area. Normalizing the CO2 uptake to the BET surface area of the samples (see Table 1) to obtain relative rather than absolute CO2 capacities, the uptake increases from 1.14 to 2.22 μmol m–2 (0–100% modification) for the amine–coCOF–H. For amine–coCOF–OH, the relative CO2 adsorption increases from 1.75 to 2.52 μmol m–2 (see Figure 4b). Whereas at 50% amine loading, the uptake is fairly similar for both systems (2.10 μmol m–2 for amine–coCOF–OH and 1.93 μmol m–2 for amine–coCOF–H), in the other amine loading regimes, the relative CO2 capacity of the more polar amine–coCOF–OH distinctly surpasses amine–coCOF–H.
Even though water is present in most applications, the behavior of COFs in water sorption experiments has rarely been studied systematically. Most studies have been performed on MOFs or porous carbons that show very different behaviors. Hydrophilicity is more pronounced in the case of MOFs because of their metal sites than on the nonpolar hydrophobic surface of carbon materials. However, MOFs are often not stable in water which leads to their degradation under humid conditions.51 COFs are expected to exhibit hydration properties that are intermediate between MOFs and carbons, where a more polar surface due to heteroatoms in the framework gives rise to a type IV sorption isotherm and a fully reversible hysteresis at lower relative pressures compared to nonpolar surfaces.
The two pristine COF systems in this study show similar behavior in water sorption measurements. The water uptake capacity is higher for amine–coCOF–H, ranging from 598 to 768 cm3 g–1 (47–61 wt %) and 416–481 cm3 g–1 (33 to 38 wt %). In both systems, the highest capacity is found for the 100%-amine samples and the lowest for the 50%-amine samples. This is likely due to a higher degree of disorder because of the distribution of modified and unmodified linkers in the 50%-amine samples. The adsorption isotherms of both systems show a step in the range of 0.30–0.45 p/p0 with a strong hysteresis; the same step is found in the desorption isotherm at 0.20–0.30 p/p0. This behavior is indicative of capillary condensation of water in the COF pores. By increasing the amine content in the samples, in both systems, the adsorption step flattens out up to an almost linear sorption isotherm in 100%-amine–coCOF–OH. This continuous pore filling might be due to a slightly higher polarity in the system. Interestingly, water sorption is not fully reversible. Fractions ranging from 6.8% (for pristine coCOF–OH) up to 11.4% (in the case of 100%-amine–coCOF–OH) of the maximum water uptake remain in the pores after desorption. The addition of amines in close proximity to the pore walls leads to higher hydrophilicity and thus increased water sorption capacities.
Isosteric heats of adsorption (Qst) at zero coverage were calculated for all samples from the CO2 sorption isotherms at 273, 288, and 298 K (see Table 1). Typical values for classical physisorption range between 8 and 25 kJ mol–1 for van der Waals forces and up to 50 kJ mol–1 for dipole–dipole interactions, whereas chemisorption is associated with heats of adsorption between 80 and 500 kJ mol–1. The values obtained in our unmodified coCOFs are 24.0 kJ mol–1 for coCOF–H and 36.7 kJ mol–1 for coCOF–OH. Upon modification, the Qst values increase drastically with maximum values of 72.4 kJ mol–1 in 100%-amine–coCOF–H and 66.0 kJ mol–1 in 75%-amine–coCOF–OH. Those values approach the chemisorption regime and are much higher than that for comparable COFs, such as COF-JLU2 (31 kJ mol–1),52 TRIPTA (56.77 kJ mol–1),53 [HO2C]100%-H2P–COF (43.5 kJ mol–1),27 ACOF-1 (27.6 kJ mol–1),54 TpPA–COF (34.1 kJ mol–1),55 or other porous materials such as FCTF-1 (35.0 kJ mol–1),56 MgMOF-74 (42 kJ mol–1),57 and imine-linked porous organic cages (20.4 kJ mol–1).58
Amine-Modification of the coCOF–H Framework
To understand the improved CO2 sorption properties of 100%-amine–coCOF–H, advanced 1D and 2D 1H, 13C, and 15N solid-state NMR techniques were used to elucidate atomic-level structures and interactions in the modified COF framework and compared with findings from quantum chemical calculations. The 15N chemical shift interaction is highly sensitive to local bonding environments;59,60 however, 15N NMR experiments are severely limited for low-density and low-nitrogen content materials by the low natural isotopic abundance (0.4%) and low gyromagnetic ratio of the 15N nuclei. These limitations are partially overcome by dynamic nuclear polarization (DNP)-enhanced NMR spectroscopy, which uses microwave excitation of nitroxide biradical polarizing agents to achieve a potential 15N sensitivity gain of γe/γ15N ≈ 6500.61 Here, DNP–NMR enables the acquisition of natural-abundance 15N spectra as demonstrated in Figure 5.
Figure 5.
