

Abstract
Background
Stroke can lead to cardiac dysfunction in patients, but the mechanisms underlying the interaction between the injured brain and the heart are poorly understood. The objective of the study is to investigate the effects of experimental murine stroke on cardiac function and molecular signalling in the heart.
Methods and results
Mice were subjected to filament‐induced left middle cerebral artery occlusion for 30 or 60 min or sham surgery and underwent repetitive micro‐echocardiography. Left ventricular contractility was reduced early (24–72 h) but not late (2 months) after brain ischaemia. Cardiac dysfunction was accompanied by a release of high‐sensitive cardiac troponin (hsTNT (ng/ml): d1: 7.0 ± 1.0 vs. 25.0 ± 3.2*; d3: 7.3 ± 1.1 vs. 52.2 ± 16.7*; d14: 5.7 ± 0.8 vs. 5.2 ± 0.3; sham vs. 60 min. MCAO; mean ± SEM; *p < 0.05); reduced heart weight (heart weight/tibia length ratio: d1: 6.9 ± 0.2 vs. 6.4 ± 0.1*; d3: 6.7 ± 0.2 vs. 5.8 ± 0.1*; d14: 6.7 ± 0.2 vs. 6.4 ± 03; sham vs. 60 min. MCAO; mean ± SEM; *p < 0.05); resulting from cardiomyocyte atrophy (cardiomyocyte size: d1: 12.8% ± 0.002**; d3: 13.5% ± 0.002**; 14d: 6.3% ± 0.003*; 60 min. MCAO vs. sham; mean ± SEM; **p < 0.01; *p < 0.05), accompanied by increased atrogin‐1 and the E3 ubiquitin ligase murf‐1. Net norepinephrine but not synthesis was increased, suggesting a reduced norepinephrine release or an increase of norepinephrine re‐uptake, resulting in a functional denervation. Transcriptome analysis in cardiac tissue identified the transcription factor peroxisome proliferator‐activated receptor gamma as a potential mediator of stroke‐induced transcriptional dysregulation involved in cardiac atrophy.
Conclusions
Stroke induces a complex molecular response in the heart muscle with immediate but transient cardiac atrophy and dysfunction.
Keywords: Ischaemic stroke, Cardiac dysfunction, Atrophy, Cardiomyocytes, Left ventricular contractility
Introduction
The heart and the brain are closely interconnected anatomically and functionally in health and disease. Cardiac diseases including arrhythmias such as atrial fibrillation cause 20–30% of all ischaemic strokes. Vice versa, acute stroke can affect cardiac function. A plethora of clinical manifestations comprising reduced heart rate,1 atrial fibrillation,2 cardiac dysfunction,3 elevated troponin,4, 5 and stress‐induced acute cardiomyopathy5, 6 can be found in stroke patients. Cardiac disease also contributes substantially to the long‐term prognosis of ischaemic stroke patients.4, 7 The conventional explanation for this is that cardiac disorders in stroke patients result from shared vascular risk factors leading to concomitant ischaemic manifestations in the heart and the brain in the same patients.3 Alternatively, brain injury may have a long‐term effect on other organs including the heart.
The mechanisms underlying the crosstalk between the acutely injured brain and the heart are not well understood at present. Current thinking is that cerebral ischaemia triggers an acute stress response that is mediated by the overactive sympathetic nervous system.8, 9 However, the signalling pathways between the brain and the heart as well as the molecular downstream processes in the heart remain to be characterized.
Herein, we show in experimental murine stroke that brain ischaemia triggers myocardial dysfunction and describe the altered molecular expression in the heart muscle.
Materials and methods
Animals
The study was conducted in accordance with national guidelines for the use of experimental animals. All experimental procedures were approved by the governmental committees (Animal Care Committee, Regierungspräsidium Karlsruhe, Germany) and were performed in accordance with the German Animal Welfare Act (directive 2010/63 EU) and the ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines. Age‐matched (8–12 weeks) male C57BL/6 mice obtained from Janvier Labs (Le Genest‐Saint‐Isle, France) were used for the experiments. In a single cage, four specific pathogen‐free mice were housed in the animal facility of the University of Heidelberg. All mice were kept on a standard 12 h light/dark cycle and had free access to food and water.
Filament model
In the ischaemia experiments, we induced transient focal cerebral ischaemia for 30 or 60 min, respectively, as described.10 Briefly, mice were anaesthetized with 2% isoflurane in O2, and middle cerebral artery occlusion (MCAO) was performed by advancing a nylon‐coated monofilament with a diameter of 0.22 mm (Doccol, Sharon, MA, USA) via the common carotid artery until a resistance was felt. Successful occlusion of the middle cerebral artery was verified by a laser Doppler flowmetry. The laser Doppler flowmetry probe (P403, Perimed, Järfälla, Sweden) was placed 3 mm lateral and 1 mm posterior to the bregma and obtained relative perfusion units (Periflux 4001, Perimed). Only animals in which relative cerebral blood flow dropped below 25% of pre‐ischaemic baseline after MCAO were included in the analysis. Mice were re‐anaesthetized to remove the filament. During surgery, a body temperature of 37°C was feedback controlled using a heating pad. The preparation for the sham surgery was identical, but the filament was not inserted into the vessel. After surgery, all mice received carprofen (5.0 mg/kg s.c.) for analgesia.
