

Abstract
DNA damage occurs on exposure to genotoxic agents and during physiological DNA transactions. DNA double-strand breaks (DSBs) are particularly dangerous lesions that activate DNA damage response (DDR) kinases, leading to initiation of a canonical DDR (cDDR). This response includes activation of cell cycle checkpoints and engagement of pathways that repair the DNA DSBs to maintain genomic integrity. In adaptive immune cells, programmed DNA DSBs are generated at precise genomic locations during the assembly and diversification of lymphocyte antigen receptor genes. In innate immune cells, the production of genotoxic agents, such as reactive nitrogen molecules, in response to pathogens can also cause genomic DNA DSBs. These DSBs in adaptive and innate immune cells activate the cDDR. However, recent studies have demonstrated that they also activate non-canonical DDRs (ncDDRs) that regulate cell type-specific processes that are important for innate and adaptive immune responses. Here, we review these ncDDRs and discuss how they integrate with other signals during immune system development and function.
The development and function of innate and adaptive immune cells are regulated by diverse extracellular cues that activate a broad variety of cell surface receptors and intracellular cues emanating from cytosolic and nuclear events. Developing and mature lymphocytes generate programmed DNA double-strand breaks (DSBs) at specific locations within the genome as necessary intermediates of physiological DNA rearrangements, such as antigen receptor gene assembly by V(D)J recombination and immunoglobulin class switch recombination (CSR) (see below)1,2. In addition, non-programmed DNA DSBs can be generated throughout the genome of immune cells, for example, during transcription, DNA replication and by genotoxic agents produced to eradicate pathogens3–5. DNA DSBs generated in these different settings, and the responses they elicit, are emerging as important signalling events in regulating immune system development and function.
DNA DSBs are dangerous genomic lesions that initiate a conserved canonical DNA damage response (cDDR) in all cells4. The cDDR promotes DNA DSB repair through either non-homologous end joining (NHEJ) or homologous recombination4,6,7. NHEJ functions to rejoin broken DNA ends at all phases of the cell cycle and frequently does so imprecisely, with nucleotides gained or lost at the join6. By contrast, homologous recombination functions only in the S and G2 phases of the cell cycle using the sister chromatid as a template for precise repair7. The cDDR also activates check-points that prevent cells with DSBs from progressing through the cell cycle and ultimately kills cells with persistent DSBs that could otherwise be resolved aberrantly leading to chromosomal rearrangements and cellular transformation4. The G2–M check-point is regulated by serine/threonine-protein kinase CHK1, whereas the G1–S checkpoint is enforced by CHK2 and p53, which also triggers cell death if DSBs persist unrepaired4.
The cDDR is initiated by phosphoinositide 3-kinase-like serine threonine kinases that are activated by DSB sensor proteins, or protein complexes, once they have bound to DNA DSBs8. These kinases include ataxia telangiectasia mutated (ATM), DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and ataxia telangiectasia and RAD3 related (ATR)8. ATM and DNA-PKcs are activated by DNA DSBs at all phases of the cell cycle, whereas ATR is primarily activated by DNA ends generated during DNA replication. Upon binding to broken DNA ends, the MRE11–RAD50–NBS1 (also known as NBN) (MRN) complex activates ATM, the heterodimer of KU70 (also known as XRCC6) and KU80 (also known as XRCC5) activates DNA-PKcs, and ATRIP activates ATR8,9. Once activated, the DDR kinases phosphory-late proteins that function in the different arms of the cDDR. However, these kinases can phosphorylate many other proteins with broad cellular activities and no known cDDR functions10. This suggests that, in some settings, in response to DSBs, DDR kinases may initiate cell type-specific non-canonical DNA damage responses (ncDDRs) that regulate normal cellular functions unrelated to DNA DSB repair. Indeed, as discussed in this Review, recent studies have shown that activation of DDR kinases by DNA DSBs in immune cells has been co-opted to initiate a variety of ncDDRs that regulate cell type-specific processes that are required for the normal development and function of innate and adaptive immune responses.
The ncDDR in developing lymphocytes
All developing B and T cells make and repair DNA DSBs as they assemble antigen receptor genes through the process of V(D)J recombination11,12. The signals that initiate this highly ordered process leading to the generation of DSBs at antigen receptor loci are well defined and, once generated, these DSBs activate an ncDDR that regulates subsequent V(D)J recombination steps. In addition, this ncDDR activates pathways that are important for normal lymphocyte development. We focus our discussion on developing B cells, as the most is known about the function of the ncDDR to DSBs generated in these cells.
V(D)J recombination.
Lymphocyte antigen receptor genes include the immunoglobulin (Ig) heavy (H) chain genes and light (L) chain κ and λ genes that are expressed in B cells and the T cell receptor (TCR) β, α, γ and δ chain genes that are expressed in T cells11. The variable (second) exon of all these genes must be assembled during development from variable (V), joining (J) and, at some loci, diversity (D) gene segments through the process of V(D)J recombination11 (FIG. 1). V(D)J recombination is initiated when two recombining gene segments are brought together in a synaptic complex with the recombination-activating gene 1 (RAG1) and RAG2 proteins, which together form the RAG endonuclease1,13. RAG proteins introduce DNA DSBs at the border of the two gene segments and the flanking recombination signal sequences (RSSs) that are recognized by RAG proteins. Each RAG-mediated DSB has a hairpin-sealed coding DNA end and blunt signal DNA end. RAG DSBs are generated in developing lymphocytes only during the G1 phase of the cell cycle and must be repaired by NHEJ, which joins the two coding ends to form a coding join and the two signal ends to form a signal join12. Signal joining is fairly precise, whereas coding join formation is imprecise, with a gain and loss of nucleotides. As a result, only one-third of the completed coding joins will be in-frame (productive) rearrangements and encode an antigen receptor chain. The generation of RAG DSBs leads to the activation of ATM and DNA-PKcs, which initiate the cDDR and are required for RAG DSB repair12,14. DNA-PKcs promotes Artemis endonuclease activity in opening hairpin-sealed coding ends so they can be joined, and ATM maintains the stability of RAG DSBs in post-cleavage complexes until the DNA ends are joined15,16. In response to RAG DSBs in developing lymphocytes, ATM also activates ncDDR programmes that regulate antigen receptor gene assembly and several pathways that are important for normal lymphocyte development (see below). It is conceivable that DNA-PKcs also participates in the activation of ncDDRs, but this has yet to be established.
Fig. 1:|. V(D)J recombination.
