

Abstract
Cardiometabolic and vascular disease, with their associated secondary complications, are the leading cause of morbidity and mortality in Western society. Chronic inflammation is a common theme that underlies initiation and progression of cardiovascular disease. In this regard, monocyte-macrophages are key players in the development of a chronic inflammatory state. Over the past decade, epigenetic modifications, such as DNA methylation and post-translational histone processing, have emerged as important regulators of immune cell phenotypes. Accumulating studies reveal the importance of epigenetic enzymes in the dynamic regulation of key signaling pathways that alter monocyte-macrophage phenotypes in response to environmental stimuli. In this review, we highlight the current paradigms of monocyte-macrophage polarization and the emerging role of epigenetic modification in the regulation of monocyte-macrophage phenotype in obesity, diabetes, atherosclerosis and abdominal aortic aneurysms.
Keywords: Macrophage, Monocyte, Inflammation, Epigenetics, Cardiometabolic, Vascular, Vascular Disease
Graphical Abstract
Introduction
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality worldwide and currently affects approximately 50% of the United States population.1,2 Further, CVD inflicts a major socioeconomic burden with both direct and indirect costs exceeding $1.1 trillion.2 The main etiologies of CVD include obesity, diabetes, hypertension, hyperlipemia, and atherosclerosis. Although these conditions are diverse, chronic inflammation with monocyte-macrophage infiltration plays a central role and represents a pathological hallmark of disease development.3–5 For example, within atherosclerotic plaques, macrophages represent major contributors to the inflammatory response through their secretion of pro-inflammatory mediators and their eventual death by necrosis or apoptosis.4 Dying macrophages release their lipid contents and tissue factors, which leads to the formation of a pro-thrombotic necrotic core, a key component of unstable plaque. Likewise, within diabetes, wound healing is altered due to chronic macrophage-mediated inflammation with excessive inflammatory cytokine production and an associated failure of tissue macrophages to transition to an anti-inflammatory phenotype.6,7 Finally in obesity, the infiltration of monocytes into adipose tissue results in increased inflammatory cytokine production, particularly tumor necrosis factor (TNF)-α, monocyte chemotactic protein (MCP)-1, and inducible nitric oxide synthase (iNOS).8 Several human and murine studies have demonstrated that the severity of adipose tissue macrophage inflammation correlates with insulin resistance.9–12
Macrophages are characterized by a remarkable degree of plasticity and are able to rapidly adapt to a wide range of environmental cues.13 The classical view presents a highly simplified M1/M2 phenotype dichotomy, based on in vitro experiments, where ‘M1’, or “classically activated,” macrophages are induced by proinflammatory mediators such as LPS and IFN-γ and secrete proinflammatory cytokines including IL-6, IL-1β, iNOS, and TNF-α. In contrast, ‘M2’, or “alternatively activated,” macrophages play a role in tissue repair, angiogenesis, and resolution of inflammation. These macrophages are induced by the anti-inflammatory mediators, IL-4 and IL-13, and secrete high levels of anti-inflammatory IL-10. Disease pathology is frequently associated with dynamic changes in macrophage phenotype, with classically activated ‘M1’ cells implicated in initiating and sustaining inflammation and ‘M2’ cells associated with resolution.14
An increasing number of studies have demonstrated the importance of epigenetics in the regulation of macrophage phenotype.15,16 Epigenetic modifications can be influenced by micro and macro-environmental factors which have been shown to induce cell-specific changes to the epigenetic landscape.17 As such, cardiovascular diseaseselicit epigenetic changes that alter gene expression and hence, cell function, in a variety of cell types, including monocytes/macrophages.18,19 Over the past decade, substantial work has been conducted on the mechanisms of epigenetic regulation of macrophage activation. Through characterization of human tissue samples and animal models, a complex picture of cellular processes have been shown to play pivotal roles in monocyte/macrophage inflammation in cardiovascular disease. In this brief review, we will discuss recent findings that provide mechanistic insight into the role of epigenetics in the regulation of macrophage phenotype and function in the setting of inflammation. Additionally, we will highlight the impact of epigenetic modifications on maladaptive inflammation in cardiometabolic and vascular disease.
Monocyte/Macrophage Plasticity
Many cardiovascular diseases have been recognized as inflammatory conditions characterized by the infiltration of monocytes that promote localized inflammation. Circulating blood monocytes originate in the bone marrow and are a heterogeneous leukocyte population with various subsets that differ in surface markers, gene-expression profile, and function. In humans, three main subsets can be distinguished on the basis of the expression of the cell surface markers CD14 and CD16. First, classical monocytes, which are defined as CD14++CD16−, are the predominant monocyte population in the blood representing up to 90%.20 Classical monocytes have high surface levels of the CC-chemokine receptor CCR2 and CD62L, and low levels of the CX3C chemokine receptor CX3CR1. This monocyte subset predominately functions in the phagocytosis of debris and infectious pathogens. Nonclassical monocytes are defined as CD14+CD16++, with low surface levels of CCR2 and high levels of CX3CR1. These monocytes predominately function in immune surveillance with vascular patrolling properties.21 Lastly, an intermediate monocyte phenotype, is characterized by high CD14 levels with low CD16 levels (CD14++CD16+) for which pro-inflammatory and phagocytic properties, have been described.20
In mice, two populations of monocytes exist and can be discriminated by variable expression of lymphocyte antigen 6C (Ly6C). Monocytes expressing high levels of Ly6C (Ly6Chi monocytes) have proinflammatory and antimicrobial functions and express high levels of C-C chemokine receptor 2 (CCR2) and low levels of CX3C chemokine receptor 1 (CX3CR1; CCR2hiCX3CR1low).22 These Ly6Chi monocytes, which most closely resemble the CD14++CD16− population in humans, are able to transport antigens to the lymph node and accumulate at sites of inflammation. In contrast, alternative monocytes, termed Ly6Clo monocytes, express high levels of CX3CR1 and low levels of CCR2 (CX3CR1hiCCR2low). These monocytes, also known as patrolling monocytes, resemble the CD14+CD16++ and survey the vasculature while being involved with early responses to inflammation and tissue repair.23
Despite the importance of circulating blood monocytes in cardiovascular disease, at the local tissue level macrophages influence inflammation and homeostasis. In-depth lineage tracking studies have demonstrated subsets of tissue macrophages are derived from multiple sources. Recent investigations have shown that tissue-resident macrophages can originate from embryonic progenitor cells that migrate into the tissues during embryonic and fetal life, and are self-maintained during adulthood.21,24 This embryonic origin and steady-state self-maintenance applies to cardiac- and vascular-resident macrophages as local proliferation of tissue macrophages are involved in several cardiovascular processes.25,26 However, during periods of inflammation or injury, circulating monocytes originating from the bone marrow and extravasating into tissues serve as the primary external source of monocyte-derived macrophages.27
Macrophages are an essential component of innate immunity and play a central role in inflammation and host defense.14 Moreover, these cells fulfill homeostatic functions beyond defense, including the regulation of metabolism and hematopoiesis. As such, macrophages maintain a high degree of functional flexibility to effectively respond to the diverse array of agents they may encounter. The classic view presents macrophage phenotypes based on their in vitro characterization with a particular focus on the dichotomy between ‘classically activated’ M1 macrophages and ‘alternatively activated’ M2 macrophages.28 ‘M1’ macrophages are induced by the Th1 cytokine IFN-γ, TLR ligands like lipopolysaccharide (LPS) and danger signals. This well-studied activation status secretes pro-inflammatory cytokines (IL-1β, IL-6, tumor necrosis factor (TNF), and IL-12) and inflammatory chemokines, including chemokine c-x-c motif ligand 1 and 2, chemokine c-c motif ligand 2–5 (CXCL1and 2, CCL2 5). Classical activated macrophages also produce reactive oxygen and nitrogen intermediates, the latter by inducible nitricoxide (iNOS) synthase.29 In contrast, M2 macrophages are induced by IL-4/IL-13 and core genes expressed by M2 macrophages include scavenger receptors, growth factors [heparin binding epidermal growth factor (HB-EGF) and insulin-like growth factor (IGF)] and suppressors of inflammation and immunity such as IL-10.30 Despite the classification of macrophage subsets being well defined in vitro, distinction of macrophage subsets in vivo is less clear as cells simultaneously express ‘M1’ and ‘M2’ markers. Indeed, the simplified M1/M2 outlook ignores the innumerable variety of macrophage subsets in vivo corresponding to a spectrum of activation and are characterized by a combination of markers. As such this has been replaced with a continuum of macrophage activation states, thus taking into account the diversity of the macrophage in tissues.28