Assessment of infarct volume
Anaesthetized mice were decapitated in anaesthesia with ketamine/xylazine, and the brains were removed. Twenty micrometre thick coronal sections were cut every 400 μm and stained with cresyl violet. Sections were scanned at 600 dpi, and infarct areas were encircled using ImageJ. The total infarct volume was obtained by integrating measured infarct areas and distance between sections. Correction for brain oedema was performed by subtraction of the ipsilateral from the contralateral hemisphere volume from the directly measured infarct volume.
Echocardiography
Transthoracic micro‐echocardiography was performed using a VisualSonics Vevo 2100 echocardiograph.11 Left ventricular parasternal short‐axis views were obtained using M‐mode imaging at the papillary muscle level. At least five consecutive beats were used for measurements of the chamber dimension with the Vevo 2100 software. Fractional shortening was calculated with the following formula: 100 × [(LVEDD − LVESD)/LVEDD], where LVEDD is the left ventricular end‐diastolic diameter and LVESD is the left ventricular end‐systolic diameter.
High‐sensitive troponin T assay
High‐sensitive troponin T (hsTNT) was measured as described.12, 13 Blood samples were taken from sacrificed animals and centrifuged to obtain serum. Further analysis was performed using an automated Cobas Troponin T hs STAT Elecsys (Roche, Mannheim, Germany).
Determination of neurohormones
For the determination of norepinephrine tissue levels in mice, the left ventricle was rinsed in ice‐cold 0.9% saline and frozen in liquid nitrogen until high‐performance liquid chromatography and electrochemical detection were performed as previously described.14
Cardiac ganglia preparation
After the sacrifice of mice, cardiac ganglia were isolated as described before15 and quickly frozen in liquid nitrogen for RNA isolation.
Heart tissue preparation
After the sacrifice of mice, the hearts were removed and weighed promptly. Relative heart weight was calculated as the ratio to the tibia as described before.15 The left ventricles were transversely dissected. Parts of the left ventricles were quickly frozen in liquid nitrogen for RNA and protein isolation. Tissue samples for histological staining were fixed in 4% paraformaldehyde and embedded in paraffin.
Quantitative RT‐PCR
Total RNA was isolated from ventricular tissue by using TRIzol (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesized from 500 ng RNA using the SuperScript first‐strand synthesis system for RT‐PCR (Invitrogen) according to the manufacturer's instructions. Quantitative RT‐PCR was performed on a 7500 Fast Cycler (Applied Biosystems, Foster City, CA, USA). Probes from the Universal Probe Library (Roche, Penzberg, Germany) and TaqMan Universal PCR Master Mix were used. Oligonucleotide primers were synthesized at LGC Genomics, Germany, and sequences of primers and the respective Universal Probe Library probe numbers are given (nppa (sense: cacagatctgatggatttcaaga; antisense: cctcatcttctaccggcatc; probe number: 25), nppb (sense: gtctggccggacactcag; antisense: tgcactggtgtcttcaacaac; probe number: 13), col3a1 (sense: tcccctggaatctgtgaatc; antisense: tgagtcgaattggggagaat; probe number: 49), col5a1 (sense: ctacatccgtgccctggt; antisense: ccagcaccgtcttctggtag; probe number: 76), atrogin‐1 (sense: agtgaggaccggctactgtg; antisense: gatcaaacgcttgcgaatct; probe number: 53), murf‐1 (sense: agagtgagctgagcgatgg; antisense: gtctgcggctgttgtcct; probe number: 25)). Data were analysed according to the ΔΔCT method as described.13
Cardiac histology
Paraffin‐embedded tissue samples were longitudinally cut into 5 μm thick cardiac sections and stained with haematoxylin and eosin or Masson's trichrome. Cardiomyocyte size was assessed on haematoxylin and eosin‐stained sections. More than 20 randomly chosen cardiomyocytes from each group were analysed to measure cross‐sectional cardiomyocyte area. To quantify cardiac fibrosis, 20 trichrome‐stained sections (magnification ×40) from the left ventricle were randomly selected, and morphometric analysis using ImageJ was performed. Photographs were acquired with an Olympus SZH zoom stereo dissection scope with an Optronics DEI‐750 CCD digital camera. All data were analysed by two observers blinded to group allocation.
Statistical analyses
All values are expressed as mean ± standard deviation. Student's t‐test was used for comparison between two groups, and analysis of variance for multiple comparisons among three or more groups with post hoc Bonferroni test. The accepted significance level was P < 0.05.