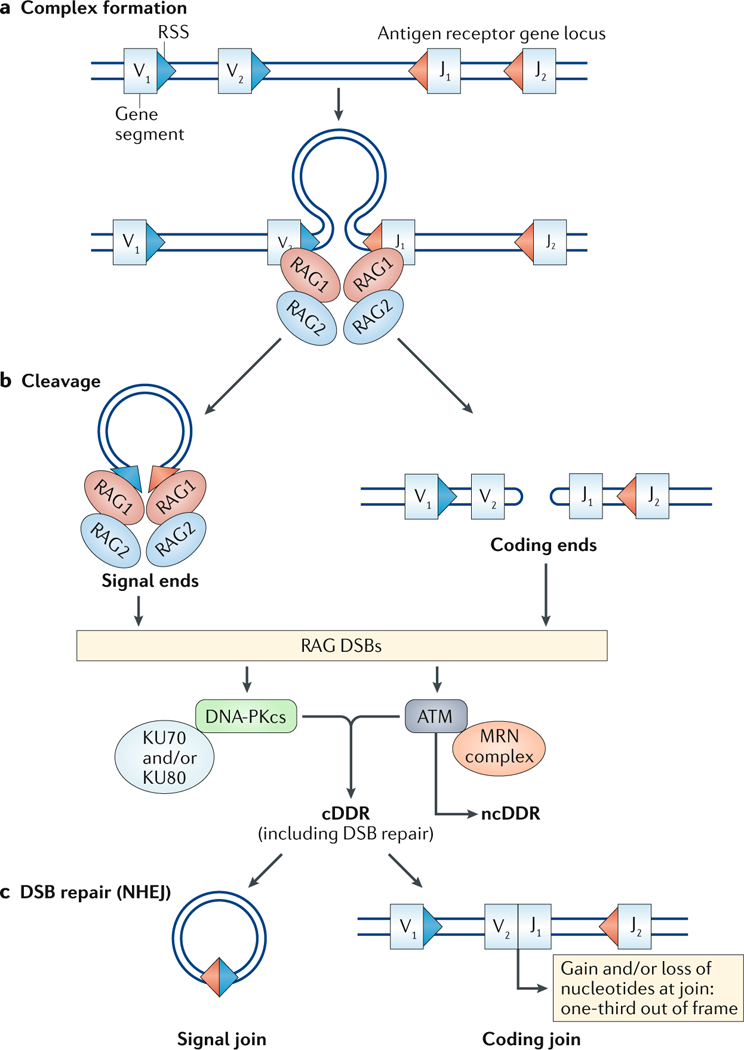
aThe initial step of the V(D)J recombination reaction is the synapsis of two gene segments (variable (V) to joining (J) recombination is shown) and their flanking recombination signal sequences (RSSs; triangles) with recombination-activating gene 1 (RAG1) and RAG2 proteins, which recognize the RSSs. b | DNA cleavage by RAG proteins leads to the generation of a blunt signal end and a hairpin-sealed coding end at each RAG-mediated DNA double-strand break (DSB). c | The two coding ends are then joined to form a coding join that completes the second exon of the antigen receptor gene. This joining is imprecise, with a gain and/or loss of nucleotides at the join leading to only one-third of completed antigen receptor genes being in-frame and productive. The signal ends are joined somewhat precisely to form a signal join. Binding of KU70, KU80 and MRE11–RAD50–NBS1 (MRN) to RAG DSBs activates the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and ataxia telangiectasia mutated (ATM) kinases, respectively. ATM and DNA-PKcs initiate a canonical DNA damage response (cDDR) to promote RAG DSB repair. ATM also activates non-canonical DNA damage response (ncDDR) signalling, which regulates B cell developmental programmes. NHEJ, non-homologous end joining.
B cell development.
B cell development proceeds through a series of discrete stages in the bone marrow, with developmental transitions orchestrated by diverse sets of signals, including ncDDRs initiated by RAG DSBs generated during lymphocyte antigen receptor gene assembly at multiple stages of B cell development17 (FIG. 2). RAG DSBs are generated in pro-B cells during Igh chain gene assembly, and a productive Igh rearrangement leads to expression of an IgH chain as a pre-B cell receptor (pre-BCR) that signals transition to the pre-B cell stage of development11,17,18. In pre-B cells, RAG DSBs are generated during Igl chain gene (Iglk and Igll) assembly, and a productive rearrangement encoding an IgL chain that pairs with the IgH chain to form a B cell receptor (BCR) drives transition to the immature B cell stage. Antigen receptor gene assembly ceases in immature B cells unless the BCR is autoreactive, in which case RAG DSBs can be generated during rearrangements that are initiated to replace the IgL chain of the auto-reactive BCR through a process termed receptor editing (see below)17,19. In response to BCR activation, mature B cells undergo CSR, leading to the generation of DNA DSBs in the constant region of the Igh locus (see below)20 (FIG. 2). Accordingly, DNA DSBs at antigen receptor loci are generated in all pro-B cells, all pre-B cells, some immature B cells and all mature B cells once they are activated (FIG. 2). Thus, ncDDRs initiated in response to these DSBs could regulate diverse processes that are required for B cell development and function. Similarly, T cells develop through pro-T and pre-T cell stages in which the TCR chain genes are assembled and likely also activate ncDDRs that regulate developmental processes, although much less is known about these responses21.
Fig. 2:|. B cell development.
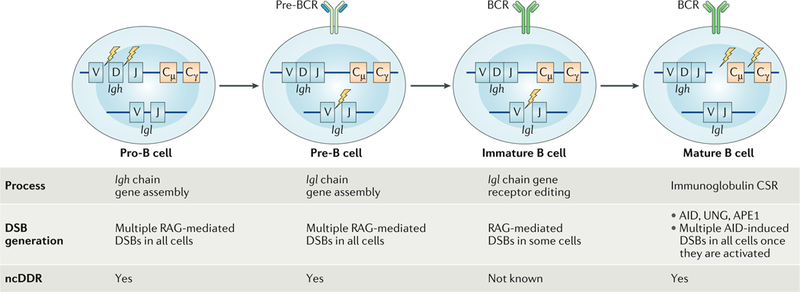
B cell development proceeds in the bone marrow through several discrete steps. From a standpoint of DNA double-strand break (DSB) generation and potential non-canonical DNA damage response (ncDDR) signalling, these stages can be generally divided into the pro-B cell stage in which Igh gene assembly occurs in all cells, the pre-B cell stage in which Igh gene assembly occurs in all cells and the immature B cell stage in which receptor editing occurs in some cells. DNA DSBs are also generated in mature B cells once they are activated and initiate class switch recombination (CSR). AID, activation-induced deaminase; APE1, apurinic/apyrimidinic endonuclease 1; BCR, B cell receptor; RAG, recombination-activating gene; UNG, uracil-DNA glycosylase.