Epigenetic modifications
Epigenetics refers to developmentally or environmentally induced modifications that do not alter the genetic code but instead control how information encoded in DNA is expressed in a tissue- and context-specific manner. Epigenetic marks have traditionally been considered to be stable, potentially transmissible to progeny, and to underlie stable differentiation into various cell types that express markedly different patterns of gene expression. Recently it has become clear that epigenetic chromatin marks are dynamically regulated in response to environmental cues. This has resulted in a shift in the usage of epigenetics to include transient changes in chromatin in response to external stimuli that control gene expression.31 Overall, epigenetic changes can be classified into three main categories: i) DNA methylation; ii) post-translational histone modifications; iii) noncoding RNA. The latter does not create heritable changes but is often grouped with epigenetic mechanisms because of its important regulatory role involving nonprotein coding regions of the genome. Regarding DNA methylation, it is predominantly associated with transcriptional repression and is characterized by transfer of a methyl group to the cytosine ring of DNA by DNA methyltransferases (DNMTs) to form 5-methyl cytosine (5mC).32 DNMT3A and DNMT3B deposit de novo methylation marks, while DNMT1 is responsible for maintaining these marks since these marks must be re-established with each cell division.33 In mammals, the vast majority of DNA methylation in somatic cells occurs at clusters of CpG dinucleotides termed CpG islands, and approximately 40% of genes contain these islands in their promoters.34
In eukaryotes, DNA is packaged into repeating units called nucleosomes by wrapping around multimeric histone proteins. When nucleosomes are organized into tightly packed bundles (heterochromatin), transcription is inhibited by barring access of transcriptional machinery. Conversely, when chromatin is relaxed (euchromatin), the nucleosomes resemble beads on a string and this state is associated with active transcription. Each histone (H) protein is an octamer comprised of 2 sets of H2A, H2B, H3, and H4 proteins with a single histone H1 linker protein between nucleosomes. Each histone subunit has an N-terminal “tail” containing a lysine (K) residue that protrudes away from the surface of the histone octamer creating an exposed surface. Here, histone modifying enzymes can regulate macrophage phenotype through the addition or removal of acetyl or methyl groups.16 Firstly, acetylation and deacetylation is facilitated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Histone acetylation is linked to transcriptional activity whereas histone deacetylation is associated with transcriptional repression.35 Bromodomain and extra-terminal proteins recognize histone acetylation marks and initiate the assembly of the transcriptional machinery.36 Similarly, methylation and demethylation of histones is achieved by histone methyltransferases (HMTs) and histone demethylases (HDMs), respectively.16 Histone methylation can induce both transcriptional activation and repression, depending on the number and location of the methyl groups. An active transcriptional state is characterized by positive marks such as di- or trimethylation(me2/me3) at H3K4, H3K36, H3K79. A repressed transcriptional state manifests itself by increased markings at H3K9me2/me3 and H3K27me3.37
Lastly, non-coding RNAs - including microRNAs (miRs) and long non-coding RNAs (lncRNAs) - do not directly affect chromatin architecture but play an essential role in post-transcriptional regulation of gene expression. lncRNAs are a large group of non-protein coding transcripts, which are more than 200 nucleotides in length. Although the mechanisms by which lncRNAs regulate gene expression are still incomplete, emerging evidence has suggested that lncRNAs can control gene expression at multiple levels including: epigenetic control, transcription, RNA processing, and translation. Many lncRNAs have been found to be significantly enriched in the chromatin fraction, and a common function of these lncRNAs is to recruit chromatin modifying complexes, including the Polycomb group (PcG) or Trithorax group (TrxG), to create a repressive chromatin state or an active chromatin state as well as affect gene expression either in cis or in trans to distant target genes.38 Alternatively, some lncRNAs can form RNA-protein complexes with transcription factors and influence the localization and activity of the transcription factors that they bind, subsequently regulating gene expression.39,40 Regarding miRs, these are evolutionary conserved, small non-coding single-stranded RNAs that are produced by multistep processes of transcription, nuclear export and cytoplasmic cleavage, and work primarily as post-transcriptional repressors via targeting the 3′-untranslated region (3′UTR) of mRNA to provoke its degradation or its translational repression.41 It is estimated that >60% of all protein-coding genes are directly regulated by miRs.42 Furthermore, a given miR may bind to and regulate >1 target, sometimes as a part of the same signaling pathway, adding multiple levels of regulation. As such, miRs are fine-tuners of gene expression patterns in response to pathophysiological stimuli.
Epigenetic regulation of monocyte/macrophage phenotype
An increasing number of studies demonstrate the importance of epigenetics in the regulation of monocyte/macrophage phenotype15,37,43,44 and are summarized in Figure 1. Briefly, DNA methylation, histone methylation and histone acetylation have been shown to influence macrophage phenotype and inflammatory gene production. One representative enzyme involved in histone methylation is SMYD3, a H3K4 methyltransferase, which has been shown to positively regulate M2 polarization. Its expression levels increase in human monocyte derived macrophages with exposure to the combination of M-CSF, IL-4, and IL-13 and decreases with exposure to ‘M1’ stimulation.44 Upregulation of SMYD3 coincides with methylation and transcriptional activation of ALOX15, a lipoxygenase ‘M2’ marker. Another epigenetic enzyme which has been shown to regulate macrophage phenotype in multiple studies is Jmjd3, an H3K27 demethylase. Jmjd3 has also been recognized as an essential regulator of ‘M2’ polarization through its induction of Irf4, Arg1, CD206, and other ‘M2’ markers in IL-4-stimulated and IL-4 + IL-13-stimulated mouse bone marrow derived macrophages.45 However, recent work by Yan et al. demonstrated that pro-inflammatory cytokine induction by the acute-phase protein serum amyloid A also depends on Jmjd3 expression.46 Further, targeting Jmjd3 with small molecule inhibitors impairs inflammatory responses in human primary macrophages.47 Overall, Jmjd3 is required for various responses to different external stimuli including IL-4, RANKL, M-CSF, SAA and LPS, but is not associated with a single macrophage polarization state likely reflecting the need for Jmjd3 to enable responses to various environmental stimuli.
Figure 1. Epigenetic enzymes that regulate macrophage phenotype.
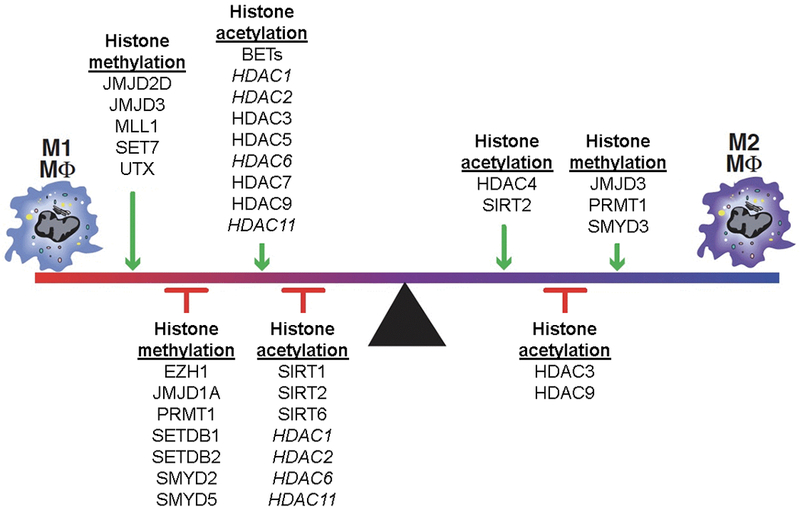
Epigenetic enzymes that regulate histone methylation and acetylation are shown with their influence on macrophage phenotype. Of note the enzymes written in italics have been shown to have both activating and repressive effects in the literature. BET, Bromodomain and extra-terminal; EZH, Enhancer of zeste homolog 2; HDAC, histone deacetylase; JMJD, Jumonji domain-containing protein; MLL, Mixed-lineage leukemia; PRMT1, Protein arginine N-methyl transferase 1; SET, SET domain containing lysine methyltransferase; SETDB, SET domain bifurcated; SIRT, Sirtulin; SMYD, SET and MYN domain; UTX, Ubiquitously transcribed tetratricopeptide repeat, X chromosome.
