

Abstract
Background
Perilipin2 (Plin2) belongs to a family of five highly conserved proteins, known for their role in lipid storage. Recent data indicate that Plin2 has an important function in cell metabolism and is involved in several human pathologies, including liver steatosis and Type II diabetes. An association between Plin2 and lower muscle mass and strength has been found in elderly and inactive people, but its function in skeletal muscle is still unclear. Here, we addressed the role of Plin2 in adult muscle by gain and loss of function experiments.
Methods
By mean of in vivo Plin2 down‐regulation (shPlin2) and overexpression (overPlin2) in murine tibialis anterior muscle, we analysed the effects of Plin2 genetic manipulations on myofiber size and lipid composition. An analysis of skeletal muscle lipid composition was also performed in vastus lateralis samples from young and old patients undergoing hip surgery.
Results
We found that Plin2 down‐regulation was sufficient to induce a 30% increase of myofiber cross‐sectional area, independently of mTOR pathway. Alterations of lipid content and modulation of genes involved in lipid synthesis occurred in hypertrophic muscles. In particular, we showed a decrease of triglycerides, ceramides, and phosphatidylcoline:phosphatidylethanolamine ratio, a condition known to impact negatively on muscle function. Plin2 overexpression did not change fibre size; however, lipid composition was strongly affected in a way that is similar to that observed in human samples from old patients.
Conclusions
Altogether these data indicate that Plin2 is a critical mediator for the control of muscle mass, likely, but maybe not exclusively, through its critical role in the regulation of intracellular lipid content and composition.
Keywords: Perilipin 2, Lipid metabolism, Muscle hypertrophy, Muscle weakness, Human ageing
Introduction
In last years, the accumulation of lipid intermediates in non‐adipose tissues has become the focus of intense study, given the effects that excess ectopic fat storage has on the development of several metabolic disorders.1 In cells, triglycerides and cholesterol are stored within lipid droplets (LDs), dynamic structures evolutionary conserved from yeast to mammals and characterized by a core of neutral lipids, mainly sterol esters and triacylglycerols (TAG), surrounded by a monolayer of phospholipids (PL). LDs membrane is decorated by key proteins involved in several metabolic processes, such as fat storage, regulation of lipid homeostasis (synthesis and degradation), and protection against lipotoxicity. Among these proteins there are: lipid‐synthesis factors, such as sterol regulatory element‐binding protein‐1 (SREBP‐1) and acyl‐CoA:diacylglycerol acyltransferase 2 (DGAT2); lipases, such as adipose tissue triacylglycerol lipase (ATGL); and structural proteins, such as Perilipins that are the most abundant. Perilipins belong to a family of five proteins (Plin1, Plin2/ADRP/adipophilin, Plin3/Tip47, Plin4/S3–12, and Plin5/OXPAT) that attracted great interest for their role in lipid metabolism.2, 3 Recent findings showed that the modification of perilipins expression dysregulates intracellular lipid deposition, accumulate diacylglycerols (DAG) or ceramides, impair cellular function, and cause lipotoxicity.4, 5
Plin2 plays a fundamental role in lipid storage and is widely expressed in active metabolic tissues, including skeletal muscle.6 We recently reported the accumulation of Plin2 in human skeletal muscles biopsies during ageing and inactivity.7 Interestingly, Plin2 expression in vastus lateralis muscle correlates with lower muscle mass and strength and mirrors the increased expression of atrophy‐related genes such as Atrogin‐1 and MuRF‐1, as well as p53.7, 8 Plin2 protein is stable only when associated to LDs; otherwise, it is targeted to proteasomal degradation.9 Therefore, the amount of Plin2 protein reflects the intracellular lipid content. Several studies showed a correlation between high levels of Plin2 and metabolic disorders, including liver steatosis, insulin resistance, type 2 diabetes, atherosclerosis, and cardiovascular diseases.6 Conversely, in mice, Plin2 deficiency attenuates hepatic steatosis and improves fatty liver metabolism. In particular, liver of Plin2‐KO mice displays a significant decrease of two important key regulators of lipid synthesis: SREBP1 and DGAT2,10 suggesting a role for Plin2 in the control of lipid homeostasis‐related genes. Indeed, Plin2 deletion dramatically reduces TAG and cholesterol levels as well as desaturation and elongation of hepatic neutral lipid species in liver.11 Moreover, a recent work in Drosophila showed that dHDAC6‐dependent Plin2 degradation reduces age‐dependent ectopic fat accumulation and protects the organism from tissue dysfunction during ageing.12 Altogether, these evidences suggest that Plin2 plays an important role in modulating lipid metabolism.
Skeletal muscle is another important player in lipid metabolism. In fact, intracellular accumulation of fatty acids has been reported to affect both muscle mass and function.13 The role of Plin2 in muscle physiopathology has not been yet dissected. Based on the evidence that Plin2 accumulates in muscles of unhealthy aged people, we exploited murine models in order to unravel the role of Plin2 in adult skeletal muscle. Here, we show the effects of Plin2 down‐regulation or overexpression on cross‐sectional area (CSA) and lipid content in adult skeletal muscles of mice. Our data show that Plin2 down‐regulation in adult tibialis anterior muscle is sufficient to increase myofibers size of 30%, while its overexpression does not elicit any effect in an acute time window. Simultaneously, lipid content and in particular TAG and ceramides were altered in hypertrophic myofibers. Finally, the down‐regulation of Plin2 expression in denervated muscles almost completely prevented muscle loss. These findings support the hypothesis that Plin2 controls muscle mass, likely through a modulation of the intramuscular lipidome.
Materials and methods
Mice
Three‐months‐old C57BL/6J male mice were purchased from Charles River Laboratories. Mice were housed in an environmentally controlled room (23°C, 12 h light/12 h dark cycle) and provided food (Mucedola srl standard diet) and water ad libitum. After a period of acclimatization, mice were subjected to surgical procedures (electroporation and denervation) as described in Milan et al.14 Mouse procedures were all approved by the Italian Ministry of Health, protocol number 1060/2015PR.
Human samples
In the present study, patients of different age, 9 young (5 males and 4 females, mean age: 31.5 ± 6.0; mean BMI: 25.9 ± 4.4) and 10 old (5 males and 5 females, mean age: 84.9 ± 4.7; mean BMI: 26.8 ± 3.3), who underwent surgery for hip dysplasia, were included. Written informed consent was obtained from all patients, and the study was approved by the responsible Ethical Committee of Istituto Ortopedico Rizzoli (protocol no. 10823, issued on 26 April 2010) (Bologna–Italy). During surgery, biopsies from vastus lateralis (VL) muscle were obtained and immediately snap frozen in liquid nitrogen and stored at −80°C. Age (>20 years) and ability to provide informed consent were inclusion criteria, while exclusion criteria were chronic kidney or liver diseases, bleeding disorders, severe diabetes mellitus, rheumatic diseases other than osteoarthritis, neuromuscular disorders, malignancies and systemic infections, chronic steroid use, major psychological problems or history of alcohol or drug abuse, and evidence of prior surgery in the involved hip.
Plasmid design and in vivo treatments
Plin2 knockdown was obtained through a RNA interference (RNAi)‐mediated strategy. Plin2 targeting (shPlin2) or scrambled oligos were purchased from Life Technologies and cloned into BLOCK‐iT Pol II miR RNAi with EmGFP expression vector (Life Technologies). This technology allows to follow transfected cells through the presence of GFP that is transcribed in the same transcript of the pre‐miRNA targeting Plin2.
For Plin2 overexpression (overPlin2), Plin2 coding sequence was inserted into a modified pBI‐CMV1 vector (Clontech). pBI‐CMV1 vector allows the cloning of two separate coding sequences within the same vector. Summarizing, one MCS 3XFlag‐Plin2 was inserted, while GFP coding sequence was cloned in the other MCS. The expression of the two proteins is driven by two different CMV minimal promoters guided by the same common enhancer, thus allowing to directly identify Plin2 overexpressing fibres by the detection of GFP expression. For controls experiments, a pBI‐CMV1 vector carrying only GFP sequences was used.
Knockdown or overexpression experiments were performed by intramuscular injection of plasmids (shPlin2 or overPlin2, respectively) in tibialis anterior (TA) muscle followed by electroporation as described by Milan et al.14 In both shPlin2 and overPlin2 experiments, after electroporation, muscles were collected 10 days after surgery.
In some experiments of Plin2 down‐regulation, mice were intraperitoneally injected with rapamycin (LC Laboratories) 2 mg/kg in 2% carboxymethylcellulose once a day for 7 days starting from the third day of transfection, while in scramble animals, only vehicle was administered.
In denervation experiments (Plin2 down‐regulation or Plin2 overexpression), immediately after transfection, the sciatic nerve was surgically cut in one side, allowing the counterpart leg to be used as control. In this case, muscles were removed after 7 days from surgery and snap frozen in liquid nitrogen for subsequent analyses.
Histology
Snap frozen TA muscles were cut into 10 μm sections with a Leica CM1950 cryostat. Sections were immediately monitored with a fluorescence microscope Leica DM5000B equipped with a Leica DFC300‐FX digital CCD camera. Cryosections of shPlin2 and overPlin2 TA, compared with their controls, were stained for succinate dehydrogenase (SDH). Cross‐sectional area (CSA) of shPlin2 or overPlin2 fibres (six mice per group) was measured and compared with an equal number of control fibres, transfected with scramble or empty vector. A minimum of 700 fibres for each muscle was analysed. Fibre CSA was measured using IMAGEJ software. All data are expressed as the mean ± SEM. Comparisons with P < 0.05 were considered statistically significant.
Gene expression analyses
Total RNA from TA muscle of six mice per group (shPlin2, overPlin2, and the respective controls) was extracted using TRIzol reagent (Invitrogen). cDNA was synthesized using iScript™ cDNA Synthesis Kit (Bio‐Rad) according to the manufacturer's instructions. Real‐time RT–PCR was performed by using iTaq™ Universal SYBR Green Supermix (Bio‐Rad) and Rotor gene Q 6000 system (QIAGEN GmbH, Germany). All data were normalized to GAPDH expression. All oligonucleotide pre‐designed primers were from Bio‐Rad. All information on these primers are available at website www.bio‐rad.com/PrimePCR.
