

Abstract
The hypercalins are dearomatized acylphloroglucinols with a pendant complex cyclopentane ring that exhibit activity against several cancer cell lines. We report the first total synthesis of (+)-hypercalin C employing a convergent strategy that enabled dissection of the essential structural features required for the observed anticancer activity. A strategic disconnection involving an unusual Csp3-Csp2 Suzuki-Miyaura coupling with an α-bromo enolether also revealed an unexpected C-H activation. Our strategy targeted designed analogs along the synthetic route to address particular biological questions. Our results support the hypothesis that hypercalin C may act as a proton shuttle with the dearomatized acylphloroglucinol moiety being essential for this activity.
Keywords: C-H insertion, β-lactone, chiral pool, Suzuki-Miyaura coupling, protonophore
Graphical Abstract
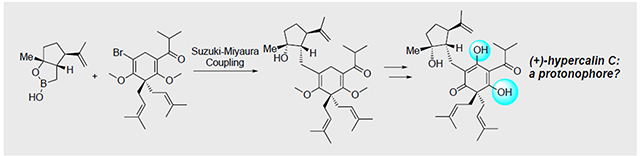
The first total synthesis of (+)-hypercalin C was achieved through a key sp3-C Suzuki coupling and a highly regioselective allylic oxidation. Several designed derivatives were targeted in the course of the synthesis to address the hypothesis that hypercalin C and congeners are cytotoxic due to their potential as protonophores. An unexpected insertion into an ether C-H bond during Pd-mediated coupling of an α-bromo, methyl enol ether was discovered.
The hypercalins and chinensins are members of the dearomatized, polyprenylated polycyclic acylphloroglucinol (PPAP) family of natural products isolated by Hostettmann from Hypericum calycinum L (Figure 1).[1] Several PPAPs undergo further oxidative cyclizations leading to polycyclic members that have attracted considerable synthetic interest;[2] however, the hypercalins and chinensins have not yet been synthesized. The hypercalins and chinensins display minor structural differences, namely, substitution at the quaternary carbon center (C4, R1) and the acyl group (C27, R2). Members of this family possess a range of biological activities including antibacterial, antiviral and cytotoxicity.[3] For example, hypercalins A-C showed growth-inhibitory activity against the Co-115 human colon carcinoma cell line with ED50 values ranging from 0.60-0.83 μM.
Figure 1.
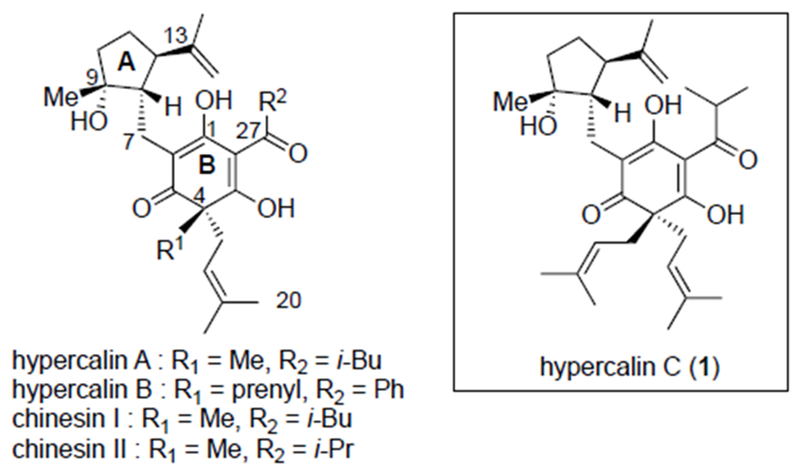
Members of the dearomatized, polyprenylated acylphloroglucinol family, the hypercalins and chinensins including hypercalin C (1)
In terms of anticancer activity, hypercalin C is one of the most potent members of this family.[1a] We therefore targeted this congener and designed analogs for synthesis aimed at interrogating its proposed mode of action as a cellular membrane proton shuttle.[4]
In our retrosynthetic analysis, we considered disconnections that would allow us to answer biological questions regarding the cytotoxicity of the hypercalins. Thus, a key disconnection involved a Csp3-Csp2 Suzuki-Miyaura coupling[5] between the boronic monoester 4,[6] derived from our previously described β-lactone 6, and bromoenol ether 5 to provide adduct 3 (Figure 2a). This disconnection enables dissection of the two principal fragments, the cyclopentyl moiety A and the dearomatized acylphloroglucinol moiety B (Figure 2b), which would allow us to separately assess their cytotoxicity. A subsequent site-selective allylic oxidation would deliver bis-methyl hypercalin C, 2. The requisite cyclopentyl moiety, given its stereochemical complexity, may play an important role in cellular target recognition or simply serve as a hydrophobic group and this could be assessed by coupling with simplified hydrophobic groups (R = alkyl group; Figure 2b) derived from boronic acid 4 accessible from our previously described β-lactone 6 in turn derived from carvone.[7] The coupling partner for the key coupling event, α-bromo enol ether 5, would be prepared from 1,3-cyclohexanedione.