Regarding histone acetylation, H3 acetylation is an important regulator of macrophage phenotypic expression.48 While current investigations into the importance of HATs in the acetylation of H3 are lacking, extensive observations have been made concerning the role of HDAC3. HDAC3 blocks NFκB signaling by deacetylation of the NFκB p65subunit and allowing its association with the IκB-α.49,50 HDAC3 is a key regulator of M1 macrophage polarization and in parallel acts as a brake for M2 macrophage polarization. HDAC3 also is an important player in the LPS-induced expression of about half of the M1 macrophages-associated inflammatory genes.51 Similarly, HDAC5 is the central regulator of the inflammatory response of macrophages.52
Epigenetic regulation targeting monocyte/macrophages in cardiovascular disease
Over the past decade, multiple investigations have demonstrated that chronic inflammation often underlies the pathogenesis of cardiometabolic and vascular disease. Inflammatory responses, based on macrophage polarization, depend on the cell’s ability to respond to environmental changes and stimuli. Given the impact of epigenetic modifications on the dynamic regulation of macrophage phenotype, recent studies have examined the role of epigenetics in disease progression. This section will focus on the role of epigenetic modifications regulating macrophage phenotype in the pathogenesis of obesity, diabetic wound healing, atherosclerosis, and aortic aneurysms.
Obesity/Metabolic Syndrome
Obesity-associated insulin resistance, diabetes, and metabolic syndrome are sustained by chronic subclinical inflammation.5 In obese subjects and mice, adipocytes release mediators such as MCP-1, TNF, or free fatty acids, which promote the recruitment and activation of adipose tissue macrophages (ATMs).53 In turn, ATMs produce inflammatory cytokines (e.g., TNF, IL-6, IL-1β) that counteract the insulin-sensitizing action of adiponectin and leptin, leading to insulin resistance.54 ATMs from obese mice and humans are polarized toward an ‘M1’ phenotype, with upregulation of TNF-α and iNOS. In contrast, “lean” ATMs express high levels of ‘M2’ genes, including IL-10, Ym1, and Arginase 1.55 Interestingly, weight loss is associated with a shift back to a ‘M2’ phenotype. Progress has been made in defining the molecular pathways that account for polarization of ATMs in obesity.56 Multiple investigations have supported the notion that epigenetics significantly impact inflammatory macrophages in patients with obesity and diabetes. Increased levels of saturated fatty acids lead to upregulation of the DNA methyltransferase DNMT3b in macrophages, thus fostering ‘M1’ polarization and adipose tissue inflammation. Targeting DNMT3b decreased inflammation and restored insulin sensitivity in adipocytes. Along the same line, inhibition of DNA methylation by myeloid deletion of DNMT1 prevented obesity-induced macrophage polarization, inflammation and insulin resistance by epigenetic regulation of the PPARγ1 promoter.57 Recently, lncRNAs have been proposed to have a regulatory role for macrophage polarization in cardiometabolic disease as ‘M1’ and ‘M2’ polarized macrophages present with distinct lncRNA profiles.58 Two lncRNAs shown to play a role in macrophage polarization in human and murine diabetic macrophages are E330013P06 and Dnm3os. Overexpression of E330013P06 lncRNA in macrophages induced inflammatory gene expression, enhanced responsiveness to inflammatory signals, and increased foam cell formation.59 Separately, expression of Dnm3os is upregulated in macrophages and monocytes under diabetic conditions via NF-κB activation. Diabetic macrophage overexpression of Dnm3os altered global histone modifications, upregulated inflammatory gene expression and phagocytosis.60
Histone specific modification has also been shown to have a role in modulating macrophage phenotype in cardiometabolic disease. Specifically, hyperglycemia decreased the inhibitory H3K9me3 mark on inflammatory genes, IL6, IL12, and MIP1α in human monocytes. Likewise, murine diabetic models noted increased activating H3K4 methylation marks at the promoter of the inflammatory NF-κB p65 subunit, mediated by the histone methyltransferases SETD7 and 9 resulting in increased expression of NF-kB-dependent genes VCAM-1, ICAM-1 and MCP-1.61 Furthermore silencing of SETD7/9 by small hairpin RNAs attenuated inflammatory gene expression elicited by advanced glycation end products seen in chronic diabetes.61 Lastly, acetylation has also been shown to impact macrophage phenotype in cardiometabolic syndrome as acetylation of H3 at the promoter of TNFα and COX2 genes was enhanced in monocytes isolated from diabetic subjects. Moreover, the H3 deacetylase SIRT1 is downregulated in the adipose tissue of obese individuals, leading to enhanced macrophage recruitment via increased chemoattractant and cytokine production.62
Wound Healing
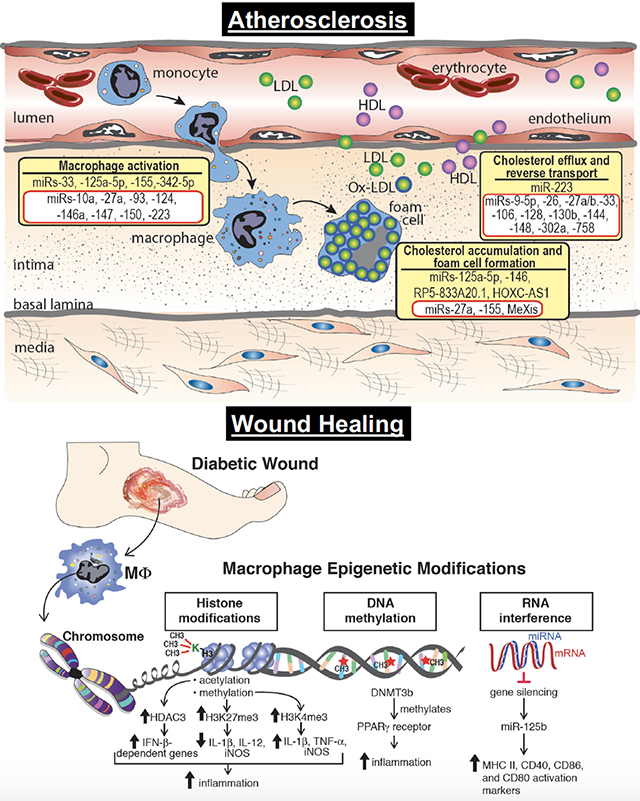
Impaired wound healing is a major secondary complication of type 2 diabetes that often results in limb loss and disability. Normal tissue repair progresses through discrete phases including hemostasis, inflammation, proliferation, and remodeling. In diabetes, progression through these phases is impaired resulting in a sustained inflammatory state and dysfunctional epithelialization in the wound. Monocytes are recruited to the wound site very early, during the inflammatory phase, where they differentiate into macrophages and dendritic cells. There is substantial evidence that infiltrating monocytes/macrophages are critical for establishing the initial inflammatory phase as well as promoting the transition from a proinflammatory to anti-inflammatory environment.63 In diabetic wounds this transition does not occur and macrophages remain in a persistent inflammatory state. Our group recently reported that Ly6Chi monocytes/macrophages, which have been shown to promote inflammation, normally transition to Ly6Clo; however, in diabetic wounds, a second wave of Ly6Chi macrophages are recruited to the wound during the reparative phase and fail to transition to the anti-inflammatory Ly6Clo cells contributing to a sustained proinflammatory environment.64 Several studies have shown a role for macrophage epigenetic modification in type 2 diabetes and diabetic wounds65 (Figure 2). Investigations using both genetic (db/db) and diet induced obesity (DIO) murine models of diabetes, showed that DNA methylation with DNMT1 or DNMT3b was elevated in bone marrow derived macrophages and promoted a proinflammatory macrophage phenotype. Moreover, DNMT1 or DNMT3b knock-down improved wound healing in db/db mice and decreased inflammation.66 Separately, histone methylation impacts macrophage phenotype in diabetic wounds. Our laboratory and others have shown that expression of Jmjd3, the histone demethylase targeting H3K27me3, is increased in wound macrophages in a DIO mouse model of diabetes.67,68 Increased JMJD3 releases the repressive H3K27me3 mark thereby promoting expression of IL-12 in wound macrophages, and this phenomenon can be reversed by JMJD3 inhibition.67 Similarly, a report by Natoli and colleagues showed that Jmjd3 expression was increased in macrophages in response to inflammatory stimuli and was responsible for regulating during bone marrow differentiation.68 Another histone methyltransferase involved in diabetic wound healing is Mixed-lineage leukemia 1 (MLL1) with site specificity for H3K4. CD14+ monocytes isolated from type 2 diabetic patients and wound macrophages isolated for DIO murine model displayed increased MLL1, increased H3K4me3 at inflammatory gene promoters, and increased inflammatory mediator production compared with controls. The use of a MLL1 inhibitor or myeloid-specific MLL1 knockout (Mll1f/fLyz2Cre+) resulted in decreased inflammatory cytokine transcription and improved wound healing.69 Another critical aspect of diabetic tissue repair is angiogenesis. Recent publications have demonstrated that following tissue ischemia, diabetic mice exhibited impaired angiogenesis and arteriogenesis. This was associated with promoter hypomethylation of multiple inflammatory genes in tissue macrophages modify macrophage phenotype toward a proinflammatory ‘M1’ as opposed to anti-inflammatory, proangiogenic ‘M2’ phenotype.70 Lastly, it has recently been demonstrated that hyperglycemia leads to changes in the miR signature in wound healing which plays a role in the dysregulated inflammation of diabetic wounds.71 For example, miR-125b epigenetically enhances macrophage activation by increasing the responsiveness to IFN-γ and concurrently inhibits the reparative phenotype by targeting the ‘M2’ transcription factor IRF4.72
Figure 2. Epigenetic modifications impact diabetic wound healing.