Protein extraction and western blot
Protein extracts were obtained starting from TA muscle of six mice per group (shPlin2, overPlin2, and the respective controls). A number of about 30 frozen slices (20 μm thick) were collected from TA muscles and lysed as previously described.14 The supernatant containing the total protein extract was quantified by BCA method (Thermo Fisher). Tissue lysates were stored at −80°C until analysis. Twenty‐five micrograms of protein per well were loaded and separated in 12% SDS‐polyacrilamide gel and transferred to a nitrocellulose membrane, as already described.6 After blocking in 5% non‐fat dry milk/0.01% Tween 20‐TBS, membranes were incubated overnight with primary antibodies, as follows: 1:1000 rabbit polyclonal anti‐Plin2 antibody (Lifespan Biosciences); 1:1000 rabbit polyclonal anti‐Plin5 (Lifespan Biosciences); 1:1000 rabbit polyclonal anti‐Plin3 (Lifespan Biosciences); 1:500 rabbit monoclonal total GSK3β and P‐GSK3β(ser9) (Cell Signaling); 1:500 rabbit monoclonal 7F5 anti‐p53 (Cell Signaling); and 1:1000 rabbit monoclonal total S6 and P‐S6(ser235/236) (Cell Signaling). Housekeeping 1:2000 goat anti‐βActin (C‐11) (Santa Cruz Biotechnology) was used as loading control. The secondary anti‐rabbit horseradish peroxidase‐conjugated antibody (Bio‐Rad) and bovine anti‐goat IgG‐HRP antibody (Santa Cruz Biotechnology) were used, followed by ECL detection (Santa Cruz Biotechnology). Densitometry analysis of bands was performed using ImageJ software.
Lipid analyses
Total lipids were extracted from TA muscle of six mice per group (shPlin2, overPlin2, and the respective controls) following the procedure described by Rodriguez‐Estrada et al.,15 with some modification, necessary to adapt the method to small quantity of the sample. Muscle sample (~60 mg) was suspended in 6 mL of chloroform/methanol (2:1) solution and homogenized for 2 min by ULTRATURRAX homogenizer (IKA®‐Werke GmbH & Co. KG, Staufen, Germany). Subsequently, samples were incubated at 50°C for 3 h and then filtrated to remove the solid phase. After the addition of 3 mL of K2CO3 1 M aqueous solution, samples were incubated at 4°C for 3 h. Apolar phase (lower phase) was transferred in a fresh tube and rotary dried. Finally, lipid fraction was resuspended with 1 mL of hexane and stored at −20°C until the analysis.
Lipid fractions [triacylglycerols (TAG), diacylglycerols (DAG), and phospholipids (PL)] were separated by thin layer chromatography as described as follow: total lipids (resuspended with 0.3 mL) were spotted onto the 10 × 20 TLC silica gel 60 F254 plate (Merck KGaA, Darmstadt, Germany). The TLC chamber was saturated beforehand with hexane/diethylether (70:30) mix solution, and the elution was performed using the same mix. Then, the plate was dried and the spot under UV light after spraying with 2′,7′‐dichlorofluorescein in ethanol. The isolated fractions were identified on thin layer plates by comparing their Rf values with that of known lipid standards. Spots corresponding to PL, TAG, and DAG were scraped and recovered in separated fresh vials. PL fraction was eluted by TLC to separate the single PL component: phosphatidylcholine (PC) and phosphatidylethanolamine (PE). Elution mix was constituted by chloroform/methanol/acetic acid/water (54:40:4:2). Spots of the single PL were scraped and recovered in fresh vials.
Fatty acids (FAs) compositions of the single fraction were obtained by gas chromatography (GC) analysis. Before GC analysis, FAs were methylated by an acid‐catalysed transmethylation procedure, as follows: all single fractions were resuspended in a tube with 2 mL of 10% acetyl chloride methanolic solution and incubated at 50°C overnight. Subsequently, 2 mL of 6% aqueous potassium carbonate solution were added to neutralize pH, and methylated FAs were extracted with 5 mL of hexane and transferred in a fresh tube. The extraction step with hexane was repeated twice. Samples were dried under nitrogen flow and, then, resuspended with 0.3 mL of hexane. Samples were collected in a vial for GC analysis.
The FA composition was determined by GC2010 Shimadzu gas chromatograph (Shimadzu, Columbia, MD, USA) equipped with a flame‐ionization detector and a high polar fused‐silica capillary column (Chrompack CP‐Sil88 Varian, 152 Middelburg, the Netherlands; 100 m, 0.25 mm i.d.; film thickness 0.20 μm). Hydrogen was used as the carrier gas at a flow of 1 mL/min. Split/splitless injector was used with a split ratio of 1:40. Two microlitres of the sample were injected under the following GC conditions: oven temperature started at 40°C and was held at that level for 1 min; then, it was increased to 163°C at a rate of 2°C/min and held at that level for 10 min, before being, once again, increased to 180°C at 1.5°C/min and held for 7 min, and, finally, to 187°C at a rate of 2°C/min; at last the temperature was increased to 220°C with a rate of 3°C/min and held for 25 min. Injector temperature was set at 270°C, and detector temperature was set at 300°C. Individual FA methyl esters were identified by comparison with a standard mixture of 52 Component FAME Mix (Nu‐Chek Prep Inc., Elysian, MN, USA). Nonadecanoic acid methyl ester was added as internal standard. The identification of isomers of 18:1 was based on commercial standard mixtures (Supelco, Bellefonte PA, USA) and published isomeric profiles.16 Total fat and FA content of single fraction was expressed as g/100 g of muscle, while FA profile was reported as g/100 g of total FA fraction.
Ceramides analyses
Ceramides were analysed in TA muscle of six mice per group (shPlin2 and scramble), following the method described by Groener et al.,17 with some modification. Briefly, an aliquot of total lipids (~30 mg) was deacylated using 0.5 mL of 0.1 M NaOH in methanol at room temperature for 1 h. After hydrolysis, 50 μL of the deacylated lipid were transferred in a fresh vials and derivatized with the addition of 25 μL OPA reagent (Sigma‐Aldrich, St. Louis, MO, USA) [5 mg OPA, 0.1 mL ethanol, 5 L 2‐mercaptoethanol, and 10 mL 3% (wt/vol) boric acid adjusted to pH 9.0] for 30 min at room temperature. OPA‐derivatized lysoglycosphingolipids were separated by use of a Prostar HPLC system (Varian) consisting of a Fluorescence Detector Spectra SYSTEM FL3000 (Thermo Finnigan, Waltham, USA) with a C18 reverse phase column (ChromSep HPLC Columns SS 250 mm × 4.6 mm including Holder with ChromSep guard column Omnispher 5 C18). The mobile phase was methanol/water 88:12 (vol/vol). The OPA‐derivatized lysoglycosphingolipids were quantified with a fluorescence detector at lambda(ex) 340 nm and lambda(em) 435 nm. Peak identification was based on comparison of the retention times with those of authentic calibrators of the lysocompounds (NHexadecanoyl‐D‐erythro‐sphingosine, Matreya LLC, USA). Peak integration was performed with Varian software. All samples were run in duplicate, and two reference samples were included in every run.
Statistical analyses
All statistical tests used are reported in each figure legend. Generally, data were analysed by two‐tailed Student's t‐test. For all graphics, data are represented as mean ± SEM.
Results
Plin2 knockdown promotes myofiber hypertrophy
Since Plin2 expression is increased in sarcopenic muscles and during denervation,8 we investigated the role of Plin2 in muscle mass by loss of function approach. Different oligos were tested to selectively down‐regulate Plin2 expression. The different shRNAs were cloned into bicistronic vectors that allow the simultaneous production of the oligos and a GFP protein. Therefore, by monitoring the GFP positive fibres, we can determine the efficiency of transfection and quantify myofiber area.
Our data showed that we efficiently reduced Plin2 transcript in vivo when compared with muscles that were transfected with scramble oligos (Figure 1A and 1B). Importantly, the down‐regulation of Plin2 is specific; indeed, the expression of Plin3 and Plin5, other two perilipins highly expressed in skeletal muscles, was unaffected ( Figure S1). Then, we transfected shRNAs into adult myofibers for 10 days and monitored the effect on muscle mass by quantifying the CSA of the transfected fibres. Surprisingly, we found that inhibition of Plin2 resulted in 30% increase of fibre size when compared with fibres that were transfected with scramble oligos (Figure 1C, left columns). The same results are represented as μm2 in Figure S2A (left columns). Interestingly, fibre type was not affected by Plin2 down‐regulation. In fact, SDH staining showed no changes in distribution of small β‐oxidative mitochondria‐rich vs. large glycolytic mitochondria‐poor fibres ( Figure S3A), despite the high transfection efficiency ( Figure S3B).
Figure 1.
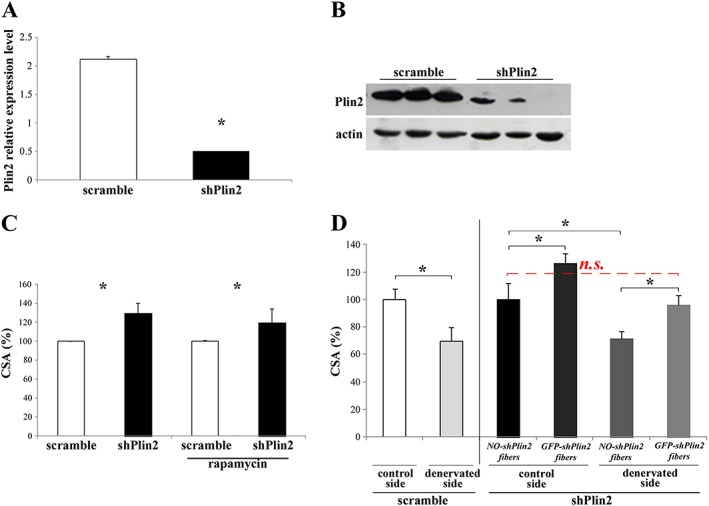
Plin2 down‐regulation in adult tibialis anterior (TA) muscle fibres. (A and B) RNAi‐mediated knockdown of Plin2 revealed by real‐time RT–PCR (* P < 0.001) (A) and immunoblotting (B). (C and D) Cross‐sectional area (CSA) of TA muscles upon knockdown of Plin2 expression. (C) CSA of adult TA muscle fibres transfected with either shPlin2 or scramble. Left columns: * P = 0.001; right columns: the same, plus treatment with rapamycin: * P = 0.02. (D) CSA of adult TA muscle fibres transfected with shPlin2 or scramble and undergone denervation performed at the same time of transfection. Left columns: CSA of TA muscle fibres transfected with scramble; control vs. denervated, * P = 0.001. Right columns: CSA of TA muscle fibres transfected with shPlin2; NO‐shPlin2 indicates fibres that remained untransfected; GFP‐shPlin2 indicates fibres transfected with the shPlin2; * P < 0.001. In all cases, transfected fibres from six mice for each group were identified by GFP fluorescence, and a minimum of 700 fibres for each muscles were measured. Data are shown as mean ± SEM. P values refer to two‐tailed Student's t‐test.