Figure 2.
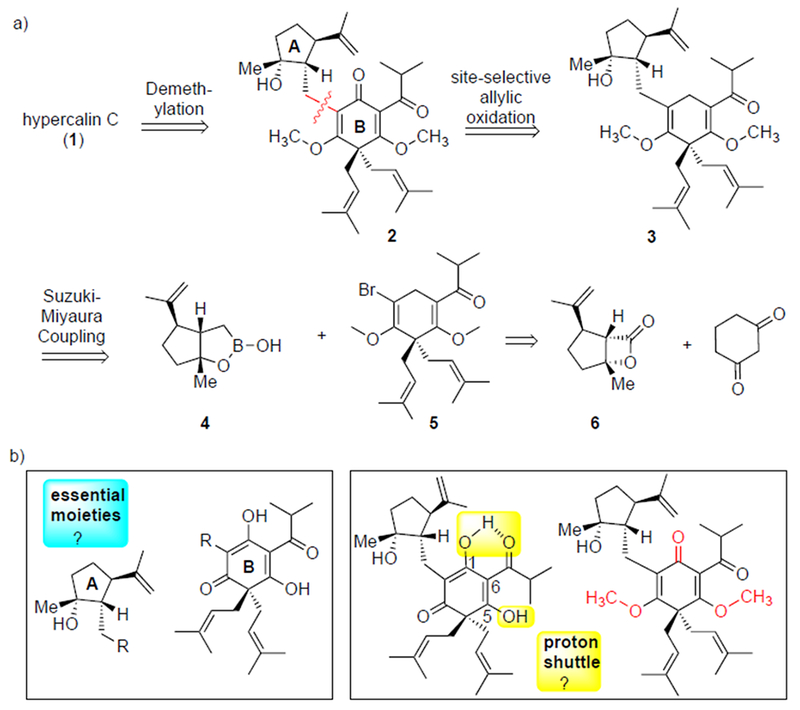
a) Retrosynthetic analysis of hypercalin C and b) targeted derivatives, including the cyclopentyl moiety A and the dearomatized acylphloroglucinol moiety B, guided by biological questions to be answered in the course of the total synthesis.
A proton shuttle, or protonophore, disrupts the pH gradient across a cell membrane by transporting protons from outside the cell to the higher pH environment inside the cell.[4, 8] We planned to test the hypothesis that hypercalin C exerts its bioactivity through this mechanism (Figure 3), which is enabled by the highlighted, intramolecularly hydrogen bonded and relatively acidic C1 and C5 enol hydroxyls (yellow boxes, Figure 2b) and the resulting highly delocalized, anionic species that would be expected to readily traverse the cell membrane (Figure 3). We therefore targeted the bis-enol ether 2, which is also a convenient synthetic precursor to the targeted bis-enol, and removes the ability of these molecules to transport protons across the cell membrane while also minimizing steric perturbations.
Figure 3.
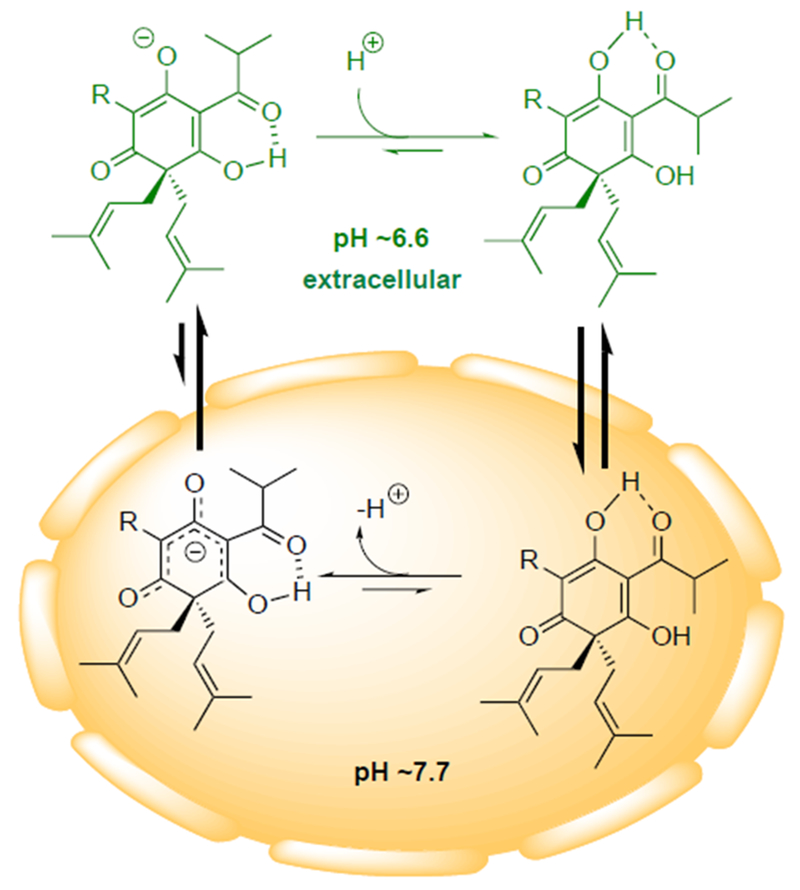
Proton shuttle hypothesis for the cytotoxicity of the hypercalins
We first targeted the synthesis of a versatile Csp3-Csp2 Suzuki-Miyaura, cyclopentyl coupling partner and chose the bicyclic boronic monoester 4 building on prior successful application of this type of coupling partner (Scheme 1).[9] The synthesis commenced with our previously described diol 8, available in four steps from (R)-carvone, through a diastereoselective nucleophile catalyzed aldol-β-lactonization (NCAL) process which proceeds with high diastereoselectivity (dr >19:1) to deliver an intermediate β-lactone 6 setting the two additional stereocenters required for the cyclopentyl moiety of the hypercalins.