Pro-B cells require IL-7 receptor (IL-7R) signalling through AKT, Janus kinases (JAKs) and signal transducer and activator of transcription 5 (STAT5) to promote proliferation, survival and transition to the large pre-B cell stage18 (FIG. 3a). Igl chain genes are assembled at the pre-B cell stage; however, IL-7R signals antagonize Igl chain gene assembly through STAT5-dependent suppression of Iglk chain germline gene transcription, which prevents accessibility of the Iglk locus to RAG11,22,23 (FIG. 3a). Furthermore, activation of AKT by IL-7R leads to phosphorylation and degradation of the transcription factor forkhead box protein O1 (FOXO1), which is required for RAG gene expression24,25 (FIG. 3a). STAT5 induces expression of the PIM family kinase PIM1, which along with AKT drives proliferation and promotes survival of large pre-B cells, in part, by antagonizing BAX-mediated cell death through phosphorylation of BCL-2-associated agonist of cell death (BAD)18,26–28 (FIG. 3a). These proliferative signals also antagonize Igl chain gene assembly, as V(D)J recombination is restricted to G1 phase cells14.
Fig. 3:|. Large pre-B cell to small pre-B cell transition.a.
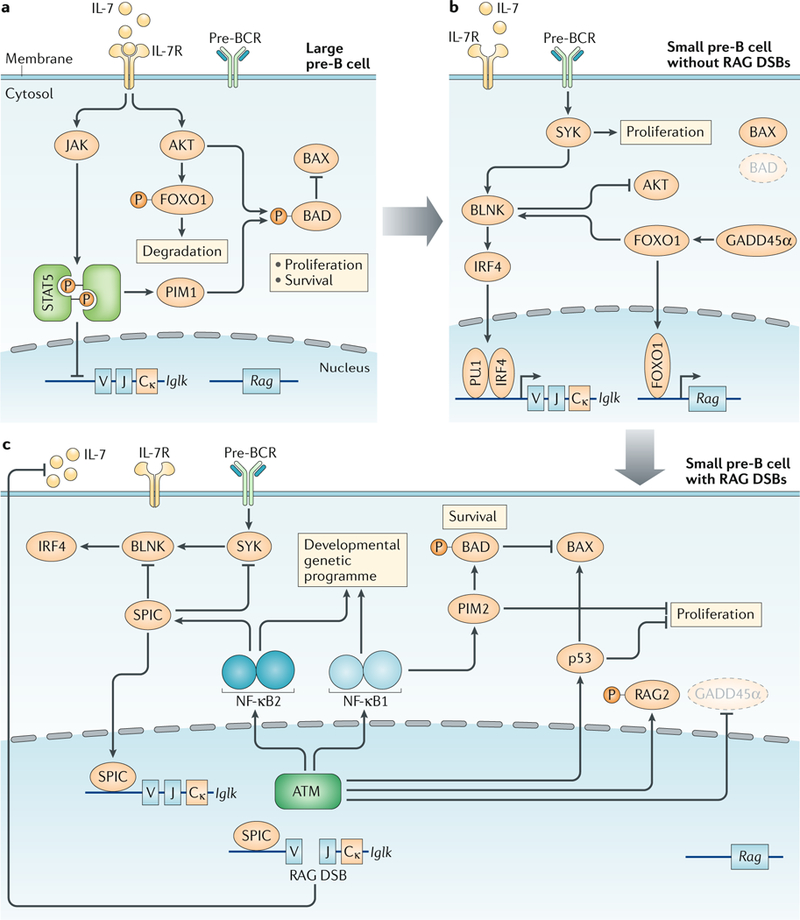
In large pre-B cells, IL-7 receptor (IL-7R) signalling activates AKT and the Janus kinase (JAK)–signal transducer and activator of transcription 5 (STAT5) pathway. This inhibits Iglk gene transcription and accessibility, inhibits recombination-activating gene (RAG) expression by causing degradation of forkhead box protein O1 (FOXO1) and promotes proliferation and survival of large pre-B cells, in part, through inhibition of the apoptosis regulator BAX by PIM1-mediated and AKT-mediated phosphorylation of BCL-2-associated agonist of cell death (BAD). b | In small pre-B cells, loss of IL-7R signalling leads to increased activity of tyrosine protein kinase SYK and B cell linker protein (BLNK), which augments signalling through the pre-B cell receptor (pre-BCR), leading to the induction of interferon regulatory factor 4 (IRF4) expression and induction of Iglk gene transcription and accessibility. Loss of AKT signals results in FOXO1-driven expression of Rag1 and Rag2 and loss of IL-7R-mediated proliferative and survival signals. Pre-BCR signals can also drive proliferation, although not as strongly as IL-7R signals. c | The initiation of Iglk gene rearrangement and the generation of RAG double-strand breaks (DSBs) leads to the activation of ataxia telangiectasia mutated (ATM), which initiates both a canonical DNA damage response (cDDR) and a non-canonical DNA damage response (ncDDR). The ncDDR is mediated, in part, by the activation of a nuclear factor-κB1 (NF-κB1)-dependent and NF-κB2-dependent genetic programme. The induction of SPIC represses Iglk gene transcription and antagonizes pre-BCR signals by inhibiting SYK and BLNK expression. This ncDDR genetic programme also promotes survival through the induction of PIM2, which phosphorylates BAD to inhibit BAX. p53 and PIM2 also enforce the G1 to S checkpoint. ATM activation leads to reduced Rag expression through inhibition of growth arrest and DNA damage-inducible protein GADD45α expression and through direct phosphorylation and inhibition of RAG2. RAG DSBs also trigger an ncDDR that inhibits the production of IL-7 by local stromal cells. Collectively, these ncDDR pathways prevent the generation of additional RAG DSBs, promote survival and block proliferation in cells with existing RAG DSBs. P, phosphorylation.
IL-7R signals in large pre-B cells must be attenuated to promote transition to the small pre-B cell stage of development, halt proliferation and promote induction of RAG1 and RAG2 expression, which depends on growth arrest and DNA damage-inducible protein GADD45α and FOXO1 (REFS18,24,25) (FIG. 3b). Furthermore, FOXO1 promotes expression of B cell linker protein (BLNK), which antagonizes AKT signalling and permits pre-BCR signalling through the tyrosine protein kinase SYK18,25. Pre-BCR signals induce expression of the transcription factor interferon regulatory factor 4 (IRF4), which along with PU.1 promotes germline Iglk chain gene transcription and accessibility to RAG, allowing for Iglk chain gene assembly18,29 (FIG. 3b). In small pre-B cells, the lost pro-survival signals following attenuation of IL-7R signals need to be replaced if these cells are to survive for several days as they undergo multiple rearrangements in an attempt to generate a productive Igl chain gene (see below)18,30.