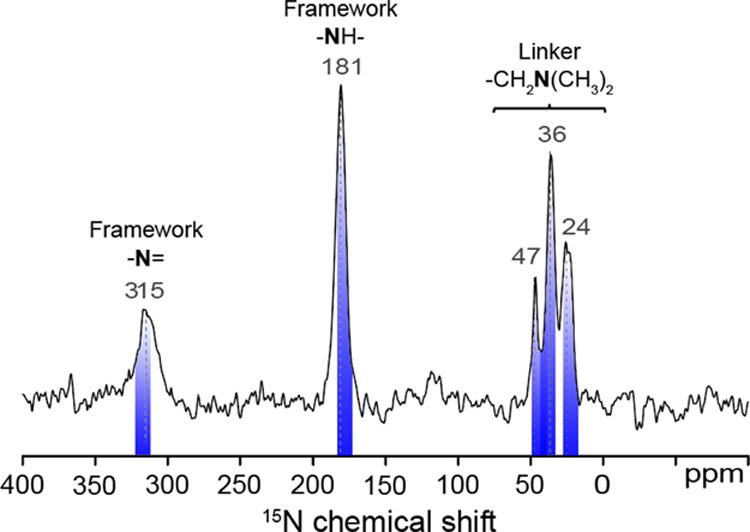
Solid-state 1D 15N{1H} DNP–CP–MAS spectra of 100%-amine–coCOF–H without CO2 exposure. The spectrum was acquired at 9.4 T, 8 kHz MAS, 95 K, in the presence of 16 mM AMUPol biradical in 60:30:10 d8-glycerol/D2O/H2O, under microwave irradiation at 263 GHz, and using cross-polarization contact times of 5 ms. Blue markings correspond to values obtained by quantum-chemical calculations (See Tables S4, S5, and S7).
Although amine-functionalized nanoporous or mesoporous solids typically exhibit broad 15N signals due to structural disorder,62−64 the 15N signals from the DtATH linker exhibit narrow 15N line shapes, which indicates relatively uniform local environments in the COF framework. The 15N signals at 315 and 181 ppm are assigned to framework hydrazone (−N=) and (−NH−) moieties, respectively, as supported by quantum chemical calculations (see Figures S11, S12 and Table S4) for a coCOF–H sub-structure and literature.59 A hydrogen bond to water causes displacement of the imine signal to lower frequency by approximately 12 ppm compared to the bare imine bond (see Figure S13 and Table S4). The presence of these signals, in addition to quantum chemical data, strongly suggest that after incorporation of DtATH into the COF framework the hydrazone (−N=) and (−NHCO−) linkages are intact and retain an atomic structure similar to unmodified coCOF–H.
In 100%-amine–coCOF–H, there are three additional 15N signals at 24, 36, and 47 ppm, which arise from the DtATH linker. For tertiary amines, the 15N chemical shift may be influenced by local bonding environments, which can be influenced by temperature, solvent effects, or hydrogen-bonding interactions to varying extents and which can displace 15N signals by as much as 40 ppm.60 In polar or acidic solvents, tertiary amines often exhibit partial deshielding of 15N nuclei, as manifested by displacement of their isotropic chemical shifts to higher values.60 Consequently, the 15N signal at 47 ppm is assigned to protonated tertiary amine linker groups, consistent with quantum chemical calculations (see Figure S14 and Table S7). The 15N signals at 24 and 36 ppm are attributed to unprotonated DtATH tertiary amine moieties also on the basis of quantum-chemical calculations (Figure S13), with the former assigned to unhydrated linkers. The 15N signal at 36 ppm is attributed to DtATH tertiary amine moieties that interact strongly with water.
CO2 Interactions with Amine-coCOF-H Moieties
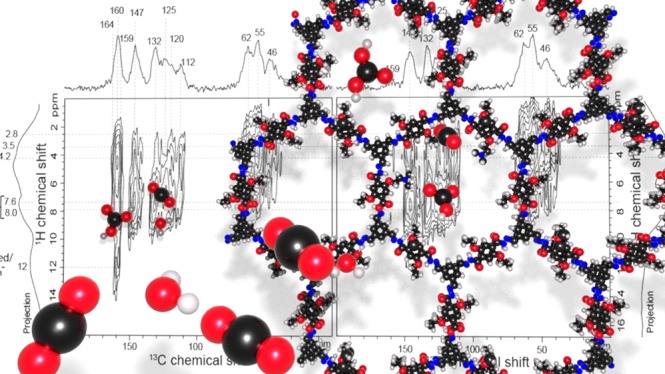
Molecular-level insights on specific interactions between CO2 and 100%-amine–coCOF–H are obtained from 2D 13C{1H}-heteronuclear correlation (HETCOR) analyses that establish spatial proximities of adsorbed CO2 and the COF sorbent. Previously, site-specific CO2 adsorption in tertiary amide (−NHCOR)-containing mesoporous materials has been investigated by inelastic neutron spectroscopy for which subtle differences in local chemical environments are difficult to resolve.65 By comparison, the 2D 13C{1H} low-temperature magic-angle-spinning (LTMAS)–HETCOR spectra (Figure 6) of 100%-amine–coCOF–H can detect and resolve atomic-level interactions of specific COF framework moieties with adsorbed water and with adsorbed CO2.