Microarray analysis
Gene expression profiling was performed using arrays of Mouse430_2 type from Affymetrix (High Wycombe, UK). A Custom CDF Version 12 with Entrez‐based gene definitions was used to annotate the arrays. The raw fluorescence intensity values were normalized applying quantile normalization. Differential gene expression was analysed on the basis of log‐linear mixed model analysis of variance,16 using a commercial software package SAS JMP8 Genomics, version 4.0, from SAS Institute (Cary, NC, USA). Hierarchical cluster analysis of differentially regulated genes in sham vs. MCAO mice was performed, and a false positive rate of a = 0.01 with false discovery rate correction was taken as the level of significance.
We performed pathway analysis from significant enriched genes using PANTHER pathway analysis.17 Motif enrichment analyses were performed using HOMER analysing tool in the promotor region of indicated genes.18 Promotor regions were determined as transcriptional start site −2 kb. Data are available on gene expression omnibus under the accession number GSE102558.
Results
Brain infarcts 24 h after 60 min MCAO were extensive and included cortical and subcortical areas (mean infarct volume: 65 ± 11.2 mm3). In contrast, 30 min MCAO caused an ischaemic lesion limited to the striatum.
Consequences of stroke on cardiac function
To assess cardiac function in a longitudinal manner, we performed sequential cardiac echocardiography. We found a significant reduction in cardiac function as reflected by reduced fractional shortening and reduced ejection fraction after 60 min but not after only 30 min MCAO compared with pre‐operative baseline and with sham‐operated animals, respectively (Figure 1A–C). Heart rate was initially decreased in both MCAO groups compared with respective sham‐operated mice. However, a persistent reduction of the heart rate, additionally limiting cardiac output, was only observed in 60 min MCAO 7 and 14 days after stroke (Figure 1 D). These changes were no longer present 60 days after MCAO (Supporting Information, Figure S1A–S1D).
Figure 1.
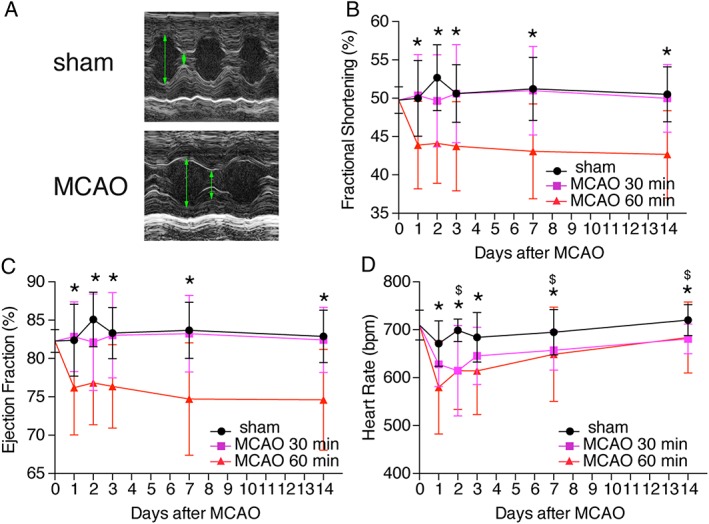
Stroke leads to cardiac dysfunction. (A) Representative echocardiographic imaging from mice 1 day after 60 min middle cerebral artery occlusion (MCAO) or sham surgery. Green arrows indicate enddiastolic diameter. (B–D) Time course of echocardiographic parameters as indicated at 1, 2, 3, 7, and 14 days. Echocardiography results at baseline (0 day) were obtained before surgery. All results are shown as mean ± standard deviation. * P < 0.05 sham vs. MCAO 60 min; $ P < 0.05 sham vs. MCAO 30 min (n = 14–28/group).
Structural changes in the heart upon stroke
To assess potential myocardial damage, we measured hsTNT repetitively in animals as a serum biomarker. We found a significant three‐fold (Day 1) to five‐fold (Day 5) increase in serum hsTNT after 60 min MCAO compared with sham‐operated animals (Figure 2A), indicating cardiomyocyte damage. This was accompanied by a reduced heart weight/tibia length ratio, which normalized again after 14 days (Figure 2B). On cardiomyocyte level, we detected a reduction in cardiomyocyte cross‐sectional diameter of up to 15% compared with sham‐operated animals already at Day 1 (Figure 2C and 2D), which was partially restored over time. We did not observe increased fibrosis on trichrome‐stained histological sections (data not shown). Sixty days after MCAO, heart weight and lung weight did not suggest cardiac dysfunction or congestion (Supporting Information, Figure S1E and S1F).
Figure 2.