IL-7R signalling is attenuated through the repositioning of pre-B cells to regions of the bone marrow that are devoid of IL-7-producing mesenchymal stromal cells29,31. Bone marrow stromal cells may produce either CXC-chemokine ligand 12 (CXCL12) or IL-7, with the expression of CXC-chemokine receptor 4 (CXCR4), the receptor for CXCL12, on large pre-B cells triggering migration away from IL-7-producing mesenchymal stromal cells and towards CXCL12-producing mesenchymal stromal cells29,32. However, recent studies have demonstrated that CXCL12 and IL-7 can be made by the same stromal cells and that pre-BCR-driven reduction in focal adhesion kinase (FAK; also known as FADK1)-mediated and α4β1 integrin-mediated adhesion to vascular cell adhesion protein 1 (VCAM1) leads to detachment and diffusion of large pre-B cells away from mesenchymal stromal cells that produce both IL-7 and CXCL12 (REF.31).
ncDDR in pre-B cells.
Once Iglk chain gene assembly is initiated, RAG DSBs activate ATM, leading to the cDDR and an ncDDR that includes the induction of a broadly functional genetic programme regulated, in part, by nuclear factor-κB1 (NF-κB1) and NF-κB2 (REFS21,33,34) (FIG. 3c). The activation of NF-κB1 in response to RAG DSBs likely occurs through the phosphorylation of NF-κB essential modulator (NEMO) in the nucleus by ATM, as occurs in response to genotoxic DSBs35,36. This genetic programme may further reinforce the extinction of IL-7R signals. In this regard, in small pre-B cells, NF-κB1 activation in response to RAG DSBs induces the expression of CD69, L-selectin (also known as CD62L) and switch-associated protein 70 (SWAP70)21. These proteins have functions in cell migration and localization that could promote the residence of small pre-B cells in regions of the bone marrow that are devoid of IL-7-producing stromal cells37–39. Moreover, RAG DSB signals in pre-B cells inhibit IL-7 production by neighbouring stromal cells31 (FIG. 3c). This intriguing ncDDR-mediated intercellular crosstalk between pre-B cells and stromal cells would be important not only for regulating Igl chain gene rearrangement but also for preserving genome stability by preventing pre-B cells with RAG DSBs from being driven into cycle by IL-7R signals.
Pre-B cell survival.
In small pre-B cells, IL-7R-mediated pro-survival signals are lost, and RAG DSBs activate a cDDR that involves p53, which can promote cell death, in part, through induction of BAX34 (FIG. 3c). However, the ncDDR activated by RAG DSBs promotes survival through the NF-κB1-dependent induction of PIM2, a kinase in the same family as PIM1 (REFS21,27,40) (FIG. 3c). Like PIM1 in large pre-B cells, PIM2 promotes survival in small pre-B cells through phosphorylation of BAD27,41 (FIG. 3c). Indeed, Pim2−/− mice exhibit a decrease in the fraction of Igll-expressing B cells, which is consistent with a survival defect in these cells, as Igll rearrangements usually occur after several failed rounds of Iglk chain gene assembly17,42.
The pro-survival effects from RAG DSB-mediated induction of the ncDDR may be important for permitting multiple Igl chain gene rearrangements in a single pre-B cell (see below). However, when DSBs persist unrepaired, the cDDR must promote cell death rather than allowing these DSBs the opportunity to be aberrantly resolved. Indeed, mice deficient in p53 and the NHEJ factors required for RAG DSB repair invariably develop B cell tumours with translocations formed by misrepaired RAG DSBs34,43,44. Thus, a balance must be maintained between ncDDR survival signals that provide a critical time window for Igl chain gene assembly and the initiation of cDDR pathways that promote the death of cells with persistent unrepaired RAG DSBs that could lead to genome instability and cellular transformation.
G1 phase arrest in pre-B cells.
Antigen receptor gene assembly occurs in developing lymphocytes at the G1 phase of the cell cycle. The transition of pre-B cells from G1 phase into S phase limits RAG activity through the degradation of RAG2 and could also promote the aberrant resolution of unrepaired RAG DSBs11,14,45. In response to RAG DSBs, the cDDR activates p53 and likely also CHK2, which enforce the G1–S checkpoint4,34,46 (FIG. 3c). However, in pre-B cells, which can receive proliferative signals from both IL-7R and the pre-BCR, the ncDDR is required to fully enforce the G1–S checkpoint27,33. In this regard, unlike PIM1, which promotes proliferation downstream of IL-7R, PIM2, which is induced by the ncDDR to RAG DSBs, promotes G1 arrest in pre-B cells by inhibiting IL-7-driven proliferation27,28. Indeed, in the presence of an intact cDDR, PIM2-deficient pre-B cells with RAG DSBs will enter S phase27.
Pre-BCR signalling through SYK and BLNK can drive pre-B cell proliferation in the absence of IL-7R signals47,48. The ncDDR to RAG DSBs in pre-B cells leads to the induction of NF-κB2, which initiates a genetic programme including SPIC, a member of the PU-box binding ETS family transcription factors that is expressed primarily in B cells and macrophages49–51 (FIG. 3c). Unlike the PU.1 and SPIB family members that are transcriptional activators required for B cell development, SPIC has a limited ability to activate transcription and may function primarily as a transcriptional repressor by competitively inhibiting PU.1 and SPIB binding to PU-box sites52,53. Indeed, in pre-B cells with RAG DSBs, SPIC binds to the promoters of both Syk and Blnk, with a concomitant decrease in PU.1 binding and transcription of these genes33,53,54. In addition to inhibiting the transcription of Syk and Blnk, ncDDR-dependent post-translational regulatory pathways could be responsible for the rapid downregulation of SYK and BLNK protein in response to RAG DSBs33 (FIG. 3c). The importance of this pathway in maintaining the G1–S checkpoint is evidenced by the ectopic expression of SYK in pre-B cells with unrepaired RAG DSBs, which drives these cells from G1 into S phase33. Thus, the ncDDR to RAG DSBs in small pre-B cells enforces the G1–S checkpoint by blocking both IL-7R and pre-BCR signals through the induction of PIM2 and SPIC, respectively (FIG. 3c).
The molecular components of the ncDDR to RAG DSBs have been best established in pre-B cells. We expect that ncDDR pathways also function in pro-B cells and developing T cells that are assembling antigen receptor genes but that these pathways may have distinct molecular components. Indeed, SPIC is not expressed in developing thymocytes49. In thymocytes, the ATM-dependent activation of the p38 mitogen-activated protein kinase (MAPK) in response to genotoxic DSBs and RAG DSBs leads to phosphorylation and inactivation of glycogen synthase kinase 3β (GSK3β), a constitutively active kinase that is known to promote cell death55. Moreover, the induction of DSBs in thymocytes by irradiation causes an ATM-independent reduction in cyclin D3, blunting proliferation56. Clearly, our understanding of the ncDDR pathways activated by RAG DSBs and their functions at different stages of B and T cell development remains incomplete.
