

SUMMARY
In order to provide a comprehensive resource for human structural variants (SVs), we generated long-read sequence data and analyzed SVs for fifteen human genomes. We sequence resolved 99,604 insertions, deletions, and inversions including 2,238 (1.6 Mbp) that are shared among all discovery genomes with an additional 13,053 (6.9 Mbp) present in the majority, indicating minor alleles or errors in the reference. Genotyping in 440 additional genomes confirms the most common SVs in unique euchromatin are now sequence resolved. We report a ninefold SV bias toward the last 5 Mbp of human chromosomes with nearly 55% of all VNTRs (variable number of tandem repeats) mapping to this portion of the genome. We identify SVs affecting coding and noncoding regulatory loci improving annotation and interpretation of functional variation. These data provide the framework to construct a canonical human reference and a resource for developing advanced representations capable of capturing allelic diversity.
Graphical Abstract
In Brief
Long-read sequencing allows generation of a large catalog of human structural variants and the development of an algorithm for genotyping SVs from shortread data, clarifying the spectrum and importance of structural variation in the human genome.
INTRODUCTION
The current human genome reference (GRCh38) is constructed from multiple individuals, and at any given locus, it is a high-quality representation of a single human haplotype. Because the bulk of the reference was derived from large-insert BAC clones (International Human Genome Sequencing Consortium et al., 2001), the sequence from a single clone insert came to represent the “human reference” by chance at these loci. For almost two decades, such sequence has served as the de facto framework for functional annotation and interpretation of biomedical results. Although not yet complete, one version of an ideal human reference genome would contain no gaps, represent a single human haplotype, and carry the most common allele at every locus—in essence, a canonical human reference genome (Schneider et al., 2017). This has not yet been achieved. Biases and errors in the reference genome affect the accuracy of sequence read alignments and the correct interpretation of human genetic variation (Brandt et al., 2015; Degner et al., 2009).
Because of its importance to biomedical research, the quality of the human genome continues to evolve since its first release in 2001. The finishing build, GRCh35 (International Human Genome Sequencing Consortium, 2004), consisted of 2.85 Gbp of sequence derived from eight individuals with a dedicated effort to target the remaining gaps. Since then, many errors have been corrected, and the primary assembly has grown to 3.1 Gbp (Schneider et al., 2017). Recent improvements have been driven by adopting decoy sequences and incorporating data from long-read sequencing technologies (Pendleton et al., 2015; Steinberg et al., 2014; Watson et al., 2013) as well as data from haploid hydatidiform moles (Chaisson et al., 2015a; Fan et al., 2002), which allowed us to better distinguish paralogous and allelic variation. While the current human reference genome build, GRCh38, is arguably the most complete mammalian genome reference constructed to date, 70% of its sequence is still derived from a single clone library, RP11 (Schneider et al., 2017), which was obtained from a single individual and assembled more than a decade ago from large-insert BAC clones propagated in E. coli.
The assembly of a single human reference genome has been particularly complicated by the discovery of widespread structural variation between and within ape species (Iafrate et al., 2004; Locke et al., 2003; Sebat et al., 2004; Tuzun et al., 2005). Widespread genome structural variation, now operationally defined as insertions, duplications, deletions, and inversions >50 bp in length (Mills et al., 2011), means that any single human haplotype may be missing or contain sequence variants that are not present in the majority of humans. As a result, a human genome reference constructed at any one location from a single haplotype must be missing genetic information or, alternatively, carry rare variants that are not common to our species.
Recent advances in sequencing technology have now allowed us to systematically whole-genome shotgun (WGS) sequence large stretches (>10 kbp) of native DNA without the need to propagate clone inserts in E. coli. This is particularly advantageous for structural variation since the long reads provide the necessary context to anchor and sequence resolve most structural variants (SVs) irrespective of sequence composition. Previously, we and others have demonstrated the utility of long-read sequencing (Chaisson et al., 2015a; Gordon et al., 2016; Pendleton et al., 2015; Seo et al., 2016; Shi et al., 2016) to improve the sensitivity of SV detection. In this study, we target a diversity panel of haploid and human genomes to create the largest collection of sequence-resolved structural variation. This resource allows us to discover fixed and major allele SVs that are common to the human species and are currently missing from the human reference genome. Sequence resolution of such major allele SVs provides an important first step for developing a canonical human reference genome.
RESULTS
Long-Read Sequencing of Human Genomes
We generated long-read sequence data from eleven human genomes using the long-read PacBio (Pacific Biosciences, Menlo Park, CA) single-molecule, real-time (SMRT) PacBio RS II and Sequel sequencing platforms (STAR Methods). In addition, we included two complete hydatidiform moles, CHM1 and CHM13, which carry a single human haplotype, that we previously sequenced and published (Huddleston et al., 2017). Eleven diploid genomes were derived primarily from HapMap samples and consisted of three African samples (Yoruban, Gambian, and Luhya), two Asian samples (Han Chinese and Vietnamese), two samples of European descent (Northwestern Europe and Finnish), three American samples (Puerto Rican, Columbian, and Peruvian), and one South Asian (Telugu) (Table 1). Because these 13 genomes are targets for the development of new human reference genomes, each was sequenced deeply achieving greater than 50-fold sequencing coverage. We also included sequence data from two recently published Asian genomes, AK1 (Seo et al., 2016) and HX1 (Shi et al., 2016), since they had been generated using the same sequencing platform. Seven of these represent new biological samples where eleven are new sequence datasets (Table 1, STAR Methods). We report SVs on all 15 of these genomes.
Table 1.
Long-Read Sample Summary
| Sample | Population | Super-population | Source | New Data | Accession | Platform | Mean Depth |
Longest Coverage |
Subread Coverage |
Longest N50 |
Subread N50 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CHM1 | Mole | NA | Reference | No | PRJNA246220 | RS II | 65x | 63x | 66x | 19,728 | 19,226 |
| CHM13 | Mole | NA | Reference | No | PRJNA269593 | RS II | 67x | 63x | 72x | 11,954 | 11,320 |
| HG00514 | CHB | EA | Reference | Yes* | PRJNA300843 | RS II | 76x | 93x | 104x | 17,472 | 16,653 |
| HG00733 | PUR | AMR | Reference | Yes* | PRJNA300840 | RS II | 55x | 63x | 69x | 16,195 | 15,461 |
| NA19240 | YRI | AFR | Reference | Yes* | PRJNA288807 | RS II | 59x | 65x | 71x | 17,343 | 16,584 |
| HG02818 | GWD | AFR | Reference | Yes | PRJNA339722 | RS II | 79x | 90x | 98x | 16,807 | 16,221 |
| NA19434 | LWK | AFR | Reference | Yes | PRJNA385272 | RS II | 59x | 62x | 71x | 17,635 | 16,853 |
| HG01352 | CLM | AMR | Reference | Yes | PRJNA339719 | RS II | 56x | 69x | 75x | 20,738 | 20,049 |
| HG02059 | KHV | SA | Reference | Yes | PRJNA339726 | RS II | 64x | 71x | 77x | 18,533 | 17,890 |
| NA12878 | CEU | EUR | Reference | Yes* | PRJNA323611 | RS II | 50x | 66x | 75x | 17,121 | 16,376 |
| HG04217 | ITU | SA | Reference | Yes | PRJNA481794 | RS II | 40x | 46x | 51x | 18,149 | 16,871 |
| HG02106 | PEL | AMR | Reference | Yes | PRJNA480858 | Sequel | 73x | 66x | 69x | 21,540 | 20,646 |
| HG00268 | FIN | EUR | Reference | Yes | PRJNA480712 | Sequel | 85x | 76x | 79x | 25,245 | 24,487 |
| AK1 | Korean | EA | Public | No | PRJNA298944 | RS II | 77x | 89x | 102x | 15,609 | 14,721 |
| HX1 | Chinese | EA | Public | No | PRJNA301527 | RS II | 98x | 79x | 103x | 13,412 | 12,002 |
Summary samples in this study. “Source” denotes reference genomes generated by authors of this paper and public genomes generated by other studies. “New Data” denotes which are newly published long-read data, and an asterisk annotates new sequence data on previously published biological samples. The coverage for each sample by mean euchromatic alignment depth excluding all regions of the genome corresponding to and mapping within 500 bp of centromeres, tandem repeats, segmental duplications (SDs), gaps, chromosome ends, or the filter applied to SV calls. Longest subread per read (or ZMW) and all subread coverage are estimated using raw sequence data throughput and a genome size of 3.2 Gbp. The longest subread measure eliminates duplicate sequence from small inserts, and thus, excludes a significant amount of mutual information in the sequence reads. RS II samples were run with P6-C4 chemistry, and Sequel was run with v2.1 chemistry.
SV Discovery
For each human genome, we identified and sequence assembled insertions, deletions, and inversions of 50 bp or greater relative to GRCh38 using SMRT-SV (Huddleston et al., 2017). We excluded pericentromeric regions with dense tandem repeat or gap structures where we found variant calls to be unreliable (STAR Methods). In total, this filter covers 254 Mbp of the primary GRCh38 assembly, and per sample, it removes an average of 7,668 SV calls. On average, we identified 22,755 SVs per sample affecting 11 Mbp. We merged these into a set of 99,604 nonredundant SVs (Figure 1A, Table S1) and classified them into four categories: shared (present in all samples), major (present in ≥ 50% of samples but not all), polymorphic (present in more than one but < 50% of samples) or singleton (present in only one sample) (STAR Methods). As expected, the African samples showed the greatest genetic diversity where each contributes 11.1% of the singleton variants versus 5.6% from non-African samples (Figure 1B). Thus, we would expect a new African sample to add approximately twice the number of novel variants compared to a non-African sample.
Figure 1. SV Discovery in 15 Human Genomes.

(A) Variants from each sample were merged using a nonredundant strategy starting with CHM1 and iteratively adding unique calls from additional samples. The growth rate of the nonredundant set declines as the number of samples increases. Variants shared among all samples are shown as red portions of each bar.
(B) The number of variants in each discovery class is shown per sample. As expected, African samples (asterisks) contribute a higher proportion of singleton variants.
(C) Discovery class frequency for each variant type: insertion (INS), deletion (DEL), and inversion (INV). Compared to deletions, a greater proportion of insertions are shared among all samples.
(D) For SVs that were genotypable in all 440 population samples and in non-repetitive loci, a discovery frequency is shown with bars colored by genotyping support based on allele frequency (AF). Generally, AF supports the genotype frequency. No support: AF = 0, Single: One allele, Poly: 0.5 > AF > 0, Major: 1 > AF ≥ 0.5, Shared: AF = 1.
(E) For each SV, the distance to the end of the chromosome arm was calculated and divided into 500 kbp bins. The number of calls within 5 Mbp of the chromosome end (dashed box) confirms a nonrandom distribution of SVs.
Growth of the nonredundant set is initially steep, but it declines as samples are added, which indicates that a large proportion of common variation is captured by this subset of 15 humans. Similarly, the set of shared variants declines quickly at first and flattens as samples are added leaving 2,238 SVs (1.6 Mbp) observed in all discovery samples (shared SVs), and the proportion of samples carrying each variant shows a similar pattern with an increase at 100% for shared variants. We modeled the growth of our nonredundant set excluding repetitive loci, and this suggests that the addition of one more human genome would likely increase this part of the callset by 2.1%, and adding 35 genomes (50 total) would increase it by 39%. To double the number of SVs in unique regions, we estimate that it would take 327 samples (STAR Methods). This model supports our conclusion that additional African samples would increase the callset more sharply. We identify 15,291 SVs (15.4%, 8.5 Mbp) that are observed in the majority of the human genomes (major and shared) indicating the current human reference genome carries a minor allele or an error at these positions. Compared to polymorphic variants, these majority variants tend to be larger and are enriched for repetitive DNA (~80%). A greater proportion of the shared variants were defined as insertions with respect to GRCh38 when compared to deletions, and this observation is consistent with the presence of “muted” gaps (i.e., regions where there is missing sequence but no indication of a gap in the reference assembly, Figure 1C) (Eichler et al., 2004).
We compared our discovery set to previously published SV calls identified from Illumina sequencing data obtained from thousands of human genomes (Francioli et al., 2014, 2015; 1000 Genomes Project Consortium et al., 2012; Mills et al., 2011; Sudmant et al., 2015a, 2015b). Consistent with earlier observations (Chaisson et al., 2015a; Huddleston et al., 2017; Shi et al., 2016), 87.3% of the SVs discovered in these 15 human genomes are novel (Figure S1A). The greatest increase in yield occurred for insertion variants where we estimate that 93.5% of the events are novel (Figure S1B), increasing the yield by an order of magnitude. Deletion variant yield was also higher but to a lesser extent than insertions (Figure S1C). Three of the samples in this study (HG00514, HG00733, and NA19240) were also recently assessed using an independent dataset and a haplotype-aware SV callset from the Human Genome Structural Variation Consortium (HGSVC) (Chaisson et al., 2017). The HGSVC set significantly increases yield from the haplotype-unaware callset (Figure S1D). Our 15 samples and the HGSVC set combined yield the most comprehensive sequence-resolved dataset of normal human structural variation to date with 113,503 insertions, deletions, and inversions. If we combine many large-scale SV analyses including short-read, long-read, dbVar data (Francioli et al., 2014, 2015; Huddleston et al., 2017; Iqbal et al., 2012; Kidd et al., 2010; Lappalainen et al., 2013a; Mills et al., 2011; Seo et al., 2016; Shi et al., 2016; Sudmant et al., 2015b, 2015a), and the HGSVC, we find that 40.8% (40,654 of 99,604) of our merged callset is novel (Figure S1E). Importantly, many of the remaining variant calls are now sequence resolved for the first time.