Figure 6.

Solid-state 2D 13C{1H} LTMAS–HETCOR spectra of vacuum-dried 100%-amine–coCOF–H (a) after exposure to 100% 13C-enriched CO2 for 12 h at 1 bar pressure and 298 K and (b) after desorption of CO2 for 48 h by vacuum heating at 0.1 bar and 363 K. The spectra were acquired at 9.4 T, 8 kHz MAS, 95 K using short cross-polarization contact times of 500 μs. 1D 13C projections are shown along the horizontal axes for comparison with the 2D spectra, and 1D 1H projections are shown along the vertical axes. Strong correlated 13C signal intensity (ca. 160 ppm) with 1H signal at 12–14 ppm establishes that CO2 chemisorbs to form a bicarbonate (HCO3–) species.
Specifically, 2D 13C{1H}-HETCOR methods rely on through-space dipole-dipole interactions to selectively detect 13C nuclei which are in molecular-level proximity (<1 nm) to 1H nuclei of both directly bound and neighboring moieties. The resulting 2D 13C{1H} spectrum represents a correlated intensity map that resolves spatially from molecularly near moieties on the basis of their isotropic 1H and 13C chemical shifts, which are sensitive to local bonding environments. For example, the 2D 13C{1H} HETCOR spectra in Figure 6a,b acquired for 100%-amine–coCOF–H, after exposure to 13C-enriched CO2 and then after subsequent degassing, respectively, both show intensity correlations arising from intramolecular correlations within the coCOF–H framework. These include the strong 13C signals at 46, 55, and 62 ppm from alkyl carbon atoms in the DtATH linker which are correlated with 1H signals from alkyl protons at 2.0–3.5 ppm; and 13C signals ranging from 112 to 147 ppm from aromatic carbon atoms in the 100%-amine–coCOF–H backbone that are strongly correlated with 1H signals at 7.0–8.0 ppm from aromatic protons. The framework amide moieties exhibit a 1H chemical shift at 11.7 ppm, which is consistent with a 2D 15N{1H} DNP–HETCOR spectrum of 100%-amine–coCOF–H (Supporting Information, Figure S10b) and a 1D solution-state 1H NMR spectrum of a small-molecular analogue (see the Supporting Information, compound 6). A correlated 2D intensity is also observed between the 13C signal at ca. 160 ppm and a new 1H signal in the range 12–14 ppm, which is assigned to intramolecular HCO3– interactions (green band).66 Nearly all of the 13C signals are correlated with 1H intensity centered at 4.2 ppm from adsorbed H2O. More interestingly, for 100%-amine–coCOF–H exposed to 13C-enriched CO2 (Figure 6a), the 13C intensity in the range 160–164 ppm is also strongly correlated with 1H signals at 4.2 and 7.0–8.0 and 11.7 ppm, which are assigned to adsorbed H2O (blue band) and hydrazone and/or aromatic 1H moieties (beige band), and amide groups (purple band), respectively. Such 2D intensity correlations unambiguously establish that chemisorption of CO2 occurs in close molecular proximities to these moieties, which are consistent with the isotropic 13C chemical shifts that have been reported for the formation of bicarbonate species in tertiary amine solutions.67 The breadth of the 13C intensity reflects a distribution of solvated neutral (160 ppm)68 and ionic bicarbonate species (171 ppm from quantum chemical calculations, see Table S6). The 2D NMR results thus establish that HCO3– strongly interacts with adsorbed H2O and amide and/or aromatic 1H moieties in the 100%-amine–coCOF–H framework.
As shown by the 1D 13C{1H} DNP-CP MAS spectra in Figure S9a,b, the 13C amide signal (orange overbar) has stronger intensity for the longest CP contact time of 5 ms. Although 13C-depleted glycerol was used in the DNP solvent formulation, there is a small intensity shoulder ranging from 65 to 80 ppm from glycerol. By comparison, the 1D 13C{1H} LTMAS–CP MAS spectra in Figure S9c,d were acquired on vacuum-dried 100%-amine–coCOF–H upon exposure to dry 100% 13C-enriched CO2 and after subsequent degassing step. As discussed in the experimental section, these materials were characterized without DNP to minimally influence adsorbed CO2. Under otherwise identical conditions, there is significantly more 13C signal at 160 ppm for the material exposed to 13C-enriched CO2. In the 1D spectra, the adsorbed bicarbonate (red overbar) and amide have overlapping signal intensity at 160 ppm. By comparison, for the short contact time (500 μs) used in Figure 6, only the strongest 13C{1H} dipole-dipole-coupled moieties are expected to yield correlated intensity, which is consistent with the reduced signals from the amide moieties, the carbon atoms of which lack a directly bonded 1H atom.