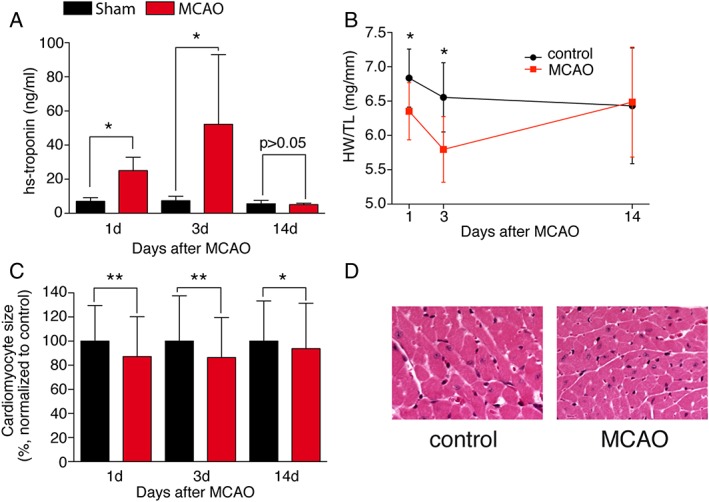
Cardiac atrophy as a consequence of ischaemic stroke. (A) Measurement of high‐sensitive troponin T in the serum of mice after sham and middle cerebral artery occlusion (MCAO) 60 min at 1, 3, and 14 days (n = 5–6/group). (B) Heart weight/tibia length ratio (WT/TL) 1, 3, and 14 days after stroke (n = 6–13/group). (C) Cardiomyocyte cross‐sectional area measured at 1, 3, and 14 days (n = 233–329/group, N > 3/group). Representative haematoxylin and eosin images of the hearts 1 day after stroke (×40). (D) Representative haematoxylin and eosin images of the hearts 1 day after stroke (×40). All results are shown as mean ± standard deviation. * P < 0.05 sham vs. MCAO.
Identification of a novel downstream signalling cascade
To elucidate potential signalling pathways, we first measured the atrial natriuretic peptide and the brain natriuretic peptide (nppa; nppb) that are normally up‐regulated upon cardiac stress.19 However, we did not found an up‐regulation upon MCAO, but rather a down‐regulation of brain natriuretic peptide at 14 days after MCAO (Figure 3A and 3B). Additionally, expression of collagens that are classically involved in pathological cardiac remodelling was not regulated (col5a1) or reduced (col3a1),20 pointing towards a process that is more linked to atrophic signalling (Figure 3C and 3D). To test whether the observed phenotype is linked to cardiac muscle wasting, we measured the expressional changes of the E3 ubiquitin ligase atrogin‐1 and the muscle ring finger protein murf‐1 that are known to regulate skeletal muscle and cardiac atrophy.21, 22, 23 Atrogin‐1 was highly up‐regulated in animals after MCAO, whereas murf‐1 was only up‐regulated at Day 3 after MCAO (Figure 3E and 3F).
Figure 3.
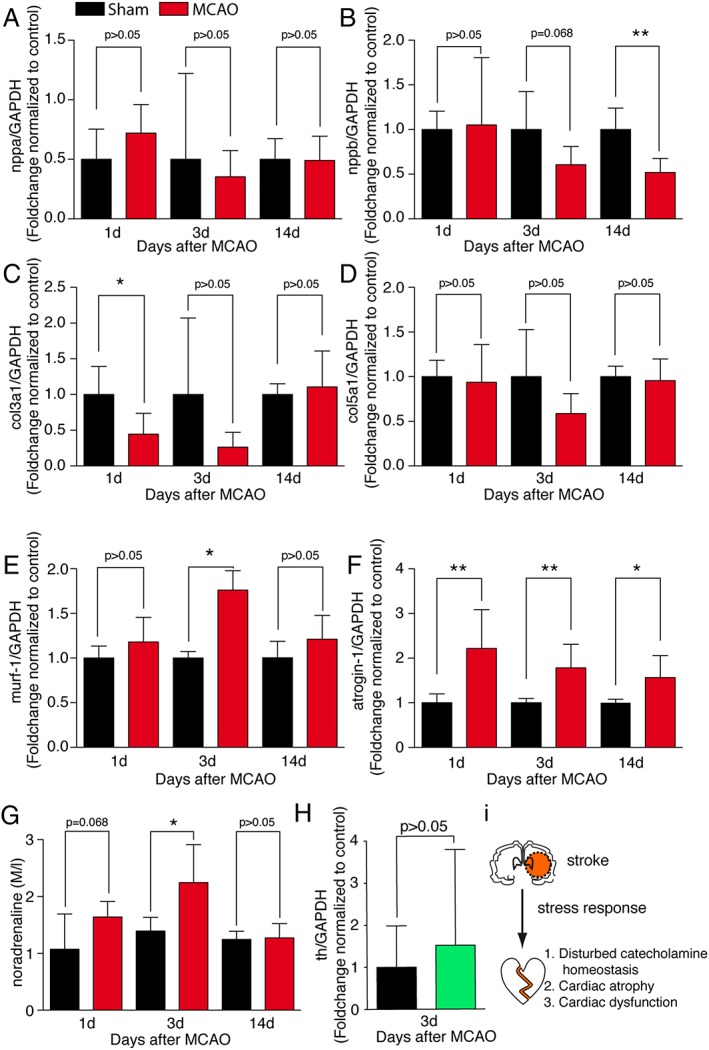
Transcriptional consequences. Transcript levels of (A) nppa, (B) nppb, (C) col3a1, (D) col5a1, (E) murf‐1, and (F) atrogin‐1 in the hearts from sham‐operated control animals and 60 min middle cerebral artery occlusion (MCAO) animals as indicated, normalized to control (n = 5–6/group) at time points 1, 3, and 14 days after 60 min MCAO and sham operation. (G) Tissue noradrenaline concentration in the hearts of sham‐operated and 60 min MCAO mice, respectively (n = 6/group). (H) Transcript levels of tyrosinhydroxylase (th) in cardiac ganglia from sham‐operated and MCAO‐operated animals, 3 days after intervention (n = 6–7/group). All results are shown as mean ± standard deviation. * P < 0.05 sham vs. MCAO. (I) Working model.