Genotyping in Additional Human Genomes
In order to better understand the population distribution of the SVs discovered in this study, we developed an updated SV genotyper (https://github.com/EichlerLab/smrtsv2) with a machine-learning approach, which uses 15 features based on mapping short-read data to the reference and sequence-resolved SVs (STAR Methods). We applied this method to a human genome diversity panel constructed from 440 human genomes generated using Illumina WGS data (Figure 1D); 55.1% of the variants successfully genotyped in at least 95% of samples, and 92.6% could be genotyped in half or more. Of those that could be successfully genotyped, we observed 97.2% of the SVs in at least one additional human genome. This suggests that the vast majority of the SVs represent true human polymorphism as opposed to private variants or somatic artifacts. As expected, among the shared and major SVs, the human reference genome represented the minor allele in 95.4% and 66.7% of the cases, respectively. For 507 (0.74%) of shared and major SVs discovered in this study, we only observed the alternate allele and never observed the human reference sequence. For these loci, the human reference genome represents either an extreme minor allele (< 0.2%) or an error. The method we have developed (SMRT-SV v2 genotyper) can now be applied to Illumina-generated WGS data to discover novel genome-wide associations.
SV Density and Chromosome Distribution
SVs are nonrandomly distributed in the genome, and while there is a general increase in regions enriched with common repeats and segmental duplications (SDs), we observe the strongest bias within the last 5 Mbp of chromosome arms (Figure 1E). We estimate a ninefold increase in SV density within subtelomeric regions (p value < 1 × 10−6, permutation) (STAR Methods). For shared variants, the bias is smaller, but it remains significant with a threefold increase (p value < 1 × 10−6, permutation). While this bias is observed for most human chromosomes, it is not uniformly distributed, and the magnitude of the effect varies by a factor of three depending on chromosome and chromosome arm. The long arms of chromosomes are more likely to show a greater subtelomeric bias than the short arms of chromosomes, but there are clear exceptions such as chromosomes 5, 19, andX. We tested whether the chromosomal bias in SV density was related to the increased mutability of subtelomeric regions by comparing the density of de novo single-nucleotide variant (SNV) mutations from a recent study of 547 families (Turner et al., 2017). We observe a modest but significant correlation (R = 0.40, Pearson; p value < 1 × 10−15, F-test) between SV and de novo SNV density suggesting that an elevated mutation rate is partially responsible for increased variant density.
In order to provide further insight into the mutational bias, we examined the subtelomeric enrichment by classifying SVs by repeat type (STAR Methods). Almost all of the SV subtelomeric bias was driven by tandem repeat sequences with no enrichment observed for SVs originating by retrotransposition (e.g., LINE, SINE, LTR, and SVA). We observe the strongest enrichment for VNTRs (variable number of tandem repeats) at a level of 4.8-fold compared to the null expectation (Figure 2A, paired Wilcoxon rank-sum test p = 2.9 × 10−9) followed by a 2.9-fold enrichment for smaller STRs (short tandem repeats) (p = 0.0017). Although the enrichment varied by chromosome, the long arms of human chromosomes generally showed a broader zone of VNTR enrichment (5–10 Mbp) when compared to the short arms (Figure 2B). Chromosome 17p is the most extreme where 85% of all VNTRs map within the last 5 Mbp. This VNTR subtelomeric enrichment persisted after adjusting for chromosome arm length differences. Both male recombination (Kong et al., 2010) and double-strand breaks are known to be particularly biased near the end of chromosome arms (Linardopoulou et al., 2005). Hence, we considered both to better understand the factors influencing this SV bias. Although we observe a moderate but significant correlation between VNTR density and male recombination rates (Kong et al., 2010), the strongest correlation exists between double-strand breaks and VNTR density (R = 0.48, p < 10−22) (Figure 2C), suggesting a relationship between regions prone to double-stranded breakage and VNTR formation.
Figure 2. VNTR Distribution and Double-Strand Break Correlation.

(A) An ideogram showing the distribution of VNTRs (green; below chromosome) and STRs (blue; above chromosome). STRs (n = 16,619) and VNTRs (n = 55,585) were defined as tandem repeats within the SV sequence with tandem motif lengths of ≤ 6 bp and ≥ 7 bp, respectively. The tick marks on the axes for each chromosome indicate a value of 20 per 500 kbp bin.
(B) A cumulative distribution of VNTRs shows that the most rapid saturation pattern for VNTRs belongs to chromosome 17p with approximately 85% of all VNTRs found in the last 5 Mbp. Windows of 500 kbp sliding from telomere ends to the centromere were used to cumulatively count STRs and VNTRs. The x axis is truncated at 20 Mbp.
(C) Abundance of STRs and VNTRs is positively correlated with the distribution of double-strand breaks with the strongest correlation occurring for larger VNTRs (R = 0.48) compared to STRs (R = 0.27).
To expand upon the factors influencing SV bias, we modeled the SV density as a function of double-strand breaks, sex-specific recombination, de novo mutation rates, and average replication timing while controlling for SD content using a multiple linear regression (STAR Methods). The total amount of variation explained by these models was the highest in the case of VNTRs (R2 = 0.43), followed by all SVs (R2 = 0.36), STRs (R2 = 0.25) and non-repeat SVs (R2 = 0.24). Of all the explanatory variables, male-specific recombination accounted for the majority of the SV density variation (R2 range of 0.042–0.069 depending on SV type) followed by double-strand breaks (R2 range of 0.034 to 0.057) (Table S2). Interestingly, the interaction of male-specific recombination rate with de novo mutation rate and male-specific recombination with double-strand breaks were the top pairwise interaction terms, explaining a range of 1.6%–4.4% of the total variation in SV density.
Properties of Shared and Major-Allele Variants
In an effort to define a canonical human reference genome, we focused on characterizing the sequence properties of all shared and major variants identified in the original 15 discovery genomes. First, we selected 112 shared SVs for validation in large-insert BAC libraries for CHM1 (CH17, n = 62) and fosmid libraries for NA12878 (ABC12, n = 50) and were able to obtain complete inserts for 99 of these clones. Of these, 96.0% (95/99) of the selected sites validated the presence of the variant, confirming that the reference genome does not accurately represent the major allele. Two of the events that failed validation were associated with a much larger insertion that was fragmented in our callset. On average, the local assemblies generated by SMRT-SV as part of the SV calling process were 99.7% identical with the BAC clones and 99.3% identical with fosmid clones. When counting each indel as a single event, sequence accuracy estimates are greater than 99.9% for BAC and fosmid clone inserts.
These shared and major SVs are enriched for most classes of repetitive DNA, and this is most clearly illustrated by STRs, VNTRs, and retroposons. Consistent with previous observations (Chaisson et al., 2015a), complex repeat insertions and mosaic Alu patterns are prevalent in sequence that is not found in the reference. Interestingly, these are also the most highly preserved insertions with 42% of complex repeat and 38% of mosaic Alu insertions appearing in the set of shared and major SVs. Conversely, the active mobile elements, L1HS and AluY, are the most prevalent in the shared and major deletions (51% and 44%, respectively), and these identify rare variant retrotransposon alleles embedded in the reference genome. Additionally, the length distribution of the shared and major variants confirms both deleted and inserted mobile element insertions with noticeable peaks at 300 bp and 6–7 kbp corresponding to Alu and L1 repeat elements. We identify approximately 580 full-length Alu repeat elements that are present in most human genomes but are missing from the current human reference genome and 268 rare full-length Alu elements that are embedded in the reference. When considering all insertions, including those that contain multiple retrotransposons, we find 223 L1 and 3,383 AluY full-length elements that may serve as a substrate for de novo retrotransposition events (Table S1). Of these, 23.4% are found in most human genomes and 30.6% are sequence resolved for the first time.
Skewed GC Composition
Because regions enriched in GC content are often difficult to clone and sequence, we examined the GC composition of the newly sequenced SVs. Compared to GRCh38, the mean GC composition was significantly higher (p < 1 × 10−6, permutation) (STAR Methods), but it was also skewed toward lower GC content with a noticeable enrichment for variants < 30% GC (p < 1 × 10−6, permutation) (Figure 3A). This bimodal distribution is driven almost entirely by SVs in tandem repeats. Excluding all classes of repeat sequence and tandem repeats, we find that the GC distribution follows the null distribution more closely (Figure 3B). Nevertheless, the mean for insertions is still shifted toward higher GC content (p < 1 × 10−6, permutation) while the mean for deletions is not as significantly affected (p = 0.01, permutation). In both cases, there is an enrichment for the proportion of variants above 70% GC (p < 1 × 10−6), but this effect begins to dissipate for deletions at 80% GC (p = 8.9 × 10−4, permutation test). This skew and the multimodal GC distribution, especially for non-repetitive shared SVs, suggests that these dropouts may represent artifacts during the assembly of the human reference genome in which extreme GC content confounded cloning, sequencing, or assembly.
Figure 3. GC Content Distribution.

(A) The mean GC composition (dashed vertical lines colored by discovery class) is greater than the reference (black dashed vertical line), but the distribution is also skewed toward lower GC content. The null distribution over the reference was computed excluding the same regions used to filter SV calls.
(B) Excluding repeats, the GC distribution follows the reference distribution more closely, but shared variants still exhibit a multi-modal distribution with peaks in GC-rich regions. Repeat content was annotated by RepeatMasker and Tandem Repeats Finder (TRF). In addition to the SV call filter, SD- and TRF-annotated loci in GRCh38 are excluded when calculating the reference distribution.
Gap Discovery and Closure
GRCh38, contains 819 gaps (annotated with N’s) over an estimated 159 Mbp of missing or unlocalized sequence. These gaps are known, and they are often the target of whole-genome assembly or directed finishing projects (Berlin et al., 2015; Chaisson et al., 2015a; Pendleton et al., 2015; Seo et al., 2016; Shi et al., 2016; Steinberg et al., 2014). In addition to known gaps, there are regions of the genome that appear to be complete but are missing sequence (muted gaps). These may arise from rare deletions during propagation of clones in E. coli, structural polymorphisms, contig misassembly, or errors when tiling contigs into a reference. Our set of shared insertion SVs is a useful resource for identifying and correcting muted gaps in the reference.
Because errors can occur where contigs are joined, we examined SVs within 200 bp of each scaffold switch-point of the human genome (STAR Methods). We identified 34 insertions (21,850 bp), 18 deletions (7,815 bp), and 1 inversion (9,419 bp) specifically at these switch-points. Of the 34 insertions, 21 (10,540 bp) intersect RefSeq annotated genes (ABCA13, ALPK2, CMC2, COL19A1, DSCR4, GABRA5, GDAP1, GLI2, IFT81, MAST4, MCTP2, MIR646HG, NCAM2, NUTM1, PPP3CA, PRDM15, RRBP1, SRPK2, TP53I11, UNC80, and ZNF573). One example of a muted gap arising from misassembled and mis-joined contigs is a 2,159 bp insertion in 15q14 that occurs at a switch-point between RP11–122P18 (AC021822.21) and RP11–602M11 (AC025678.7) (Figures S2A and S2B). Mapping BAC end sequences for RP11–122P18 predicts that this contig should be ~65 kbp longer than its assembly, and this suggests that the assembly was fragmented. The truncated end of RP11–122P18 (Figure S2C) is stitched together with a misassembly in RP11–602M11 (Figure S2D) leaving missing sequence without a gap annotation. After sequencing both clones from an RP11 BAC library with PacBio reads to high depth and assembling them, we confirm that RP11–122P18 and RP11–602M11 actually contain this missing 2 kbp of sequence (Figures S2E and S2F).
Genic and Potential Regulatory SVs
We intersected our shared and major variants with RefSeq annotations and identified 86 events affecting coding sequence, 47 events in untranslated regions (UTRs), and 7,417 events in introns or within 2 kbp flanking any gene. Additionally, we specifically identify 1,033 events affecting putative noncoding regulatory sequence, which is defined here as the union of annotated DNase I hypersensitive, H3K27Ac, H3K4Me1, and H3K4Me3 sites (Table 2). Many of these events are embedded in regions of GC-rich or low-complexity DNA and would potentially affect the gene structure. For example, at the 5’ end of UBEQ2L1, we identify a 1.6 kbp insertion comprised primarily of di- and tri-nucleotide CACA repeat units (Figure 4A) adjacent to a 94 bp GC-rich sequence. The breakpoint of the insertion maps precisely to the first base of the 5’ UTR, likely extending the length of the UBEQ2L1 promoter. AT-rich sequences are also resolved, such as the 458 bp repeat mapping within the 3’ UTR of the apolipoprotein APOOL (Figure 4B). A similar, expanded 1.06 kbp GC-rich insertion is observed in the 3’ UTR of the RNA editing gene ADARB1 where a degenerate 40-mer predicted to form quadruplex DNA is expanded to approximately 31 copies compared to the reference, which predicts seven copies (Figure 4C). This UTR repeat structure is largest in humans compared to other great apes, and it is interesting to note that adenosine editing in the human brain is also reported to be the highest (Paz-Yaacov et al., 2010).
Table 2.
SVs Intersect Genes and Regulatory Elements
| Class | CDS | UTR | NC Regulatory | Intron/2kbp Flank | All Gene/Reg | All | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | N | % | N | |
| Shared | 5 | 0.22% | 6 | 0.27% | 160 | 7.15% | 1,111 | 49.64% | 1,180 | 52.73% | 2,238 |
| Major | 81 | 0.62% | 41 | 0.31 % | 873 | 6.69% | 6,306 | 48.31 % | 6,712 | 51.42% | 13,053 |
| Polymorphic | 326 | 0.82% | 161 | 0.41 % | 2,410 | 6.07% | 18,969 | 47.81 % | 20,262 | 51.07% | 39,676 |
| Singleton | 429 | 0.96% | 286 | 0.64% | 2,838 | 6.36% | 20,653 | 46.27% | 22,232 | 49.81 % | 44,637 |
| All | 841 | 0.84% | 494 | 0.50% | 6,281 | 6.31 % | 47,039 | 47.23% | 50,386 | 50.59% | 99,604 |
Number and percent of SVs affecting a coding region (CDS), untranslated region (UTR), noncoding regulatory element (NC Regulatory), or any noncoding base in introns or within a 2 kbp flank of an annotated gene (Intron/2kbp Flank). Noncoding regulatory is defined as SVs intersecting H3KMe1, H3KMe3, H3K27Ac, or open chromatin by DNase hypersensitivity.