[10] Selective mesylation of the primary alcohol followed by a Finkelstein reaction[11] provided iodide 9 readied for alkylithium generation. Since lithium-halogen exchange is known to be faster than deprotonation,[12] iodide 9 was pre-treated with PhLi to deprotonate the tertiary alcohol and avoid intramolecular quenching. Subsequent addition of t-BuLi and quenching with B(OMe)3 delivered the desired cyclopentyl cyclic boronic monoester 4.
Scheme 1.
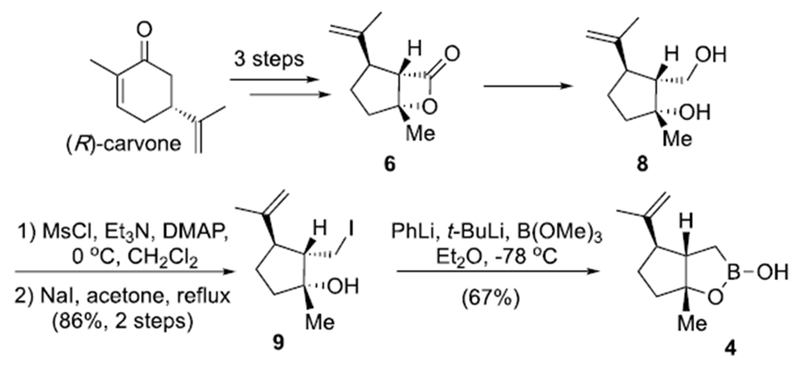
Synthesis of the cyclopentyl, cyclic boronic monoester 4
Since transition metal-couplings with ortho-disubstituted arenes can be challenging,[13] model studies utilizing boronic monoester 4 and various aryl bromides were undertaken (Scheme 2). Use of less substituted halogenated arenes would also provide simplified ring A hypercalin C derivatives to inform structure-activity relationships (SAR). Screening of >50 different reaction conditions with tris-methoxy aryl bromide 10, failed to provide the desired adduct 11, a species containing most of the structural features of the natural product. The steric hindrance caused by the bis-ortho substitution of the aromatic substrate is known to dramatically impede the reductive elimination step,[14] leading to a competing β-hydride elimination as evidenced through isolation of the exocylic alkene resulting from dehydroboronation of boronic monoester 4, and the reduced arene derived from bromoarene 10. We anticipated that removal of one of the ortho-substituents would facilitate the reductive elimination step by minimizing β-hydride elimination to enable subsequent cross-coupling. Indeed, boronic monoester 4 coupled efficiently under conditions reported by Buchwald and coworkers[15] with the less substituted aromatic bromides 12 and 14, with at least one open ortho position.
Scheme 2.