Such a dramatic hypertrophy resembles the growth reported when IGF1–AKT–mTOR pathway is acutely activated in adult muscles.18 Therefore, to address whether this pathway is affected by Plin2 down‐regulation, we treated transfected mice with rapamycin to block mTORC1‐dependent protein synthesis. Evaluation of phospho‐S6 confirmed the efficient inhibition of the pathway after rapamycin treatment ( Figure S4A). Importantly, mTORC1 inhibition did not reduce the hypertrophic effect of Plin2 down‐regulation (Figure 1C, right columns). The same results are represented as μm2 in Figure S2A (right columns). Therefore, in a short time frame (10 days), Plin2 down‐regulation is sufficient to promote muscle growth independently of mTORC1.
Plin2 down‐regulation counteracts myofiber atrophy after denervation
Because of the important hypertrophic effect after Plin2 down‐regulation, we asked whether Plin2 down‐regulation was able to counteract denervation‐induced myofiber atrophy. For this purpose, we down‐regulated Plin2 expression in denervated muscles. After 7 days from surgery (transfection and denervation), we collected muscles and measured fibre size. Remarkably, Plin2 knockdown protected denervated myofibers from muscle atrophy when shPlin2 fibres were compared with not‐transfected ones (Figure 1D). The same results are represented as μm2 in Figure S2B. The experimental design of Plin2 down‐regulation and denervation‐mediated atrophy is represented in Figure S4B.
Plin2 overexpression does not affect muscle CSA
We next asked whether Plin2 overexpression elicits the opposite effect of Plin2 down‐regulation. We therefore transfected for 10 days adult TA muscles with an expression vector containing two different promoters to simultaneously encode Plin2 and a fluorescent tracker. Quantitative RT–PCR and western blot analyses confirmed Plin2 overexpression (Figure 2A and 2B) in adult TA. To deeply investigate Plin2 overexpression phenotype, we analysed and measured the area of transfected myofibers identified through GFP fluorescence. Interestingly, in an acute time window (10 days), the overexpression of Plin2 did not alter myofiber CSA when compared with controls (Figure 2C). Consistently, in denervated muscles, the overexpression of Plin2 did not further worsen muscle loss (Figure 2D). The same results are represented as μm2 in Figure S2C and S2D.
Figure 2.
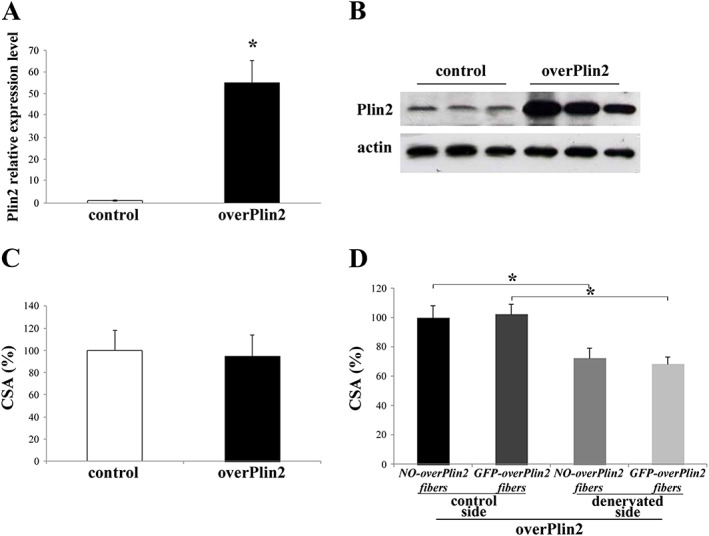
Plin2 overexpression in adult tibialis anterior (TA) muscle fibres. (A and B) Plin2 overexpression revealed by real‐time RT–PCR and immunoblotting. TA muscles were transfected with vectors expressing Plin2 gene. (C) Cross‐sectional area (CSA) of adult TA muscle fibres transfected with either overPlin2 or control. (D) CSA of adult TA muscle fibres transfected with overPlin2 and undergone denervation at the same time of transfection. In all cases, transfected fibres from six mice for each group were identified by GFP fluorescence, and a minimum of 700 fibres for each muscles were measured. Data are shown as mean ± SEM. * P < 0.001, two‐tailed Student's t‐test.
Plin2 modulation affects lipid content and composition of TA muscle
It is known that muscle mass is modulated by lipids such as ceramides, DAG, and specific phospholipid species. To get insights into the mechanisms that promote muscle growth, we quantified lipid composition in Plin2 knocked‐down or overexpressing muscles (Table 1). Plin2 down‐regulation caused a dramatic decrease of total TAG when compared with scramble, while DAG remained unchanged. On the contrary, overexpression of Plin2 caused an increase of TAG and a decrease of DAG content as compared with control.
Table 1.
Effect of Plin2 modulation on lipid fractions content in tibialis anterior muscle (g/100 g of muscle)
| Plin2 down‐regulation experiment | Plin2 overexpression experiment | |||||||
|---|---|---|---|---|---|---|---|---|
| Scramble | shPLIN2 | SEM | P‐value | Control | overPLIN2 | SEM | P‐value | |
| Triacylglycerols | 1.08 | 0.63 | 0.16 | 0.04 | 0.76 | 1.68 | 0.14 | 0.04 |
| Diacylglycerols | 0.39 | 0.44 | 0.14 | 0.46 | 0.49 | 0.05 | 0.02 | <0.01 |
| Phosphatidylethanolamine | 0.13 | 1.08 | 0.21 | <0.01 | 0.06 | 0.02 | 0.00 | <0.01 |
| Phosphatidylcholine | 0.63 | 0.11 | 0.23 | 0.16 | 0.90 | 0.02 | 0.00 | <0.01 |
SEM, standard error of the mean.
The TAG composition was also analysed. FA profile was modified by Plin2 down‐regulation resulting in a decrease of palmitoleic acid (C16:1c9) and oleic acid (C18:1c9) and in an increase of the palmitic acid (C16:0) and stearic acid (C18:0) as compared with controls (Table 2). These FAs are products (C16:1c9 and C18:1c9) and substrates (C16:0 and C18:0) of Stearoyl‐CoA Desaturase (SCD1), which inserts a cis‐double bond in Δ‐9 position. It is therefore reasonable to expect that that Plin2 modulation in some way modify the activity of this enzyme. A useful parameter to estimate SCD1 activity is the desaturation index (DI).19 The analysis of C16:1/C16:0 and C18:1/C18:0 DI showed that these ratios were significantly lower in shPlin2 muscle compared with controls (Figure 3A). At the same time, shPlin2 muscle showed a significantly lower percentage of linoleic (C18:2n6, −53%) and α‐linolenic acid (C18:3n3, −47%). The reduction of these FAs was accompanied by a concomitant increase of some very‐long‐chain FA (VLCFA), in particular C20:3n6, C20:4n6, C20:5n3, and C22:5n3. The first two VLFAs are derived from the elongation of linoleic acid while the latter two from α‐linolenic acid.
Table 2.
Effect of Plin2 modulation on triacylglycerol FA profile in tibialis anterior muscle (g/100 g of total triacylglycerol fatty acids)
| Plin2 down‐regulation experiment | Plin2 overexpression experiment | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Scramble | shPLIN2 | SEM | P‐value | Control | overPLIN2 | SEM | P‐value | ||
| C10:0 | 0.27 | 0.90 | 0.09 | <0.01 | 0.44 | 0.37 | 0.11 | 0.40 | |
| C12:0 | 0.72 | 3.46 | 1.33 | 0.19 | 3.34 | 1.03 | 0.27 | 0.25 | |
| C14:0 | 2.88 | 5.59 | 0.53 | 0.01 | 3.82 | 4.52 | 0.26 | 0.08 | |
| C14:1t9 | 0.28 | 0.61 | 0.05 | <0.01 | 0.24 | 0.20 | 0.12 | 0.62 | |
| C15:0anteiso | 0.15 | 0.40 | 0.08 | 0.07 | 0.13 | 0.33 | 0.01 | 0.01 | |
| C15:0 | 0.64 | 1.11 | 0.14 | 0.05 | 0.68 | 0.70 | 0.05 | 0.12 | |
| C16:0 | 25.45 | 31.96 | 0.81 | <0.01 | 30.63 | 29.35 | 0.17 | 0.05 | |
| C16:1c7 | 0.96 | 1.36 | 0.32 | 0.41 | 0.53 | 1.07 | 0.01 | 0.02 | |
| C16:1c9 | 8.22 | 4.34 | 0.41 | <0.01 | 2.89 | 5.15 | 0.04 | 0.02 | |
| C17:0anteiso | 0.12 | 0.23 | 0.02 | <0.01 | 0.58 | 0.40 | 0.06 | 0.11 | |
| C17:0 | 1.46 | 1.90 | 0.26 | 0.28 | 1.69 | 1.19 | 0.28 | 0.49 | |
| C18:0 | 5.58 | 13.47 | 1.15 | <0.01 | 14.53 | 10.03 | 0.19 | 0.10 | |
| C18:1t6–8 | 0.18 | 0.16 | 0.08 | 0.83 | 0.38 | 0.37 | 0.05 | 0.01 | |
| C18:1t9 | 0.32 | 0.41 | 0.16 | 0.70 | 0.20 | 0.19 | 0.04 | 0.03 | |
| C18:1t10 | 0.26 | 0.28 | 0.06 | 0.82 | 0.20 | 0.17 | 0.05 | 0.26 | |
| C18:1t11 | 0.14 | 0.49 | 0.19 | 0.24 | 0.46 | 0.41 | 0.21 | 0.51 | |
| C18:1t12 | 0.27 | 0.77 | 0.32 | 0.32 | 0.36 | 0.46 | 0.11 | 0.15 | |
| C18:1c9 | 25.50 | 16.21 | 1.46 | <0.01 | 30.32 | 36.52 | 0.70 | 0.02 | |
| C18:1c11 | 2.12 | 1.16 | 0.12 | <0.01 | 1.22 | 1.64 | 0.04 | 0.04 | |
| C18:2n6 | 19.80 | 9.25 | 1.24 | <0.01 | 2.48 | 2.88 | 0.12 | 0.03 | |
| C20:0 | 0.04 | 0.21 | 0.06 | 0.05 | 0.38 | 0.23 | 0.03 | 0.01 | |
| C18:3n6 | 0.18 | 0.39 | 0.06 | 0.05 | 0.10 | 0.07 | 0.02 | 0.36 | |
| C18:3n3 | 1.17 | 0.62 | 0.05 | <0.01 | 0.40 | 0.53 | 0.01 | 0.03 | |
| C20:3n6 | 0.43 | 1.59 | 0.18 | <0.01 | 0.21 | 0.27 | 0.11 | 0.41 | |
| C20:4n6 | 0.18 | 0.96 | 0.26 | 0.05 | 2.07 | 1.14 | 0.04 | 0.03 | |
| C20:5n3 | 0.13 | 0.25 | 0.07 | 0.04 | 0.19 | 0.19 | 0.05 | 0.21 | |
| C24:0 | 0.87 | 0.47 | 0.13 | 0.05 | 0.40 | 0.46 | 0.22 | 0.45 | |
| C22:5n3 | 0.25 | 0.47 | 0.08 | 0.02 | 1.04 | 0.90 | 0.08 | 0.15 | |
| SFA | 38.45 | 60.12 | 3.10 | <0.01 | 56.63 | 48.18 | 0.83 | 0.08 | |
| PUFA | 24.49 | 14.74 | 1.45 | <0.01 | 6.49 | 5.57 | 0.18 | 0.11 | |
| MUFA | 38.24 | 25.77 | 2.01 | <0.01 | 36.78 | 46.16 | 0.91 | 0.02 | |
| MCFA | 41.15 | 51.85 | 1.79 | <0.01 | 44.97 | 44.28 | 1.04 | 0.07 | |
| LCFA | 55.51 | 43.19 | 1.83 | <0.01 | 50.64 | 52.85 | 1.35 | 0.02 | |
| VLCFA | 4.51 | 5.57 | 0.56 | 0.22 | 4.30 | 2.78 | 0.28 | 0.41 | |
LCFA, long‐chain fatty acid (17 < C < 20); MCFA, medium‐chain fatty acid (10 < C < 18); MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acid; SEM, standard error of the mean; SFA, saturated fatty acid; VLCFA, very‐long‐chain fatty acid (C < 19).