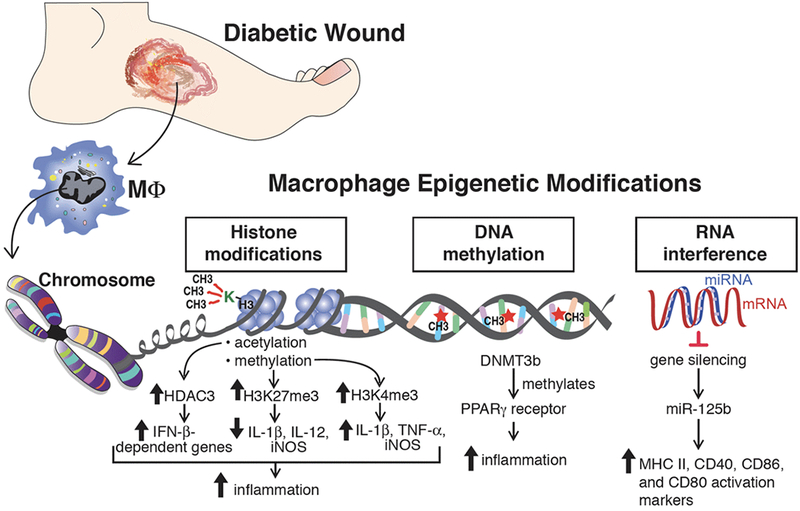
Histone modifications (acetylation and methylation), DNA methylation, and RNA interference are common epigenetic alterations that have been associated with changes in gene expression that influence inflammation during wound healing.
Atherosclerosis
One of the earliest pathogenic events in atherosclerosis is the recruitment of circulating monocytes to the artery wall in areas of endothelial dysfunction and lipoprotein retention. On differentiation into macrophages, these cells play central roles in the pathophysiology of atherosclerosis by maintaining lipid homeostasis in the vessel wall and secreting inflammatory mediators. Lipoprotein uptake by macrophages in the nascent plaque results in the formation of lipid-laden macrophage foam cells that are hallmarks of atherosclerosis.4 These foam cells persist in the artery wall, setting off a maladaptive immune response that mediate the recruitment and activation of other immune cells, thereby chronically sustaining the inflammation that fuels plaque progression. Macrophage cholesterol homeostasis is maintained by the balance between cholesterol uptake, endogenous synthesis, and efflux. Modifications to histones, both methylation and acetylation, have been shown to play a role in macrophage cholesterol metabolism (Figure 3A). Recently, the histone methyltransferase EZH2, which trimethylates H3K27, was shown to exacerbate atherosclerosis in a murine model by inhibiting macrophage cholesterol efflux via ABCA1 thereby promoting foam cell formation.73 In contrast, alterations by histone deacetylases, including SIRT1, and SIRT6, increase cholesterol efflux by the activation of ABCA1 and ABCG1 resulting in a reduction in macrophage derived foam cell formation and its associated inflammation.15 Beyond cholesterol homeostasis, histone modifications also influence macrophage phenotype within the atherosclerotic plaque as alterations in the levels of methylation and acetylation have been observed on H3K9 and H3K27 in macrophages from human advanced atherosclerotic plaques, compared with healthy controls.74 Using a high-throughput RNA-sequencing approach, Neele et al. demonstrated that absence of JMJD3, a H3K27 demethylase, resulted in exacerbation of atherosclerotic plaque morphology and decreased expression of gene sets responsible for ‘M2’ macrophages polarization.75 Further, the expression of several histone deacetylases, HDAC3 and HDAC9, are induced upon monocyte differentiation to macrophages in LDLr−/− mice fed an atherogenic diet. Systemic deletion or myeloid-specific deletion of HDAC9 and HDAC3 respectively attenuated atherosclerosis by an increase in ‘M2’ macrophage polarization and reduction in pro-inflammatory gene expression.51 Separately, recent genome-wide association studies revealed that a genetic variant in the loci corresponding to HDAC9 is associated with coronary artery and atherosclerotic disease.76,77 HDAC9 has been shown to play a vital role in cholesterol homeostasis and inflammation as systemic and bone marrow cell deletion of HDAC9 resulted in upregulation of lipid homeostatic genes, downregulation of inflammatory genes, and polarization toward an ‘M2’ phenotype via increased accumulation of total acetylated H3 and H3K9 at the promoters of ABCA1 and ABCG1 in macrophages.78,79
Figure 3. Histone modifications, miRs, and lncRNAs implicated in atherosclerosis.