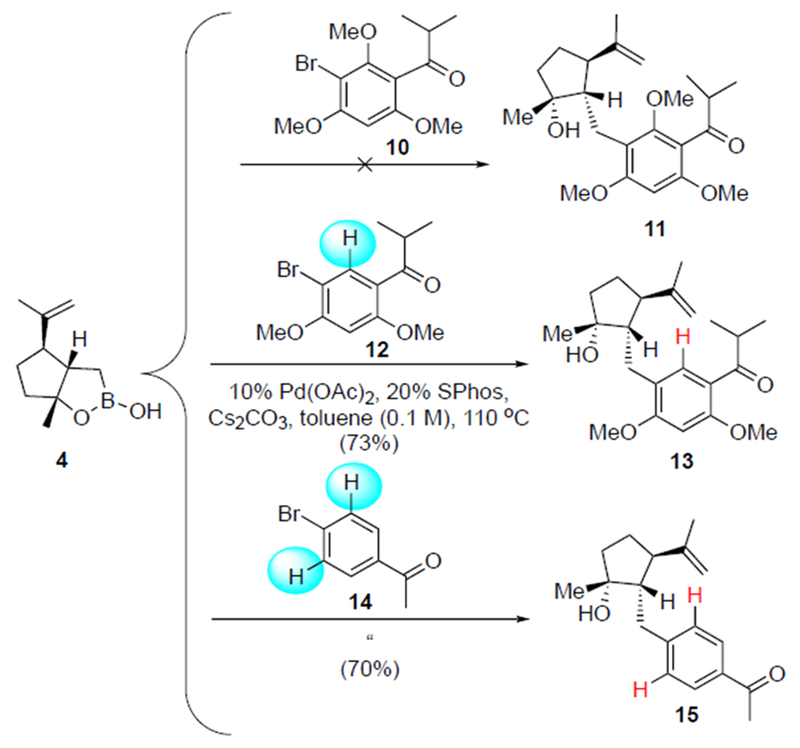
Model studies for the cross-coupling with cyclopentyl boronic monoester 4 leading to simplified, aromatic variants of the cyclohexyl trione core
Having identified successful cross-coupling conditions, we targeted the synthesis of an appropriately substituted coupling partner (i.e. vinyl bromide 5), cognizant of steric issues identified during model studies (Scheme 3). Alkylative bis-prenylation of 1,3-cyclohexanedione provided dialkylated dione 16.[16] Subsequent conversion to the highly unstable dibromo diketone 17 was achieved through bromination (freshly recrystallized NBS) of the derived bis-silyl enol ether (freshly distilled TMSCl) and immediate conversion to the bis-enolether 18 through a bis-α-deprotonation, bis-O-methylation sequence. This process required extensive optimization since the light sensitive dibromide 17 upon deprotonation was, not surprisingly, highly susceptible to side reactions likely stemming from α-elimination leading to carbene generation. Following extensive experimentation, slow, inverse addition of a THF solution of dibromide 17 to a mixture of high quality KHMDS (a relatively new/recently opened bottle) and MeOTf in DMPU/THF (1:20) at −78oC was found to be key for reproducible production of bis-enolether 18. Finally, a mono lithium-halogen exchange was achieved by careful control of n-BuLi stoichiometry at low temperature to deliver a vinyllithium species which was reacted with the Weinreb amide derived from 2-methyl propionic acid to afford the desired keto vinyl bromide 5.
Scheme 3.
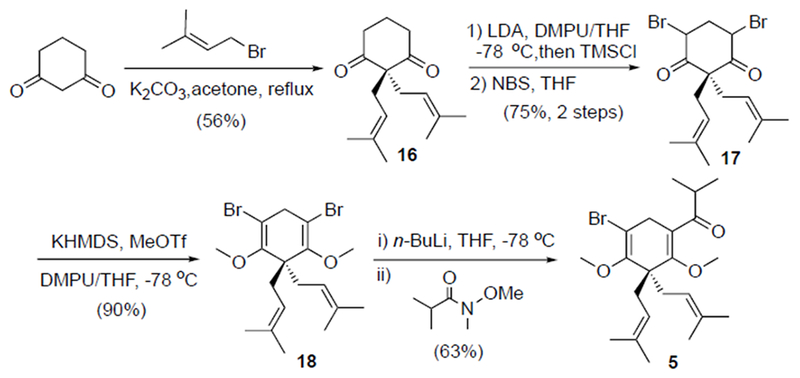
Synthesis of the Suzuki-Miyaura coupling partner, bis-α-bromo enol ether 5
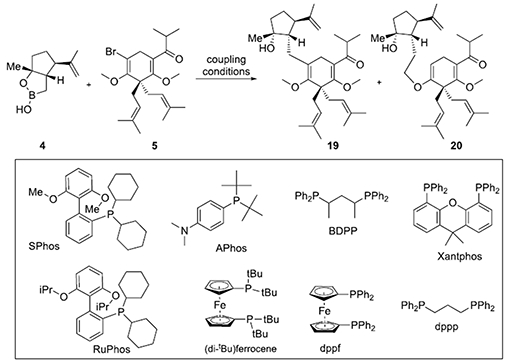
Our initial studies of the key Suzuki-Miyaura coupling with vinyl bromide 5 and boronic monoester 4 employing the conditions previously optimized for arene 12 did not deliver the expected adduct 19 but rather the Csp3-Csp3 cross-coupled product 20 (30% yield) via a presumed C-H insertion into the adjacent methoxy group (Table 1, entry 1). The observed C-H activation of an enol ether is unprecedented, to the best of our knowledge, and this may be due to the fact that α-bromo enol ethers are not frequently used as substrates for cross-coupling reactions. However, insertion into C-H bonds adjacent to oxygen atoms are well-precedented.[15b]
Table 1.