Ordering antigen receptor gene assembly.
Most lymphocytes express a single heterodimeric antigen receptor, with cells generating only a single in-frame rearrangement for each antigen receptor chain. This occurs through the process termed allelic exclusion, which requires that rearrangements be initiated on one allele at a time and that newly completed antigen receptor genes have time to be tested to determine whether they are in-frame before another rearrangement is initiated on the alternative allele11,57. To achieve allelic exclusion, antigen receptor gene assembly is inter-allelically ordered, which depends on the differential accessibility of the two antigen receptor alleles so that RAG cleavage is initiated on a single allele58–60 (FIG. 4). However, inter-allelic ordering also depends on activation of the ncDDR by the newly generated RAG DSBs, which prevents additional RAG DSBs from being generated on the alternative allele (FIG. 4). Indeed, ATM-deficient B cells, which cannot initiate an ncDDR to RAG DSBs, exhibit a higher frequency of simultaneous RAG-mediated cleavage events at both Igh alleles (pro-B cells) and both Igl alleles (pre-B cells) than wild-type B cells61. In pre-B cells with NHEJ deficiencies, RAG DSBs are generated but not repaired, leading to persistent activation of ATM and the ncDDR. In these pre-B cells, RAG DSBs are found at approximately half of the Iglk alleles, which is consistent with RAG cleavage at a single allele in each cell62. However, loss of ATM or inhibition of ATM kinase activity in NHEJ-deficient pre-B cells leads to RAG cleavage at both Iglk alleles62. These findings are consistent with the notion that activation of ATM, and the ncDDR, by RAG DSBs functions to enforce allelic exclusion by limiting RAG cleavage to one Iglk allele at a time in pre-B cells. In agreement, developing B and T cells from ATM-deficient mice exhibit a partial loss of allelic exclusion62,63.
Fig. 4:|. ordered assembly of antigen receptor genes.

Antigen receptor gene assembly at several loci is ordered inter-allelically, such that one allele rearranges at a time. Once completed, the rearrangement is tested to determine whether it is productive before rearrangement at the alternative allele occurs. This inter-allelic ordering is critical for enforcing allelic exclusion. Antigen receptor gene assembly is also intra-allelically ordered. After a rearrangement is initiated on one allele, the initiation of additional rearrangements on the same allele must be prevented. Cleavage events at an antigen receptor loci mediated by recombination-activating gene (RAG) proteins are regulated by accessibility of the loci. However, once RAG double-strand breaks (DSBs) are made, the non-canonical DNA damage response (ncDDR) participates in enforcing both inter-allelic and intra-allelic ordering to prevent additional RAG DSBs from being generated until the initial rearrangement has been completed and tested to determine whether it encodes a functional antigen receptor gene. ATM, ataxia telangiectasia mutated.
Some antigen receptor loci (Iglk and Tcra) are assembled from clusters of V and J gene segments that are organized such that a VJ rearrangement can be replaced by rearrangement of an upstream V to a downstream J on the same allele64–66 (FIG. 4). This allele structure is advantageous, as it permits individual cells multiple sequential opportunities to successfully complete a productive antigen receptor gene on each allele. However, these sequential rearrangements must be intra-allelically ordered so that a VJ rearrangement can be completed and tested to determine whether it encodes a functional antigen receptor chain before it is deleted by another rearrangement on the same allele (FIG. 4). Promoters upstream of the J gene segment clusters in the Iglk and Tcra loci function to target initial rearrangements at these loci to the most upstream J gene segment67,68. Indeed, in NHEJ-deficient pre-B cells, RAG cleavage occurs primarily at the most 5′ Jk segment33,62. However, in pre-B cells deficient in NHEJ and ATM, RAG cleavage progresses to more 3′ Jk gene segments33,62. Moreover, Tcra rearrangements in ATM-deficient thymocytes use more 3′ Ja gene segments69. Thus, once a RAG DSB is generated, the ncDDR limits subsequent RAG DSBs on the same allele.
The ncDDR can limit RAG DSBs through multiple mechanisms that impact both RAG activity and antigen receptor locus accessibility. In pro-B cells, RAG cleavage at one Igh allele causes an ATM-dependent repositioning of the other Igh allele to heterochromatic regions, presumably making it less accessible to RAG cleavage61. In pre-B cells, ATM antagonism of pre-BCR signals leads to diminished levels of IRF4 and a decrease in Iglk germline transcripts and Iglk locus accessibility33 (FIG. 3c). Moreover, SPIC displaces PU.1 from the Iglk 3′ enhancer, which would further reduce Iglk germline transcription and accessibility33,70,71 (FIG. 3c). The ncDDR also feeds back directly to inhibit RAG expression and activity. In pre-B cells, the ncDDR causes a reduction in GADD45α, which is required for RAG gene expression24,62 (FIG. 3c). NF-κB1, which is activated by RAG DSBs, leads to downregulation of RAG gene expression in response to genotoxic DSBs in developing lymphocytes21,72. Moreover, genotoxic DSBs also lead to an ATM-dependent reduction in FOXO1 binding to the RAG enhancer, ERAG73. ATM-dependent phosphorylation of RAG2 also leads to diminished RAG cleavage activity74 (FIG. 3c). Thus, ncDDR signals to RAG DSBs likely feed back through multiple mechanisms that reduce RAG gene expression and RAG protein activity.
The toggle model.
How is it that a small pre-B cell undergoes iterative Iglk chain gene rearrangements in an ordered fashion while allowing a single rearrangement to be tested to determine whether it is productive before another rearrangement is initiated? We propose that this occurs through a toggling between pre-BCR signalling and RAG DSB-mediated ncDDR signalling (FIG. 5). In small pre-B cells, the two Iglk alleles are differentially accessible such that in response to pre-BCR signalling, RAG DSBs are efficiently generated on a single allele58–60. The generation of a pair of RAG DSBs at an Iglk allele leads to activation of ATM and the ncDDR, which antagonizes pre-BCR signalling, Iglk locus accessibility and RAG activity, preventing initiation of additional Iglk rearrangements. Completion of Iglk chain gene assembly through NHEJ-mediated repair of the RAG DSBs leads to silencing of ATM activity and the ncDDR. If the completed Iglk chain gene is productive, encoding an IgLκ chain that can pair with the IgH chain to generate a BCR, then the BCR signals promote transition to the immature B cell stage, where RAG activity and Iglk chain gene rearrangement cease if the BCR is non-autoreactive17. If the rearrangement does not encode a functional IgLκ chain, then loss of ATM signals, and the ncDDR, would presumably lead to re-initiation of pre-BCR signalling, Iglk locus accessibility and RAG activity, which together would trigger another Iglk chain gene rearrangement. This rearrangement could be on the alternative Iglk allele or the same allele, leading to deletion of the previous non-functional Iglk rearrangement. It is tempting to speculate that reactivation of pre-BCR signals in these cells may drive them through the cell cycle, stochastically resetting the accessibility of the two Iglk alleles during DNA replication and rendering a single Iglk allele accessible in each of the daughter cells. In immature B cells, autoreactive BCRs can generate signals that maintain RAG expression and lead to new Iglk chain rearrangements that replace the Iglk chain gene responsible for the autoreactive BCR, a process termed receptor editing17,19. Thus, in addition to iteratively toggling between the ncDDR and pre-BCR signals in pre-B cells, immature B cells with autoreactive BCRs may toggle between ncDDR and BCR signals as they attempt to replace this BCR. In this regard, the ncDDR may antagonize BCR signalling to regulate receptor editing in immature cells through pathways similar to those used to antagonize pre-BCR signalling in pre-B cells.