As muscle atrophy and expression of atrogin‐1 and murf‐1 are closely linked to autonomic innervation,23, 24 we investigated whether catecholamines are involved in the atrophic process. Norepinephrine was significantly increased in cardiac tissue 3 days after MCAO (Figure 3G). However, tyrosinhydroxylase expression, the rate limiting enzyme of norepinephrine synthesis,25 was not increased in isolated cardiac ganglia on Day 3 after MCAO (Figure 3H), indicating an involvement of other mechanisms, such as norepinephrine re‐uptake or release, rather than norepinephrine synthesis.
Next, we performed a genome‐wide unbiased analysis of the transcriptome of cardiac tissue harvested 24 and 72 h after 60 min MCAO or sham surgery. Bioinformatical analysis showed a strong regulation of 3494 genes in the heart 1 day after 60 min MCAO compared with sham surgery. Interestingly, when we generated a heat map with hierarchical clustering of genes that were either significantly regulated at Day 1 or Day 3 after MCAO, we found a pronounced regulation on Day 1 (Supporting Information, Figure S2A) but much less difference at Day 3 compared with the sham‐operated control group. Pathway annotation showed a high number of genes involved in metabolic processes (Supporting Information, Figure S2B). Correspondingly, pathway enrichment analysis revealed a strong and significant enrichment in metabolic pathways (Supporting Information, Figure S2C).
To get insights into transcription factors that may be involved in the regulation of these up‐regulated genes and pathways, we focused on promotor regions that were already early up‐regulated (Day 1). Motif enrichment showed a strong association to the peroxisome proliferator‐activated receptor gamma (Pparg) binding site within these promotors, suggesting a regulatory role of this transcription factor (Supporting Information, Figure S2D).
Discussion
Our study provides three major new findings: (i) cerebral infarction induces transient contractile cardiac dysfunction in mice; (ii) cardiac damage and subsequent restructuring occur after experimental stroke resulting in an atrophic cardiac phenotype; and (iii) stroke triggers a functional denervation with activation of an atrophic signalling cascade in the heart including a Pparg‐dependent gene programme, which is known to be involved in mitochondrial remodelling, regulation of catabolism, and hypertrophic gene programmes.
In echocardiography, performed after acute stroke, 20–30% of stroke patients have focal or generalized contractile abnormalities, and 7% suffer from overt heart failure with left ventricular ejection fraction below 30%.3 These features are commonly interpreted as a reflection of pre‐existing heart disease rather than as a consequence of the stroke. While there is a documented history of pre‐existing heart disease in some patients, longitudinal clinical studies monitoring heart function before and after stroke do not exist. In our longitudinal experimental study, we found a substantial reduction of echocardiographic contractility in the early and subacute phase after extensive stroke in young, previously healthy mice that had intact cardiac function before MCAO. Similar observations have been reported using other methodologies such as endovascular catheterization.26 The observed echocardiographic phenotype did not result in overt heart failure in our study as evidenced by normal lung weight, but this might have been different if aged animals with pre‐existing reduced function had been measured.