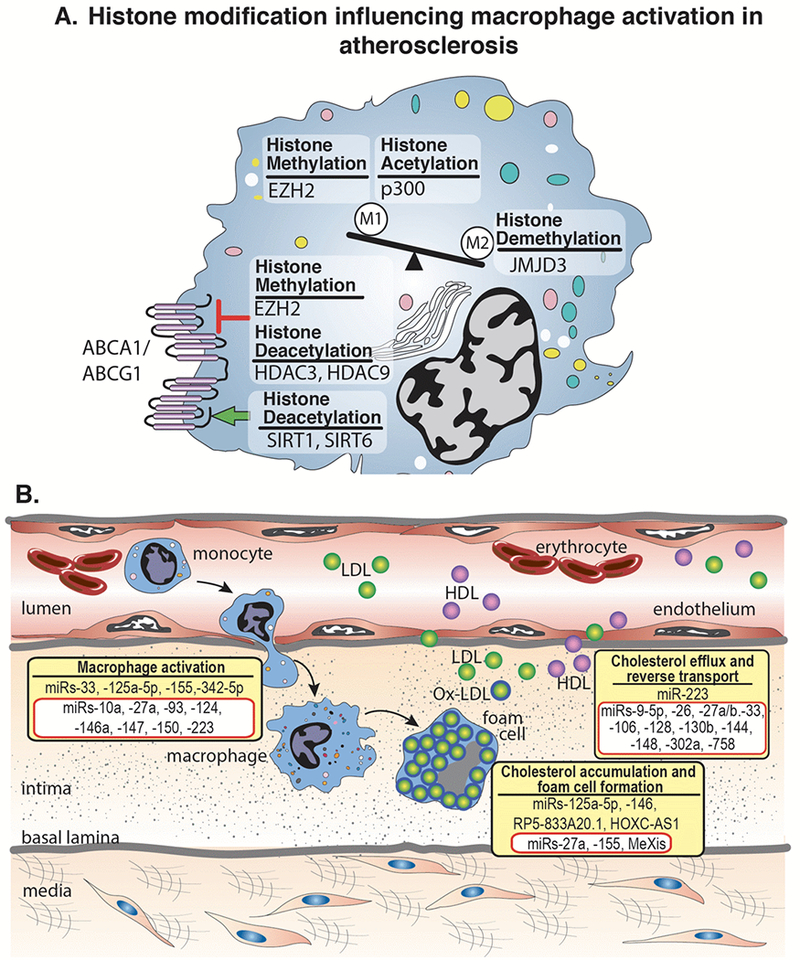
A. Histone modifying enzymes regulate the polarization of macrophages toward classical ‘M1’ or alternative ‘M2’ inflammatory activation. Within atherosclerosis specifically, studies have shown the histone methyl transferase, EZH2, and the histone acetytransferase, p300, polarize macrophages toward an ‘M1’ phenotype. In contrast, the histone demethylase, JMJD3, polarizes toward an ‘M2’ phenotype In addition, histone modifying enzymes can have stimulatory (green arrow) or inhibitory (red arrow) effects on ABCA1/ABCG1 altering cholesterol efflux and reverse cholesterol transport. B. The initial stages of atherosclerosis include adhesion of blood monocytes to the activated endothelium, their maturation into macrophages and their activation and uptake of ox-LDL to form macrophage-derived foam cells. Cholesterol efflux mechanisms, aided by ABCA1/ABCG1 and other factors, help to regress lesion development. miRs and lncRNAs exhibit diverse roles in atherosclerotic progression through the involvement of macrophage activation, foam cell formation, and cholesterol efflux. Representative miRs and lncRNAs that stimulate these processes are shown in yellow boxes. The miRs and lncRNAs highlighted in red boxes have been shown to inhibit macrophage activation, foam cell formation, or cholesterol efflux.
One of the most important prognostic factor for atherosclerotic cardiovascular disease is age, and the accumulation of genomic DNA mutations is a well-known feature of cellular aging. Recently it has been demonstrated that somatic mutations leading to clonal expansion in the absence of other hematologic abnormalities, or clonal hematopoiesis of indeterminate potential (CHIP) predisposed to increase risk of atherosclerotic cardiovascular disease. A pioneering study demonstrated that somatic mutations in DNMT3A, TET2, ASXL1, and JAK2 were each individually associated with coronary heart disease.80 Supporting a major role of mutations in the DNA methyltransferase, DNMT3A, in clonal hematopoiesis in humans, mouse HSPCs exhibiting heterozygotic DNMT3A loss of function develop a competitive advantage and myeloid skewing over time.81 Importantly, DNMT3A deficiency has also been reported to lead to several potentially proatherogenic phenotypes in immune cells, including exacerbated proinflammatory activation of mast cells, and restrained immunosuppressive function in myeloid-derived suppressor cells.82 However, DNMT3A inhibition has also been shown to limit the production of type I interferons in macrophages, which could potentially protective.83 Therefore, given the complexity of the immunomodulatory functions of DNMT3A, carefully designed experimental studies will be required to determine the potential contribution of somatic mutations in this gene to CVD.
Alterations in miRs have also been implicated in aberrant macrophage cholesterol homeostasis and polarization during atherosclerosis. Many miRs have been implicated in macrophage cholesterol metabolism and have been previously described in detail84–86 and summarized in Figure 3B. To briefly review, miR-125a-5p and miR-146 decrease lipid uptake and cytokine release in oxidized LDL–stimulated macrophages, in part, by targeting the genes oxysterol binding protein-like 9 and TLR4, respectively.87 Furthermore, numerous miRNAs have been identified that inhibit macrophage cholesterol efflux via ABCA1 thereby promoting foam cell formation, including miR- 26, miR-33, miR-106, miR-128-1, miR-130b, miR-144, and miR-148, and miR-302a.84 Beyond cholesterol homeostasis, miRs also influence macrophage phenotype during progression of atherosclerosis as in response to microenvironmental signals, macrophages can initiate different activation programs, including the classical proinflammatory and anti-inflammatory phenotype. One miR involved in regulation of macrophage phenotype is miR-33 which regulates macrophage cellular metabolism to alter the cell’s inflammatory phenotype. miR-33 reduces fatty acid oxidation, the metabolic program that fuels ‘M2’ macrophages, and promotes aerobic glycolysis, which in turn sustains the inflammatory ‘M1’ macrophage phenotype.88 Inhibition of miR-33 metabolically reprograms plaque macrophages to the ‘M2’ phenotype involved in resolving inflammation and tissue repair. Further, miR-155 and miR-342-5p can also reprogram macrophages from the ‘M2’ to ‘M1’ phenotype, thereby impacting accumulation of ‘M1’ macrophages.89,90 miR-155 expression is significantly higher in CD14+ monocytes from patients with coronary artery disease than from healthy controls and is induced by oxidized forms of LDL in macrophages. miR-155 acts to repress negative regulators of inflammatory cytokine signaling, such as SOCS, and thereby promoting the release of proinflammatory cytokines.91 On the other hand, miR-146a exerts atheroprotective effects on macrophages by inhibiting the activation and secretion of pro-inflammatory cytokines via suppressing NF-κB.92 Notably, expression of miR-146a in hematopoietic cells is crucial in restraining the chronic inflammatory response in vivo.93 miR-27a can promote markers of anti-inflammatory macrophages and secretion of IL-10.94 Lastly, recent literature has demonstrated an interesting interaction between miRs and histone modifying enzymes with miRs regulating the expression of epigenetic enzymes.95,96 The histone methylase, EZH2, has been shown to be regulated by expression of miR-26a, miR-101, miR-205 and miR-21 in a variety of disease states.97–100 Further, members of the miR-29 and miR-148 family directly target DNMTs 3A and 3B in nonsmall cell lung cancer cells and acute myeloid leukemia.101–103 Thus, miRs may be involved in the establishment and ⁄ or maintenance of DNA methylation. At this time it is undefined if miRs have similar roles regulating expression of epigenetic enymes in macrophages, but it is hypothesized that parallel miR and epigenetic enzyme interactions exist in innate immune cells during atherosclerotic development.
A number of studies have shown important contributions of lncRNAs in atherosclerosis and have been recently summarized.104 In addition, accumulating evidence suggests that lncRNAs guide chromatin-modifying enzymes to specific loci contributing to the epigenetic regulation of many autosomal genes.105,106 Regarding macrophages specifically, lncRNAs are key drivers of sterol regulation during artherogenesis. RP5-833A20.1 is an intronic lncRNA that regulates the transcription factor NFIA in human foam-cell macrophages by modulating the miR, hsa-miR-382-5p.107 Further, recent work has identified the lncRNA, MeXis, which influences chromatin architecture at the locus of ABCA1 resulting in enhanced ABCA1 expression.108 This effect is dependent on the transcriptional coactivator DDX17. Genetic deletion of MeXis from immune cells in a murine model markedly enhances foam cell formation and accelerates atherosclerosis. In addition, perturbing the human orthologue of MeXis influences ABCA1 levels and human macrophage function. Finally, it has been noted that HOXC-AS1 is downregulated in atherosclerotic plaques and that overexpression of HOXC-AS1 reduces ox-LDL-induced cholesterol accumulation in macrophages by activating homeobox C6 (HOXC6).109
Abdominal Aortic Aneurysm
Abdominal aortic aneurysms (AAAs) are a degenerative cardiovascular disease resulting in the progressive dilation of the aorta that may lead to rupture which if occurs is fatal in 80% of cases. This disease represents a global health concern as AAAs are responsible for between 2 and 4% of deaths in white males over the age of 65.110 Currently, surgical repair is the only therapeutic option in order to treat AAA rupture with a lack of proven medical therapy to prevent AAA progression. The exact etiological basis of AAA is still not fully understood, however macrophage inflammation represents a pathological hallmark of disease development with the majority of macrophages within the aortic wall originate from circulating monocytes.111,112 At this time, the contribution of tissue resident macrophages remains poorly defined. Recently, through the characterization of surgical samples and animal models investigations have demonstrated a role of epigenetic modification in monocytes/macrophages within the pathogenesis of aortic aneurysm formation. Global methylation levels within peripheral blood mononuclear cells are significantly altered on CpG islands in AAA patients in comparison to controls and exhibit a positive correlation with increased aortic diameter.113 Further within human AAA samples, expression of HDACs, specifically class I and IIa, were found to be increased in comparison to control samples and colocalized with macrophages within the aortic wall. Using murine models, treatment with HDAC class I (MS-275) or class IIa (MC-1568) inhibitors reduced AAA incidence, decreased macrophage inflammation, and reduce proinflammatory mediators.114 Lastly, miRs have also been shown to regulate macrophage function in murine models of AAAs as miR-33 deficiency resulted in decreased ‘M1’ gene expression, protease activity, and macrophage infiltration into the aortic wall. Further, bone marrow transplant of miR-33 deficient cells decreased AAA formation suggesting myeloid-specific miR-33 may be instrumental in AAA pathogenesis.115 Although additional research is necessary to further uncover the role of epigenetic regulation of macrophage phenotype in AAA pathogenesis, given the impact of epigenetic modification in other cardiovascular diseases this represents a promising field of discovery.