Fig. 5:|. The toggle model.
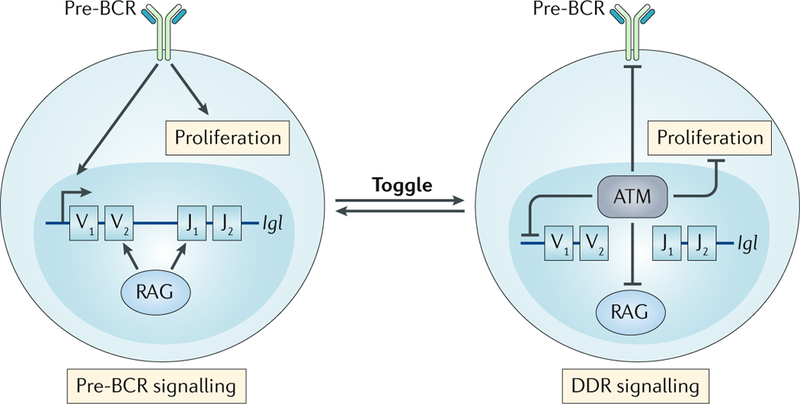
Small pre-B cells have two signalling states; first, pre-B cell receptor (pre-BCR) signalling that promotes Igl gene rearrangement and, second, signalling via ataxia telangiectasia mutated (ATM), in response to double-strand breaks (DSBs) generated by recombination-activating gene (RAG) proteins, that activates a non-canonical DNA damage response (ncDDR). The ncDDR suppresses additional RAG DSBs until the Igl gene assembly is complete and is tested to determine whether it encodes a functional IgL chain. Small pre-B cells may iteratively toggle between these two signalling states as they undergo multiple rounds of Igl chain gene rearrangements in an ordered manner as they attempt to generate a functional IgL chain.
The ncDDR in mature B cells
Once activated, mature B cells generate antibodies of different isotypes through the process of CSR20 (FIG. 6). Activation of a naive IgM-expressing B cell leads to transcription of the switch region (Sμ) upstream of the Cμ exon and the switch region associated with the target constant region gene (FIG. 6). This transcription leads to the formation of DNA–RNA R-loops with non-template single-strand DNA, which serves as a substrate for activation-induced deaminase (AID). AID converts cytosine to uridine, with the resulting mismatch processed primarily by the base excision repair pathway and also by the mismatch repair pathway20. Uracil-DNA glycosylase (UNG) of the base excision repair pathway forms an abasic site, and apurinic/apyrimidinic endonuclease 1 (APE1) excises this abasic site to form a single-strand nick. In conjunction with the RNA exosome complex, AID also targets the template strand, although less efficiently, and it is thought that DSBs form through closely staggered single-strand nicks on the alternative strands75. DNA ends in two switch regions are then joined with deletion of the intervening sequence to place the new C region exons downstream of the V region exons20 (FIG. 6). During CSR, the DNA ends can join by either classical or alternative NHEJ6,76.
Fig. 6:|. Immunoglobulin class switch recombination.
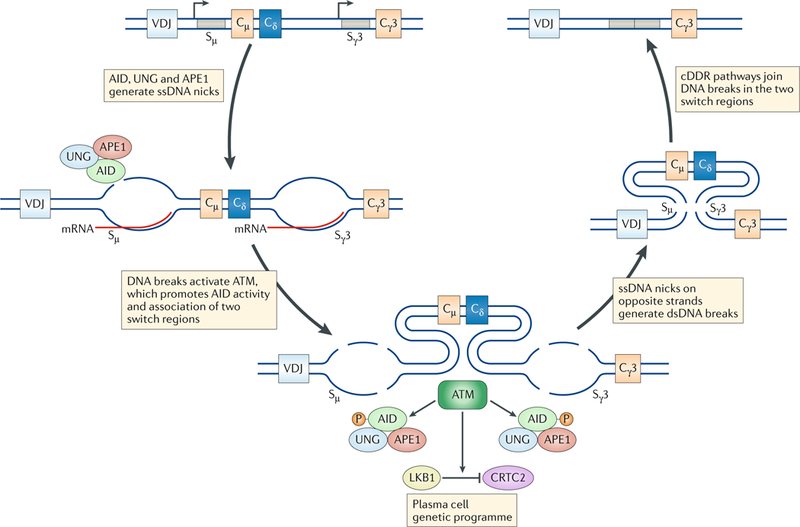
Immunoglobulin class switch recombination (CSR) is initiated by transcription through the switch μ (Sμ) region and a downstream switch region to which switching will occur (shown here to be Sγ3). Activation-induced deaminase (AID) targets the non-template single-strand DNA (ssDNA), deaminating cytosine to form uridine. The proteins uracil-DNA glycosylase (UNG) and apurinic/apyrimidinic endonuclease 1 (APE1) of the base excision repair pathway then deglycosylate and excise the uridine to generate a single-strand nick. The same reaction occurs on the template strand, although less efficiently, and the formation of closely staggered nicks on the two DNA strands leads to a DNA double-strand break (DSB). Broken DNA ends from the two switch regions are then joined by either classical or alternative non-homologous end joining. CSR DSBs activate ataxia telangiectasia mutated (ATM), initiating a non-canonical DNA damage response (ncDDR) that regulates CSR and B cell differentiation. Phosphorylation of AID by ATM in response to CSR DSBs promotes the association of APE1 with AID, leading to more efficient nick formation. ATM activity promotes the association of the two switch regions with DSBs. ATM activates the kinase LKB1, which inhibits CREB-regulated transcription co-activator 2 (CRTC2), a transcriptional co-activator of cAMP-responsive element-binding protein 1 (CREB1), promoting a plasma cell genetic programme. cDDR, canonical DNA damage response; dsDNA, double-strand DNA; P, phosphorylation.