Figure 3.
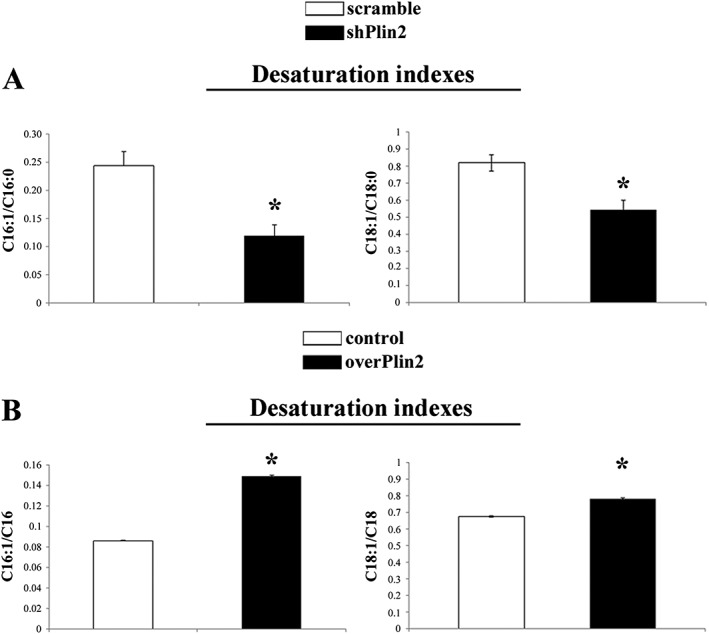
Stearoyl‐CoA desaturase 1 (SCD1) saturation indexes in tibialis anterior muscle after Plin2 modulation. Desaturation indexes of C16:1/C16:0 (* P = 0.002) and C18:1/C18:0 (* P = 0.003) in Plin2 down‐regulated muscle (shPlin2) vs. scramble. Desaturation indexes of C16:1/C16:0 (* P < 0.001) and C18:1/C18:0 (* P = 0.001) in Plin2 overexpressed muscle (overPlin2) vs. control (B, n = 4). Data refer to six samples for each group. Data are shown as mean ± SEM. P values refer to two‐tailed Student's t‐test.
The overexpression of Plin2 also modified the FA profile (Table 2). Stearic and palmitic acid was significantly reduced by Plin2 overexpression because of SCD1 activity. In fact, the higher C16:1/C16:0 and C18:1/C18:0 DI supports the idea that SCD1 likely is activated when Plin2 is overexpressed (Figure 3B). Moreover, Plin2 overexpression triggered high levels of linoleic (+16%) and α‐linolenic acid (+32%) and lower levels of VLCFA as a possible consequence of a reduced elongase activity.
Finally, we quantified phosphatidylcholine (PC) and phosphatidylethanolamine (PE), the two most abundant phospholipid species in mammalian cell membranes. We found a significant decrease of PC upon overexpression of Plin2, while Plin2 down‐regulation did not trigger significant changes. Conversely, PE was highly increased after Plin2 down‐regulation and decreased when Plin2 was overexpressed (Table 1).
Plin2 modulation also affected FA profile of PE fraction (Table 3). PE is constituted principally by C16:0, C18:0, and C20:4n6. Plin2 overexpression caused a significant modification of PE acidic profile, while no effect was observed after Plin2 down‐regulation. Particularly, in overPlin2 TA, we observed a higher content of C16:0 and C18:0 and a dramatic reduction of C20:4n6.
Table 3.
Effect of Plin2 modulation on phosphatidylethanolamine FA profile in tibialis anterior muscle (g/100 g of total phosphatidylethanolamine fatty acids)
| Plin2 down‐regulation experiment | Plin2 overexpression experiment | |||||||
|---|---|---|---|---|---|---|---|---|
| Scramble | shPLIN2 | SEM | P‐value | Control | overPLIN2 | SEM | P‐value | |
| C14:0 | 7.42 | 10.93 | 2.13 | 0.29 | 5.77 | 7.68 | 0.47 | 0.10 |
| C15:0 | 1.98 | 1.54 | 0.60 | 0.26 | 1.35 | 1.09 | 0.03 | 0.02 |
| C16:0 | 32.76 | 24.04 | 7.11 | 0.31 | 23.00 | 44.07 | 0.74 | 0.00 |
| C16:1c7 | 0.01 | 0.77 | 0.14 | 0.01 | 0.00 | 1.08 | 0.68 | 0.38 |
| C16:1c9 | 0.02 | 0.38 | 0.15 | 0.15 | 0.00 | 0.40 | 0.05 | 0.02 |
| C17:0anteiso | 0.02 | 0.38 | 0.09 | 0.03 | 0.00 | 1.03 | 0.04 | 0.00 |
| C17:0 | 2.83 | 0.73 | 0.49 | 0.02 | 5.42 | 2.53 | 0.47 | 0.05 |
| C18:0 | 23.96 | 25.66 | 4.60 | 0.80 | 23.47 | 27.07 | 0.05 | <0.01 |
| C18:1c9 | 6.82 | 9.55 | 3.53 | 0.04 | 6.63 | 6.59 | 0.20 | 0.92 |
| C18:2n6 | 0.69 | 0.65 | 0.23 | 0.91 | 1.80 | 0.76 | 0.06 | 0.01 |
| C20:3n6 | 0.39 | 1.47 | 0.46 | 0.90 | 0.51 | 1.36 | 0.70 | 0.48 |
| C20:4n6 | 19.85 | 20.82 | 1.39 | 0.34 | 29.10 | 6.33 | 0.57 | 0.01 |
| C22:5n3 | 3.24 | 3.07 | 1.38 | 0.93 | 2.95 | 0.00 | 0.11 | <0.01 |
| SFA | 68.98 | 63.28 | 3.26 | 0.28 | 59.02 | 83.47 | 1.01 | <0.01 |
| PUFA | 24.17 | 26.02 | 1.85 | 0.32 | 34.36 | 8.45 | 1.48 | 0.01 |
| MUFA | 6.85 | 10.70 | 3.62 | 0.05 | 6.63 | 8.08 | 0.55 | 0.21 |
| MCFA | 45.05 | 58.76 | 4.96 | 0.31 | 35.54 | 57.88 | 2.04 | 0.02 |
| LCFA | 31.47 | 35.87 | 6.20 | 0.58 | 31.90 | 34.42 | 1.35 | 0.03 |
| VLCFA | 23.48 | 5.37 | 1.84 | <0.01 | 32.56 | 7.69 | 0.68 | <0.01 |
LCFA, long‐chain fatty acid (17 < C < 20); MCFA, medium‐chain fatty acid (10 < C < 18); MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acid; SEM, standard error of the mean; SFA, saturated fatty acid; VLCFA, very‐long‐chain fatty acid (C < 19).
Since PC:PE ratio can influence energy metabolism20 and plays a role in skeletal muscle contractile function,21 we determined the PC:PE ratio in shPlin2 and overPlin2. A significant decrease in PC:PE ratio was observed in shPlin2 muscles compared with controls, but not in overPlin2 muscles (Figure 4), suggesting that Plin2 could affect muscle mass through a modulation of the PC:PE ratio.
Figure 4.