Conclusion
During the past several decades, significant progress has been achieved in the understanding and treatment of cardiometabolic and vascular disease. However, the persistent clinical impact of these conditions suggest more work remains to be done. To improve treatment of cardiovascular patients, targeting inflammatory pathways offers additional benefits over and in combination with traditional therapies.116 Evidence discussed here suggests that epigenetic processing plays a central role in the balance of macrophage phenotypes as well as the regulation of inflammation during the pathogenesis of cardiometabolic and vascular disorders. Inflammatory responses and macrophage polarization depend on the cell’s ability to respond to environmental changes and stimuli. Epigenetic mechanisms allow such ‘adaptive’ responses and enhance macrophage diversity and plasticity. However, maladaptive epigenetic changes contribute to the persistence of disease inflammation. Therefore, pharmacological intervention to reprogram a ‘diseased’ epigenetic landscape is an attractive therapeutic target for the treatment of cardiovascular disorders. The understanding of chromatin architecture has led to the design of specific molecules to modulate chromatin accessibility by enhancing or repressing epigenetic marks on DNA/histone complexes.117 New small molecules that reduce the polarization and activation of inflammatory cells and in particular macrophages at inflammatory sites are considered to be the next generation of anti-inflammatory drugs. Noteworthy, some of these drugs have been already approved for the treatment of several conditions including cancer. The challenge for future investigations is how to achieve tissue-specific modulation of chromatin in monocytes/macrophages since systemic inhibition or activation may lead to adverse side effects. With future research, epigenetics will serve as a useful framework through which to gain new insights into the role of myeloid cells in cardiometabolic and vascular disease.
Highlights.
Monocytes and macrophages have distinct phenotypes during the development and progression of cardiovascular disease and these phenotypes can be regulated by epigenetic modifications.
Histone modifications (acetylation and methylation) and DNA methylation underlies the chronic inflammatory macrophage phenotype observed in obesity and diabetic wound healing.
Non-coding RNAs - including microRNAs (miRs) and long non-coding RNAs (lncRNAs)-alter monocytes and macrophage function during the development and progression of atherosclerosis.
ACKNOWLEDGEMENTS
We would like to thank Robin G. Kunkel, Research Associate in the Pathology Department, University of Michigan, for her artistic work.
SOURCES OF FUNDING
This laboratory is supported by National Institutes of Health (NIH) RO1- HL137919, NIH-F32 DK-117545-01, American College of Surgeons, Vascular and Endovascular Surgery Society, and the Doris Duke Foundation.
Footnotes
DISCLOSURES
The authors report no conflicts of interest
References
- 1.Timmis A, Townsend N, Gale C, et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur Heart J. 2018;39(7):508–579. doi: 10.1093/eurheartj/ehx628. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137(12):e67–e492. [DOI] [PubMed] [Google Scholar]
- 3.Libby P Inflammation in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. [DOI] [PubMed] [Google Scholar]
- 6.Davis FM, Kimball A, Boniakowski A, Gallagher K. Dysfunctional Wound Healing in Diabetic Foot Ulcers: New Crossroads. Curr Diab Rep. 2018;18(1):2. [DOI] [PubMed] [Google Scholar]
- 7.Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL, Gallagher KA. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J Immunol. 2017;199(1):17–24. [DOI] [PubMed] [Google Scholar]
- 8.Bourlier V, Bouloumie A. Role of macrophage tissue infiltration in obesity and insulin resistance. Diabetes Metab. 2009;35(4):251–260. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest. 2017;127(1):5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. 2008;4(11):619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE, Harrison LC. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59(7):1648–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11(11):750–761. [DOI] [PubMed] [Google Scholar]
- 14.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27(1):451–483. [DOI] [PubMed] [Google Scholar]
- 15.Gosselin D, Glass CK. Epigenomics of macrophages. Immunol Rev. 2014;262(1):96–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van den Bossche J, Neele AE, Hoeksema MA, de Winther MPJ. Macrophage polarization: the epigenetic point of view. Curr Opin Lipidol. 2014;25(5):367–373. [DOI] [PubMed] [Google Scholar]
- 17.Baccarelli A, Ghosh S. Environmental exposures, epigenetics and cardiovascular disease. Curr Opin Clin Nutr Metab Care. 2012;15(4):323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanson MA, Godfrey KM. Epigenetic mechanisms underlying type 2 diabetes mellitus. Nat Rev Endocrinol. 2015;11(5):261–263. [DOI] [PubMed] [Google Scholar]
- 19.Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, Bhatt K, Leung A, Wu X, Putta S, Saetrom P, Devaraj S, Natarajan R. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes. 2014;63(12):4249–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJM, Liu Y-J, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–80. [DOI] [PubMed] [Google Scholar]
- 21.Idzkowska E, Eljaszewicz A, Miklasz P, Musial WJ, Tycinska AM, Moniuszko M. The Role of Different Monocyte Subsets in the Pathogenesis of Atherosclerosis and Acute Coronary Syndromes. Scand J Immunol. 2015;82(3):163–173. [DOI] [PubMed] [Google Scholar]
- 22.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. [DOI] [PubMed] [Google Scholar]
- 23.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DWH, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ensan S, Li A, Besla R, et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. 2016;17(2):159–168. [DOI] [PubMed] [Google Scholar]
- 25.Lhoták Š, Gyulay G, Cutz J-C, Al-Hashimi A, Trigatti BL, Richards CD, Igdoura SA, Steinberg GR, Bramson J, Ask K, Austin RC. Characterization of Proliferating Lesion-Resident Cells During All Stages of Atherosclerotic Growth. J Am Heart Assoc. 2016;5(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS, Epelman S. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20(1):29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. 2016;17(1):2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van den Bossche J, Lamers WH, Koehler ES, Geuns JMC, Alhonen L, Uimari A, Pirnes-Karhu S, Van Overmeire E, Morias Y, Brys L, Vereecke L, De Baetselier P, Van Ginderachter JA. Pivotal Advance: Arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J Leukoc Biol. 2012;91(5):685–699. [DOI] [PubMed] [Google Scholar]
- 30.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. [DOI] [PubMed] [Google Scholar]
- 31.Natoli G Maintaining cell identity through global control of genomic organization. Immunity. 2010;33(1):12–24. [DOI] [PubMed] [Google Scholar]
- 32.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187(4173):226–232. [PubMed] [Google Scholar]
- 33.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. [DOI] [PubMed] [Google Scholar]
- 34.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci. 2002;99(6):3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64(2):435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 37.Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013;34(5):216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Wu Z, Fu X, Han W. lncRNAs: Insights into their function and mechanics in underlying disorders. Mutat Res Mutat Res. 2014;762:1–21. [DOI] [PubMed] [Google Scholar]
- 40.Angrand P-O, Vennin C, Le Bourhis X, Adriaenssens E. The role of long non-coding RNAs in genome formatting and expression. Front Genet. 2015;6:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8(1):23–36. [DOI] [PubMed] [Google Scholar]