Despite degassing and drying the sorbent prior to CO2 adsorption, 100%-amine–coCOF–H strongly retains adsorbed H2O, which favors the formation of bicarbonates. Evidence for hydrogen-bonding interactions between H2O and the framework amide (−NH−) moieties was also observed in a 2D 15N{1H} DNP–HETCOR spectrum (see the Supporting Information, Figure S8). After desorption of the CO2 100% 13C-enriched CO2, the 13C signal (ca. 160 ppm) from bicarbonate completely disappears. However, there are still strong intensity correlations associated with 13C moieties in 100%-amine–coCOF–H and 1H moieties from adsorbed H2O at ca. 4.2 ppm (blue band) and only very weak correlated intensity associated with the amide 13C signal remains at ca. 159 ppm, which is consistent with the observed water desorption behavior of the samples. The retention of H2O in 100%-amine–coCOF–H likely contributes to the reduction in the apparent BET surface area. Stronger interactions with CO2 are usually attributed to a higher amount of heteroatoms, mostly nitrogen and oxygen, on the pore walls of porous framework materials, because of the higher interaction affinity of the heteroatoms to CO2.3,69,70 In the context of the aminated COF materials, the interaction with water cannot be neglected. In addition to CO2 adsorption functionality, the amine groups impart hydrophilicity that leads to increased water uptake by the framework. The increased network hydrophilicity promotes H2O adsorption and deprotonation near the basic amine side chains, which promotes CO2 coadsorption as bicarbonate species in the 100%-amine–coCOF–H pores. Note that increasing amounts of adsorbed water in the pores with increasing degree of amine functionalization is consistent with the water isotherms discussed above.
Conclusions
In this study, a mixed linker strategy was used to modify coCOF–H and coCOF–OH with a tertiary amine functionality by copolymerization of isostructural linkers. Addition of the functionalized linker species yields a higher affinity to CO2 as shown by an increased relative CO2 adsorption capacity, along with an increase of the heat of adsorption at zero coverage up to a value of 72.4 kJ mol–1. We demonstrate by solid-state 2D 13C{1H} NMR analyses, supplemented by quantum chemical NMR calculations, that CO2 sorption in the 100%-amine–coCOF–H pores proceeds via formation of a bicarbonate species adsorbed within the COF–CO2 pores, along with water which is strongly retained. Thus, the hydrophilicity of the COF framework appears to promote increased CO2 sorption capacity, with different hydrophilicities leading to distinctly different adsorption behaviors of water in the pores. This is likely the reason that increased extents of framework functionalization with amine species lead to increased CO2 affinity because of the formation of bicarbonate species. Such effects are partially offset by decreased surface area because of steric effects associated with the linkers, as well as strongly retained water in the pores. Tuning the COF’s inherent water sorption properties by introducing functional groups such as tertiary amines or amides that promote CO2 solvation71 is expected to further enhance CO2 adsorption in porous systems.
Acknowledgments
Financial support is gratefully acknowledged from the Max Planck Society, the ERC Starting Grant (project COF Leaf, grant number 639233), the cluster of excellence “Nanosystems Initiative München” (NIM), the Center for NanoScience (CeNS), and for the UCSB group, the U.S. National Science Foundation (award no. CBET-1335694). We thank Prof. T. Bein and Prof. W. Schnick (University of Munich, LMU) for granting access to the XRD facility and V. Duppel for the assistance with material analysis. The solid-state NMR measurements were conducted using the MRL Shared Experimental Facilities supported by the MRSEC Program of the U.S. NSF under award no. DMR 1720256, a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org). Z.J.B. acknowledges funding from the BASF corporation. C.O. acknowledges also support as Max-Planck-Fellow at MPI-FKF Stuttgart.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater.8b04643.