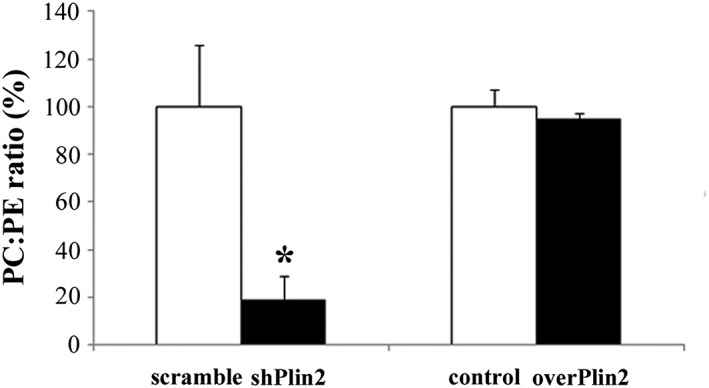
Phosphatidylcholine (PC) and phosphatidylethanolamine (PE) ratio in tibialis anterior muscle after Plin2 modulation. Left columns: shPlin2 muscles compared with scramble (n = 6 mice per group), * P = 0.001. Right columns: overPlin2 muscles compared with controls (n = 6 per group). Data are shown as mean ± SEM. P values refer to two‐tailed Student's t‐test.
Modulation of Plin2 expression affects lipogenesis
The above reported results suggest that modulation of Plin2 expression impacts on intramuscular lipid content and composition. In order to get insights on such effect, we investigated the expression of genes involved in lipid metabolism: DAG acyltransferase 2 (DGAT2) that catalyses the conversion of DAG into TAG; stearoyl CoA desaturase 1 (SCD1); sterol regulatory element‐binding protein 1 (SREBP1), a key transcription factor regulating the expression of genes required for the de novo lipogenesis; and adipose triglyceride lipase (ATGL) that is involved in lipolysis of TAG into DAG. We performed quantitative RT–PCR comparing shPlin2 and overPlin2 muscles with their respective controls and found that DGAT2 significantly decreased when Plin2 was knocked‐down and increased after Plin2 overexpression (Figure 5A and 5B). The expression of SCD1 and SREBP1 decreased upon Plin2 down‐regulation and did not change upon Plin2 overexpression (Figure 5C–5F). Furthermore, the expression of ATGL was not altered by either Plin2 down‐regulation or Plin2 overexpression (Figure 5G–5H). Altogether, these results suggest that Plin2 controls lipid content by regulating master genes of lipogenesis.
Figure 5.
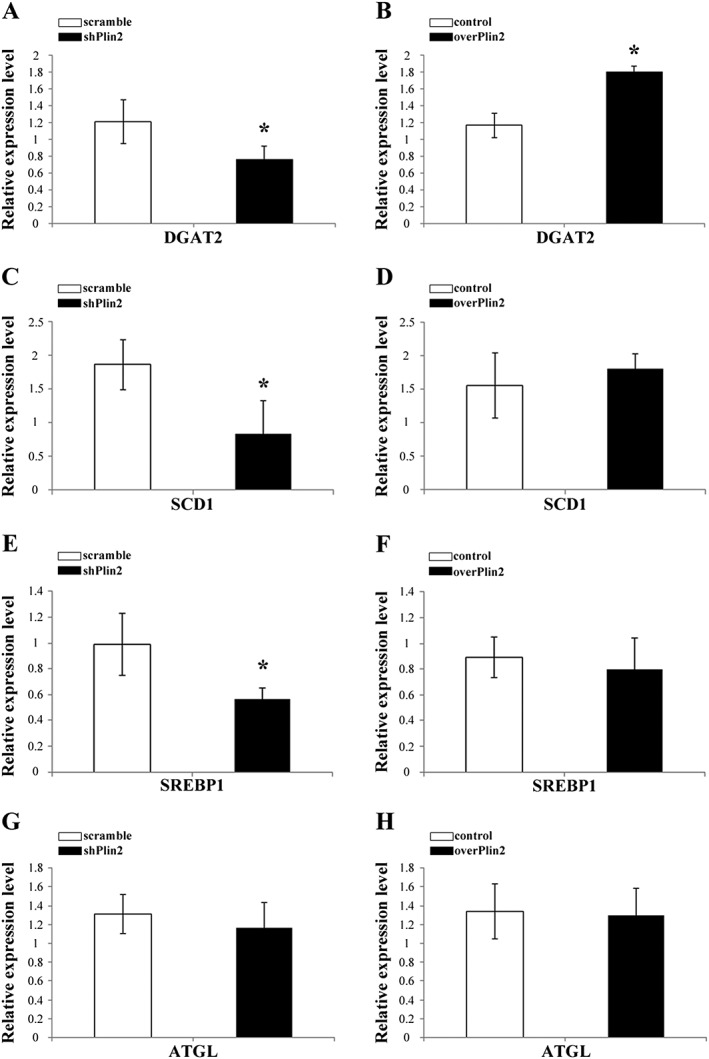
Relative expression of relevant genes involved in lipid metabolism in tibialis muscle after Plin2 modulation. (A, C, E, and G) Plin2 down‐regulation. (B, D, F, and H) Plin2 overexpression. (A and B) DAG acyltransferase 2 (DGAT2). (C and D) Stearoyl CoA desaturase 1 (SCD1). (E and F) Sterol regulatory element‐binding protein 1 (SREBP1). (G and H) Adipose triglyceride lipase (ATGL). Results were normalized to GAPDH RNA expression. Data refer to six samples for each group. Data are shown as mean ± SEM. * P < 0.02, two‐tailed Student's t‐test.
Plin2 down‐regulation affects ceramide content
Recent data indicate that oleic acid, the major product of SCD1, plays an important role in the regulation of the intracellular ceramide synthesis. Since Plin2 down‐regulation alters SCD1 expression and the levels of oleic acid, we checked ceramides content in shPlin2. The total content of ceramide was reduced by 80% in shPlin2 TA muscles as compared with controls (Figure 6A). Moreover, since ceramides are precursors of sphingolipids, including sphingomyelin, sphingosine, and glucosylceramide, we checked the content of these lipids and found that they were significantly decreased upon Plin2 down‐regulation (Figure 6B–6D). Moreover, previous studies demonstrated that ceramides indirectly activate GSK3β by promoting its dephosphorylation at ser922 and increase p53 protein.23, 24 Consistently, we observed an increase of the phosphorylated form of GSK3β and a reduction of p53 expression in shPlin2 muscle (Figure 6E and 6F). As a whole, shPlin2 muscle displays a dramatic decrease of ceramide content that could be at least in part responsible for CSA increase via inhibition of GSK3β.
Figure 6.
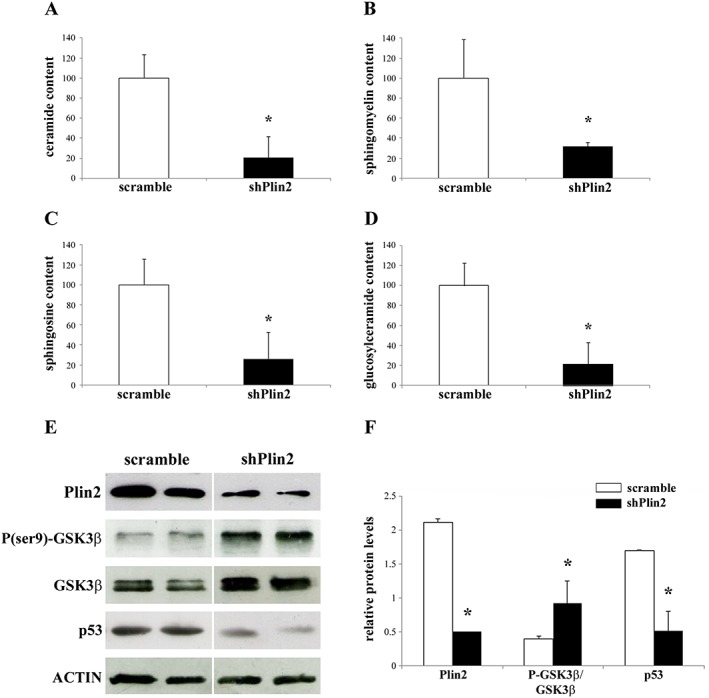
Ceramide content and protein expression of GSK3β and p53 in tibialis anterior (TA) muscles after Plin2 modulation. (A–D) Total ceramide (A), sphingomyelin (B), sphingosine (C), and glucosylceramide (D) contents in shPlin2 muscles compared with scramble. (E) Representative immunoblots of protein extracts from scramble and shPlin2 muscle for the indicated antibodies. (F) Relative expression levels of Plin2, ratio between phosphorylated and total GSK3β, and p53 proteins in scramble and shPlin2 TA muscles. Data are normalized to actin expression and refer to six samples for each group. Data are shown as mean ± SEM. * P < 0.001, two‐tailed Student's t‐test.
Plin2 level increases with ageing in human muscles and affects lipid composition
Our recent data indicate that Plin2 expression increases with ageing in human skeletal muscle.7, 8 To check whether the results obtained in our animal model have a counterpart in humans, we analysed lipid composition in VL muscle biopsies of young (<40 years) and old (>70 years) patients undergoing hip surgery. Consistent with the above results, muscle samples from old patients (which display higher levels of Plin2, Figure 7A) showed a significant increase of TAG and DAG when compared with young (Figure 7B). We also measured desaturation index (C16:1/C16:0 and C18:1/C18:0 DI) and phospholipid composition in these samples. In agreement with animal data, the C16:1/C16:0 and C18:1/C18:0 DI were higher in old subjects compared with young (Figure 7C). PE levels were similar between young and old, while PC levels were significantly lower in old compared with young (Figure 7D). However, the PC:PE ratio remained unchanged in the two different age groups (Figure 7E).
Figure 7.