Figure 4. Missing Genic and Regulatory Sequence.

(A) A shared 1.6 kbp insertion in the 5’ UTR of UBEQ2L1 is almost completely comprised of simple repeat units (CACA) or low-complexity, GC-rich sequences. The breakpoints lie precisely at the start position of the 5’ UTR, and the missing sequence is largely conserved among chimpanzee and orangutan haplotypes.
(B) A 458 bp insertion is detected in 50% of the discovery samples in the large 5.63 kbp 3’ UTR of APOOL. The insertion is comprised of an AT-rich repeat array consisting of 30 bp units for a total of 24 tandem copies. Because of its AT-rich sequence composition, analysis with RNA-seq is inconclusive (“ind human” is a brain sample from a single anonymous individual). Comparison with nonhuman primates reveals that the repeat array is largely absent.
(C) A 1.1 kbp shared insertion in the 3’ UTR of the ADARB1 corresponds to a large VNTR comprised primarily of GC-rich sequence. Each repeat unit is 42 bp with a variable number of copies present in CHM13, chimpanzee, and orangutan. We detect 31 tandem copies in CHM13 compared to only 7 in the GRCh38 reference assembly.
(D) A 13.8 kbp inversion in intron 32 of DSCAM. The shared inversion is flanked by inverted, complete LINE-L1 repeat sequences.
(E) A 480 bp shared insertion detected in the first exon of RRBP1 (transcript ENST00000246043.8) is associated with gaps in RefSeq and UCSC gene annotations (top). Mapping human IPS-derived PacBio Iso-Seq data to the GRCh38 reference assembly identifies discordant read alignments at the insertion site (Iso-Seq alignments, left). Analysis of the insertion and adjacent flanking sequence identifies a large VNTR (1,380 bp) comprised of 30 bp repeat units. In our discovery set, the number of copies varies between 15 (450 bp) and 16 (480 bp). Translation of the newly assembled haplotype sequence from CHM13 (15 copies, 450 bp) shows that the insertion maintains the open reading frame and adds an additional 150 amino acids (Iso-Seq alignments right).
For each panel: regions of shared or major allele structural variation are annotated and compared between GRCh38, alternate human reference assemblies (CHM1/CHM13), and nonhuman primates. Multiple sequence alignments were generated using MAFFT or visualized using Miropeats against sequenced large-insert clones. Additional functional annotations are shown using short-read Illumina RNA-seq data, PolyA-seq, and PacBio long-read Iso-Seq data.
In other cases, the change to the human reference is more subtle, such as a 13.8 kbp inversion mediated by inverted L1 elements changing the orientation of a portion of intron 32 of DSCAM (Figure 4D). We note that many of the improvements in protein-coding sequence involve repetitive amino acid motifs, which we deduce are often incomplete or contracted in the reference. Specific gene families are particularly affected, such as the mucin and KRAB C2H2 zinc finger (ZNF) gene families, which are known to carry long carboxy repeat motifs. We sequence resolve, for example, the full-length protein-encoding sequence of the N-terminal decapeptide repeats of the ribosome-binding protein RRBP1, which is incompletely annotated in both RefSeq and UCSC gene annotations (Figure 4E). The resolved repeats are 65% GC-rich, and alternative splicing produces distinct protein isoforms thought to be important in altering ribosome-binding affinity and secretory function (Langley et al., 1998). As a result, complete RRBP1 annotation improves full-length cDNA mapping with Iso-Seq datasets. Additionally, we note several SVs affecting regulatory loci including a shared 1.2 kbp insertion immediately upstream of KDM6B (Figure 5A) and a major 1.5 kbp insertion upstream of FGFR1OP (Figure 5B) intersecting H3K27Ac and H3K4Me3 sites. These SVs may have an impact on regulatory sequences or their annotations.
Figure 5. Correcting Regulatory Elements and the FOXO6 Reading Frame.

(A) A high-GC 1.2 kbp insertion immediately upstream of KDM6B was discovered in all but one sample (HG04217, Telugu). This variant is proximal to an AGP switch-point in GRCh38, and it was genotypable in 16% of Illumina samples with an allele frequency of 1.0 suggesting that observed variation among humans and nonhuman primates may be a technical artifact.
(B) A high-GC 1.5 kbp insertion proximal to the FGFR1OP promoter appears to be present in nonhuman primates but has become variable in humans with a discovery frequency of 0.66 and genotype allele frequency of 0.76.
(C) A 200 bp shared insertion (80% GC) in the final exon of FOXO6 is surrounded by low-complexity, GC-rich (> 70%) repeat sequences. Translation of the complete open reading frame (ORF) demonstrates a 67 amino acid deletion in the reference (ENST00000641094.1).
(D) Using the gnomAD database, we identified loss-of-function (LoF) variation in FOXO6 (red points) and show their coding positions (x axis) and their allele count (y axis) with a dashed line representing the SV insertion. The LoF variants with the highest allele counts (6, 10, and 38) were no longer annotated as LoF when translated in the corrected reading frame. Two frameshift variants at the breakpoint of the insertion are a 32 bp and a 200 bp insertion with an allele count of 3 and 2, respectively, and the inserted sequence for both is > 99% identical with our SV call.
SVs and Expression Analysis
To further enhance the utility of this resource, we assessed what impact the SVs identified in this study might have on RNA expression and transcript splicing. We searched specifically for eQTLs (expression quantitative trait loci) in lymphoblastoid RNA-seq data from 376 European cell lines developed as part of the GEUVADIS Consortium (Lappalainen et al., 2013b). We first applied our genotyper to low-coverage GEUVADIS WGS samples and successfully genotyped 61,244 SVs. We identified 379 SV eQTLs affecting 411 genes and 244 SV sQTLs (splicing QTLs) affecting 197 genes (Table S3) at a 5% FDR (false discovery rate) (STAR Methods). Among the former set, we identified 30 genes for which the expression was more strongly associated with an SV than any nearby single-nucleotide polymorphism or indel highlighting potential causal SVs. It should be noted, however, that the SV genotyping error is substantially higher than that of SNVs and we estimate that ~49% of the SV eQTLs will be more significantly associated with gene expression levels if we account for this difference in genotyping accuracy (STAR Methods). These associations, thus, should be regarded as a starting point for future studies focusing on the effect of SVs on gene expression.
Improved Mapping and Variant Discovery
Improvements in genome annotation and an understanding of structural differences between human haplotypes have important consequences in variant discovery and interpretation. A more comprehensive representation of the human genome including these additional sequences will recover more of the unmapped sequence reads and improve overall mapping quality (Novak et al., 2017). Among 30 Illumina WGS samples, we find that 2.62% of unmapped reads (674,000 reads per sample) can now be recovered if we add our SV contigs to the human reference genome and its alternate contigs (STAR Methods). Moreover, 1.24% of the reads mapping to those contigs increased in mapping quality. For reads mapping to SV insertions, we find that 25.68% have an improved mapping quality with 14.53% showing a dramatic improvement of 1,000-fold or greater (MAPQ increase of 30+). Moreover, these new mapped reads enable discovery of SNVs and indels among the SV insertions. Using the GATK (McKenna et al., 2010) HaplotypeCaller, for example, we identify 21,969 unique variants totaling 68,656 alternate alleles of those variants (Table S4). This variation was not ascertainable with short-read sequences and a simple linear reference.
These differences can have a dramatic effect on variant interpretation, especially if the missing sequence maps to coding sequence. We identify, for example, a 200 bp insertion in exon 2 of FOXO6, which is associated with memory consolidation and dendritic spine density in the hippocampus (Salih et al., 2012) (Figure 5C). The missing sequence is reflected in RefSeq and Ensembl gene annotations, where the second (and final) exon was split at the location of this insertion. RefSeq joins the exons with a 0 bp intron and with a third exon of 1,391 bp, but Ensembl joins them with a 1 bp intron, which may have been introduced to maintain the reading frame, and with a third exon of 477 bp. Our analysis suggests that including this 200 bp sequence creates one continuous coding exon, adds 67 amino acids to the open reading frame, corrects the downstream frame, and alters location of the gene’s stop codon compared to its RefSeq annotation. The Genome Aggregation Database (gnomAD) (Lek et al., 2016) reports seven loss-of-function (LoF) variants discovered in FOXO6 based on the GRCh38 annotation. With the corrected FOXO6 reading frame, two of these putative LoF variants become synonymous SNVs (10/147,554 and 38/160,854 alleles), and one becomes a variant in the 3’ UTR (6/102,960 alleles) (Figure 5D). It is interesting that gnomAD reports two variant insertions (32 bp and 200 bp) that align to our SV insertion with >99% identity. Our analysis, however, suggests that this insertion is common to all human genomes and that the reported gnomAD rare insertion “variants” are in fact reference errors discovered with alternate sequence technologies, such as SMRT sequencing.
DISCUSSION
Over the last few years, improvements in the human genome have gone far beyond simply closing gaps to creating a better representation of both the diversity and complexity of human genetic variation. Instead of a linear representation of a single haplotype, researchers have proposed the development of “pan genomes” (Nguyen et al., 2014; Paten et al., 2017), where all the genomic content and variation of a species could be captured, which may be represented as graph-based structures at the haplotypic level. Initial studies suggest that such “pan references” have the potential to significantly improve sequence read mapping, variant discovery, and genetic disease associations. While GRCh38 alternate loci add diversity to a linear reference, this approach cannot scale to full human diversity because read alignments over multiple similar haplotypes will be duplicated, which impacts analysis time and the size of alignment files.
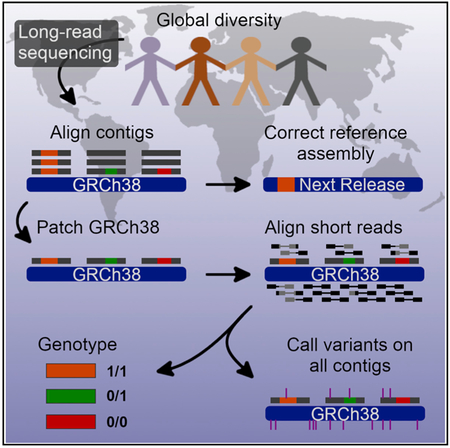
The human reference genome poses a particular challenge because it is structurally polymorphic, but in its current form, the bulk of the reference is only derived from a single haplotype originating from large-insert clones propagated in E. coli. In this study, we have sequenced native DNA, as opposed to clonally propagated DNA, from 13 human genomes using long-read sequencing technology. Combining these data with AK1 and HX1, we systematically analyzed almost all euchromatin for insertions, deletions, and inversions that are present in a majority of human genomes but not represented by the reference. Our analysis identified 15,291 distinct sites (8.5 Mbp) of structural variation found in more than half of our samples. Discovery and genotyping suggest a fraction of these (507 sites, 0.74% of genotypable variants) demarcate likely errors in the assembly, and our genotyping results suggest the human reference genome simply carries a minor alternate allele in a majority of cases. Importantly, the breakpoints and content of these major alleles are now resolved at the single-base-pair level providing the requisite sequence specificity on a GRCh38 coordinate system to begin to develop not only alternate haplotypes, but also to develop a more comprehensive graph-based assembly representation of the human genome (Schneider et al., 2017). The sequence resolution of these SV alleles means that graph content can be completely represented instead of encoding variables to summarize breakpoint variability, sequence composition, or approximate length uncertainty.
While a pan-reference genome has not yet been fully realized, critical infrastructure tools, such as vg, are now being developed (Garrison et al., 2018). To facilitate the use of this resource for these and other efforts, we are distributing data more widely to the genomics community. First, we generated and released a patched human reference containing SVs as alternate loci. This has been placed on an EBI ftp site dedicated to SV research (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1). The patched reference will allow ALT-aware aligners, such as BWA-MEM, to utilize the new sequence and improve mapping of short reads. A VCF of SVs annotating breakpoints on primary and SV contigs is shared on the FTP along with a BAM file of contigs. The VCF and BAM can be applied to additional Illumina samples with the SMRT-SV genotyper (https://github.com/EichlerLab/smrtsv2). All sequence contigs from our 13 samples have also been released to NCBI (PRJN481779), and their SVs are shared in dbVar (nstd162) to increase the utility and availability for those interested in SVs. Finally, a VCF of the high-coverage genotype calls from 440 samples is shared on the FTP site providing a standard for those interested in SV genotyping and analysis with short-read sequencing technology.
We find that these SVs are nonrandomly distributed, being particularly biased (three to ninefold depending on variant frequency) to the last 5 Mbp of most human chromosomes. This enrichment partially correlates with increased rates of de novo SNV mutations and is consistent with increased SNV divergence observed between chimpanzees and humans within 10 Mbp of the telomeres of metacentric chromosomes and the long arms of acrocentric chromosomes (Mikkelsen et al., 2005). Although the genetic basis for this enrichment is not known, elevated rates of double-strand breaks (Pratto et al., 2014), increased male recombination (Lynn et al., 2002), and biased gene conversion (Mikkelsen et al., 2005) have all been proposed to account for subtelomeric mutational biases. Although our analysis supports this observation, we also find a strong correlation with VNTR formation and an enrichment for larger mutational events (> 50 bp). We observe this for all SV classes irrespective of allele frequency. In this regard, it is interesting that a spike in the total SVs is also observed for the ancestral subtelomeric position at human chromosome 2q21, which suggests that the genomic sequence as well as chromosomal architecture of these regions may make them particularly prone to mutation.