The DSBs generated during CSR activate ATM, leading to an ncDDR that, like RAG DSBs in developing B cells, regulates a broadly functional genetic programme. CSR, and the resulting DSBs, occur in rapidly proliferating germinal centre B cells. BCL-6, a key transcription factor that regulates the B cell germinal centre programme, suppresses the expression of several DDR factors, including ATR, p53 and p21, which potentially limits the cDDR to CSR DSBs77–79. However, CSR DSBs activate ATM, initiating an ncDDR that includes activation of the kinase LKB1 (also known as STK11)80. LKB1 inactivates CREB-regulated transcription co-activator 2 (CRTC2), which leads to the acquisition of a genetic programme that promotes the differentiation of activated B cells into plasma cells80. Thus, through activation of an ncDDR, CSR DSBs provide the signals that are required for the differentiation of activated B cells into plasma cells.
Unlike RAG DSBs, which are inhibited by ATM during V(D)J recombination, during CSR, ATM augments AID-induced DSB generation at switch regions. In this regard, ATM-mediated phosphorylation of AID promotes its association with the endonuclease APE1, which is required for the generation of single-strand nicks81. Moreover, ATM activity promotes the association of two switch regions with DSBs82. The intriguing contrast between the activity of ATM in inhibiting RAG DSBs and its activity in promoting AID-induced DSBs could be related, in part, to the different manner in which the paired DSBs are generated by RAG and AID. RAG DSBs are generated in a synaptic complex that contains RAG and two recombining gene segments, and, as discussed above, ATM-mediated activation of the ncDDR prevents the formation of additional RAG DSBs. By contrast, AID-induced DSBs are generated independently at two switch regions that are not necessarily associated with each other20,83–86. Here, ATM signalling in response to AID-induced DSBs at one switch region is used to stimulate AID-induced DSBs at another switch region and to drive association of the two switch regions with DSBs, thereby promoting CSR. Indeed, in ATM-deficient B cells, DSBs are observed at increased frequency at the most upstream switch region, Sμ, but at decreased frequency in down-stream switch regions87. This contrast in the regulation of RAG-induced and AID-induced DSB generation by ATM provides an important example of how the ncDDR has evolved to regulate similar processes in different ways on the basis of cell type-specific and context-specific requirements.
The ncDDR in macrophages
Activated macrophages produce reactive oxygen intermediates (ROIs) and reactive nitrogen intermediates, such as nitric oxide (NO), to eradicate pathogens5. These agents could also damage host cellular DNA, leading to DNA DSBs that activate the cDDR and ncDDR. Indeed, bone marrow-derived macrophages (BMDMs) activated with Toll-like receptor agonists, such as bacterial lipopolysaccharide (LPS), and IFNγ or infected with the intracellular bacterial pathogen Listeria monocytogenes activate both the cDDR and ncDDR88 (FIG. 7). ROIs can activate ATM directly without the generation of DNA DSBs89. However, the cDDR in activated BMDMs does not depend on ROIs; it depends on NO and the generation of DNA DSBs by NO88. Unlike ROI production, which occurs immediately upon macrophage activation, NO production is delayed owing to the requirement for inducible NO synthase (NOS2) gene induction90. Thus, activation of an ncDDR will be delayed relative to the initial activation of the macrophage. In BMDMs, an optimal cDDR and ncDDR depend on both ATM and DNA-PKcs activation as well as on type I interferon signals88. The basis for the requirement for type I interferons is not known but is not due solely to the role of type I interferons in stimulating NO production or to a requirement for type I interferon signals in promoting expression of key DDR proteins88. The ncDDR in macrophages regulates a functional genetic programme that includes several cytokine and chemokine genes in addition to genes encoding cell surface receptors with functions in phagocytosis88. Irradiation of macrophages also leads to ATM-dependent induction of an ncDDR that regulates the expression of many genes that are involved in innate immune responses91,92. The type I interferons required for this response may come from the genotoxic DNA damage caused by irradiation93. In BMDMs, ATM and DNA-PKcs also promote optimal caspase 1 activation and cleavage of pro-IL-1β and pro-IL-18 to form IL-1β and IL-18 during AIM2 inflammasome activation in response to L. monocytogenes infection88,94. Indeed, ATM-deficient mice exhibit defects in innate immunity owing to impaired inflammasome activation95. NBS1, a component of the MRN complex, is required for normal macrophage proliferation and differentiation, which could reflect MRN function in DNA replication in addition to its role in activating ATM and the ncDDR4,88,96. In the setting of chronic inflammatory signals in vivo, ATR regulates pathways that promote the generation of polyploid macrophages in granuloma lesions97. Thus, all three DDR kinases, ATM, DNA-PKcs and ATR, promote ncDDRs that regulate macrophage differentiation and function.
Fig. 7:|. Activation of the ncDDR in macrophages.
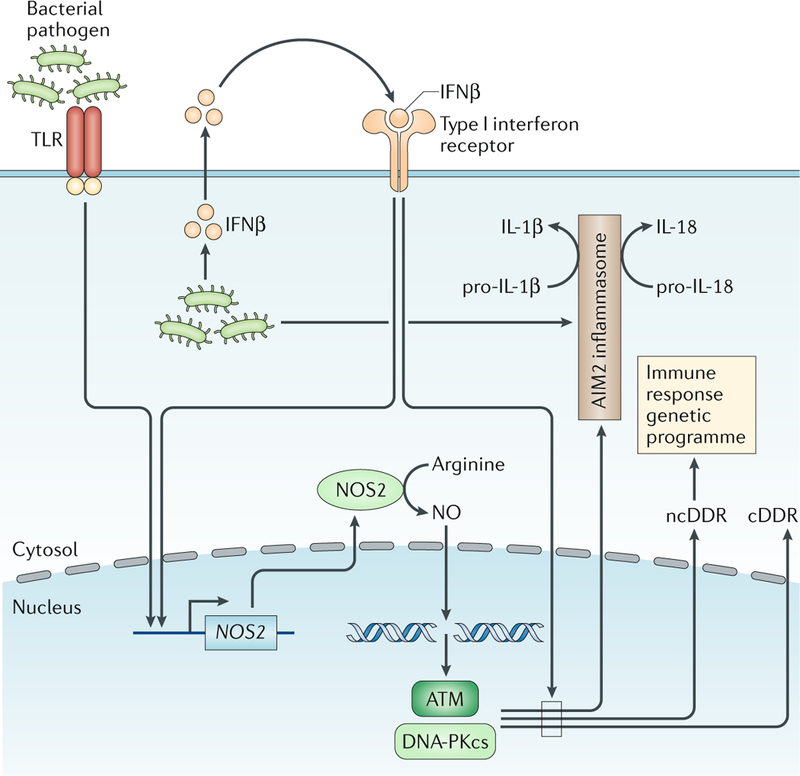
Macrophages activated by signals through Toll-like receptors (TLRs) and interferon receptors produce nitric oxide (NO), which causes genomic DNA double-strand breaks (DSBs) that activate ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), leading to a canonical DNA damage response (cDDR) and a non-canonical DNA damage response (ncDDR). The ncDDR promotes a broadly functional genetic programme in these cells and regulates inflammasome activation. Type I interferon signals are required for optimal cDDR and ncDDR activation. NOS2, nitric oxide synthase.