Synthesis and methods, additional measurements and characterization, and details of quantum-chemical calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hug S.; Mesch M. B.; Oh H.; Popp N.; Hirscher M.; Senker J.; Lotsch B. V. A fluorene based covalent triazine framework with high CO2 and H2 capture and storage capacities. J. Mater. Chem. A 2014, 2, 5928–5936. 10.1039/c3ta15417c. [DOI] [Google Scholar]
- Doonan C. J.; Tranchemontagne D. J.; Glover T. G.; Hunt J. R.; Yaghi O. M. Exceptional ammonia uptake by a covalent organic framework. Nat. Chem. 2010, 2, 235–238. 10.1038/nchem.548. [DOI] [PubMed] [Google Scholar]
- Li Z.; Feng X.; Zou Y.; Zhang Y.; Xia H.; Liu X.; Mu Y. A 2D azine-linked covalent organic framework for gas storage applications. Chem. Commun. 2014, 50, 13825–13828. 10.1039/c4cc05665e. [DOI] [PubMed] [Google Scholar]
- Wan S.; Guo J.; Kim J.; Ihee H.; Jiang D. A Belt-Shaped, Blue Luminescent, and Semiconducting Covalent Organic Framework. Angew. Chem., Int. Ed. 2008, 47, 8826–8830. 10.1002/anie.200803826. [DOI] [PubMed] [Google Scholar]
- Wan S.; Guo J.; Kim J.; Ihee H.; Jiang D. A Photoconductive Covalent Organic Framework: Self-Condensed Arene Cubes Composed of Eclipsed 2D Polypyrene Sheets for Photocurrent Generation. Angew. Chem., Int. Ed. 2009, 48, 5439–5442. 10.1002/anie.200900881. [DOI] [PubMed] [Google Scholar]
- Dogru M.; Handloser M.; Auras F.; Kunz T.; Medina D.; Hartschuh A.; Knochel P.; Bein T. A Photoconductive Thienothiophene-Based Covalent Organic Framework Showing Charge Transfer Towards Included Fullerene. Angew. Chem., Int. Ed. 2013, 52, 2920–2924. 10.1002/anie.201208514. [DOI] [PubMed] [Google Scholar]
- Stegbauer L.; Schwinghammer K.; Lotsch B. V. A hydrazone-based covalent organic framework for photocatalytic hydrogen production. Chem. Sci. 2014, 5, 2789–2793. 10.1039/c4sc00016a. [DOI] [Google Scholar]
- Cote A. P.; Benin A. I.; Ockwig N. W.; O’Keeffe M.; Matzger A. J.; Yaghi O. M. <Porous, Crystalline, Covalent Organic Frameworks Yaghi 2005.pdf>. Science 2005, 310, 1166–1170. 10.1126/science.1120411. [DOI] [PubMed] [Google Scholar]
- Ding S.-Y.; Wang W. Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 2013, 42, 548–568. 10.1039/c2cs35072f. [DOI] [PubMed] [Google Scholar]
- Tilford R. W.; Mugavero S. J.; Pellechia P. J.; Lavigne J. J. Tailoring Microporosity in Covalent Organic Frameworks. Adv. Mater. 2008, 20, 2741–2746. 10.1002/adma.200800030. [DOI] [PubMed] [Google Scholar]
- Stegbauer L.; Zech S.; Savasci G.; Banerjee T.; Podjaski F.; Schwinghammer K.; Ochsenfeld C.; Lotsch B. V. Tailor-Made Photoconductive Pyrene-Based Covalent Organic Frameworks for Visible-Light Driven Hydrogen Generation. Adv. Energy Mater. 2018, 8, 1703278. 10.1002/aenm.201703278. [DOI] [Google Scholar]
- Nagai A.; Guo Z.; Feng X.; Jin S.; Chen X.; Ding X.; Jiang D. Pore surface engineering in covalent organic frameworks. Nat. Commun. 2011, 2, 536. 10.1038/ncomms1542. [DOI] [PubMed] [Google Scholar]
- Li Z.-J.; Ding S.-Y.; Xue H.-D.; Cao W.; Wang W. Synthesis of -C=N- linked covalent organic frameworks via the direct condensation of acetals and amines. Chem. Commun. 2016, 52, 7217–7220. 10.1039/c6cc00947f. [DOI] [PubMed] [Google Scholar]
- Chen X.; Addicoat M.; Jin E.; Xu H.; Hayashi T.; Xu F.; Huang N.; Irle S.; Jiang D. Designed synthesis of double-stage two-dimensional covalent organic frameworks. Sci. Rep. 2015, 5, 14650. 10.1038/srep14650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Shen X.; Feng X.; Xia H.; Mu Y.; Liu X. Covalent organic frameworks as pH responsive signaling scaffolds. Chem. Commun. 2016, 52, 11088–11091. 10.1039/c6cc05748a. [DOI] [PubMed] [Google Scholar]
- Kang Z.; Peng Y.; Qian Y.; Yuan D.; Addicoat M. A.; Heine T.; Hu Z.; Tee L.; Guo Z.; Zhao D. Mixed Matrix Membranes (MMMs) Comprising Exfoliated 2D Covalent Organic Frameworks (COFs) for Efficient CO2 Separation. Chem. Mater. 2016, 28, 1277–1285. 10.1021/acs.chemmater.5b02902. [DOI] [Google Scholar]
- Olajire A. A. Recent advances in the synthesis of covalent organic frameworks for CO2 capture. J. CO2 Util. 2017, 17, 137–161. 10.1016/j.jcou.2016.12.003. [DOI] [Google Scholar]
- Chowdhury F. A.; Yamada H.; Higashii T.; Goto K.; Onoda M. CO2 Capture by Tertiary Amine Absorbents: A Performance Comparison Study. Ind. Eng. Chem. Res. 2013, 52, 8323–8331. 10.1021/ie400825u. [DOI] [Google Scholar]
- Ko Y. G.; Shin S. S.; Choi U. S. Primary, secondary, and tertiary amines for CO2 capture: Designing for mesoporous CO2 adsorbents. J. Colloid Interface Sci. 2011, 361, 594–602. 10.1016/j.jcis.2011.03.045. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Liang Z.; Liu H.; Rongwong W.; Luo X.; Idem R.; Yang Q. Study of Formation of Bicarbonate Ions in CO2-Loaded Aqueous Single 1DMA2P and MDEA Tertiary Amines and Blended MEA–1DMA2P and MEA–MDEA Amines for Low Heat of Regeneration. Ind. Eng. Chem. Res. 2016, 55, 3710–3717. 10.1021/acs.iecr.5b03097. [DOI] [Google Scholar]