In terms of content, SVs are enriched for various classes of repetitive of DNA. Our analysis produces a comprehensive catalog with thousands of sequence-resolved, full-length L1, Alu and HERV elements that are absent and polymorphic within human populations. Such full-length data can be particularly valuable for defining new “hot” source elements capable of both germline and somatic retrotransposition (Beck et al., 2010; Coufal et al., 2009). We also find that the SVs are particularly biased toward GC-rich and GC-poor sequences, which were likely problematic to clone, sequence, and assemble using large-insert BAC clones (International Human Genome Sequencing Consortium et al., 2001). Indeed, a small fraction of “muted” gaps appear at assembly switch-points where large-insert clone assemblies were joined since earliest builds of the human genome reference. Large, degenerate VNTRs also figure prominently, but even after excluding all mobile element insertions and tandem repeats, 2,684 sites remain that appear essentially unique. From a functional perspective, we estimate that these major alleles alter the structure of 86 protein-coding sequences, 47 genic UTRs, and as many as 1,033 regulatory sequences, as determined by DNase I hypersensitive structures and ENCODE ChIP-seq data. Many of these genes belong to families that have stretches of variable amino-acid motifs at their carboxy terminus (e.g., NPIP, ZNF, NBPF, KRTAP and mucins). We also observe large expansions of either GC-rich or AT-rich DNA within the UTRs of human genes. Many of these GC- or AT-rich repeat sequences (ADARB1, DBET, APOOL, etc.) consist of both short and large tandem repeat structures that are candidates for expansions associated with repeat instability and potential phenotypic consequences (Gatchel and Zoghbi, 2005). Importantly, the sequences we now add to the human genome provide the necessary substrate to discover new disease associations, especially as they relate to repeat instability.
Despite these advances, there remain two limitations of this work. First, in this study, we focused only on resolving the canonical structure of major allele SVs (≥50 bp). In principle, the same approach could be applied to systematically identify other forms of genetic variation, including SNVs and indels. Like any sequencing technology, long-read sequencing platforms present their own biases, specifically an excess of small insertion and deletion (1–2 bp) errors (Chaisson et al., 2015a). While smaller genetic variants could be investigated using this dataset, other sequencing platforms with higher individual read accuracy and that are less prone to small insertion errors would be more appropriate. Emerging small variation databases, such as dbSNP (Sherry et al., 2001) and gnomAD (Lek et al., 2016), already carry much of that information. Second, current long-read sequencing technology is insufficient to access and assemble all regions of the human genome, and this is particularly problematic in larger repetitive regions including centromeric, acrocentric, and SD blocks where structural variation (> 20 kbp) is known to be enriched 10- to 20-fold (Chaisson et al., 2015b; Henikoff et al., 2015). Because inversions are often flanked by large inverted SDs, long-read technology is still limited to detect them. We estimate that ~308 Mbp of especially copy number polymorphic DNA is still inaccessible by this technology. Resolving the diversity of this 10% of the genome will require even longer reads, continued dependence on large-insert clones, and/or the development of computational algorithms that can accurately assemble these larger and more complex regions of the human genome (Vollger et al., 2019).
STAR★METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Evan E. Eichler (eee@gs.washington.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
| Sample | Sex | Age | Provider | Material | Consent |
|---|---|---|---|---|---|
| CHM1 | N/A | N/A | Dr. Urvashi Surti | Placental TERT | UPMC† |
| CHM13 | F | N/A | Dr. Urvashi Surti | Placental TERT | UPMC† |
| HG00514 | F | N/A | Coriell | LCL | Coriell‡ |
| HG00733 | F | N/A | Coriell | LCL | Coriell‡ |
| NA19240 | F | N/A | Coriell | LCL | Coriell‡ |
| HG02818 | F | N/A | Coriell | LCL | Coriell‡ |
| NA19434 | F | N/A | Coriell | LCL | Coriell‡ |
| HG01352 | F | N/A | Coriell | LCL | Coriell‡ |
| HG02059 | F | N/A | Coriell | LCL | Coriell‡ |
| NA12878 | F | N/A | Coriell | LCL | Coriell‡ |
| HG04217 | F | N/A | Coriell | LCL | Coriell‡ |
| HG02106 | F | N/A | Coriell | LCL | Coriell‡ |
| HG00268 | F | N/A | Coriell | LCL | Coriell‡ |
| AK1 | M | N/A | Seo et al.(2016) | LCL | IRB of Seoul National University |
| HX1 | M | N/A | Shi et al. (2016) | Lymphocyte | IRB of Jinan University |
† Consent was reviewed by an IRB affiliated with the University of Pittsburgh Medical Center.
‡ Coriell samples must be reviewed by the submitting organization’s IRB or the Coriell University IRB.
N/A: Data not available.
METHOD DETAILS
Sample selection
We present structural variant (SV) calls on 15 samples. Of these, 11 are newly sequenced for this study (HG00514, HG00733, NA19240, HG02818, NA19434, HG01352, HG02059, NA12878, HG04217, HG02106, and HG00268), and the remaining samples were obtained from published sources, including two we previously sequenced (CHM1 and CHM13).
Thirteen samples were selected to represent diversity across four continents, and each genome is the target for the construction of new human genome references. We placed special emphasis on African samples (3 of the 13) because of their genetic diversity. Genomic material was obtained through lymphoblastoid cell lines (LCLs) available from Coriell. Data for the two hydatidiform moles (CHM1 and CHM13) was previously published as part of other manuscripts (Chaisson et al., 2015a; Huddleston et al., 2017). The remaining 11 are new data samples.
A separate publication on three of the biological samples (HG00514, HG00733, and NA19240) was prepared by the Human Genome Structural Variation Consortium (HGSVC) and submitted to bioRxiv (Chaisson et al., 2017). These data were lower coverage (40x), and we chose to sequence them to high coverage (60x) for this study; thus, the sequence data and methods to ascertain variation differ. The motivation of the HGSVC paper was to develop a method to increase sensitivity of SV detection per genome using a suite of methods, including 10X Genomics, Strand-seq, PacBio sequencing, and parent–child trio data. The focus of this paper was to explore human SV diversity in a larger sample set of genomes. Both efforts were pursued contemporaneously, and thus the optimum methodologies did not become apparent until 2018. Orthogonal data, parent–child trios and deeper PacBio coverage are not available for a majority of the samples studied here. Also, the PacBio long-read data used in our study was generated by a single center that was not used by the HGSVC, which generated their data for the three children from three different centers. Nevertheless, because we do not have the 10X and Strand-seq data to phase, we call SVs in a diplotype manner, which results in some reduction in sensitivity.
NA12878 was long-read sequenced with an older technology (RS II P5-C3) (Pendleton et al., 2015). We use this biological sample, but new sequence data. Since they were sequenced to high depth with similar technology, we do include two published samples and their data: HX1 (Shi et al., 2016) and AK1 (Seo et al., 2016). Our 13 genomes and these two published datasets comprise the 15 deep-coverage genomes we analyze. Table 1 provides a summary of all samples.
SMRT genome sequencing
Samples were selected to represent diversity across four continents, and each genome is a target for the construction of new human genome references. DNA samples were obtained from Coriell LCLs. DNA was fragmented using the Diagenode Megaruptor with the 50 kbp setting, and libraries were processed using the PacBio SMRTbell Template Prep kit following the protocol ‘Procedure & Checklist - Preparing > 30 kb SMRTbell Libraries Using Megaruptor Shearing and BluePippin Size-Selection for PacBio RS II and Sequel Systems’. Libraries were size-selected on the Sage BluePippin with a BP Start value of 18000 and a BP End value of 50000. Current DNA/Polymerase Binding Kit was used to bind DNA template to DNA polymerase and the MagBead kit was used to capture DNA polymerase/template complexes for loading. Libraries were sequenced on the PacBio RS II platform. Sequencing yielded 64-fold coverage per sample on average.
In each sample, at least 26% of reads are 10 kbp or greater, which yields at least 37-fold throughput per sample in these 10+ kbp reads (Figure S3A, Table S5A).
Sequence read mapping
The original version of SMRT-SV was used to analyze RS II samples (https://github.com/EichlerLab/pacbio_variant_caller), and an updated version (SMRT-SV v2) was used for Sequel samples (https://github.com/EichlerLab/smrtsv2). The SV calling method is the same with some updates for handling Sequel data.
Sequences were mapped to GRCh38 human reference distributed by the University of California, Santa Cruz (UCSC) (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz) without alternate sequences. Mapping was performed with BLASR (Chaisson and Tesler, 2012) (1.MC.rc43 for RS II samples, 5.3 for Sequel samples) with SMRT-SV default parameters (“–bestn 2 –maxAnchorsPerPosition 100 –advanceExactMatches 10 –affineAlign–affineOpen 100 –affineExtend 0 –insertion 5 –deletion 5 –extend –maxExtendDropoff 50”).
We examined regions outside of the 254 Mbp centromeric, pericentromeric, and acrocentric filter where long reads did not map (5x or less) and found an average of 369 kbp per sample (Figure S3B). When we merged these loci and by selecting regions 10 kbp or greater, we find 2.2 Mbp of the reference where alignment depth was lost in at least one sample, and 266 kbp (12.1%) of these regions intersected large deletion SVs by 50% reciprocal overlap (RO). Of all merged regions, including those intersecting deletions, 365 kbp (16.2%) mapped to segmental duplications (SDs), 174 kbp (7.7%) mapped to tandem repeats, less than 2% mapped to both, and 1.7 Mbp (77.3%) mapped to neither.
We also examined regions of excess read depth (2+ standard deviations) outside the centromeric and pericentromeric filter (Figure S3B). Per sample, we noted an average of 664 regions greater than 10 kbp covering an average of 19.9 Mbp. Through 50% RO merging, we note that 3.2 Mbp (6.0%) were found in all samples, and 35.9 Mbp (66.9%) were in a minority of samples, which indicates that most of these regions are likely copy number variable and not systematic mapping biases. In total, we find 53.4 Mbp in 2,265 distinct regions with excess read depth in at least one sample. Of these merged regions of copy number, we find that 96.7% (2,191 of 2,265) intersect SDs by 50% overlap and, as expected, there is an enrichment for SDs sharing high identity with another SD in the reference (Figure S3C).
Unmapped read analysis
We selected all unmappable PacBio reads from two individual genomes, Yoruban (NA19240) and Finnish (HG00268), representing the RS II and Sequel platforms, respectively. For each genome, there are typically 1–2 million unmappable reads, but the quality of these is significantly lower (Wilcoxon rank-sum test p < 2.2 × 10–16, Figures S3D and S3E), and many of these correspond to uncalled bases (N’s). We first removed low-quality unmappable reads, which were defined as reads with a QV score percentile < 75%, < 10 kbp in length, or 25% uncalled bases. Of the 793,157 and 1,447,477 unmappable reads from NA19240 and HG00268, respectively, only 47,115 (5.9%) and 107,899 (7.5%) passed these quality filters. The remaining reads were annotated to better understand their origin.
Of the high-quality unmappable reads, 47.0% and 30.5% for NA19240 and HG00268, respectively, were classified as repetitive DNA, and the vast majority of these consisted of tandem repeats as detected by Tandem Repeats Finder (TRF) (Benson, 1999). We also found that 6.0% and 2.7% of the remainder mapped to insertion polymorphisms from our final SV callset (Table S5B).
This left approximately 22,421 (2.8%) and 68,356 (4.7%) high-quality long reads from NA19240 and HG00268 genomes for further analysis. We found that 16,317 (2.1%) and 31,490 (2.2%) reads successfully mapped to sequences in the NCBI nucleotide database, for NA19240 and HG00268, respectively. As expected, these hits included alignments to the EBV virus, which was used to transform lymphoblastoid cells into the cell lines we sequenced. We searched for contiguous DNA sequences in NCBI hit by five or more unmapped reads where each read mapped to at least 5 kbp of the sequence. From this, we found 138 accessions (15.7 Mbp), and 25% or more of each NCBI contig sequence length was represented by the unmapped reads.
To gain further insight on these BAC and fosmid sequences, we annotated all 138 NCBI contigs using RepeatMasker. We found that 130 (94%) of these sequences contained over 50% satellite DNA with centromeric satellites being the most abundant. Interestingly, the only fosmid clone that was identified in both samples (AC188786) had a total repeat content of 23.6%, making this primarily euchromatic DNA sequence. Thus, these are important seed points for future characterization of pericentromeric transition regions that are still incomplete.
Local assemblies
SMRT-SV defines assembly regions by first tiling 60 kbp sliding windows offset by 20 kbp. Signatures of SVs are identified in the alignments, and additional windows are added around likely SV-containing regions. For each window, all reads with any base mapping in the window are extracted and assembled. RS II samples were assembled Celera assembler 8.3rc2 with default SMRT-SV parameters (“ -length 1000 -partitions 50 -l local -s SPEC_FILE genomeSize=WINDOW_SIZE assembleMinCoverage=5” with SPEC_FILE containing “useGrid=0, scriptOnGrid=0, ovlMemory=32, ovlStoreMemory=32000, ovlConcurrency=1, cnsConcurrency=8, merylThreads=32, merylMemory=32000, frgCorrThreads=2, frgCorrBatchSize=100000, ovlCorrBatch-Size=100000”). Sequel samples were assembled with Canu 1.7 using SMRT-SV default parameters (“genomeSize=WINDOW_SIZE -p asm useGrid=false corMhapSensitivity=high corMinCoverage=2 correctedErrorRate=0.045”). Reads are mapped back to the assemblies and polished (quiver for RS II, arrow for Sequel). Assemblies are aligned back to the reference with BLASR (same version used to map reads for each sample) with SMRT-SV default parameters (“–affineAlign –affineOpen 8 –affineExtend 0 –bestn 1 –maxMatch 30 –sdpTupleSize 13”).
Variant calls
Variants are called with SMRT-SV using the contig alignments. Insertions and deletions were annotated with RepeatMasker 3.3.0 (WU-BLAST engine) (Table S5C).