An important finding relates to the morphological cardiac phenotype observed in mice after stroke. Previous studies reported cardiac hypertrophy,27 which was also present after 30 min of right transient focal cerebral ischaemia accompanied by sympathetic overactivity weeks after the experimental stroke.28 In contrast, mice in our study (left MCAO, 60 min ischaemia) showed rapid loss of heart weight and cardiomyocyte atrophy. In our experiments, the expression of collagens (col3a1 and col5a1) that are normally up‐regulated in pathological cardiac remodelling was less affected. This difference might be explained by different duration of brain ischaemia (30 vs. 60 min) and resulting infarct size and by the side of brain ischaemia (right vs. left). Our data with 30 min left MCAO are comparable with those of Bieber et al., who reported only a mild cardiac phenotype 8 weeks after left MCAO with 30 min ischaemia.28 Previously, we have described an atrophic cardiac phenotype in an experimental model of cancer.29 In both instances (cancer and left MCAO 60 min), metabolic pathways and cardiac dysfunction are closely connected to structural changes. Using an unbiased approach, we found many dysregulated genes, which participate in metabolic pathways. A link between murf‐1, protein degradation, the basis of muscle atrophy, and metabolic changes has been described for metabolic pathways.21, 22, 24, 30, 31
To identify other potential modifiers linking metabolic pathways to muscle atrophy, we performed a promoter analysis of regulated genes. We identified the up‐regulation of Pparg‐dependent genes. Pparg is a regulator of cardiomyocyte metabolic and structural remodelling‐ Other family members are also strongly linked to muscle atrophy whereas the binding motif of ppa‐receptors share a high similarity.32 Models of cardiomyocyte‐specific loss‐of‐function have shown that Pparg can inhibit cardiomyocyte hypertrophy and supresses cardiac growth.33 Overexpression of Pparg in cardiomyocytes in turn leads to reduced cardiac function accompanied by multiple metabolic changes.34 Although we describe a strong correlation to metabolic pathways and Pparg, future studies need to address if Pparg is required and sufficient to mediate stroke‐induced cardiac dysfunction and how this depends on the E3 ubiquitin ligase atrogin‐1. If confirmed, Pparg activators that are already in clinical use could be an option for prevention or treatment of stroke‐induced cardiac damage and dysfunction in patients. Clinical data point towards beneficial effects of peroxisome proliferator‐activated receptor inhibitors for treatment of patients with stroke which supports a mechanistic interplay with cardiomyocyte signalling.35, 36
In addition to morphological and molecular alterations inside the heart muscle, the signalling pathways between the injured brain and the heart deserve further attention. Some of our findings suggest that the effect is mediated via humoral factors in the blood. Stress hormones including catecholamines have been implicated in the Takotsubo cardiomyopathy syndrome in patients after a variety of conditions including stroke.37 Using the same stroke models that were used in the present study, we have previously shown a sharp rise of catecholamines after extensive but not circumscribed murine brain ischaemia.38 Interestingly, mice in the present study had a profound bradycardic response after brain ischaemia that is not expected in catecholamine‐mediated heart injury. This was accompanied by increased tissue noradrenaline, pointing towards a reduced catecholamine release or an increase of noradrenaline re‐uptake. Down‐regulation of the β‐receptor as described in chronic heart failure could also be a possible explanation. However, an increase of catecholamine release would result in increased synthesis, which we ruled out by investigating neuronal norepinephrine synthesis. Reduced post‐synaptic norepinephrine that can be summarized as a functional denervation would explain most of the MCAO‐induced changes.
In conclusion, we propose a model of profound stroke‐induced cardiac alterations where brain ischaemia triggers a disturbance of catecholamine homeostasis in the heart. This leads to up‐regulation of the E3‐ligase atrogin‐1 and up‐regulation of Pparg‐dependent genes resulting in cardiomyocyte atrophy and transient cardiac dysfunction (Figure 3I). Future experimental and clinical studies are needed to evaluate the contribution of this brain–heart interaction to cardiac morbidity in stroke patients.
Conflict of Interest
All except H.A.K. declared that they have no conflict of interest. H.A.K. has invented the high‐sensitive troponin T assay and receives royalties from Roche Diagnostics.
Authors' Contributions
R.V., J.B., and L.H.L designed the study. L.H.L., S.U., and M.M. carried out the experiments. R.V., L.H.L., S.U., C.S., D.F., N.G., and J.B. analysed and interpreted the data. H.A.K., J.B., H.‐J.G, and R.V. provided research support and conceptual advice. R.V. and L.H.L. wrote the first draft of the paper. All authors critically revised the paper.
Supporting information
Figure S1. (a) Ejection fraction, (b) heart rate, (c) left ventricular enddiastolic diameter (LVEDD) and (d) left ventricular endsystolic diameter (LVESD) 60 days after MCAO compared with sham operated controls (n = 8–11/group). (e) Heart weight (HW) and (f) lung weight (LW) to tibial length (TL) ratio 60 days after MCAO (n = 7–9/group). All results are shown as mean ± SD; * indicates p < 0.05 Sham vs. MCAO
Figure S2. (a) Hierarchical clustering of significantly regulated genes in hearts of 60 min MCAO and sham operated mice (n = 6/group) and top 20 significant upregulated genes 1d after MCAO sorted by the foldchange MCAO vs. Sham group. (b) Pathway analysis of genes, upregulated at day1 after MCAO. (c) Pathway enrichment analyses of genes upregulated at day 1 after MCAO. (d) Motif enrichment analysis of all genes upregulated at day 1. TSS, Transcriptional start site; kb, kilobase.