- Rochelle G. T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652–1654. 10.1126/science.1176731. [DOI] [PubMed] [Google Scholar]
- Wang J.; Huang L.; Yang R.; Zhang Z.; Wu J.; Gao Y.; Wang Q.; O’Hare D.; Zhong Z. Recent advances in solid sorbents for CO2 capture and new development trends. Energy Environ. Sci. 2014, 7, 3478–3518. 10.1039/c4ee01647e. [DOI] [Google Scholar]
- Titirici M.-M.; White R. J.; Brun N.; Budarin V. L.; Su D. S.; del Monte F.; Clark J. H.; MacLachlan M. J. Sustainable carbon materials. Chem. Soc. Rev. 2015, 44, 250–290. 10.1039/c4cs00232f. [DOI] [PubMed] [Google Scholar]
- Liu J.; Thallapally P. K.; McGrail B. P.; Brown D. R.; Liu J. Progress in adsorption-based CO2 capture by metal–organic frameworks. Chem. Soc. Rev. 2012, 41, 2308–2322. 10.1039/c1cs15221a. [DOI] [PubMed] [Google Scholar]
- Sumida K.; Rogow D. L.; Mason J. A.; McDonald T. M.; Bloch E. D.; Herm Z. R.; Bae T.-H.; Long J. R. Carbon Dioxide Capture in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 724–781. 10.1021/cr2003272. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Lu J.; Zhang J. Porosity Enhancement of Carbazolic Porous Organic Frameworks Using Dendritic Building Blocks for Gas Storage and Separation. Chem. Mater. 2014, 26, 4023–4029. 10.1021/cm501717c. [DOI] [Google Scholar]
- Huang N.; Chen X.; Krishna R.; Jiang D. Two-Dimensional Covalent Organic Frameworks for Carbon Dioxide Capture through Channel-Wall Functionalization. Angew. Chem. 2015, 127, 3029–3033. 10.1002/ange.201411262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizzie A. C.; Wong-Foy A. G.; Matzger A. J. Effect of Humidity on the Performance of Microporous Coordination Polymers as Adsorbents for CO2 Capture. Langmuir 2011, 27, 6368–6373. 10.1021/la200547k. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang Y.; Benin A. I.; Jakubczak P.; Willis R. R.; LeVan M. D. CO2/H2O Adsorption Equilibrium and Rates on Metal–Organic Frameworks: HKUST-1 and Ni/DOBDC. Langmuir 2010, 26, 14301–14307. 10.1021/la102359q. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Marshall M.; Chaffee A. L. CO2 Adsorption-Based Separation by Metal Organic Framework (Cu-BTC) versus Zeolite (13X). Energy Fuels 2009, 23, 2785–2789. 10.1021/ef800938e. [DOI] [Google Scholar]
- Krishnamurthy S.; Rao V. R.; Guntuka S.; Sharratt P.; Haghpanah R.; Rajendran A.; Amanullah M.; Karimi I. A.; Farooq S. CO2 capture from dry flue gas by vacuum swing adsorption: A pilot plant study. AIChE J. 2014, 60, 1830–1842. 10.1002/aic.14435. [DOI] [Google Scholar]
- Webley P. A. Adsorption technology for CO2 separation and capture: a perspective. Adsorption 2014, 20, 225–231. 10.1007/s10450-014-9603-2. [DOI] [Google Scholar]
- Kong X.; Scott E.; Ding W.; Mason J. A.; Long J. R.; Reimer J. A. CO2 Dynamics in a Metal–Organic Framework with Open Metal Sites. J. Am. Chem. Soc. 2012, 134, 14341–14344. 10.1021/ja306822p. [DOI] [PubMed] [Google Scholar]
- Lin L.-C.; Kim J.; Kong X.; Scott E.; McDonald T. M.; Long J. R.; Reimer J. A.; Smit B. Understanding CO2 Dynamics in Metal–Organic Frameworks with Open Metal Sites. Angew. Chem., Int. Ed. 2013, 52, 4410–4413. 10.1002/anie.201300446. [DOI] [PubMed] [Google Scholar]
- McDonald T. M.; Mason J. A.; Kong X.; Bloch E. D.; Gygi D.; Dani A.; Crocellà V.; Giordanino F.; Odoh S. O.; Drisdell W. S.; Vlaisavljevich B.; Dzubak A. L.; Poloni R.; Schnell S. K.; Planas N.; Lee K.; Pascal T.; Wan L. F.; Prendergast D.; Neaton J. B.; Smit B.; Kortright J. B.; Gagliardi L.; Bordiga S.; Reimer J. A.; Long J. R. Cooperative insertion of CO2 in diamine-appended metal-organic frameworks. Nature 2015, 519, 303. 10.1038/nature14327. [DOI] [PubMed] [Google Scholar]
- Wilson P. J.; Bradley T. J.; Tozer D. J. Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials. J. Chem. Phys. 2001, 115, 9233–9242. 10.1063/1.1412605. [DOI] [Google Scholar]
- Jensen F. The Basis Set Convergence of Spin–Spin Coupling Constants Calculated by Density Functional Methods. J. Chem. Theory Comput. 2006, 2, 1360–1369. 10.1021/ct600166u. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/physrevlett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Häser M.; Patzelt H.; Ahlrichs R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. 10.1016/s0009-2614(98)00862-8. [DOI] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- TURBOMOLE V7.1 2017. http://www.turbomole.com (accessed Jan 30, 2019), a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. TURBOMOLE GmbH, since 2007.