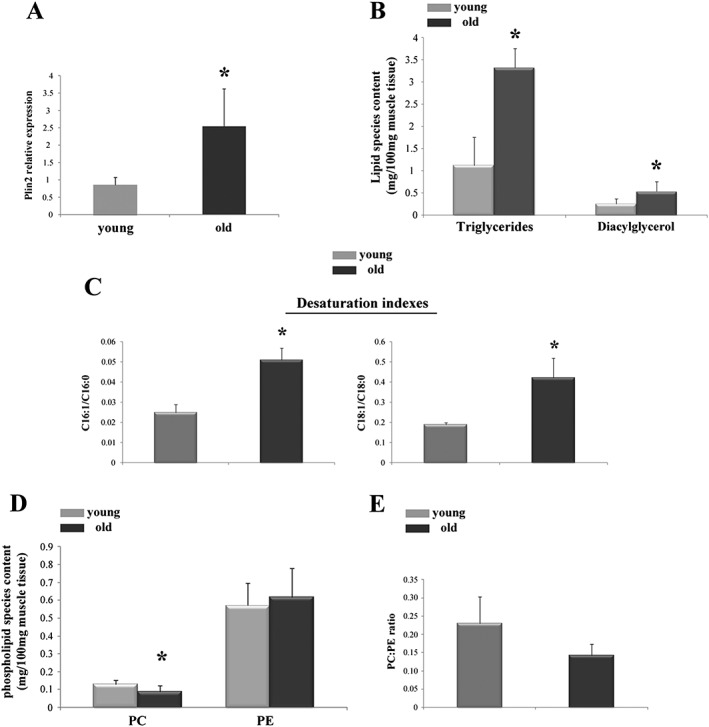
Plin2 expression and lipid content and composition in vastus lateralis muscle of young and old patients. (A) Relative expression level of Plin2, * P = 0.001. (B) Total triglycerides (TAG) and diacylglycerols (DAG), * P = 0.04 and * P = 0.002, respectively. (C) Desaturation indexes of C16:1/C16:0 and C18:1/C18:0, * P = 0.02 and * P = 0.03, respectively. (D) Phosphatidylcholine (PC), * P = 0.04, and phosphatidylethanolamine (PE) content. (E) PC/PE ratio. Data refer to 10 samples for young group and 9 samples for old group. Data are shown as mean ± SEM. P values refer to two‐tailed Student's t‐test.
FA composition of TAG in human samples is summarized in Table 4. Young and old subjects showed a different composition of FA profile. In particular, the muscles of old subjects are characterized by lower SFA and higher MUFA. These data are very similar to those obtained in mice suggesting a possible conserved function of Plin2 in lipids regulation.
Table 4.
Triacylglycerol content (g/100 g of muscle) and FA profile (g/100 g of total triglyceride fatty acids) in vastus lateralis muscle biopsies from subjects of different age
| Young | SEM | Old | SEM | P‐value | |
|---|---|---|---|---|---|
| Total | 1.12 | 0.81 | 3.32 | 0.78 | <0.01 |
| C10:0 | 4.03 | 0.87 | 1.06 | 0.82 | 0.02 |
| C10:1c9 | 1.03 | 0.14 | 0.65 | 0.13 | 0.05 |
| C12:0 | 1.77 | 0.25 | 1.00 | 0.23 | 0.04 |
| C14:0 | 6.49 | 0.81 | 4.73 | 0.77 | 0.13 |
| C14:1c9 | 1.42 | 0.21 | 1.25 | 0.20 | 0.56 |
| C15:0 | 0.92 | 0.12 | 0.60 | 0.11 | 0.06 |
| C16:0 | 32.13 | 2.62 | 28.97 | 2.48 | 0.39 |
| C16:1c9 | 0.94 | 0.44 | 1.43 | 0.42 | 0.43 |
| C17:0anteiso | 2.38 | 1.92 | 7.21 | 1.83 | 0.09 |
| C17:0 | 1.19 | 1.19 | 0.88 | 0.22 | 0.35 |
| C18:0 | 13.98 | 1.15 | 9.08 | 1.09 | 0.01 |
| C18:1c9 | 1.68 | 4.66 | 16.78 | 4.42 | 0.03 |
| C18:1c11 | 1.23 | 0.42 | 1.54 | 0.40 | 0.59 |
| C18:2c9c12 | 8.25 | 2.19 | 10.86 | 2.08 | 0.40 |
| C20:0 | 1.31 | 0.26 | 0.66 | 0.25 | 0.05 |
| C18:3c9c12c15 | 0.58 | 0.09 | 0.48 | 0.09 | 0.44 |
| C20:3n3 | 0.44 | 0.09 | 0.22 | 0.08 | 0.05 |
| C20:4n6 | 17.08 | 2.81 | 11.76 | 2.67 | 0.19 |
| C22:5n3 | 3.16 | 0.81 | 0.85 | 0.77 | 0.04 |
| SFA | 64.20 | 3.27 | 54.18 | 3.11 | 0.04 |
| MUFA | 6.30 | 4.14 | 22.65 | 3.94 | 0.02 |
| PUFA | 29.51 | 0.15 | 24.17 | 2.44 | 0.15 |
| MCFA | 52.30 | 2.62 | 47.78 | 2.49 | 0.23 |
| LCFA | 25.72 | 3.62 | 38.74 | 3.50 | 0.02 |
| VLCFA | 21.99 | 3.51 | 13.49 | 3.01 | 0.05 |
LCFA, long‐chain fatty acid (17 < C < 20); MCFA, medium‐chain fatty acid (10 < C < 18); MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acid; SEM, standard error of the mean; SFA, saturated fatty acid; VLCFA, very‐long‐chain fatty acid (C < 19).
Discussion
It is well established that alterations of lipid metabolism in non‐adipose tissue lead to an excess of lipid metabolites (including ceramides) that trigger lipotoxicity, cellular dysfunction, and metabolic disorders. Recent findings indicate that high levels of Plins are associated with deregulation of intracellular lipid deposition causing disease onset. In particular, high levels of Plin2 have been found in patients affected by liver steatosis, atherosclerosis, obesity, cardiovascular diseases, type 2 diabetes, and muscle weakness.6 Consistently, Plin2 down‐regulation ameliorates the above described diseases in mice.6 In previous studies, we reported that Plin2 is differently expressed in young and old subjects, being particularly elevated in muscles of inactive old people.7 In these subjects, we observed that Plin2 accumulation is associated with lower muscle mass and strength and paralleled by increased expression of p53 and atrophy‐related genes such as Atrogin‐1 and MuRF‐1.7, 8 In a recent study, obese type 2 diabetes STD fatty rats were reported to have high content of Plin2 and lower CSA in type IIb muscle fibres.25 However, the role of Plin2 in skeletal muscle homeostasis is still not completely understood. In the present study, we took advantage of in vivo genetic manipulations to dissect the function of Plin2 in adult muscles. Acute Plin2 down‐regulation was sufficient to increase the CSA of TA, while its overexpression did not change muscle mass. This shPlin2‐mediated hypertrophy appears largely mTORC1 independent, suggesting that Plin2 may modulate muscle growth in an unusual way.
The role of Plin2 in determining lipid droplet fate is well known, being the central core protein regulating intracellular lipid accumulation and metabolism. Moreover, changes of Plin2 greatly affect lipid composition in several tissues including skeletal muscle.26, 27 Accumulation of intramuscular lipids plays a pathogenic role in insulin resistance and type 2 diabetes.28 Finally, several recent studies highlighted the involvement of fatty acids and their metabolites, such as DAG and ceramides, in blocking muscle growth and altering myofiber metabolism.13, 29, 30 For this reason, we investigated how the modulation of Plin2 expression could affect the lipid metabolites in adult skeletal muscle and whether this can account for the observed effect on myofiber size. Our gain and loss of function experiments confirmed that Plin2 control lipid composition and expression of genes related to lipid biosynthesis. In agreement with previous studies,26, 27 we found that SREBP1 and DGAT2 are greatly modulated by Plin2. SREBP1 is the master regulator of genes involved in de novo lipogenesis,31 while DGAT2 is the enzyme of the last step in the pathway of TAG synthesis.32, 33, 34 Different studies showed that these two genes are involved in lipid accumulation and in the control of muscle mass. DGAT2 overexpression increases TAG, ceramide, and DAG content in muscle and liver,31, 35 while SREBP1 promotes skeletal muscle atrophy of both differentiated myotubes in vitro and tibialis muscle in vivo.36, 37
Contrary to our expectations based on previous data,8 the overexpression of Plin2 in our mouse model did not yield any variation of CSA, although lipid profile resulted affected. In the present experimental setting, DGAT2, SCD1, and SREBP1 expression decreases upon Plin2 down‐regulation, while DGAT2 but neither SREBP1 nor SCD1 expression increases upon Plin2 overexpression. It is thus likely that the observed shPlin2‐mediated muscular hypertrophy relies on the down‐regulation of SCD1 and SREBP1 but not DGAT2. It is possible that this lack of effect of Plin2 overexpression on myofiber size is due to the short time course (mice sacrificed after 10 days from transfection). Further studies are needed to clarify this point.
Besides lipid content, the regulation of Plin2 expression also affects the FA profile, especially of TAG fraction. We observed a higher level of C18:0 and C16:0 and a decrease of the relative MUFA (C18:1c9 and C16:1c9) when Plin2 was down‐regulated, while opposite results were obtained when Plin2 was overexpressed. It is unknown whether changes in TAG FA composition affect muscle growth; nevertheless, C18:0 and C16:0 represent the substrates of SCD1 enzyme, while C18:1c9 and C16:1c9 are the products.38, 39 Both SCD1 expression and DI, which represents a useful tool to estimate SCD1 activity,40 are significantly affected by Plin2. High levels of SCD1 has been reported in patients affected by obesity as well as type 2 diabetes41 and has been suggested to impact muscle structure and quality.13, 42, 43 Moreover, SCD1 activity regulates ceramide synthesis, and in particular, SCD1 deficiency reduces total ceramide content in skeletal muscle.44 Importantly, Plin2 down‐regulation reduced ceramide content by decreasing SCD1 expression. The reduction of ceramides releases the anabolic pathways from their inhibitory action45 and therefore promotes protein synthesis and muscle hypertrophy. Moreover, ceramides promote muscle wasting through the activation of apoptotic pathways.13 In particular, apoptosis can be activated via dephosphorylation at serine 9 of GSK3β, which in turn interacts with p53 in response to various cellular stresses.21, 46, 47, 48 In agreement with these findings, in shPlin2 muscle, we observed an increase of phosphorylated, inactive GSK3β form and a decrease of p53 level. p53 is known to play a role in muscle wasting, leading to skeletal muscle atrophy and muscle stem cell perturbation.49, 50 As our previous data suggested an involvement of muscle wasting,8 we investigated the expression of atrophy‐related genes Myostatin, Atrogin‐1, and MUSA‐1 in shPlin2 muscles, but no changes were detected (data not shown). However, GSK3β is also a negative regulator of skeletal muscle growth, as it inhibits protein synthesis.51, 52 The inhibition of GSK3β is in fact a potential strategy for the prevention or treatment of muscle atrophy.51 In agreement with literature data,53, 54, 55 the inhibition of GSK3β resulted independent from the AKT–mTOR pathway, as the level of the phosphorylated form of AKT (P‐AKT‐ser473) did not change in shPlin2 muscles (data not shown). Another pathway should therefore exist that can inhibit GSK3β.