Acknowledgements
We thank Jutta Krebs for excellent technical assistant. R.V. is an investigator of the Imperial Biomedical Research Centre. L.H.L. is recipient of the Clinician‐Scientist Program (CSP) of the German Cardiac Society (DGK). H.A.K., J.B., and L.H.L. were supported by grants from the DZHK (Deutsches Zentrum für Herz‐Kreislauf‐Forschung—German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research). The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017.39
Veltkamp, R. , Uhlmann, S. , Marinescu, M. , Sticht, C. , Finke, D. , Gretz, N. , Gröne, H.‐J. , Katus, H. A. , Backs, J. , and Lehmann, L. H. (2019) Experimental ischaemic stroke induces transient cardiac atrophy and dysfunction. Journal of Cachexia, Sarcopenia and Muscle, 10: 54–62. 10.1002/jcsm.12335.
References
- 1. Erdur H, Scheitz JF, Grittner U, Laufs U, Endres M, Nolte CH. Heart rate on admission independently predicts in‐hospital mortality in acute ischemic stroke patients. Int J Cardiol 2014;176:206–210. [DOI] [PubMed] [Google Scholar]
- 2. Rizos T, Rasch C, Jenetzky E, Hametner C, Kathoefer S, Reinhardt R, et al. Detection of paroxysmal atrial fibrillation in acute stroke patients. Cerebrovasc Dis 2010;30:410–417. [DOI] [PubMed] [Google Scholar]
- 3. Holmstrom A, Fu ML, Hjalmarsson C, Bokemark L, Andersson B. Heart dysfunction in patients with acute ischemic stroke or TIA does not predict all‐cause mortality at long‐term follow‐up. BMC Neurol 2013;13:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thalin C, Rudberg AS, Johansson F, Jonsson F, Laska AC, Nygren AT, et al. Elevated troponin levels in acute stroke patients predict long‐term mortality. J Stroke Cerebrovasc Dis 2015;24:2390–2396. [DOI] [PubMed] [Google Scholar]
- 5. Scheitz JF, Mochmann HC, Nolte CH, Haeusler KG, Audebert HJ, Heuschmann PU, et al. Troponin elevation in acute ischemic stroke (TRELAS)—protocol of a prospective observational trial. BMC Neurol 2011;11:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porto I, Della Bona R, Leo A, Proietti R, Pieroni M, Caltagirone C, et al. Stress cardiomyopathy (tako‐tsubo) triggered by nervous system diseases: a systematic review of the reported cases. Int J Cardiol 2013;167:2441–2448. [DOI] [PubMed] [Google Scholar]
- 7. Gattringer T, Niederkorn K, Seyfang L, Seifert‐Held T, Simmet N, Ferrari J, et al. Myocardial infarction as a complication in acute stroke: results from the austrian stroke unit registry. Cerebrovasc Dis 2014;37:147–152. [DOI] [PubMed] [Google Scholar]
- 8. Sander D, Winbeck K, Klingelhofer J, Etgen T, Conrad B. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology 2001;57:833–838. [DOI] [PubMed] [Google Scholar]
- 9. Dorrance AM, Fink G. Effects of stroke on the autonomic nervous system. Compr Physiol 2015;5:1241–1263. [DOI] [PubMed] [Google Scholar]
- 10. Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke 2009;40:2849–2858. [DOI] [PubMed] [Google Scholar]
- 11. Moreth K, Afonso LC, Fuchs H, Gailus‐Durner V, Katus HA, Bekeredjian R, et al. High throughput phenotyping of left and right ventricular cardiomyopathy in calcineurin transgene mice. Int J Cardiovasc Imaging 2015;31:669–679. [DOI] [PubMed] [Google Scholar]
- 12. Giannitsis E, Kurz K, Hallermayer K, Jarausch J, Jaffe AS, Katus HA. Analytical validation of a high‐sensitivity cardiac troponin T assay. Clin Chem 2010;56:254–261. [DOI] [PubMed] [Google Scholar]
- 13. Weinreuter M, Kreusser MM, Beckendorf J, Schreiter FC, Leuschner F, Lehmann LH, et al. CaM kinase II mediates maladaptive post‐infarct remodeling and pro‐inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med 2014;6:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Touyz RM, Tabet F, Schiffrin EL. Redox‐dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol 2003;30:860–866. [DOI] [PubMed] [Google Scholar]
- 15. Werfel S, Jungmann A, Lehmann L, Ksienzyk J, Bekeredjian R, Kaya Z, et al. Rapid and highly efficient inducible cardiac gene knockout in adult mice using AAV‐mediated expression of Cre recombinase. Cardiovasc Res 2014;104:15–23. [DOI] [PubMed] [Google Scholar]