SMRT-SV uses several tools, including BLASR 1.MC.rc43, Celera Assembler 8.3rc2, and RepeatMasker 3.3.0 (WU-BLAST engine). Sequel samples were run with SMRT-SV v2 using BLASR 5.3 and Canu 1.7 (https://github.com/EichlerLab/smrtsv2). Calls were filtered by support (2+ local assembly contigs) and SVs inside inversions were removed. Each SV is given a unique name within its callset by “CHR-POS-TYPE-LENGTH” where POS is the 1-based location of the first affected base, TYPE is INS, DEL, or INV, and LENGTH is the size of the SV.
Variants were further filtered by low-confidence regions, which include centromeres (CEN), regions of dense pericentromeric tandem repeats, gaps immediately adjacent to pericentromeric repeats, and chrY (Figure S3B). In total, this filter covers 254 Mbp of the primary GRCh38 assembly, and per sample, it removes 7,668 SV calls on average.
This filter was developed based on reproducibility of SMRT-SV calling on the CHM1 genome whole-genome shotgun (WGS) sequence. Because CHM1 lacks allelic variation, it eliminates the complexity of trying to phase allele variants such that all SV differences represent paralogous sequence variation. We performed an analysis where we compared reproducibility of our initial callset (Huddleston et al., 2017) with a second run using SMRT-SV with the same parameters. Using the UCSC Genome Browser, we first observed specific regions where reproducibility was low (Figure S4A). Examination of the regions showed that these discrepancies frequently corresponded to pericentromeric regions flanked by reference gaps and enriched for alpha-satellite sequence. While it is likely that considerable structural variation occurs here, the results are not reproducible likely because of the paralogy and differences in the BLASR seed alignments or the PBcR assembler. We used these discrepant regions to develop an exclusion filter, which ultimately includes all centromeres, and extended to other tandem repeat loci, pericentromeric regions and regions flanking and including gaps.
Variant merging
We constructed a nonredundant set of discovery SVs from the 341,331 SV calls from all samples using a merging strategy where CHM1 served as the initial callset and new sites were added per sample. We excluded any variants in the sample that have 50% RO with an existing discovery variant. This merging was done per variant type (i.e., insertions are only merged with other insertions; same for deletions and inversions), and these sets are concatenated to yield 99,604 nonredundant SVs in the final merged set. Samples were added in the same order as they are listed in Table 1. Each merged call is represented by a single SV call in a single sample, but it is annotated with its discovery sample along with a list of all samples it was found in and the ID of the variant in each of those samples.
Annotations, such as SDs, tandem repeats, RefSeq, and cytoband, were determined by using annotation tracks obtained from the UCSC Genome Browser for hg38 (GRCh38). Variants in the original callsets are annotated, and the annotations are carried into the nonredundant set after merging using the representative SV call to find the correct annotation for the merged variant. These variants and their annotations are distributed in Table S1. The distribution of variants by discovery frequency follows a distribution expected of neutral variation but with an increase for variants found in all samples (Figure S4B).
Based on our 15 genomes, we modeled the growth of the nonredundant merged SV set outside of tandem repeats and SDs with a logarithmic function as the number of samples increases. We separately modeled insertions (R2 = 0.9832) and deletions (R2 = 0.9801). We expected the African curve would outpace the curve of all non-African samples, but this was only clear when we omitted tandem repeats and SDs from the model (Figure S4C).
With 15 samples, three of them African, there remains a large amount of structural variation left to be identified and sequence resolved with these methods. We note that each African sample contributes 11.1% of the singleton variants versus 5.6% from non-African samples (singletons are SVs found in exactly one sample) (Table S5D). We would expect a new African sample to add approximately twice the number of novel variants compared to a non-African sample. This trend holds whether we consider SVs within or outside of tandem repeats and SDs. We also modeled the reduction in shared variants we observed by adding each sample and note that African samples disproportionately reduce the shared variant set (Table S5E). The hydatidiform moles, CHM1 and CHM13, also disproportionately reduce the shared set, which might be expected considering they were some of the first samples sequenced.
We tested the effect of modifying the overlap percentage. With 90% RO, small variations in size or placement inflate the number of SVs in the merged callset (Table S5F). By decreasing it to any overlap (1 bp), the size of the callset shrinks significantly because variants are over-merged. With 50% RO, our genotyping results follow closely with the expected allele frequency based on the merged discovery set. Therefore, we selected 50% RO as an acceptable balance of these extremes in the manuscript.
Additionally, we permuted the sample order 50 times and created a merged set of SVs for each permutation. On average, our 50% RO callset of merged INS, DEL and INV differed from the 50 permutations by 260.4, 67.8, and 0.3 variants, respectively (Table S5G). The size of the SV callset we report did not differ significantly from the permuted size distribution (p > 0.05). The inconsistent loci (i.e., the complement set between the reported and permuted callset) contained a higher proportion of tandem repeats and retrotransposable elements than expected by approximately 8% and 12% for INS and DEL, respectively.
Variant distributions
A visual inspection of locations reveals that SVs are not distributed uniformly over the genome (Figure S4D). Both insertions and deletions are enriched in telomeres and SDs.
We grouped SVs into 500 kbp bins and note this enrichment within 5 Mbp of chromosome ends. Other than shared variants, which plot with low resolution because of their small number, the discovery class does not have a large effect on this trend (Figure S5A); however, the correlation with de novo variants does increase (Figure S5B).
We observe an expected size distribution for SVs with peaks for ALU (300 bp) and LINE (6–7 kbp) elements (Figures S5C and S5D). However, the size distribution by SV class differs with more common SVs affecting more bases than less conserved SVs (Table S5H). We also find that shared and major SV insertions are enriched in complex and mosaic Alu elements, and SV deletions are enriched in L1HS and AluY transposable elements (Table S5I).
The distribution of GC content shows a long tail toward low GC (Figure 3, main text), which is largely explainable by tandem repeats (Figure S5E). This effect appears to be stronger for insertions than deletions, which may indicate collapsed low-GC tandem repeats in GRCh38 or a bias toward low-GC expansions.
Tandem duplications
To estimate the number of our insertion calls that are tandem duplications, we took all inserted sequences and mapped them to GRCh38 with BLASR and calculated the distance from the SV insertion to its mapping location. We required that alignment regions match the SV insertion size by ± 10% and that the point of insertion be within 2x the SV length from the mapping location. From this analysis, we find that 16,787 (28.9% of 57,995 insertions) are possible tandem duplications. Of these, 12,120 (72.2%) are annotated as tandem repeats by TRF and 3,558 (21.2%) are not tandem repeats or known repeat classes (TRF and RepeatMasker). The remaining insertions (6.6%) are associated with interspersed repeats. These annotations are shared in Table S1.
Comparing GRC patches
We searched our alignments for loci with lost read coverage (depth ≤ 5 over 1 kbp or more) and found 4.1 Mbp where read mapping was especially low in one or more samples. We find that GRCh38.12 patches (fix and novel patches) intersect 1.1 Mbp (26.8%) of these loci, and this suggests that variation within patches may be affecting PacBio read alignments or that they contain large deletions.
Within these loci, we find 19,419 insertions and 13,990 deletions from our merged set indicating that long reads are capable of mapping and sequence resolving SVs in these variable loci. Furthermore, we called SVs from the patch sequences and find that 964 insertions and 433 deletions in the merged callset are consistent with variation in those patches, which accounts for 5.0% and 3.1% of the insertion and deletion variants in these patched loci.
Full-length transposons
We analyzed SV insertion and deletion calls for full-length transposable elements with RepeatMasker, including large variants with multiple repeat elements (Table S5J). These retrotransposons in missing sequence may be capable of de novo retrotransposition events. Complete annotations for each SV, including insertions and deletions, can be found in Table S1.
Viral integrations
We searched for potential viral integrations within our insertions against the RefSeq virus records and filtered for hits where 85% of the inserted sequence corresponded to virus. There were two unique hits among our SVs. The first (2,865 SVs) corresponded to a viral genome (BeAN 58058, NC_032111.1) where an AluY element is incorporated into the assembly (Wanzeller et al., 2017), and so AluY SVs were the basis for this homology. The virus may have acquired an AluY in an infected human (Piskurek and Okada, 2007), or it may be human contamination in the sequence assembly. The second hit (48 SVs) corresponded to an HERV element (NC_022518.1, Human endogenous retrovirus K113). We had already annotated these 48 inserts as HERV, LTR, or complex insertion polymorphisms (containing more than one repeat). We did not sequence assemble complete insertions corresponding to EBV, which was used during the transformation of some of the LCLs. Unmapped sequence reads corresponding to EBV, however, were recovered suggesting that the EBV insertions (which typically form larger multi-kbp tandem arrays) were not completely sequence resolved.
STR and VNTR distribution
We assessed levels of subtelomeric enrichment and association with double-strand breaks for both STRs and VNTRs. The subtelomeric abundances and their associations to double-strand breaks are shown in Figure S6A and Figure S6B for STRs and Figure S6C and Figure S6D for VNTRs. As shown in Table S5K, subtelomeric enrichment at a distance of 5 Mbp from the telomere ends was significantly higher for VNTRs (4.8-fold) compared to STRs (2.9-fold). The chromosome arm length differences did not explain the variation in VNTR subtelomeric enrichment, particularly in the p-arm (Figure S6D). We also observed that double-strand break density is significantly associated with this subtelomeric enrichment, particularly in the case of VNTRs (Figure S6C).
We annotated the repeat content of each SV sequence using TRF and RepeatMasker (v 4.0.7) and grouped the repeat-annotated SVs into STRs (repeat unit length < 7 bp), VNTRs (repeat unit length ≥ 7 bp), and interspersed retrotransposable element repeats. We employed two annotation approaches for the tandem- and interspersed-repeat discovery from our SV sequences: (i) we required a unique repeat label to be assigned to each SV record, and (ii) we allowed for designation of multiple repeat classes to a single SV sequence (e.g., if both STRs and VNTRs are contained within the SV sequence, they are both reported). For the first approach, we required ≥ 50% of the entire SV length to be composed of the specific repeat type, while prioritizing for interspersed repeats over tandem repeats. For example, if an insertion sequence was composed of ≥ 50% AluY elements and also ≥ 50% STRs, the entire SV was designated as an AluY. Otherwise, the entire SV was labeled as a non-repeat SV. We used the first annotation approach in the multiple regression modeling of the genome-wide SV density. For the second approach, we retained all the STR, VNTR and retrotransposable element annotations for all SV sequences. To avoid ambiguity, TRF annotation was conducted outside of the interspersed repeat elements sequence space. The second annotation approach was used when estimating the subtelomeric enrichments. The subtelomeric enrichment coefficient of different SV types was determined by comparing the observed relative abundance of each event class in the last 5 Mbp of a chromosome’s arm against the expectation. The expected null relative abundance was determined by assuming a uniform distribution of events across chromosome arms, resulting in approximately 11% expectation in the last 5 Mbp of the chromosome arms.
The genome-wide tracks of replication timing represented by the smoothed wavelet values were obtained from GRCh37 UCSC Genome Browser for the ENCODE LCL GM06990. The male- and female-specific genetic maps were obtained from the Icelandic study that fine-mapped meiotic recombination events using 15,257 parent–offspring pairs (Kong et al., 2010). The map of double-strand breaks was previously generated using chromatin immunoprecipitation sequencing of five male samples to map genomic locations of the PRDM9 protein, known to accumulate at sites of meiotic double-strand breaks (Pratto et al., 2014). The de novo SNV mutation rate was obtained from a previous study on Simons Simplex Collection autism quad families (Turner et al., 2017).
Using a multiple linear regression model, we modeled SV density as a function of the density of SDs, double-strand breaks, the averages of replication timing, sex-specific recombination rate and de novo mutation rate in windows of 500 kbp across GRCh38. We allowed for pairwise interactions between variables. The final model was:
where SVd represents the density of SVs, DNM is the de novo mutation rate, Rmale and Rfemale are the sex-specific recombination rates, DSB represents the double-strand breaks (Pratto et al., 2014), RT is the replication timing, and SD is segmental duplication density. The squared summation represents all the pairwise interaction terms, while the error term represents a random sampling from the normal distribution ~N(0,1) of the same sample size as the SV type under consideration. In order to (i) identify the simplest models containing only the most informative independent variables and (ii) account for potential colinearity between our interacting terms, we constructed parsimonious regression models for each SV type.
To identify the most parsimonious multiple regression models explaining our SV density variation, we updated our initial model in a stepwise fashion. To satisfy the principle of parsimony, we removed non-significant parameters from the model, prioritizing for pairwise interaction terms, followed by squared explanatory terms, and lastly non-significant single explanatory variables. The most parsimonious models for the four SV types were:
The total explained variance for each of the parsimonious models above were 33%, 21.5%, 18.5% and 30% for the distributions of VNTR, STR, non-repeat SVs and all SVs, respectively. We wish to note that these estimates should be viewed as upper bounds of explained variance, since the known colinearity between some explanatory terms, e.g., DSB and Rmale, may lead to inflated variance estimates.
SMRT-SV genotyper training set
Using CHM1, CHM13, and NA19240 variant calls and Illumina data from CHM1 and CHM13, a pseudodiploid sample was created to train and test the genotyping model. The Illumina data was emulated by merging reads from CHM1 and CHM13. The original sequence data for each has approximately 40-fold coverage in aligned BAMs of 151bp paired-end reads. An equal number of reads were randomly selected from CHM1 and CHM13 and then merged. Reads were selected by name so that all alignment records of the randomly selected read were retrieved. This process was repeated to create samples with fold coverages of 4, 5, 6, 7, 8, 10, 15, 20, 30, 40, 50, and 60.
A set of SV calls was constructed by merging SV calls generated with PacBio reads over CHM1, CHM13, and NA19240. To ensure quality data for building a model, densely packed SVs were identified and removed because a true genotype is difficult to determine for these SVs. Regions of dense SVs were identified by finding all 500 bp windows over the reference genome that contained four or more SVs in any one sample. That set of windows from all samples was merged and used to filter the SV calls for all three genomes. The filter covered 2.4 Mbp of the reference and removed 23% of the calls from all samples (4,682, 4,715, and 5,820 from CHM1, CHM13, and NA19240, respectively).