Finally, also phospholipids may contribute to the muscle phenotype because PE has been reported to affect skeletal muscle growth20, 21 and PC:PE ratio is inversely correlated to insulin sensitivity.21, 56, 57 Recent studies in mice showed an impaired calcium uptake and muscle weakness when PE is reduced and consequently, the PC:PE ratio is increased.58, 59 Interestingly, we found that Plin2 down‐regulation, but not Plin2 overexpression, decreased PC:PE ratio due to a strong increase in PE. This phospholipid change may also contribute to the growth‐promoting action of Plin2 down‐regulation.
Altogether, our data suggest that Plin2 is involved in the regulation of intracellular content of lipid species, some of which are involved in muscle mass regulation. However, inhibition of the degradation pathways is not sufficient to promote muscle growth. In fact, both muscle‐specific autophagy deficient mice and knockout for the ubiquitin ligases such as Atrogin‐1 or MuRF1 do not show muscle hypertrophy.60, 61 Muscle growth instead depends on the increase of protein synthesis as shown by the inducible muscle‐specific AKT transgenic mice that displayed muscle growth.61 Therefore, it is possible that Plin2 could be a still unrecognized negative regulator of protein synthesis and therefore of muscle growth, likely acting through its control on intracellular lipid content and composition, in particular on ceramides and PE. Further studies are needed to clarify this aspect.
Human ageing is characterized by a progressive loss of skeletal muscle mass and regenerative capacity.62, 63 In previous studies, we demonstrated that VL muscle biopsies of elderly people contain higher Plin2 content when compared with young patients.7, 8 In addition, the level of Plin2 positively correlates with lower quadriceps strength and increased expression of atrophy markers.7, 8 For this reason, we investigated whether the lipid profile found on muscle biopsies from old subjects was at least in part similar to that observed on mice muscles. Consistent with our mouse data on overPlin2 muscles, old subjects displayed higher muscular TAG and DAG levels as well as increased DI and lower levels of PC with respect to young people. Moreover, the FA profile of TAG also recapitulates the findings obtained in mice. In fact, biopsies of old patients displayed a decrease of C18:0, an increase of C18:1c9, and a decrease of C20:3n3 and C22:5n3. Therefore, a certain degree of similarity exists between mouse and human muscle lipid profile, notwithstanding the fact that the two muscles investigated (TA in mice and VL in humans) have a different fibre type composition and that the first experimental model (mouse) entailed an acute stress phenomenon (transfection or denervation), while the second (patients) reflected a chronic situation (advanced age or prolonged inactivity). Further studies are granted to clarify the relationship among Plin2 accumulation, muscle mass modulation, and human ageing.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Representative immunoblot showing the specificity of Plin2 downregulation by in vivo RNAi technique in adult Tibialis anterior (TA) muscle fibers. TA muscles were transfected with vectors expressing two different short harpin RNAs (shRNAs) specific for Plin2. The expression of Plin3 and Plin5 (two Plins present in skeletal muscle fibers) in the same samples resulted unchanged, confirming the specificity of shRNA only for Plin2.
Figure S2. A‐B: Cross Sectional Area (CSA) of TA muscles upon knockdown of Plin2 expression. A: CSA of adult TA muscle fibers transfected with either shPlin2 or scramble. Left columns: *p = 0.001; right columns: the same, plus treatment with rapamycin: *p = 0.02. B: CSA of adult TA muscle fibers transfected with shPlin2 or scramble and undergone denervation; muscles were collected after 7 days from transfection; NO‐shPlin2 indicates fibers that remained untransfected, GFP‐shPlin2 indicates fibers transfected with the shPlin2; *p < 0.001. C‐D: CSA of TA muscles upon overexpression of Plin2 expression. C: CSA of adult TA muscle fibers transfected with either overPlin2 or control. D: CSA of adult TA muscle fibers transfected with overPlin2 and undergone denervation; muscles were collected after 7 days from transfection. In all cases, transfected fibers from 6 mice for each group were identified by GFP fluorescence, and CSA was measured after 7 days from transfection. Data are expressed as mean ± SEM. p values refer to two‐tailed Student's t test.
Figure S3. A: Representative images of succinate dehydrogenase (SDH) from Tibials anterior (TA) transversal sections isolated from scramble and shPlin2 mice analyzed by SDH staining. B: Fluorescence microscope images (in 5x magnification) of a whole TA section of mouse transfected with shPlin2, and the SDH respectively staining.
Figure S4. A: Western blot analysis showing the reduction in S6 phosphorylation, demonstrating the corrected mTORC1 inhibition by rapamycin treatment; CMC: carboxymethylcellulose. B: Schematic representation of Plin2 downregulation and denervation experiments performed in the same mouse.
Acknowledgements
This work has been partially supported by the EU 7FP Project ‘MYOAGE’ (GA no. 223576) to C.F. and M.S., ERC (282310‐MyoPHAGY), Foundation Leducq, and CARIPARO to M.S. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.64
Conte, M. , Armani, A. , Conte, G. , Serra, A. , Franceschi, C. , Mele, M. , Sandri, M. , and Salvioli, S. (2019) Muscle‐specific Perilipin2 down‐regulation affects lipid metabolism and induces myofiber hypertrophy. Journal of Cachexia, Sarcopenia and Muscle, 10: 95–110. 10.1002/jcsm.12355.
References
- 1. Shimabukuro M, Kozuka C, Taira S, Yabiku K, Dagvasumberel M, Ishida M, et al. Ectopic fat deposition and global cardiometabolic risk: new paradigm in cardiovascular medicine. J Med Invest 2013;60:1–14. [DOI] [PubMed] [Google Scholar]
- 2. Carr RM, Ahima RS. Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res 2016;340:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itabe H, Yamaguchi T, Nimura S, Sasabe N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis 2017;16:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krahmer N, Farese RV, Walther TC. Balancing the fat: lipid droplets and human disease. EMBO Mol Med 2013;5:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perreault L, Starling AP, Glueck D, Brozinick JT, Sanders P, Siddall P, et al. Biomarkers of ectopic fat deposition: the next frontier in serum lipidomics. J Clin Endocrinol Metab 2016;101:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conte M, Franceschi C, Sandri M, Salvioli S. Perilipin 2 and age‐related metabolic diseases: a new perspective. Trends Endocrinol Metab 2016;27:893–903. [DOI] [PubMed] [Google Scholar]
- 7. Conte M, Vasuri F, Trisolino G, Bellavista E, Santoro A, Degiovanni A, et al. Increased Plin2 expression in human skeletal muscle is associated with sarcopenia and muscle weakness. PLoS One 2013;8:e73709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conte M, Vasuri F, Bertaggia E, Armani A, Santoro A, Bellavista E, et al. Differential expression of perilipin 2 and 5 in human skeletal muscle during aging and their association with atrophy‐related genes. Biogerontology 2015;16:329–340, Erratum in: Biogerontology 16: 341. [DOI] [PubMed] [Google Scholar]
- 9. Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, et al. Post‐translational regulation of adipose differentiation‐related protein by the ubiquitin/proteasome pathway. J Biol Chem 2005;280:42841–42847. [DOI] [PubMed] [Google Scholar]
- 10. Carr RM, Peralta G, Yin X, Ahima RS. Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol‐fed mice. PLoS One 2014;9:e97118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Libby AE, Bales E, Orlicky DJ, McManaman JL. Perilipin‐2 deletion impairs hepatic lipid accumulation by interfering with sterol regulatory element‐binding protein (SREBP) activation and altering the hepatic lipidome. J Biol Chem 2016;291:24231–24246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan Y, Wang H, Hu M, Jiang L, Wang Y, Liu P, et al. HDAC6 suppresses age‐dependent ectopic fat accumulation by maintaining the proteostasis of PLIN2 in Drosophila. Dev Cell 2017;43:99–111. [DOI] [PubMed] [Google Scholar]
- 13. Lipina C, Hundal HS. Lipid modulation of skeletal muscle mass and function. J Cachexia Sarcopenia Muscle 2017;8:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez‐Estrada MT, Penazzi G, Caboni MF, Bertacco G, Lercker G. Effect of different cooking methods on some lipid and protein components of hamburgers. Meat Sci 1997. Mar;45:365–375. PubMed. [DOI] [PubMed] [Google Scholar]
- 16. Kramer JK, Hernandez M, Cruz‐Hernandez C, Kraft J, Dugan ME. Combining results of two GC separations partly achieves determination of all cis and trans 16:1, 18:1, 18:2 and 18:3 except CLA isomers of milk fat as demonstrated using Agion SPE fractionation. Lipids 2008;43:259–273. [DOI] [PubMed] [Google Scholar]
- 17. Groener JE, Poorthuis BJ, Kuiper S, Helmond MT, Hollak CE, Aerts JM. HPLC for simultaneous quantification of total ceramide, glucosylceramide, and ceramide trihexoside concentrations in plasma. Clin Chem 2007;53:742–747. [DOI] [PubMed] [Google Scholar]
- 18. Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J 2009;23:3896–3905. [DOI] [PubMed] [Google Scholar]
- 19. Miyazaki M, Dobrzyn A, Elias PM, Ntambi JM. Stearoyl‐CoA desaturase‐2 gene expression is required for lipid synthesis during early skin and liver development. PNAS 2005;102:12501–12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta 2017;1859:1558–1572. [DOI] [PubMed] [Google Scholar]
- 21. Newsom SA, Brozinick JT, Kiseljak‐Vassiliades K, Strauss AN, Bacon SD, Kerege AA, et al. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine are related to insulin sensitivity and respond to acute exercise in humans. J Appl Physiol (1985) 2016;120:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK‐3beta acts downstream of PP2A and the PI 3‐kinase‐Akt pathway, and upstream of caspase‐2 in ceramide‐induced mitochondrial apoptosis. J Cell Sci 2007;120:2935–2943. [DOI] [PubMed] [Google Scholar]
- 23. Brás A, Albar JP, Leonardo E, de Buitrago GG, Martínez‐A C. Ceramide‐induced cell death is independent of the Fas/Fas ligand pathway and is prevented by Nur77 overexpression in A20 B cells. Cell Death Differ 2000;7:262–271. [DOI] [PubMed] [Google Scholar]