- 16. Hsieh WP, Chu TM, Wolfinger RD, Gibson G. Mixed‐model reanalysis of primate data suggests tissue and species biases in oligonucleotide‐based gene expression profiles. Genetics 2003;165:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 2013;8:1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. einz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hohl M, Wagner M, Reil JC, Muller SA, Tauchnitz M, Zimmer AM, et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest 2013;123:1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kreusser MM, Lehmann LH, Keranov S, Hoting MO, Kohlhaas M, Reil JC, et al. The cardiac CaMKII genes delta and gamma contribute redundantly to adverse remodeling but inhibit calcineurin‐induced myocardial hypertrophy. Circulation 2014; doi:CIRCULATIONAHA.114.006185 [pii] 10.1161/CIRCULATIONAHA.114.006185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Willis MS, Schisler JC, Li L, Rodriguez JE, Hilliard EG, Charles PC, et al. Cardiac muscle ring finger‐1 increases susceptibility to heart failure in vivo . Circ Res 2009;105:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willis MS, Rojas M, Li L, Selzman CH, Tang RH, Stansfield WE, et al. Muscle ring finger 1 mediates cardiac atrophy in vivo . Am J Physiol Heart Circ Physiol 2009;296:H997–H1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010;143:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zaglia T, Milan G, Franzoso M, Bertaggia E, Pianca N, Piasentini E, et al. Cardiac sympathetic neurons provide trophic signal to the heart via beta2‐adrenoceptor‐dependent regulation of proteolysis. Cardiovasc Res 2013;97:240–250. [DOI] [PubMed] [Google Scholar]
- 25. Kreusser MM, Lehmann LH, Haass M, Buss SJ, Katus HA, Lossnitzer D. Depletion of cardiac catecholamine stores impairs cardiac norepinephrine re‐uptake by downregulation of the norepinephrine transporter. PLoS One 2017;12:e0172070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Min J, Farooq MU, Greenberg E, Aloka F, Bhatt A, Kassab M, et al. Cardiac dysfunction after left permanent cerebral focal ischemia: the brain and heart connection. Stroke 2009;40:2560–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J, Cui C, Yang X, Xu J, Venkat P, Zacharek A, et al. miR‐126 affects brain‐heart interaction after cerebral ischemic stroke. Transl Stroke Res 2017;8:374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bieber M, Werner RA, Tanai E, Hofmann U, Higuchi T, Schuh K, et al. Stroke‐induced chronic systolic dysfunction driven by sympathetic overactivity. Ann Neurol 2017;82:729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schafer M, Oeing CU, Rohm M, Baysal‐Temel E, Lehmann LH, Bauer R, et al. Ataxin‐10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol Metab 2016;5:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang F, Paterson AJ, Huang P, Wang K, Kudlow JE. Metabolic control of proteasome function. Physiology (Bethesda) 2007;22:373–379. [DOI] [PubMed] [Google Scholar]
- 31. Willis MS, Zungu M, Patterson C. Cardiac muscle ring finger‐1—friend or foe? Trends Cardiovasc Med 2010;20:12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Castillero E, Alamdari N, Aversa Z, Gurav A, Hasselgren PO. PPARbeta/delta regulates glucocorticoid‐ and sepsis‐induced FOXO1 activation and muscle wasting. PLoS One 2013;8:e59726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte‐specific knockout and agonist of peroxisome proliferator‐activated receptor‐gamma both induce cardiac hypertrophy in mice. Circ Res 2005;97:372–379. [DOI] [PubMed] [Google Scholar]
- 34. Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest 2007;117:2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu J, Wang LN. Peroxisome proliferator‐activated receptor gamma agonists for preventing recurrent stroke and other vascular events in patients with stroke or transient ischaemic attack. Cochrane Database Syst Rev 2015;10:CD010693. [DOI] [PubMed] [Google Scholar]
- 36. Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 2016;374:1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Templin C, Ghadri JR, Napp LC. Takotsubo (stress) cardiomyopathy. N Engl J Med 2015;373:2689–2691. [DOI] [PubMed] [Google Scholar]
- 38. Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp R. Differential effects of sympathetic nervous system and hypothalamic‐pituitary‐adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun 2014;41:200–209. [DOI] [PubMed] [Google Scholar]
- 39. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Ejection fraction, (b) heart rate, (c) left ventricular enddiastolic diameter (LVEDD) and (d) left ventricular endsystolic diameter (LVESD) 60 days after MCAO compared with sham operated controls (n = 8–11/group). (e) Heart weight (HW) and (f) lung weight (LW) to tibial length (TL) ratio 60 days after MCAO (n = 7–9/group). All results are shown as mean ± SD; * indicates p < 0.05 Sham vs. MCAO
Figure S2. (a) Hierarchical clustering of significantly regulated genes in hearts of 60 min MCAO and sham operated mice (n = 6/group) and top 20 significant upregulated genes 1d after MCAO sorted by the foldchange MCAO vs. Sham group. (b) Pathway analysis of genes, upregulated at day1 after MCAO. (c) Pathway enrichment analyses of genes upregulated at day 1 after MCAO. (d) Motif enrichment analysis of all genes upregulated at day 1. TSS, Transcriptional start site; kb, kilobase.