Optimization of the key Suzuki-Miyaura coupling of vinyl bromide 5 and boronic monoester 4.a
 | ||||
|---|---|---|---|---|
| entry | Ligand (0.2 equiv) | Base (3 equiv) | Suzuki-Miyaura couplingb | C-H insertionb |
| 1 | SPhosc | Cs2CO3 | trace | 30% |
| 2 | SPhosd | Cs2CO3 | trace | 51% |
| 3 | PPh3 | Cs2CO3 | trace | 2% |
| 4 | Xantphos | Cs2CO3 | trace | 21% |
| 5 | dppp | Cs2CO3 | trace | trace |
| 6 | dppf | Cs2CO3 | trace | 17% |
| 7 | RuPhos | CsF | 47% | trace |
| 8 | SPhos | K2CO3 | 34% | 22% |
| 9 | SPhos | K3PO4 | 10% | 17% |
| 10 | APhos | Cs2CO3 | 40% | 12% |
| 11 | APhose,f | Cs2CO3 | 74% | trace |
Standard conditions: 10% Pd(OAc)2, ligand (0.2 equiv), base (3 equiv), PhMe (0.1M), 65 °C.
Yields refer to purified, isolated products.
Reaction temperature was 110 °C.
Concentration: 0.01M.
Slow addition of the catalyst (0.1 mL/h, over 3 h) as a toluene solution.
Concentration: 0.2 M.
Building on literature precedent for insertions into C-H bonds adjacent to oxygen atoms,[15b] we propose that a Pd(II) intermediate II is formed following oxidative addition [17] and due to sterics induced by the adjacent quaternary center and the bulky nature of the SPhos ligand, the methoxy group is forced closer to the Pd-center facilitating an agostic interaction with the C-H bonds of the methyl group which at elevated temperatures can lead to C-H insertion, generating a Pd (IV) species III. The Csp3 nature of the boronic monoester 4, may also retard the rate of transmetalation, enabling C-H insertion to be competitive with transmetalation. Reductive elimination would presumably be facilitated by release of severe steric compression through insertion leading to the Pd(II)-metallocycle III. Since β-hydride elimination is not possible at this stage, the longevity and relative stability of intermediate III enables subsequent transmetalation with boronic monoester 4 which following reductive elimination delivers the observed CH-insertion adduct 20.
To find optimal conditions for both the desired Suzuki cross coupling and unexpected C-H insertion, we considered the factors likely impacting these reaction pathways. Most importantly, since C-H activation of the methoxy group is likely driven by the extreme steric environment caused by both the quaternary center in the substrate and the ligand, we anticipated that replacement of SPhos with a smaller ligand would drive the reaction to the desired Csp3 Suzuki coupling. On the other hand, to promote oxidative addition of palladium to the rather electron rich bromo enol ether, the ligand employed should be electron rich and bulky, which in turn also promotes the final reductive elimination. At the same time, a considerable energy barrier is present for C-H insertion, thus the high temperature favors this side reaction. Indeed, when the temperature was lowered from 110 to 65 °C, the desired Suzuki-Miyaura coupled product 19 was obtained in 20-30% yield, while the C-H insertion product, ether 20, was still obtained in 30% yield (Table1, entry 2). Furthermore, the intramolecular nature of the C-H insertion process makes reaction concentration a key factor and it was surmised that lower concentration would favor the C-H insertion pathway. With the above considerations and extensive screening including variation of ligands and base (Table1, entries 3-11; for other conditions examined, see SI, Table S1), we ultimately found reaction conditions that favored the desired Suzuki-Miyaura coupling using APhos as ligand (74%, Table 1, entry 11) while alternative conditions employing SPhos at low concentration gave primarily the C-H insertion product 20 (51% yield, Table 1, entry 2).
The final steps to the targeted dimethyl hypercalin C and hypercalin C required a selective allylic oxidation followed by demethylation for the latter product. This oxidation required site-selectivity given the presence of two prenyl groups and an isobutenyl moiety presenting nine potential oxidizable allylic positions with most positions being less hindered than the targeted allylic position. However, electronic effects of the targeted bis-allylic position were expected to favor a site-selective oxidation. This led us to oxidative methods that involved initial hydrogen atom abstraction since the resulting bis-allylic radical would be stabilized.[18] We were gratified to find that conditions reported by Shair in their synthesis of hyperforin[19], involving generation of a peroxide radical for hydrogen atom abstraction at the most reactive allylic site (weakest C-H bond) and subsequent oxidation, provided a 43% yield of the desired ketone 2 (Scheme 5). Demethylation of dimethyl hypercalin C (2) was achieved with LiCl in DMSO at elevated temperature providing clean conversion to hypercalin C (1) in 90% yield.