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Pre-selective screening for matrix elements in linear-scaling exact exchange calculations. J. Chem. Phys. 2013, 138, 134114. 10.1063/1.4796441. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Preselective Screening for Linear-Scaling Exact Exchange-Gradient Calculations for Graphics Processing Units and General Strong-Scaling Massively Parallel Calculations. J. Chem. Theory Comput. 2015, 11, 918–922. 10.1021/ct501189u. [DOI] [PubMed] [Google Scholar]
- Uribe-Romo F. J.; Doonan C. J.; Furukawa H.; Oisaki K.; Yaghi O. M. Crystalline Covalent Organic Frameworks with Hydrazone Linkages. J. Am. Chem. Soc. 2011, 133, 11478–11481. 10.1021/ja204728y. [DOI] [PubMed] [Google Scholar]
- Spitler E. L.; Koo B. T.; Novotney J. L.; Colson J. W.; Uribe-Romo F. J.; Gutierrez G. D.; Clancy P.; Dichtel W. R. A 2D Covalent Organic Framework with 4.7-nm Pores and Insight into Its Interlayer Stacking. J. Am. Chem. Soc. 2011, 133, 19416–19421. 10.1021/ja206242v. [DOI] [PubMed] [Google Scholar]
- Haase F.; Gottschling K.; Stegbauer L.; Germann L. S.; Gutzler R.; Duppel V.; Vyas V. S.; Kern K.; Dinnebier R. E.; Lotsch B. V. Tuning the stacking behaviour of a 2D covalent organic framework through non-covalent interactions. Mater. Chem. Front. 2017, 1, 1354–1361. 10.1039/c6qm00378h. [DOI] [Google Scholar]
- Stegbauer L.; Hahn M. W.; Jentys A.; Savasci G.; Ochsenfeld C.; Lercher J. A.; Lotsch B. V. Tunable Water and CO2 Sorption Properties in Isostructural Azine-Based Covalent Organic Frameworks through Polarity Engineering. Chem. Mater. 2015, 27, 7874–7881. 10.1021/acs.chemmater.5b02151. [DOI] [Google Scholar]
- Lowell S.; Shields J. E.; Thomas M. A.; Thommes M.. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Springer, 2012. [Google Scholar]
- Datta S. J.; Khumnoon C.; Lee Z. H.; Moon W. K.; Docao S.; Nguyen T. H.; Hwang I. C.; Moon D.; Oleynikov P.; Terasaki O.; Yoon K. B. CO2 capture from humid flue gases and humid atmosphere using a microporous coppersilicate. Science 2015, 350, 302–306. 10.1126/science.aab1680. [DOI] [PubMed] [Google Scholar]
- Zhongping L.; Yongfeng Z.; Xiao F.; Xuesong D.; Yongcun Z.; Xiaoming L.; Ying M. An Azine-Linked Covalent Organic Framework: Synthesis, Characterization and Efficient Gas Storage. Chem.—Eur. J. 2015, 21, 12079–12084. 10.1002/chem.201501206. [DOI] [PubMed] [Google Scholar]
- Gomes R.; Bhaumik A. A new triazine functionalized luminescent covalent organic framework for nitroaromatic sensing and CO2 storage. RSC Adv. 2016, 6, 28047–28054. 10.1039/c6ra01717g. [DOI] [Google Scholar]
- Shan M.; Seoane B.; Rozhko E.; Dikhtiarenko A.; Clet G.; Kapteijn F.; Gascon J. Azine-Linked Covalent Organic Framework (COF)-Based Mixed-Matrix Membranes for CO2/CH4 Separation. Chem.—Eur. J. 2016, 22, 14467–14470. 10.1002/chem.. [DOI] [PubMed] [Google Scholar]
- Kandambeth S.; Mallick A.; Lukose B.; Mane M. V.; Heine T.; Banerjee R. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 2012, 134, 19524–19527. 10.1021/ja308278w. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Yao K. X.; Teng B.; Zhang T.; Han Y. A perfluorinated covalent triazine-based framework for highly selective and water–tolerant CO2 capture. Energy Environ. Sci. 2013, 6, 3684–3692. 10.1039/c3ee42548g. [DOI] [Google Scholar]
- Mason J. A.; Sumida K.; Herm Z. R.; Krishna R.; Long J. R. Evaluating metal–organic frameworks for post-combustion carbon dioxide capture via temperature swing adsorption. Energy Environ. Sci. 2011, 4, 3030–3040. 10.1039/c1ee01720a. [DOI] [Google Scholar]