- 42.Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoeksema MA, Gijbels MJ, Van den Bossche J, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6(9):1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, Carson WF, Mukherjee S, Grembecka J, Cierpicki T, Jarai G, Westwick J, Kunkel SL, Hogaboam CM. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. Zissel G, ed. PLoS One. 2013;8(10):e78045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11(10):936–944. [DOI] [PubMed] [Google Scholar]
- 46.Xia B, Xu B, Sun Y, Xiao L, Pan J, Jin H, Tong P. The effects of Liuwei Dihuang on canonical Wnt/β-catenin signaling pathway in osteoporosis. J Ethnopharmacol. 2014;153(1):133–141. [DOI] [PubMed] [Google Scholar]
- 47.Kruidenier L, Chung C, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488(7411):404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feng D, Sangster-Guity N, Stone R, Korczeniewska J, Mancl ME, Fitzgerald-Bocarsly P, Barnes BJ. Differential requirement of histone acetylase and deacetylase activities for IRF5-mediated proinflammatory cytokine expression. J Immunol. 2010;185(10):6003–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–691. [DOI] [PubMed] [Google Scholar]
- 50.Leus NG, Zwinderman MR, Dekker FJ. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-κB-mediated inflammation. Curr Opin Chem Biol. 2016;33:160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, Feng D, Steger DJ, Schug J, Artis D, Lazar MA. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011;25(23):2480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poralla L, Stroh T, Erben U, Sittig M, Liebig S, Siegmund B, Glauben R. Histone deacetylase 5 regulates the inflammatory response of macrophages. J Cell Mol Med. 2015;19(9):2162–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olefsky JM, Glass CK. Macrophages, Inflammation, and Insulin Resistance. Annu Rev Physiol. 2010;72(1):219–246. [DOI] [PubMed] [Google Scholar]
- 54.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6(1):275–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Cao Q, Yu L, Shi H, Xue B, Shi H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI insight. 2016;1(19):e87748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li C, Xu MM, Wang K, Adler AJ, Vella AT, Zhou B. Macrophage polarization and meta-inflammation. Transl Res. 2018;191:29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, Bhatt K, Leung A, Wu X, Putta S, Saetrom P, Devaraj S, Natarajan R. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes. 2014;63(12):4249–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das S, Reddy MA, Senapati P, Stapleton K, Lanting L, Wang M, Amaram V, Ganguly R, Zhang L, Devaraj S, Schones DE, Natarajan R. Diabetes Mellitus-Induced Long Noncoding RNA Dnm3os Regulates Macrophage Functions and Inflammation via Nuclear Mechanisms. Arterioscler Thromb Vasc Biol. 2018;38(8):1806–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Reddy MA, Miao F, Shanmugam N, Yee J-K, Hawkins D, Ren B, Natarajan R. Role of the Histone H3 Lysine 4 Methyltransferase, SET7/9, in the Regulation of NF-κB-dependent Inflammatory Genes. J Biol Chem. 2008;283(39):26771–26781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, Muise ES, Hsiao JJ, Frederick DW, Yonemitsu S, Banks AS, Qiang L, Bhanot S, Olefsky JM, Sears DD, Caprio S, Shulman GI. SirT1 Regulates Adipose Tissue Inflammation. Diabetes. 2011;60(12):3235–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44(3):450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kimball A, Schaller M, Joshi A, Davis FM, denDekker A, Boniakowski A, Bermick J, Obi A, Moore B, Henke PK, Kunkel SL, Gallagher KA. Ly6CHi Blood Monocyte/Macrophage Drive Chronic Inflammation and Impair Wound Healing in Diabetes Mellitus. Arterioscler Thromb Vasc Biol. 2018;38(5):1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ahmed M, de Winther MPJ, Van den Bossche J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology. 2017;222(10):937–943. [DOI] [PubMed] [Google Scholar]
- 66.Yan J, Tie G, Wang S, Tutto A, DeMarco N, Khair L, Fazzio TG, Messina LM. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat Commun. 2018;9(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gallagher K a., Joshi A, Carson WF, Schaller M, Allen R, Mukerjee S, Kittan N, Feldman EL, Henke PK, Hogaboam C, Burant CF, Kunkel SL. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes. 2015;64(4):1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130(6):1083–1094. [DOI] [PubMed] [Google Scholar]
- 69.Kimball AS, Joshi A, Carson WF, Boniakowski AE, Schaller M, Allen R, Bermick J, Davis FM, Henke PK, Burant CF, Kunkel SL, Gallagher KA. The Histone Methyltransferase MLL1 Directs Macrophage-Mediated Inflammation in Wound Healing and Is Altered in a Murine Model of Obesity and Type 2 Diabetes. Diabetes. 2017;66(9):2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Babu M, Durga Devi T, Mäkinen P, Kaikkonen M, Lesch HP, Junttila S, Laiho A, Ghimire B, Gyenesei A, Ylä-Herttuala S. Differential Promoter Methylation of Macrophage Genes Is Associated With Impaired Vascular Growth in Ischemic Muscles of Hyperlipidemic and Type 2 Diabetic Mice: Genome-Wide Promoter Methylation Study. Circ Res. 2015;117(3):289–299. [DOI] [PubMed] [Google Scholar]
- 71.Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes Complications: The MicroRNA Perspective. Diabetes. 2011;60(7):1832–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chaudhuri AA, So AY-L, Sinha N, Gibson WSJ, Taganov KD, O’Connell RM, Baltimore D. MicroRNA-125b potentiates macrophage activation. J Immunol. 2011;187(10):5062–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lv Y-C, Tang Y-Y, Zhang P, Wan W, Yao F, He P-P, Xie W, Mo Z-C, Shi J-F, Wu J-F, Peng J, Liu D, Cayabyab FS, Zheng X-L, Tang X-Y, Ouyang X-P, Tang C-K. Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. Feng Y-M, ed. PLoS One. 2016;11(6):e0157265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greißel A, Culmes M, Burgkart R, Zimmermann A, Eckstein H-H, Zernecke A, Pelisek J. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol. 2016;25(2):79–86. [DOI] [PubMed] [Google Scholar]
- 75.Neele AE, Prange KH, Hoeksema MA, van der Velden S, Lucas T, Dimmeler S, Lutgens E, Van den Bossche J, de Winther MP. Macrophage Kdm6b controls the pro-fibrotic transcriptome signature of foam cells. Epigenomics. 2017;9(4):383–391. [DOI] [PubMed] [Google Scholar]
- 76.Dichgans M, Malik R, König IR, et al. Shared Genetic Susceptibility to Ischemic Stroke and Coronary Artery Disease. Stroke. 2014;45(1):24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malik R, Chauhan G, Traylor M, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cao Q, Rong S, Repa JJ, St. Clair R, Parks JS, Mishra N. Histone Deacetylase 9 Represses Cholesterol Efflux and Alternatively Activated Macrophages in Atherosclerosis Development. Arterioscler Thromb Vasc Biol. 2014;34(9):1871–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Azghandi S, Prell C, van der Laan SW, Schneider M, Malik R, Berer K, Gerdes N, Pasterkamp G, Weber C, Haffner C, Dichgans M. Deficiency of the Stroke Relevant HDAC9 Gene Attenuates Atherosclerosis in Accord With Allele-Specific Effects at 7p21.1. Stroke. 2015;46(1):197–202. [DOI] [PubMed] [Google Scholar]
- 80.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, Helton NM, Guo M, Klco JM, O’Laughlin S, Fronick C, Fulton R, Chang GS, Petti AA, Miller CA, Ley TJ. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J Clin Invest. 2017;127(10):3657–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, Monticelli S. Dnmt3a restrains mast cell inflammatory responses. Proc Natl Acad Sci U S A. 2017;114(8):E1490–E1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, Shen Q, Liu X, Zhu X, Li N, Cheng Z, Fan G, Wang Q, Cao X. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat Immunol. 2016;17(7):806–815. [DOI] [PubMed] [Google Scholar]