Scheme 5.
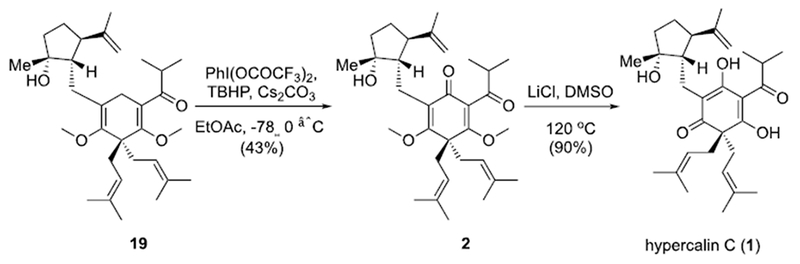
Completion of the total synthesis of hypercalin C (1)
We measured the cytotoxicity of synthetic hypercalin C and derivatives against HCT-116 colon cancer and MDA-MB-231 breast cancer cell lines (Table 2). The bare cyclopentyl moiety bearing an additional alcohol 8 or aromatic moieties in place of the cyclohexanetrione, namely arene derivatives 15 and 13, were not cytotoxic up to 100 μM. These results suggest that the cyclopentyl moiety alone, though rich in stereochemical information, does not contribute significantly to the observed cytotoxicity. This is further supported by derivative 21,[20] which retains some of the original cytotoxicity (6.5-7.7X decrease) while only possessing a simple isopentyl chain in place of the complex cyclopentane found in hypercalin C. Interestingly, the presence of oxygen in the arene ring, cf. derivative 13, but devoid of acidic protons led to the initial observed cytotoxicity (at <100 μM). The cytotoxicity of synthetic hypercalin C was found to be in the 3-4 μM range, consistent with previous reports[1a] however dimethyl hypercalin C (2) showed a measurable decrease in cytotoxicity (~5-7X). While this lends some credence to the proposed requirement of acidic enols for cytotoxicity, the difference is not significant enough to exclude alternative hypotheses regarding the mode of action of the hypercalins. On the other hand, the hydrogenated derivative 22 showed similar activity as the natural product, indicating that unsaturation of the prenyl side chains has minimal effect on cytotoxicity.
Table 2.
Cytotoxicity of hypercalin C and derivatives against HCT-116 (in black) and MDA-MB-231 (in red) cell lines (IC50, μM).
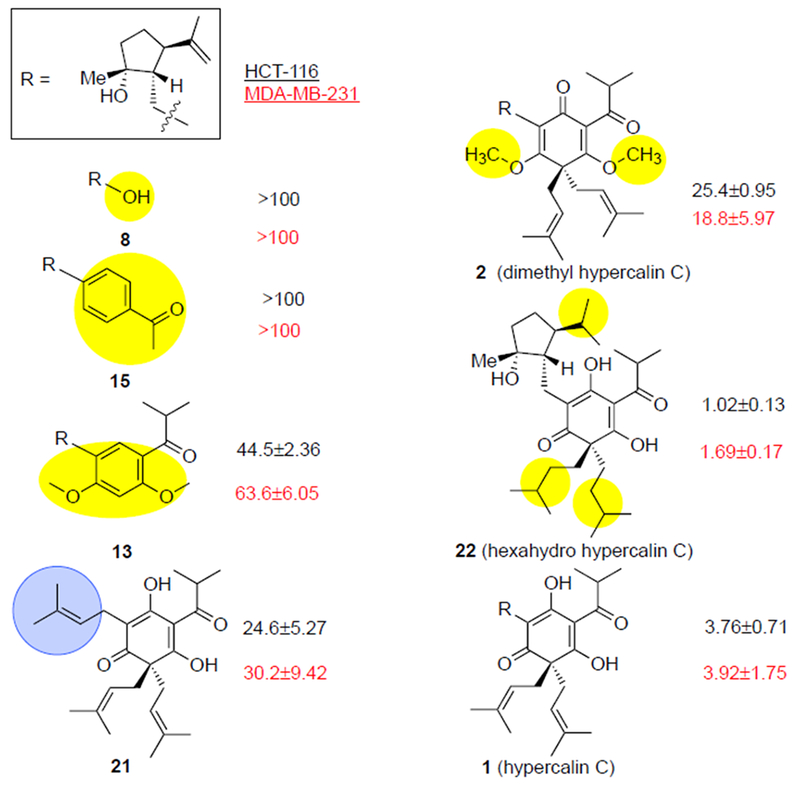 |
IC50 values were determined for the indicated compounds with MDA-MB-231 and HCT-116 cells in vitro. Standard error of the mean represents the variation of three biological replicates, each with at least three technical replicates
In conclusion, the first total synthesis of hypercalin C was achieved in 10 steps (longest linear sequence from carvone) with an overall yield of 8%. The synthetic sequence featured a key Csp3-Csp2 Suzuki coupling with an uncommon α-bromo enol ether coupling partner and a boronic monoester derived from carvone. This disconnection represents a formal α-alkylation of a ketone via transition metal coupling, offering a unique perspective for ketone α-functionalization. In addition, we discovered an unexpected and conceptually novel C-H insertion/Suzuki coupling process employing the α-bromo enol ether substrate. Guided by the desire to answer particular biological questions regarding SAR enabled by the designed synthetic strategy, several hypercalin C analogs were accessed and tested against two cancer cell lines for cytotoxicity. Some support for the proton shuttle hypothesis for hypercalin C was garnered through these studies however the modest decrease in cytotoxicity when the acidic enol protons are substituted for methyl groups suggests that other cellular mechanisms for the observed cytotoxicity of the hypercalins are likely operative.