SMRT-SV variants discovered in CHM1 and CHM13 were merged and annotated by expected genotype. Homozygous alternate (HOM-ALT) variants were determined by using a 50% RO between CHM1 and CHM13. HOM-ALT variants were merged by taking 100 SVs from each sample in turn (100 from CHM1, then 100 from CHM13, then 100 from CHM1, etc.). Heterozygous (HET) variants were found by taking a 500 bp window around each variant in one sample and using it to filter variants in the other so that HET variants are not within 500 bp of a variant in the other mole. To avoid overrepresenting HET calls, these variants were merged by taking 50% of the HET variants from each sample. Lastly, homozygous reference (HOM-REF) variants were found by using the same 500 bp windows around variants in CHM1 and CHM13 to filter calls in NA19240 and including variants not in CHM1 or CHM13. The result is a balanced set of SV calls with confident genotype annotations for all genotype classes.
SMRT-SV genotyper feature extraction
The SMRT-SV genotyper attempts to genotype insertion and deletion SVs in a sample given Illumina sequence data for the sample and a set of known sequence-resolved SVs. Each SV must be located on a contig and have known breakpoints on the reference and the contig. Using the reference, contigs, and breakpoints, the genotyper searches for patterns to determine if the SV is in the sample and whether it is heterozygous or homozygous.
A reference is constructed using the primary contigs from the same reference SVs were called against and adding contigs containing with SVs. A reference FASTA is constructed from these assemblies and indexed with SAMtools faidx (1.4) (Li et al., 2009). A reference “.alt” file is constructed by extracting SAM records from a BAM file of contigs aligned to the reference. To save space, the sequence and sequence quality fields are removed (replaced with “*”) and optional tags are removed. These fields are not needed by the aligner.
Reads are extracted from sample BAMs containing Illumina reads using a combination of SAMtools collate, SAMtools bam2fq, and Seqtk dropse, to produce a FASTQ file of the reads, and the reads are piped into BWA-MEM.7.15 (from bwa-kit 0.7.15) to be aligned against the reference with alternates. Alignments are then processed with bwa-postalt.js (bwa-kit 0.7.15) to copy alignments to alternate contigs and adjust quality scores; bwa-postalt.js sometimes outputs garbage lines with “NaN,” and these were removed. Low-quality alignments with a MAPQ of less than 20 are filtered out, and records that do not map near an SV or SV breakpoint are removed to reduce the size of the alignment output. The remaining alignments are sorted, saved as a CRAM file, and indexed.
For each variant call, a set of 15 features is extracted from the sample alignment to the reference with SV contigs. Features include the SV type (INS or DEL), the number of bases inserted or deleted, and 13 other features related to read alignments and include read-depth, split-read, and paired-end features on SV breakpoints, in SV sequences, and regions near SV breakpoints (Table S5L). This set of features is extracted for each pseudodiploid Illumina sample (4- to 60-fold).
Since a machine learning model is trained on this data, features are engineered to avoid over-training on sample-specific characteristics such as read depth. Instead, relative proportions are used (e.g., proportion of clipped reads instead of the total number of clipped reads). The feature table itself does include absolute read counts over breakpoints so that no-call criteria may be applied later, but the model ignores those attributes.
SMRT-SV genotyper model training
Features from one pseudodiploid sample, such as 30-fold, are selected for training the model. No-call criteria are applied to the features by choosing a no-call cutoff based on the read depths over reference and alternate breakpoints (summed) and removing features with depth less than the cutoff value. The model is then trained with the remaining features.
A stratified eightfold cross-validation method was applied to test the model training process and to choose parameters for each model. First, the set of SVs with known genotypes is partitioned into eight parts stratifying over SV type (INS or DEL) and genotype call (HOM-REF, HET, and HOM-ALT) so that the proportion of SVs and genotype calls is preserved in each partition. A model is trained on seven out of eight partitions and tested against the held-out data. This process is iterated eight times so that each partition is the hold-out set once. Test statistics are averaged, and the genotype results from each partition are merged into one complete set with genotype calls.
A machine learning model was trained using Python (version 3.6.2) and scikit-learn (version 0.19.0). Training features are first scaled to unit variance and to center the mean at 0 (sklearn.preprocessing.StandardScaler), and the scaler object is saved so that the same transformation can be applied to future data.
A support vector machine (SVM) with a radial basis function (RBF, aka “Gaussian”) kernel is trained on the training set of data. The training data, which is 7/8 of the full dataset (see above), was partitioned again using the same stratified eightfold strategy as described above. This round of cross-validation was used to tune hyper-parameters controlling the width of the RBF kernel (gamma) and the error tolerance of the SVM model (C) with a grid-search over all combinations of parameters (gamma in [1 × 10−1, 5.5 × 10−2, 1 × 10−2, 5.5 × 10−3, 1 × 10−3] and C in [10, 100, 500, 1000, 5000, 5500]). Attempts to further refine these parameters yielded only marginal improvements of accuracy (< 1%). Each combination of parameters trains a model, the model is tested on the test set, and the training is repeated on all eight hold-out sets. The average is merged, and the best parameters are used to train the final model.
Eight models were generated and tested, and the accuracy of the training process was estimated. A final model was then generated using the same eightfold cross-validation process to tune hyper-parameters.
SMRT-SV genotyper model selection
We trained 36 models using a variety of pseudodiploid coverages and no-call cutoffs and tested their accuracy on 30-fold and 6-fold (Figure S7A) sequence data using a variety of no-call cutoff values. For 30-fold genomes, we chose a model trained on the 30-fold pseudodiploid with a no-call cutoff of 4 for training, and we applied the model to data with a no-call cutoff of 8. In this model, we find it is beneficial to allow for more noise in the training data by using a lower no-call cutoff than we allow for genotype samples. For 6-fold data, we chose a model trained on the 8-fold pseudodiploid with a no-call cutoff of 2, and we applied it to data with a no-call cutoff of 2. We found that models generated on 8-fold generalized much better than models trained on 6-fold, and the accuracy of this model is better for low-coverage data than the 30-fold model.
Using cross-validation, we estimate genotype accuracy of 79.9% for insertions, which grows to 91.3% outside repetitive loci. For deletions, we observe an accuracy of 84.2% at all loci and 95.3% outside repetitive loci (Table S5M). Insertions have been historically difficult to detect or genotype with short-read data, and accuracy can be greatly improved by using sequence-resolved variants to guide genotyping.
Since machine learning models are prone to overfitting, we tested how well the model generalizes with varying read depths. Accuracy of the 30-fold model increases monotonically with read depth, and so it may be generally applied to high-coverage samples (Figure S7B). The 6-fold model has a higher accuracy for low-coverage samples; however, it peaks at 15-fold and declines with greater read depth. We also examined misclassified genotype calls, and as expected, most of these errors involve misclassifying homozygous as heterozygous calls (or the inverse), which is lower for the 30-fold model than the 6-fold model (Figure S7C). Very few homozygous references were called homozygous alternate (or the inverse).
Genotyping population samples
Insertion and deletion SVs (99,382) in each original sample were genotyped using Illumina WGS obtained from 174 samples from the 1KG Phase 3 (The 1000 Genomes Project Consortium et al., 2015), where 150 of those were released by the recent Polaris project (https://github.com/Illumina/Polaris) and 266 genomes sequenced as part of the Simons Genome Diversity Project or SGDP (Mallick et al., 2016; Sudmant et al., 2015b) (Table S5N). Global alignment depth estimation averaged 25-fold, 18-fold, and 15-fold for the 1KG, 1KG Polaris, and SGDP samples, respectively. These samples were genotyped with the high-coverage model.
We examined the proportion of the population samples each SV is callable in and note that there it is clearly multimodal around 0% and 100%, which is more extreme for SVs outside tandem repeats and SDs.
Outside of tandem repeats and SDs, which are known to confound short-read methods, we find a typical allele frequency distribution where density declines with allele frequency, but there is an enrichment for shared variants (Figure S7D). The proportion of heterozygous calls follows Hardy-Weinberg equilibrium, which is an indication that the genotyper is functioning as expected (Figure S7E).
Of insertion and deletion variants, genotyping supported the variant calls. For example, 92.9% of variants callable in all 440 samples were supported by at least one sample (Table S5O). Genotype frequency also supports discovery frequency (Figure 1D, Table S5P).
Expression quantitative trait loci
We asked whether the novel SVs we identified impact gene expression levels and splicing in a panel of LCLs derived from 376 European individuals from the GEUVADIS Consortium (Lappalainen et al., 2013b). To answer this question in the context of gene expression, we first quantified the association between gene expression levels and SVs within a 1 Mbp window, centered around the gene’s annotated transcription start site. We focused our analysis on 61,244 SVs for which we had genotype information from whole-genome sequencing data in at least 50 individuals at minor allele frequency (MAF) above 5%. After correcting for confounding effects and multiple testing, we identified 411 SV eQTLs at a false discovery rate (FDR) of 5%. We also utilized the same pipeline to identify LCL eQTLs within the set of single-nucleotide polymorphisms (SNPs) and small indels that were previously imputed by the GEUVADIS Consortium. This resulted in 3,317 SNP eQTLs at 5% FDR.
To identify SV eQTLs in LCLs derived from 376 European individuals from the GEUVADIS Consortium, we first obtained three genotype principal components (PCs) from the genotype matrix and ten PCs from the expression matrix. We then tested the association between gene expression levels, as measured by GEUVADIS, and genotypes of SVs within a 1 Mbp window centered around each gene’s annotated transcription start site, using fastQTL (Ongen et al., 2016) and the 13 PCs as covariates. As fastQTL computes association p value adjusted for multiple testing of variants, we obtained the calibrated (or adjusted) p values for all associations. We next used the Benjamini-Hochberg procedure to identify all SV-gene eQTL pairs at 5% FDR, which resulted in 411 SV eQTLs. The same approach was taken to identify SNP eQTLs using genotypes obtained from the GEUVADIS Consortium, which resulted in 3,014 SNP/indel eQTLs.
Next, we investigated whether variation in expression levels of specific genes may be caused by a subset of the SV eQTLs we identified. Because there is extensive linkage disequilibrium between SVs and SNPs, this question is not easily addressable. Nevertheless, we posited that if the association between an SV and the expression level of a gene remains significant after considering the effects of all nearby SNPs and indels, then there is suggestive evidence that the SV causally affects gene expression. To test this hypothesis, we quantified whether the association between the top SV eQTL and gene expression for every gene remained when we replaced the genotype of the top SV eQTL with the genotypes of all nearby SNPs or indels. More precisely, the genotypes of nearby SNPs or short indels were used as additional explanatory variables in our linear regression. After this conditioning step, we identified 34 genes for which SVs appear to be driving differences in gene expression levels. For 30 of these we found that the SV was more strongly associated with the gene expression level than any nearby SNP or indel (Table S5Q). It should be noted that many of these 34 eQTLs lack any signal from nearby SNPs, and for these, it is likely some are due to genotyping error rather than true SV eQTLs.
While only a small number of SVs may appear to affect gene expression levels, we note that our estimated genotyping error for SVs in low-coverage data is approximately 25%, which substantially reduces our ability to identify SVs as the lead variants. To obtain a better estimate of the number of SVs that affect gene expression levels, we identified 379 genes that had both a significant SNP eQTL and an SV eQTL at 5% FDR. We compared the association strength of the top SV eQTL to that of the lead SNP/indel eQTL with simulated genotyping errors and found that for 186 of 379 (49.1%) genes, the SV eQTL was more significantly associated with gene expression level than the SNP eQTL (Figure S7F). Thus, we estimate that up to one half of our SV eQTLs are likely to affect gene expression levels when we account for the high genotyping error rates for our SVs.
To simulate SNP genotyping errors representative of our real SV genotyping errors, we used our estimated SV genotyping errors for all SVs identified in this study. For each top SNP/indel eQTL, we replaced the genotype according to the empirical error distribution we computed (e.g., replaced a homozygous reference call with a heterozygous call or a homozygous reference call with probabilities 0.235 and 0.048, respectively). We used these updated genotypes to compute association to gene expression levels using linear regression.
We also quantified the loss of power due to genotyping errors and simulated genotyping errors for the top SNP eQTL for all 3,013 genes for which we identified eQTLs. We then asked whether we were still able to detect the top eQTL as significant when considering other SNP eQTLs, as we did for our SV eQTLs. We observed that only 43 of all 3,317 lead eQTLs (1.3%) remained significant. Thus, even if the lead SVs were the top most associated variants (i.e., the variants most likely to be causal) for all 411 genes with an SV eQTL, only about 1% (~4) would be detected using our approach. This analysis suggests that our approach is highly conservative and that more than 34 SV eQTLs are likely to be causal.
We note that 16/34 of these eQTLs show corresponding SNP support consistent with strong linkage disequilibrium between the SNPs and the SVs. Another four SV events showed some evidence of SNP support although not as strong. We further note that the majority (32/34) of the regions occur within or adjacent to large blocks of repetitive DNA or tandem duplications where there is a paucity of SNP calls. For example, HG00733_chr19–36349851-DEL-6321 is a large deletion surrounded by high-copy repeats that would confound SNP calling, but the genotype calls can be made with more confidence. This deletion contains a tandem-repeat locus and a full-length AluY element, which are both known to affect gene expression. HG02818_chr18–26167843-DEL-3517 deletes an entire SD flanked by both high-copy repeats and H3K4Me3, H3K27Ac, and DNase hypersensitivity sites.
Splicing quantitative trait loci
Similarly, to address this question in the context of splicing, we quantified the association between intron splicing levels and SVs, again focusing our analysis on the same 61,244 SVs for which we had genotype information. Previous work (Li et al., 2016) suggested that over 70% of common SNPs that affect RNA splicing do not affect gene expression levels. Thus, we reasoned that SVs may also affect RNA splicing without affecting gene expression levels. We performed splicing quantitative trait loci (sQTL) mapping using Leaf-Cutter quantifications (Li et al., 2018) of intron splicing. This resulted in 244 SV sQTLs affecting 197 genes and 3,119 SNP sQTLs affecting 2,123 genes, both at 5% FDR.