- 84.Feinberg MW, Moore KJ. MicroRNA Regulation of Atherosclerosis. Circ Res. 2016;118(4):703–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Donaldson CJ, Lao KH, Zeng L. The salient role of microRNAs in atherogenesis. J Mol Cell Cardiol. 2018;122:98–113. [DOI] [PubMed] [Google Scholar]
- 86.Schober A, Weber C. Mechanisms of MicroRNAs in Atherosclerosis. Annu Rev Pathol. 2016;11(1):583–616. [DOI] [PubMed] [Google Scholar]
- 87.Yang K, He YS, Wang XQ, Lu L, Chen QJ, Liu J, Sun Z, Shen WF. MiR-146a inhibits oxidized low-density lipoprotein-induced lipid accumulation and inflammatory response via targeting toll-like receptor 4. FEBS Lett. 2011;585(6):854–860. [DOI] [PubMed] [Google Scholar]
- 88.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K, Rayner KJ, Steinberg GR, Zamore PD, Fisher EA, Loke P, Moore KJ. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125(12):4334–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang C-Y, Zen K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012;4(5):341–343. [DOI] [PubMed] [Google Scholar]
- 90.Wei Y, Nazari-Jahantigh M, Chan L, Zhu M, Heyll K, Corbalán-Campos J, Hartmann P, Thiemann A, Weber C, Schober A. The microRNA-342-5p fosters inflammatory macrophage activation through an Akt1- and microRNA-155-dependent pathway during atherosclerosis. Circulation. 2013;127(15):1609–1619. [DOI] [PubMed] [Google Scholar]
- 91.Sun H-X, Zeng D-Y, Li R-T, Pang R-P, Yang H, Hu Y-L, Zhang Q, Jiang Y, Huang L-Y, Tang Y-B, Yan G-J, Zhou J-G. Essential Role of MicroRNA-155 in Regulating Endothelium-Dependent Vasorelaxation by Targeting Endothelial Nitric Oxide Synthase. Hypertension. 2012;60(6):1407–1414. [DOI] [PubMed] [Google Scholar]
- 92.Li K, Ching D, Luk FS, Raffai RL. Apolipoprotein E enhances microRNA-146a in monocytes and macrophages to suppress nuclear factor-κB-driven inflammation and atherosclerosis. Circ Res. 2015;117(1):e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheng HS, Besla R, Li A, Chen Z, Shikatani EA, Nazari-Jahantigh M, Hammoutène A, Nguyen M-A, Geoffrion M, Cai L, Khyzha N, Li T, MacParland SA, Husain M, Cybulsky MI, Boulanger CM, Temel RE, Schober A, Rayner KJ, Robbins CS, Fish JE. Paradoxical Suppression of Atherosclerosis in the Absence of microRNA-146a. Circ Res. 2017;121(4):354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saha B, Bruneau JC, Kodys K, Szabo G. Alcohol-induced miR-27a regulates differentiation and M2 macrophage polarization of normal human monocytes. J Immunol. 2015;194(7):3079–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van den Hove DL, Kompotis K, Lardenoije R, Kenis G, Mill J, Steinbusch HW, Lesch K-P, Fitzsimons CP, De Strooper B, Rutten BPF. Epigenetically regulated microRNAs in Alzheimer’s disease. Neurobiol Aging. 2014;35(4):731–745. [DOI] [PubMed] [Google Scholar]
- 96.Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011;278(10):1598–1609. [DOI] [PubMed] [Google Scholar]
- 97.Friedman JM, Liang G, Liu C-C, Wolff EM, Tsai YC, Ye W, Zhou X, Jones PA. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009;69(6):2623–2629. [DOI] [PubMed] [Google Scholar]
- 98.Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TFE, Möller P, Stilgenbauer S, Pollack JR, Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112(10):4202–4212 [DOI] [PubMed] [Google Scholar]
- 99.Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M, Salvioni R, Supino R, Moretti R, Limonta P, Valdagni R, Daidone MG, Zaffaroni N. miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res. 2009;69(6):2287–2295. [DOI] [PubMed] [Google Scholar]
- 100.Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell. 2009;36(1):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104(40):15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CEA, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V, Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD, Byrd JC, Chan K, Wu L-C, Croce CM, Marcucci G. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113(25):6411–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Duursma AM, Kedde M, Schrier M, le Sage C, Agami R. miR-148 targets human DNMT3b protein coding region. RNA. 2008;14(5):872–877. doi: 10.1261/rna.972008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Z, Salisbury D, Sallam T. Long Noncoding RNAs in Atherosclerosis. J Am Coll Cardiol. 2018;72(19):2380–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mathiyalagan P, Keating ST, Du X-J, El-Osta A. Interplay of chromatin modifications and non-coding RNAs in the heart. Epigenetics. 2014;9(1):101–112. doi: 10.4161/epi.26405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Magistri M, Faghihi MA, St Laurent G, Wahlestedt C. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 2012;28(8):389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu Y-W, Zhao J-Y, Li S-F, Huang J-L, Qiu Y-R, Ma X, Wu S-G, Chen Z-P, Hu Y-R, Yang J-Y, Wang Y-C, Gao J-J, Sha Y-H, Zheng L, Wang Q. RP5-833A20.1/miR-382-5p/NFIA–Dependent Signal Transduction Pathway Contributes to the Regulation of Cholesterol Homeostasis and Inflammatory Reaction. Arterioscler Thromb Vasc Biol. 2015;35(1):87–101. [DOI] [PubMed] [Google Scholar]
- 108.Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, Eskin A, Casero D, Zhang Z, Sandhu J, Salisbury D, Rajbhandari P, Civelek M, Hong C, Ito A, Liu X, Daniel B, Lusis AJ, Whitelegge J, Nagy L, Castrillo A, Smale S, Tontonoz P. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat Med. 2018;24(3):304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang C, Hu Y-W, Zhao J-J, Ma X, Zhang Y, Guo F-X, Kang C-M, Lu J-B, Xiu J-C, Sha Y-H, Gao J-J, Wang Y-C, Li P, Xu B-M, Zheng L, Wang Q. Long Noncoding RNA HOXC-AS1 Suppresses Ox-LDL-Induced Cholesterol Accumulation Through Promoting HOXC6 Expression in THP-1 Macrophages. DNA Cell Biol. 2016;35(11):722–729. [DOI] [PubMed] [Google Scholar]
- 110.Sidloff D, Stather P, Dattani N, Bown M, Thompson J, Sayers R, Choke E. Aneurysm Global Epidemiology Study. Circulation. 2014;129(7):747–753. [DOI] [PubMed] [Google Scholar]
- 111.Davis FMM, Rateri DLL, Daugherty A. Mechanisms of aortic aneurysm formation: translating preclinical studies into clinical therapies. Heart. 2014;100(19):1498–1505. [DOI] [PubMed] [Google Scholar]
- 112.Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14(8):457–471. [DOI] [PubMed] [Google Scholar]
- 113.Toghill BJ, Saratzis A, Freeman PJ, Sylvius N, UKAGS collaborators MJ, Bown MJ. SMYD2 promoter DNA methylation is associated with abdominal aortic aneurysm (AAA) and SMYD2 expression in vascular smooth muscle cells. Clin Epigenetics. 2018;10(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Galán M, Varona S, Orriols M, Rodríguez JA, Aguiló S, Dilmé J, Camacho M, Martínez-González J, Rodriguez C. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: therapeutic potential of HDAC inhibitors. Dis Model Mech. 2016;9(5):541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nakao T, Horie T, Baba O, Nishiga M, Nishino T, Izuhara M, Kuwabara Y, Nishi H, Usami S, Nakazeki F, Ide Y, Koyama S, Kimura M, Sowa N, Ohno S, Aoki H, Hasegawa K, Sakamoto K, Minatoya K, Kimura T, Ono K. Genetic Ablation of MicroRNA-33 Attenuates Inflammation and Abdominal Aortic Aneurysm Formation via Several Anti-Inflammatory Pathways. Arterioscler Thromb Vasc Biol. 2017;37(11):2161–2170. [DOI] [PubMed] [Google Scholar]
- 116.Ridker PM, Lüscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J. 2014;35(27):1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Neele AE, Van den Bossche J, Hoeksema MA, de Winther MPJ. Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol. 2015;763:79–89. [DOI] [PubMed] [Google Scholar]