- 24. Kim SS, Chae HS, Bach JH, Lee MW, Kim KY, Lee WB, et al. P53 mediates ceramide‐induced apoptosis in SKN‐SH cells. Oncogene 2002;21:2020–2028. [DOI] [PubMed] [Google Scholar]
- 25. Kemmochi Y, Ohta T, Motohashi Y, Kaneshige A, Katsumi S, Kakimoto K, et al. Pathophysiological analyses of skeletal muscle in obese type 2 diabetes SDT fatty rats. J Toxicol Pathol 2018;31:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bosma M, Hesselink MK, Sparks LM, Timmers S, Ferraz MJ, Mattijssen F, et al. Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes 2012;61:2679–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng YZ, Lund J, Li Y, Knabenes IK, Bakke SS, Kase ET, et al. Loss of perilipin 2 in cultured myotubes enhances lipolysis and redirects the metabolic energy balance from glucose oxidation towards fatty acid oxidation. J Lipid Res 2017;58:2147–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goto‐Inoue N, Yamada K, Inagaki A, Furuichi Y, Ogino S, Manabe Y, et al. Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high‐fat diet. Sci Rep 2013;3:3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rivas DA, McDonald DJ, Rice NP, Haran PH, Dolnikowski GG, Fielding RA. Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am J Physiol Regul Integr Comp Physiol 2016;310:R561–R569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akhmedov D, Berdeaux R. The effects of obesity on skeletal muscle regeneration. Front Physiol 2013;4:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruiz R, Jideonwo V, Ahn M, Surendran S, Tagliabracci VS, Hou Y, et al. Sterol regulatory element‐binding protein‐1 (SREBP‐1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem 2014;289:5510–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tomoda H, Omura S. Potential therapeutics for obesity and atherosclerosis: inhibitors of neutral lipid metabolism from microorganisms. Pharmacol Ther 2007;115:375–389. [DOI] [PubMed] [Google Scholar]
- 33. Ying F, Gu H, Xiong Y, Zuo B. Analysis of differentially expressed genes in gastrocnemius muscle between DGAT1 transgenic mice and wild‐type mice. Biomed Res Int 2017;2017:5404682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zammit VA. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. Biochem J 2013;451:1–12. [DOI] [PubMed] [Google Scholar]
- 35. Levin MC, Monetti M, Watt MJ, Sajan MP, Stevens RD, Bain JR, et al. Increased lipid accumulation and insulin resistance in transgenic mice expressing DGAT2 in glycolytic (type II) muscle. Am J Physiol Endocrinol Metab 2007;293:E1772–E1781. [DOI] [PubMed] [Google Scholar]
- 36. Lecomte V, Meugnier E, Euthine V, Durand C, Freyssenet D, Nemoz G, et al. A new role for sterol regulatory element binding protein 1 transcription factors in the regulation of muscle mass and muscle cell differentiation. Mol Cell Biol 2010;30:1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dessalle K, Euthine V, Chanon S, Delarichaudy J, Fujii I, Rome S, et al. SREBP‐1 transcription factors regulate skeletal muscle cell size by controlling protein synthesis through myogenic regulatory factors. PLoS One 2012;7:e50878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dobrzyń A, Dobrzyń P. Stearoyl‐CoA desaturase—a new player in skeletal muscle metabolism regulation. J Physiol Pharmacol 2006;57:31–42. [PubMed] [Google Scholar]
- 39. Conte G, Jeronimo E, Serra A, Bessa Rui JB, Mele M. Effect of dietary polyunsaturated fatty acids on Stearoyl CoA‐Desaturase gene expression in intramuscular lipids of lamb. Ital J Anim Sci 2012;11:453–458. [Google Scholar]
- 40. Ntambi JM, Miyazaki M, Dobrzyn A. Regulation of stearoyl‐CoA desaturase expression. Lipids 2004;39:1061–1065. [DOI] [PubMed] [Google Scholar]
- 41. Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, et al. Elevated stearoyl‐CoA desaturase‐1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab 2005;2:251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park SW, Goodpaster BH, Lee JS, Kuller LH, Boudreau R, de Rekeneire N, et al. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 2009;32:1993–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tomlinson DJ, Erskine RM, Morse CI, Winwood K, Onambélé‐Pearson G. The impact of obesity on skeletal muscle strength and structure through adolescence to old age. Biogerontology 2016;17:467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Cohen P, Asilmaz E, et al. Stearoyl‐CoA desaturase‐1 deficiency reduces ceramide synthesis by downregulating serine palmitoyltransferase and increasing beta‐oxidation in skeletal muscle. Am J Physiol Endocrinol Metab 2005;288:E599–E607. [DOI] [PubMed] [Google Scholar]
- 45. Rivas DA, Morris EP, Haran PH, Pasha EP, Morais Mda S, Dolnikowski GG, et al. Increased ceramide content and NFκB signaling may contribute to the attenuation of anabolic signaling after resistance exercise in aged males. J Appl Physiol (1985) 2012;113:1727–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase‐3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem 2003;278:48872–48879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kulikov R, Boehme KA, Blattner C. Glycogen synthase kinase 3‐dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol 2005;25:7170–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hage‐Sleiman R, Esmerian MO, Kobeissy H, Dbaibo G. p53 and ceramide as collaborators in the stress response. Int J Mol Sci 2013;14:4982–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schwarzkopf M, Coletti D, Marazzi G, Sassoon D. Chronic p53 activity leads to skeletal muscle atrophy and muscle stem cell perturbation. Basic Appl Myol 2008;18:131–138. [Google Scholar]
- 50. Dirks‐Naylor AJ, Lennon‐Edwards S. Cellular and molecular mechanisms of apoptosis in age‐related muscle atrophy. Curr Aging Sci 2011;4:269–278. [PubMed] [Google Scholar]
- 51. Verhees KJ, Pansters NA, Schols AM, Langen RC. Regulation of skeletal muscle plasticity by glycogen synthase kinase‐3β: a potential target for the treatment of muscle wasting. Curr Pharm Des 2013;19:3276–3298, Review. [DOI] [PubMed] [Google Scholar]
- 52. Theeuwes WF, Gosker HR, Langen RCJ, Verhees KJP, Pansters NAM, Schols AMWJ, et al. Inactivation of glycogen synthase kinase‐3β (GSK‐3β) enhances skeletal muscle oxidative metabolism. Biochim Biophys Acta 2017;1863:3075–3086. [DOI] [PubMed] [Google Scholar]
- 53. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, et al. Mediation of IGF‐1‐induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 2001;3:1009–1013. [DOI] [PubMed] [Google Scholar]
- 54. Minetti GC, Feige JN, Bombard F, Heier A, Morvan F, Nürnberg B, et al. Gαi2 signaling is required for skeletal muscle growth, regeneration, and satellite cell proliferation and differentiation. Mol Cell Biol 2014;34:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Funai K, Parkington JD, Carambula S, Fielding RA. Age‐associated decrease in contraction‐induced activation of downstream targets of Akt/mTor signaling in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 2006;290:R1080–R1086. [DOI] [PubMed] [Google Scholar]
- 56. Abbatecola AM, Paolisso G, Fattoretti P, Evans WJ, Fiore V, Dicioccio L, et al. Discovering pathways of sarcopenia in older adults: a role for insulin resistance on mitochondria dysfunction. J Nutr Health Aging 2011;15:890–895. [DOI] [PubMed] [Google Scholar]
- 57. Cleasby ME, Jamieson PM, Atherton PJ. Insulin resistance and sarcopenia: mechanistic links between common co‐morbidities. J Endocrinol 2016;229:R67–R81. [DOI] [PubMed] [Google Scholar]
- 58. Funai K, Song H, Yin L, Lodhi IJ, Wei X, Yoshino J, et al. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J Clin Invest 2013;123:1229–1240, Erratum in: J Clin Invest 2013;123:3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Funai K, Lodhi IJ, Spears LD, Yin L, Song H, Klein S, et al. Skeletal muscle phospholipid metabolism regulates insulin sensitivity and contractile function. Diabetes 2016;65:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009;10:507–515. [DOI] [PubMed] [Google Scholar]
- 61. Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 2014;8:1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin‐1. Am J Physiol Endocrinol Metab 2014;307:E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Petersen KF, Morino K, Alves TC, Kibbey RG, Dufour S, Sono S, et al. Effect of aging on muscle mitochondrial substrate utilization in humans. Proc Natl Acad Sci U S A 2015;112:11330–11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative immunoblot showing the specificity of Plin2 downregulation by in vivo RNAi technique in adult Tibialis anterior (TA) muscle fibers. TA muscles were transfected with vectors expressing two different short harpin RNAs (shRNAs) specific for Plin2. The expression of Plin3 and Plin5 (two Plins present in skeletal muscle fibers) in the same samples resulted unchanged, confirming the specificity of shRNA only for Plin2.
Figure S2. A‐B: Cross Sectional Area (CSA) of TA muscles upon knockdown of Plin2 expression. A: CSA of adult TA muscle fibers transfected with either shPlin2 or scramble. Left columns: *p = 0.001; right columns: the same, plus treatment with rapamycin: *p = 0.02. B: CSA of adult TA muscle fibers transfected with shPlin2 or scramble and undergone denervation; muscles were collected after 7 days from transfection; NO‐shPlin2 indicates fibers that remained untransfected, GFP‐shPlin2 indicates fibers transfected with the shPlin2; *p < 0.001. C‐D: CSA of TA muscles upon overexpression of Plin2 expression. C: CSA of adult TA muscle fibers transfected with either overPlin2 or control. D: CSA of adult TA muscle fibers transfected with overPlin2 and undergone denervation; muscles were collected after 7 days from transfection. In all cases, transfected fibers from 6 mice for each group were identified by GFP fluorescence, and CSA was measured after 7 days from transfection. Data are expressed as mean ± SEM. p values refer to two‐tailed Student's t test.
Figure S3. A: Representative images of succinate dehydrogenase (SDH) from Tibials anterior (TA) transversal sections isolated from scramble and shPlin2 mice analyzed by SDH staining. B: Fluorescence microscope images (in 5x magnification) of a whole TA section of mouse transfected with shPlin2, and the SDH respectively staining.
Figure S4. A: Western blot analysis showing the reduction in S6 phosphorylation, demonstrating the corrected mTORC1 inhibition by rapamycin treatment; CMC: carboxymethylcellulose. B: Schematic representation of Plin2 downregulation and denervation experiments performed in the same mouse.