To identify sQTLs in the same GEUVADIS dataset as above, we used a similar approach to eQTL mapping. In brief, we used LeafCutter (Li et al., 2018) to identify and quantify annotated and novel splicing events by focusing on intron excision events. LeafCutter uses the junction reads that are captured from RNA-seq data, which are representative of intron splicing or intron excision events, to identify all possible junctions. To avoid read mapping biases, we removed all clusters harboring split reads that overlap common genetic variants (MAF ≥ 5%). This is a stringent filtering step to avoid false positive sQTL mapping using LeafCutter and removes about 30% of all clusters. We tested the association between intron splicing levels, as identified by LeafCutter, and genotypes of SVs, using fastQTL and five PCs as covariates. We then used the Benjamini-Hochberg procedure to identify all SV-sQTL pairs and SNP-sQTL pairs at 5% FDR, which resulted in 244 SV sQTLs and 3,119 SNP/indel sQTLs.
Comparing published variants
We compared our SV calls to several published callsets, including HGSVC (Chaisson et al., 2017), Huddleston 2017 (Huddleston et al., 2017), HX1 (Shi et al., 2016), AK1 (Seo et al., 2016), 1KG Phases 1 and 3 (1000 Genomes Project Consortium et al., 2012; Sudmant et al., 2015a), Simons Genome Diversity Project or SGDP (Sudmant et al., 2015b), Mills 2011 (Mills et al., 2011), Kidd 2010 (Kidd et al., 2010), Genome of the Netherlands or GoNL (Francioli et al., 2014, 2015), and dbVar (Lappalainen et al., 2013a).
We convert SVs from these studies into BED files, lift over variants to GRCh38 if they were called against another reference (UCSC liftOver tool), and apply the same pericentromeric filter applied to our SVs. For comparisons against a subset of the data, such as those outside tandem repeats and SDs, we apply the same filters to the published calls as applied to our discovery set. When making a comparison to multiple published sets, the sets are merged using the same 50% RO strategy used to merge discovery variants. dbVar contains many redundant variant calls, and so we merge this set on itself with a 50% RO strategy before merging with other sets or making comparisons. Since insertions cover a 1 bp breakpoint of the reference, we add the variant length to its end position to create BED records that can be intersected. With BEDTools 2.26.0 (Quinlan and Hall, 2010), variants in our discovery set are then compared to the equivalently merged and filtered published variants using a 50% RO strategy by SV type (e.g., insertions versus insertions, deletions versus deletions, etc.).
We compared our discovery set to Illumina studies (Francioli et al., 2014; 1000 Genomes Project Consortium et al., 2012; Mills et al., 2011; Sudmant et al., 2015a). With long-read sequencing of 15 genomes, we discovered an equivalent number of SVs (99,604 versus 89,927) as 2,740 population samples and 250 Dutch families (0.5% of the sample size) and observe 16.7% of the variants from those diverse large-scale studies (Figure S1A). As expected, short-read methods are less sensitive to insertions (Figure S1B), where we detect 173% more insertions, but they are more sensitive to deletions (Figure S1C), which can now be more accurately genotyped in Illumina-sequenced samples (SMRT-SV v2 genotyper).
Although SMRT-SV is limited in its ability to detect inversions, we note that given the number of human genomes in this manuscript versus 1KG Phase 3 (15 versus 2,504, < 1%) the yield of inversion calls is actually very good (222 versus 783, 28%). We find that 33 of our inversions are consistent with the 1KG callset yielding 189 new inversions as part of this study. Compared with HGSVC, 109 are new (95 against both HGSVC and 1KG Phase 3). Both platforms are biased to the detection of smaller inversions although the yield per genome is far greater for long-read data.
The HGSVC released variant calls on three of the same samples as our set (HG00514, HG00733, and NA19240), but with different sequence data at lower coverage. We merged our variant calls from these three samples and compared to HGSVC. Phasing in HGSVC improved sensitivity, but we expect that our set recovered additional variation due to its deeper coverage (Figure S1D).
We compared our discovery set to a large collection of population-based SVs from PacBio data (Chaisson et al., 2017; Huddleston et al., 2017; Seo et al., 2016; Shi et al., 2016), Illumina data (Francioli et al., 2014; 1000 Genomes Project Consortium et al., 2012; Mills et al., 2011; Sudmant et al., 2015a), a Sanger/BAC experiment (Kidd et al., 2010), and dbVar (Lappalainen et al., 2013a), which is a collection of SVs from multiple data types including sequencing and arrays (Figure S1E). Compared to this diverse collection, approximately half of our SV calls (40,654 of 99,604) are new and the majority of the remaining calls (58,950 of 99,604) are now sequence resolved for the first time.
SV validations
We prepared DNA from human BAC (CH17) and fosmid (ABC12) clones corresponding to the same genomes that had been WGS sequenced (CHM1 and NA12878, respectively) in order to estimate the SV validation rate (Huddleston et al., 2016; Kidd et al., 2008). We selected clones corresponding to specific SV sites, prepared SMRTbell libraries, and sequenced each clone to high depth (> 50-fold) using PacBio (Pacific Biosciences, Inc., Menlo Park, CA) RS II P6-C4 or Sequel v2.1 chemistry. Inserts were assembled using CANU v1.5 (Koren et al., 2017) and error corrected using Quiver or Arrow (Chin et al., 2013). We compared assembled inserts to sequence-resolved SVs using Miropeats (Parsons, 1995), BLASR (Chaisson and Tesler, 2012), and dot matrix analysis scripts.
We find that the number of bases between the SV contigs and the BAC differs by less than 10% in all samples with fewer differences in CH17 (CHM1 BAC library), which is likely attributable to its lack of allelic variation (Table S5R). If we compute by the number of mismatch events allowing many bases per event, we find that the contigs diverge by less than 1%.
In Huddleston 2017 (Huddleston et al., 2017), SVs were called against CHM1, and 214 random variants were selected for validation using PCR and Sanger sequencing. Of these, we found 12 shared variants from our study, and 10 of the 12 validated (83%). The two that failed to validate are insertions on chr12 (131 and 197 bp) that exactly match our calls by both location and size. We note that these variants were discovered in all three HGSVC samples (Chaisson et al., 2017), which used a different variant discovery method, and they both had PacBio and Illumina support. We assert these are more likely failures of validation than they are false SV calls or systematic discovery errors.
Reference integration
We tested the impact of long-read SVs on short-read mapping by integrating SV-containing contigs with the human genome reference. We first obtained primary contigs from GRCh38, alternate contigs from GRCh38 (ALT), and SMRT-SV contigs for all major, shared, and polymorphic variants in the merged callset from the first 10 samples in Table 1 (additional samples were later added to make the final callset). We merged these contigs into two references, one with SMRT-SV contigs (GRCh38-ALT-Ext) and one without SMRT-SV contigs (GRCh38-ALT). Both GRCh38 alternate and SMRT-SV contigs were marked as alternate contigs for ALT-aware mapping. We obtained 30 short-read samples from SGDP (Mallick et al., 2016) and applied a quality filter where all read pairs were removed if one end contained less than 70% base quality 20+. Filtered reads were mapped to the reference with and without SMRT-SV contigs using BWA-MEM (Li and Durbin, 2009) version 0.7.15 with default parameters. Finally, “bwa-postalt.js” (bwakit 0.7.15) was applied to copy reads to alternate contigs and adjust quality scores.
We then measured the effect of our contigs on read mapping by comparing alignments (GRCh38-ALT versus GRCh38-ALT-Ext) using two approaches. First, by comparing alignment flags in both alignments, we find that we recover 2.62% of unmapped reads (673,555 per sample) (Table S5S).
Second, we summarized the impact of extended contigs on mapping quality (MAPQ) by finding the highest quality value for each read pair mapped to one of the extended contigs and match it to the best MAPQ for the same read pair in the alignments without extended contigs and find considerable gains in MAPQ (Table S5T; see also main text).
We applied the GATK (McKenna et al., 2010) HaplotypeCaller to the alignments restricting variant calls to regions of SMRT-SV contigs containing an SV insertion and selected all variants with a minimum quality score of 20. From this, we identify 21,969 unique variants and 68,656 alternate alleles of those variants (Table S4).
QUANTIFICATION AND STATISTICAL ANALYSIS
GC content
The GC composition of each SV is determined by dividing the number of C and G bases in the sequence by the length of the SV. Over a given set of variants, we summarize GC composition by taking the mean of the composition or calculating the proportion of variants with an extreme GC composition using some threshold, such as < 30%, > 70%, or > 80%. Permutation tests were performed by randomly shuffling windows the size of each SV 1,000,000 times and calculating the same summary statistics. To obtain a relevant null distribution, any regions of the genome where variant calls were not considered were also removed from the permutation test. Summary statistics were calculated for each permutation and the p value calculated as the sum of permutations with an equal or more extreme statistic than the unpermuted statistic (e.g., a lower proportion of variants with < 30% GC). This test was repeated for non-repetitive SVs by removing both SVs and regions in SDs and tandem repeats using the UCSC tracks on build hg38 (GRCh38) to identify these loci. Additionally, any variant annotated as a tandem repeat by TRF or common repeat by RepeatMasker was also removed. Permutations and summary statistics were calculated as already described. These tests were also repeated for variant types (insertions or deletions) as necessary.
Subtelomeric enrichment
All the chromosome arms, with exception of the acrocentric arms 13p, 14p, 15p, 21p and 22p, were assessed for subtelomeric enrichment individually. The SV density was defined as the number of events from a certain SV class (i.e., STR, VNTR, interspersed repeat, and non-repeat SVs) in windows of 500 kbp. These counts were added cumulatively starting at the telomere and moving toward the centromere. See METHOD DETAILS for additional details.
Telomere distance
On each chromosome arm, variants were divided into 500 kbp bins ordered from the end of the chromosome to the centromere, and the number of variants in each bin was summed. The first 10 bins on each arm (outermost 5 Mbp of each arm) are compared to all other bins on the chromosome. The fold increase is calculated by dividing the mean in the first 10 bins by the mean of all other bins. A 1,000,000 round permutation test was performed by randomly exchanging the bin sums and calculating the fold increase for each permutation. The p value is calculated by dividing the sum of bins with a fold increase greater or equal to the unpermuted distribution. This analysis was performed on the set of all variants and the set of shared variants.
De novo correlation
The same bins used for telomere distance calculations were applied to SVs and de novo SNVs (500 kbp bins from telomere to centromere on each chromosome arm). For each bin, a sum for both SVs and SNVs was obtained creating a set of paired sums. Bins were intersected with SD and TRF (Benson, 1999) tracks obtained from the UCSC Genome Browser (build hg38) using BED-Tools (Quinlan and Hall, 2010). Because SNV calls are likely absent in repetitive regions, any bin with 20% SD or 20% TRF content was discarded for this analysis. A linear model was generated over the paired points using R (version 3.4.3) and its lm function with default parameters (QR decomposition). An F-test was performed using standard R functions and summary data from the linear model.
Modeling of SV density
Using a multiple linear regression model, we modeled SV density as a function of the density of SDs, double-strand breaks as well as the average values of replication timing, sex-specific recombination rate and de novo mutation rate in windows of 500 kbp across GRCh38. We modeled the full SV callset as well as each specific class of tandem repeats, interspersed repeats, and non-repeat SVs. We used R (version 3.4.3) and its lm function with default parameters. See METHOD DETAILS for additional information.
Expression quantitative trait loci
We tested for association between gene expression levels and genotypes of neighboring SVs (≤1 Mbp away from the transcription start site), using fastQTL (Ongen et al., 2016) and 13 principal components as covariates, using the GEUVADIS cohort of 376 European samples. As fastQTL computes the association p value adjusted for multiple testing of variants, we obtained the calibrated (or adjusted) p values for all associations. We next used the Benjamini-Hochberg procedure to identify all SV-gene eQTL pairs at 5% FDR. To test for the SV as a cause of expression changes, a linear regression was run again including SNPs and indels as explanatory variables.
Lastly, we tested the effect of genotyping error on SV eQTL analysis by correcting the association strength of the top SNP or indel eQTL for the same gene with the expected genotyping error rate of the SV, as estimated by the SMRT-SV genotyper. We replaced a homozygous reference call with a heterozygous call or a homozygous reference call with probabilities 0.235 and 0.048, respectively, and then we used the updated genotypes to recompute the SNP or indel eQTL. See METHOD DETAILS for additional information.
Splicing quantitative trait loci
We quantified the association between intron splicing levels and SVs by performing sQTL mapping with LeafCutter. To avoid read mapping biases, we removed all clusters harboring split reads that overlap common genetic variants (MAF ≥ 5%). We then used the Benjamini-Hochberg procedure to identify all SV-sQTL pairs and SNP-sQTL pairs at 5% FDR. See METHOD DETAILS for additional information.
DATA AND SOFTWARE AVAILABILITY
Long-read data used in this study can be obtained through the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) with accessions NCBI:PRJNA246220 (CHM1), NCBI:PRJNA269593 (CHM13), NCBI:PRJNA300843 (HG00514), NCBI:PRJNA300840 (HG00733), NCBI:PRJNA288807 (NA19240), NCBI:PRJNA339722 (HG02818), NCBI:PRJNA385272 (NA19434), NCBI:PRJNA339719 (HG01352), NCBI:PRJNA339726 (HG02059), NCBI:PRJNA323611 (NA12878), NCBI:PRJNA481794 (HG04217), NCBI:PRJNA480858 (HG02106), NCBI:PRJNA480712 (HG00268), NCBI:PRJNA298944 (AK1), and NCBI:PRJNA301527 (HX1).