- Lei Z.; Long X.; Cheng H.; Wenlong L.; Wei H.; Yichang P. From Discrete Molecular Cages to a Network of Cages Exhibiting Enhanced CO2 Adsorption Capacity. Angew. Chem., Int. Ed. 2017, 56, 7787–7791. 10.1002/anie.201702399. [DOI] [PubMed] [Google Scholar]
- Radek M.; Antonin L.; Erkki K.; Elina S.; Jaromir T. 15N NMR Spectroscopy in Structural Analysis: An Update (2001–2005). Curr. Org. Chem. 2007, 11, 1154–1205. 10.2174/138527207781662519. [DOI] [Google Scholar]
- Martin G. J.; Martin M. L.; Gouesnard J.-P.. 15N-NMR Spectroscopy; Springer-Verlag, 1981. [Google Scholar]
- Kobayashi T.; Gupta S.; Caporini M. A.; Pecharsky V. K.; Pruski M. Mechanism of Solid-State Thermolysis of Ammonia Borane: A 15N NMR Study Using Fast Magic-Angle Spinning and Dynamic Nuclear Polarization. J. Phys. Chem. C 2014, 118, 19548–19555. 10.1021/jp504328x. [DOI] [Google Scholar]
- Pinto M. L.; Mafra L.; Guil J. M.; Pires J.; Rocha J. Adsorption and Activation of CO2 by Amine-Modified Nanoporous Materials Studied by Solid-State NMR and 13CO2 Adsorption. Chem. Mater. 2011, 23, 1387–1395. 10.1021/cm1029563. [DOI] [Google Scholar]
- Chen C.-H.; Shimon D.; Lee J. J.; Didas S. A.; Mehta A. K.; Sievers C.; Jones C. W.; Hayes S. E. Spectroscopic Characterization of Adsorbed 13CO2 on 3-Aminopropylsilyl-Modified SBA15 Mesoporous Silica. Environ. Sci. Technol. 2017, 51, 6553–6559. 10.1021/acs.est.6b06605. [DOI] [PubMed] [Google Scholar]
- Moschetta E. G.; Sakwa-Novak M. A.; Greenfield J. L.; Jones C. W. Post-Grafting Amination of Alkyl Halide-Functionalized Silica for Applications in Catalysis, Adsorption, and 15N NMR Spectroscopy. Langmuir 2015, 31, 2218–2227. 10.1021/la5046817. [DOI] [PubMed] [Google Scholar]
- Benson O.; da Silva I.; Argent S. P.; Cabot R.; Savage M.; Godfrey H. G. W.; Yan Y.; Parker S. F.; Manuel P.; Lennox M. J.; Mitra T.; Easun T. L.; Lewis W.; Blake A. J.; Besley E.; Yang S.; Schröder M. Amides Do Not Always Work: Observation of Guest Binding in an Amide-Functionalized Porous Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 14828–14831. 10.1021/jacs.6b08059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel H.; Neumann M.; Mayer C.; Epple M. On the Structure of Amorphous Calcium Carbonate—A Detailed Study by Solid-State NMR Spectroscopy. Inorg. Chem. 2008, 47, 7874–7879. 10.1021/ic8007409. [DOI] [PubMed] [Google Scholar]
- Kortunov P. V.; Siskin M.; Baugh L. S.; Calabro D. C. In Situ Nuclear Magnetic Resonance Mechanistic Studies of Carbon Dioxide Reactions with Liquid Amines in Non-aqueous Systems: Evidence for the Formation of Carbamic Acids and Zwitterionic Species. Energy Fuels 2015, 29, 5940–5966. 10.1021/acs.energyfuels.5b00985. [DOI] [Google Scholar]
- Seravalli J.; Ragsdale S. W. 13C NMR Characterization of an Exchange Reaction between CO and CO2 Catalyzed by Carbon Monoxide Dehydrogenase. Biochemistry 2008, 47, 6770–6781. 10.1021/bi8004522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yongfei Z.; Ruqiang Z.; Yanli Z. Covalent Organic Frameworks for CO2 Capture. Adv. Mater. 2016, 28, 2855–2873. 10.1002/adma.201505004. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Malani A.; Medhekar N. V.; Babarao R. CO2 adsorption and separation in covalent organic frameworks with interlayer slipping. CrystEngComm 2017, 19, 6950–6963. 10.1039/c7ce01647f. [DOI] [Google Scholar]
- Lee H. M.; Youn I. S.; Saleh M.; Lee J. W.; Kim K. S. Interactions of CO2 with various functional molecules. Phys. Chem. Chem. Phys. 2015, 17, 10925–10933. 10.1039/c5cp00673b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.