Supplementary Material
Scheme 4.
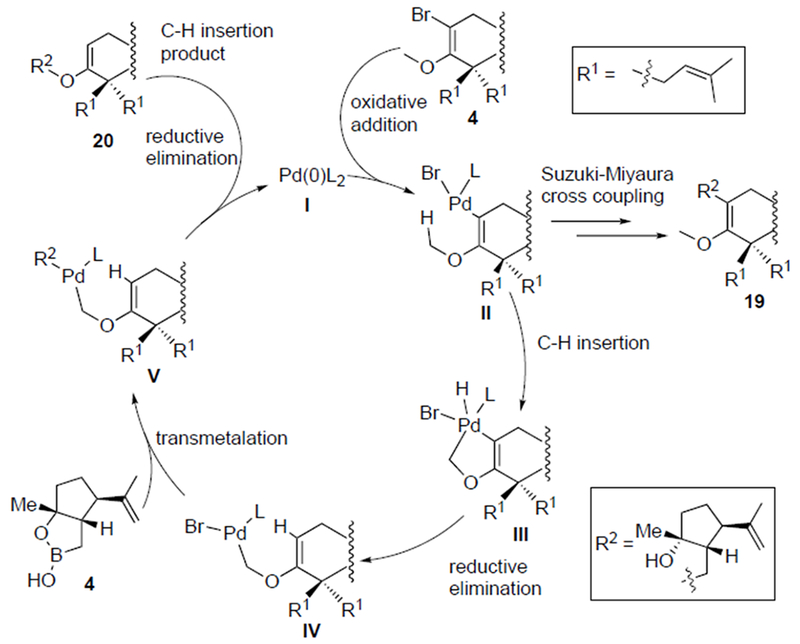
Proposed catalytic cycle leading to typical Suzuki-Miyaura and an unexpected C-H insertion into an enol ether
Acknowledgements
Support from the Welch Foundation (AA-1280) and partial support from NIH (R37 GM052964) is gratefully acknowledged. We acknowledge Dr. Morgan A. Shirley for early synthetic studies toward hypercalin C in the Romo Group and Tim Philip for technical support in the Taube Group. HCT-116 and MDA-MB-231 cells were generous gifts from Dr. Ajay Goel and Dr. Sendurai Mani, respectively.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References and Notes
- [1].a) Decosterd LA, Stoeckli‐Evans H, Chapuis JC, Sordat B, Hostettmann K, Helv. Chim. Acta 1989, 72, 1833–1845; [Google Scholar]; b) Aramaki Y, Chiba K, Tada M, J. Chem. Soc., Perkin Trans 1 1995, 683–688; [Google Scholar]; c) Nagai M, Tada M, Chem. Lett 1987, 16, 1337–1340. [Google Scholar]
- [2].a) Mitasev B, Porco JA Jr, Org. Lett 2009, 11, 2285–2288; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tsukano C, Siegel DR, Danishefsky SJ, Angew. Chem. Int. Ed 2007, 46, 8840–8844; [DOI] [PubMed] [Google Scholar]; c) Narender T, Reddy KP, Shweta, Srivastava K, Mishra D, Puri S, Org. Lett 2007, 9, 5369–5372; [DOI] [PubMed] [Google Scholar]; d) Uwamori M, Nakada M, Tetrahedron Lett 2013, 54, 2022–2025; [Google Scholar]; e) Ting C, Maimone T, Synlett 2016, 27; [Google Scholar]; f) Usuda H, Kanai M, Shibasaki M, Org. Lett 2002, 4, 859–862; [DOI] [PubMed] [Google Scholar]; g) Kuramochi A, Usuda H, Yamatsugu K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2005, 127, 14200–14201; [DOI] [PubMed] [Google Scholar]; h) Bellavance G, Barriault L, Angew. Chem. Int. Ed 2014, 53, 6701–6704. [DOI] [PubMed] [Google Scholar]
- [3].a) Tada M, Chiba K, Takakuwa T, Kojima E, J. Med. Chem 1992, 35, 1209–1212; [DOI] [PubMed] [Google Scholar]; b) Zhang J-J, Yang X-W, Ma J-Z, Ye Y, Shen X-L, Xu G, Tetrahedron 2015, 71, 8315–8319; [Google Scholar]; c) Osman K, Evangelopoulos D, Basavannacharya C, Gupta A, McHugh TD, Bhakta S, Gibbons S, Int J Antimicrob Agents 2012, 39, 124–129; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tala MF, Tchakam PD, Wabo HK, Talontsi FM, Tane P, Kuiate JR, Tapondjou LA, Laatsch H, Rec. Nat. Prod 2013, 7, 65–68. [Google Scholar]
- [4].Wu S-B, Long C, Kennelly EJ, Nat. Prod. Res 2014, 31, 1158–1174. [DOI] [PubMed] [Google Scholar]
- [5].Miyaura N, Yamada K, Suzuki A, Tetrahedron Lett 1979, 20, 3437–3440. [Google Scholar]