SV-containing contigs generated by this study can be found on BioProject (https://www.ncbi.nlm.nih.gov/bioproject) with accession NCBI:PRJNA481779. SV calls are shared on dbVar (https://www.ncbi.nlm.nih.gov/dbvar) under accession dbVar:nstd162. We could not obtain permission from AK1 authors to submit contigs and SV calls to NCBI, and so these AK1 data are not available in BioProject or dbVar.
A patched GRCh38 reference including SV-containing contigs as alternates is available through EBI (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1). AK1 contigs and variants are also not available in these resources.
SMRT-SV v2 and the genotyper are available on GitHub (https://github.com/EichlerLab/smrtsv2).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| HG00514 | This paper | NCBI: PRJNA300843 |
| HG00733 | This paper | NCBI: PRJNA300840 |
| NA19240 | This paper | NCBI: PRJNA288807 |
| HG02818 | This paper | NCBI: PRJNA339722 |
| NA19434 | This paper | NCBI: PRJNA385272 |
| HG01352 | This paper | NCBI: PRJNA339719 |
| HG02059 | This paper | NCBI: PRJNA339726 |
| NA12878 | This paper | NCBI: PRJNA323611 |
| HG04217 | This paper | NCBI: PRJNA481794 |
| HG02106 | This paper | NCBI: PRJNA480858 |
| HG 00268 | This paper | NCBI: PRJNA480712 |
| SV calls (VCF) | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1, dbVar: nstd162 |
| SV contigs | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1NCBI: PRJN481779 |
| Patched reference as ALTs (20181025_EEE_SV-Pop.1) | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1 |
| Genotypes on 440 population samples | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1 |
| Recombinant DNA | ||
| CHM1 (BAC library CH17) | Steinberg et al., 2014 | AssemblyDB: GCA_000306695.2 |
| NA12878(FOSMID library ABC12) | Agencourt | https://ftp.ncbi.nih.gov/repository/clone/archive/Homo_sapiens ABC12 |
| Software and Algorithms | ||
| SMRT-SVv2 | This paper | https://github.com/paudano/smrtsv2 |
| SMRT-SV | Huddleston et. al.,2017 | https://github.com/EichlerLab/pacbio_variant_caller |
| karyoploteR | Gel and Serra, 2017 | http://bioconductor.org/packages/release/bioc/html/karyoploteR.html |
| BLASR 1.MC.rc43 | PacBio | https://github.com/mchaisso/blasr |
| BLASR 5.3 | PacBio | https://github.com/PacificBiosciences/blasr |
| BWA-kit 0.7.15 | Li et. al. 2009 | http://bio-bwa.sourceforge.net/ |
| SAMTools 1.4 | Li et. al. 2009 | http://samtools.sourceforge.net/ |
| BEDTools 2.26.0 | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| Other | ||
| CHM1 | Chaisson et. al., 2015a | NCBI: PRJNA246220 |
| CHM13 | Huddleston et. al.,2017 | NCBI: PRJNA269593 |
| AK1 | Seo et. al.,2016 | NCBI: PRJNA298944 |
| HX1 | Shi et. al., 2016 | NCBI: PRJNA301527 |
Highlights.
We sequence resolve and annotate 99,604 common human structural variants
55% of VNTRs map to the end of chromosomes and correlate with double-strand breaks
Alternate alleles facilitate accurate genotyping with short reads and new associations
We patch the reference and add diversity needed for developing a pan human genome
ACKNOWLEDGMENTS
We thank N. Arang, V. Dang, and A. Lewis for technical assistance and QC in generating sequencing data, A. Ly for helping with library construction and sequencing, J. R. Fitch for data management. We also thank Z. Kronenberg and P. Hsieh for population genetics advice. The authors thank J. Huddleston for assistance with troubleshooting and implementation of SMRT-SV and T. Brown for assistance in editing this manuscript. This work was supported, in part, by grants from the U.S. National Institutes of Health (NIH grants HG002385 to E.E.E.; HG007635 to R.K.W. and E.E.E.; HG009081 to S.K.D. and E.E.E.; HG003079 to R.K.W.; HG009478 to M.L.D.). S.C. was supported by a National Health and Medical Research Council (NHMRC) CJ Martin Biomedical Fellowship (#1073726). A.Sulovari was supported by the U.S. National Institutes of Health (T32 HG000035–23). E.E.E is an investigator of the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and five tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.12.019.
DECLARATION OF INTERESTS
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc.
REFERENCES
- 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA, Flicek P, et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, and Moran JV (2010). LINE-1 retrotransposition activity in human genomes. Cell 141, 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin C-S, Drake JP, Landolin JM, and Phillippy AM (2015). Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol 33, 623–630. [DOI] [PubMed] [Google Scholar]
- Brandt DYC, Aguiar VRC, Bitarello BD, Nunes K, Goudet J, and Meyer D (2015). Mapping Bias Overestimates Reference Allele Frequencies at the HLA Genes in the 1000 Genomes Project Phase I Data. G3 (Bethesda) 5, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, and Tesler G (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. (2015a). Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Wilson RK, and Eichler EE (2015b). Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet 16, 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez O, Guo L, Collins RL, et al. (2017). Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10.1038/s41467-018-08148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, and Gage FH (2009). L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, and Pritchard JK (2009). Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25, 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Clark RA, and She X(2004). An assessment of the sequence gaps: unfinished business in a finished human genome. Nat. Rev. Genet 5, 345–354. [DOI] [PubMed] [Google Scholar]
- Fan JB, Surti U, Taillon-Miller P, Hsie L, Kennedy GC, Hoffner L, Ryder T, Mutch DG, and Kwok PY (2002). Paternal origins of complete hydatidiform moles proven by whole genome single-nucleotide polymorphism haplotyping. Genomics 79, 58–62. [DOI] [PubMed] [Google Scholar]
- Francioli LC, Menelaou A, Pulit SL, Van Dijk F, Palamara PF, Elbers CC, Neerincx PBT, Ye K, Guryev V, Kloosterman WP, et al. ; Genome of the Netherlands Consortium (2014). Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet 46, 818–825. [DOI] [PubMed] [Google Scholar]
- Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, van Duijn CM, Swertz M, Wijmenga C, van Ommen G, et al. ; Genome of the Netherlands Consortium (2015). Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet 47, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Sirén J, Novak AM, Hickey G, Eizenga JM, Dawson ET, Jones W, Garg S, Markello C, Lin MF, et al. (2018). Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol 36, 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchel JR, and Zoghbi HY (2005). Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet 6, 743–755. [DOI] [PubMed] [Google Scholar]
- Gel B, and Serra E (2017). KaryoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Huddleston J, Chaisson MJP, Hill CM, Kronenberg ZN, Munson KM, Malig M, Raja A, Fiddes I, Hillier LW, et al. (2016). Long-read sequence assembly of the gorilla genome. Science 352, aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff JG, Thakur J, Kasinathan S, and Henikoff S (2015). A unique chromatin complex occupies young a-satellite arrays of human centromeres. Sci. Adv 1, e1400234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Chaisson MJ, Meltz Steinberg K, Warren W, Hoekzema K, Gordon DS, Graves-Lindsay TA, Munson KM, Kronenberg ZN, Vives L, et al. (2016). Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res, gr.214007.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, Graves-Lindsay TA, Munson KM, Kronenberg ZN, Vives L, et al. (2017). Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, and Lee C (2004). Detection of large-scale variation in the human genome. Nat. Genet 36, 949–951. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium, Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium (2004). Finishing the euchromatic sequence of the human genome. Nature 431, 931–945. [DOI] [PubMed] [Google Scholar]
- Iqbal Z, Caccamo M, Turner I, Flicek P, and McVean G (2012). De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet 44, 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. (2008). Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, and Eichler EE (2010). A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, et al. (2010). Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103. [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley R, Leung E, Morris C, Berg R, McDonald M, Weaver A, Parry DA, Ni J, Su J, Gentz R, et al. (1998). Identification of multiple forms of 180-kDa ribosome receptor in human cells. DNA Cell Biol. 17, 449–460. [DOI] [PubMed] [Google Scholar]
- Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M, Zhou G, et al. (2013a). DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 41, D936–D941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. ; Geuvadis Consortium (2013b). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, and Pritchard JK (2016). RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, and Pritchard JK (2018). Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet 50, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, and Trask BJ (2005). Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature 437, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke DP, Segraves R, Carbone L, Archidiacono N, Albertson DG, Pinkel D, and Eichler EE (2003). Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome Res. 13, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn A, Koehler KE, Judis L, Chan ER, Cherry JP, Schwartz S, Seftel A, Hunt PA, and Hassold TJ (2002). Covariation of synaptonemal complex length and mammalian meiotic exchange rates. Science 296, 2222–2225. [DOI] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, et al. (2016). The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hillier LW, Eichler EE, Zody MC, Jaffe DB, Yang SP, Enard W, Hellmann I, Lindblad-Toh K, Altheide TK, et al. ; Chimpanzee Sequencing and Analysis Consortium (2005). Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87. [DOI] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. ; 1000 Genomes Project (2011). Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N, Hickey G, Zerbino DR, Raney B, Earl D, Armstrong J, Haussler D, and Paten B (2014). Building a pangenome reference for a population. J. Comput. Biol 22, 387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak AM, Hickey G, Garrison E, Blum S, Connelly A, Dilthey A, Eizenga J, Elmohamed MS, Guthrie S, Kahles A, et al. (2017). Genome Graphs. bioRxiv. 10.1101/101378. [DOI] [Google Scholar]
- Ongen H, Buil A, Brown AA, Dermitzakis ET, and Delaneau O (2016). Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 32, 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JD (1995). Miropeats: graphical DNA sequence comparisons. Comput. Appl. Biosci 11, 615–619. [DOI] [PubMed] [Google Scholar]
- Paten B, Novak AM, Eizenga JM, and Garrison E (2017). Genome graphs and the evolution of genome inference. Genome Res. 27, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, and Rechavi G (2010). Adenosineto-inosine RNA editing shapes transcriptome diversity in primates. Proc. Natl. Acad. Sci. USA 107, 12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, Stütz AM, Stedman W, Anantharaman T, Hastie A, et al. (2015). Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskurek O, and Okada N (2007). Poxviruses as possible vectors for horizontal transfer of retroposons from reptiles to mammals. Proc. Natl. Acad. Sci 104, 12046–12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratto F, Brick K, Khil P, Smagulova F, Petukhova GV, and Camerini-Otero RD (2014). DNA recombination. Recombination initiation maps of individual human genomes. Science 346, 1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DAM, Rashid AJ, Colas D, Torre-ubieta L De, Zhu RP, Madison DV, Shamloo M, Butte AJ, Bonni A, and Josselyn SA (2012). FoxO6 regulates memory consolidation and synaptic function FoxO6 regulates memory consolidation and synaptic function. 2780–2801. [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. (2017). Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, et al. (2004). Large-scale copy number polymorphism in the human genome. Science 305, 525–528. [DOI] [PubMed] [Google Scholar]
- Seo J-S, Rhie A, Kim J, Lee S, Sohn M-H, Kim C-U, Hastie A, Cao H, Yun J-Y, Kim J, et al. (2016). De novo assembly and phasing of a Korean human genome. Nature 538, 243–247. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, and Sirotkin K (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Guo Y, Dong C, Huddleston J, Yang H, Han X, Fu A, Li Q, Li N, Gong S, et al. (2016). Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun 7, 12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Schneider VK, Graves-lindsay TA, Schneider VA, Robert S, Agarwala R, and Huddleston J (2014). Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res. 24, 2066–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, et al. ; 1000 Genomes Project Consortium (2015a). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, et al. (2015b). Global diversity, population stratification, and selection of human copy-number variation. Science 349, aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, Kronenberg ZN, Hormozdiari F, Raja A, Pennacchio LA, et al. (2017). Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. (2005). Fine-scale structural variation of the human genome. Nat. Genet 37, 727–732. [DOI] [PubMed] [Google Scholar]
- Vollger MR, Dishuck PC, Sorensen M, Welch AE, Dang V, Dougherty ML, Graves-Lindsay TA, Wilson RK, Chaisson MJP, and Eichler EE (2019). Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanzeller ALM, Souza ALP, Azevedo RSS, Sousa EC, Filho LCF, Oliveira RS, Lemos PS, Júnior JV, and Vasconcelos PFC (2017). Complete genome sequence of the BeAn 58058 virus isolated from Oryzomys sp. rodents in the Amazon Region of Brazil. Genome Announc. 5, 4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CT, Steinberg KM, Huddleston J, Warren RL, Malig M, Schein J, Willsey AJ, Joy JB, Scott JK, Graves TA, et al. (2013). Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am. J. Hum. Genet 92, 530–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Long-read data used in this study can be obtained through the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) with accessions NCBI:PRJNA246220 (CHM1), NCBI:PRJNA269593 (CHM13), NCBI:PRJNA300843 (HG00514), NCBI:PRJNA300840 (HG00733), NCBI:PRJNA288807 (NA19240), NCBI:PRJNA339722 (HG02818), NCBI:PRJNA385272 (NA19434), NCBI:PRJNA339719 (HG01352), NCBI:PRJNA339726 (HG02059), NCBI:PRJNA323611 (NA12878), NCBI:PRJNA481794 (HG04217), NCBI:PRJNA480858 (HG02106), NCBI:PRJNA480712 (HG00268), NCBI:PRJNA298944 (AK1), and NCBI:PRJNA301527 (HX1).
SV-containing contigs generated by this study can be found on BioProject (https://www.ncbi.nlm.nih.gov/bioproject) with accession NCBI:PRJNA481779. SV calls are shared on dbVar (https://www.ncbi.nlm.nih.gov/dbvar) under accession dbVar:nstd162. We could not obtain permission from AK1 authors to submit contigs and SV calls to NCBI, and so these AK1 data are not available in BioProject or dbVar.
A patched GRCh38 reference including SV-containing contigs as alternates is available through EBI (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/hgsv_sv_discovery/working/20181025_EEE_SV-Pop_1). AK1 contigs and variants are also not available in these resources.
SMRT-SV v2 and the genotyper are available on GitHub (https://github.com/EichlerLab/smrtsv2).