- [6].Miyaura N, Ishiyama T, Ishikawa M, Suzuki A, Tetrahedron Lett 1986, 27, 6369–6372. [Google Scholar]
- [7].Liu G, Romo D, Angew. Chem. Int. Ed 2011, 50, 7537–7540. [DOI] [PubMed] [Google Scholar]
- [8].Sell TS, Belkacemi T, Flockerzi V, Beck A, Sci Rep 2014, 4, 7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dixon DD, Lockner JW, Zhou Q, Baran PS, J. Am. Chem. Soc 2012, 134, 8432–8435. [DOI] [PubMed] [Google Scholar]
- [10].a) Liu G, Shirley ME, Romo D, J.Org. Chem 2012, 77, 2496–2500; [DOI] [PubMed] [Google Scholar]; b) Liu G, Shirley ME, Van KN, McFarlin RL, Romo D, Nat. Chem 2013, 5, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Finkelstein H, Ber. Dtsch. Chem. Ges 1910, 43, 1528–1532. [Google Scholar]
- [12].Bailey WF, Patricia JJ, Nurmi TT, Wang W, Tetrahedron Lett 1986, 27, 1861–1864. [Google Scholar]
- [13].Martin R, Buchwald SL, Acc. Chem. Res 2008, 41, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Maestri G, Motti E, Della Ca’ N, Malacria M, Derat E, Catellani M, J. Am. Chem. Soc 2011, 133, 8574–8585. [DOI] [PubMed] [Google Scholar]
- [15].a) Walker SD, Barder TE, Martinelli JR, Buchwald SL, Angew. Chem 2004, 116, 1907–1912; [DOI] [PubMed] [Google Scholar]; b) Barder TE, Walker SD, Martinelli JR, Buchwald SL, J. Am. Chem. Soc 2005, 127, 4685–4696; [DOI] [PubMed] [Google Scholar]; c) Martin R, Buchwald SL, Acc. Chem. Res 2008, 41, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kotha S, Ali R, Srinivas V, Krishna NG, Tetrahedron 2015, 71, 129–138. [Google Scholar]
- [17].a) Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Desai LV, Hull KL, Sanford MS, J. Am. Chem. Soc 2004, 126, 9542–9543. [DOI] [PubMed] [Google Scholar]
- [18].a) Stultz LK, Huynh MHV, Binstead RA, Curry M, Meyer TJ, J. Am. Chem. Soc 2000, 122, 5984–5996; [Google Scholar]; b) Yu J-Q, Corey E, Org. Lett 2002, 4, 2727–2730; [DOI] [PubMed] [Google Scholar]; c) Catino AJ, Forslund RE, Doyle MP, J. Am. Chem. Soc 2004, 126, 13622–13623; [DOI] [PubMed] [Google Scholar]; d) Yu J-Q, Wu H-C, Corey E, Org. Lett 2005, 7, 1415–1417; [DOI] [PubMed] [Google Scholar]; e) Choi H, Doyle MP, Org. Lett 2007, 9, 5349–5352; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhao Y, Yeung Y-Y, Org. Lett 2010, 12, 2128–2131. [DOI] [PubMed] [Google Scholar]
- [19].Sparling BA, Moebius DC, Shair MD, J. Am. Chem. Soc 2013, 135, 644–647. [DOI] [PubMed] [Google Scholar]
- [20].George JH, Hesse MD, Baldwin JE, Adlington RM, Org. Lett 2010, 12, 3532–3535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.