Heritable effects of ncDDR signalling
DNA DSBs generated during V(D)J recombination and CSR impact the cellular events that are coincident with DSB generation through initiation of ncDDRs. However, recent studies suggest that DNA DSBs generated early in immune cell development may influence the function of mature immune cells long after the DNA damage has been resolved and any damage response signalling has been extinguished. Lymphocytes and natural killer (NK) cells differentiate from common progenitors that can express RAG98–101. In response to viral pathogens, NK cells derived from progenitors that expressed RAG exhibit greater fitness, proliferation and better survival than NK cells derived from progenitors that failed to express RAG102,103. Although it was not established that this requires DDR kinase activity and an ncDDR, the endonuclease activity of RAG1 is required for this effect, indicative of a requirement for RAG DSB generation102. Moreover, antigen receptor gene rearrangements can be found in mature NK cells, which demonstrates that RAG-generated DNA DSBs are produced and repaired in progenitors that give rise to NK cells101. Thus, activation of an ncDDR by these DSBs may have long-term effects on mature NK cells, presumably through heritable alterations in their genetic programmes. In this regard, NK cells derived from progenitors that expressed RAG exhibit increased expression of several genes encoding DDR proteins102.
Determinants of ncDDRs
The unique pathways that are activated by ncDDRs in different cell types may be related, in part, to the nature of the DSB (for example, RAG DSBs versus CSR DSBs). However, in developing pre-B cells, DSBs induced by ionizing radiation trigger many of the gene expression changes induced by RAG DSBs21,104. Indeed, NF-κB1, a major determinant of this genetic programme, can be activated through the phosphorylation of NEMO by ATM in the nucleus in response to genotoxic DSBs35,36. Thus, cells that undergo programmed DNA DSBs may be poised to express a genetic programme in response to DSB signals regardless of how the DSBs are generated. Activation of ATM provides the cellular cue that a DSB has been generated, initiating an ncDDR that regulates cell type-specific processes. Implied in this model is the idea that exposure of immune cells to genotoxic agents, as in the setting of cancer therapy, could alter the genetic programme of these cells, impacting immune system development and function21.
Conclusion
We are now just beginning to understand how ncDDRs to DNA DSBs generated in different physiological settings signal events that are important for the development and function of cells of the innate and adaptive immune system. Although most of what we know comes from the analysis of programmed DSBs generated during V(D)J recombination and CSR, DSBs made in other settings, such as during transcription and DNA replication in immune cells that have been stimulated to divide, may also initiate important ncDDRs. In addition, fragments of damaged genomic DNA that gain access to the cytosol can also regulate immune responses through activation of the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), which leads to type I interferon production105. This pathway, which evolved to respond to pathogen DNA, may also functionally intersect with the cDDR and ncDDR106. The full spectrum of ncDDR pathways, how they integrate with other cellular pathways and the processes that they regulate in innate and adaptive immune cells remain to be determined. The regulation of diverse immune responses by ncDDRs raises the intriguing possibility that therapeutic agents that target DDR proteins may be used to manipulate immune responses in different disease settings.
Programmed DNA double-strand breaks
DNA double-strand breaks made at specific genomic regions as required intermediates of physiological processes, such as V(D)J recombination or immunoglobulin class switch recombination.
Non-programmed DNA DSBs
DNA double-strand breaks (DSBs) made by genotoxic agents or nucleases throughout the genome.
Canonical DNA damage response
(cDDR). A cellular response to DNA double-strand breaks (DSBs) initiated by DNA damage response kinases in all cells that activates the pathways required for DNA DSB repair and genome stability.
Non-homologous end joining
(NHEJ). A DNA repair process that joins broken DNA ends (double-strand breaks) at all phases of the cell cycle without using homologous DNA as a template. The core components of this pathway include the proteins KU70, KU80, X-ray repair cross-complementing protein 4 (XRCC4) and DNA ligase IV.
Non-canonical DNA damage responses
(ncDDRs). Cellular responses to DNA double-strand breaks (DSBs) activated by DNA damage response kinases but that are not involved in DNA DSB repair or in maintaining genome integrity. Rather, these DSB-dependent programmes regulate cell type-specific pathways that are involved in a broad variety of cellular functions.
Pre-B cell receptor
(Pre-BCR). A receptor composed of immunoglobulin heavy chains and surrogate light chains, VpreB and λ5, that associates with the transmembrane Igα and Igβ proteins that have cytoplasmic domains that transduce intracellular signals through the tyrosine kinase SYK and adaptor protein B cell linker protein (BLNK).
IL-7 receptor
(IL-7R). A receptor composed of the IL-7Rα chain and the cytokine receptor common γ-chain. Upon binding to IL-7, IL-7R activates the kinase AKT and the kinases Janus kinase 1 (JAK1) and JAK3, which phosphorylate and activate signal transducer and activator of transcription 5 (STAT5), to regulate cellular proliferation and survival.
Accessibility
Refers to the structure of chromatin. When chromatin is more loosely packed (or open), it is accessible for transcription, whereas tightly packed or closed chromatin is refractory to factors that need to gain access to the DNA template. Recombination-activating gene (RAG) is expressed in developing B and T cells, but it can only carry out V(D)J recombination at antigen receptor loci with an accessible chromatin structure.
ETS family transcription factors
A large family of proteins with conserved DNA binding domains that bind to GGA(A/T) (PU-box) motifs. Family members have distinct transactivation domains that dictate binding partners and regulate transcription. The PU.1 (also known as SPI1), SPIB and SPIC ETS family members all have roles in regulating transcription during B cell development.
Acknowledgements
This work was supported by US National Institutes of Health grants AI047829 (B.P.S.), AI074953 (B.P.S.) and K08AI102946 (J.J.B.). J.J.B. was supported by an Alex’s Lemonade Stand Foundation A Award, an American Society of Hematology Scholar Award, the Foundation for Barnes-Jewish Hospital, the Cancer Frontier Fund and the Barnard Trust.
Footnotes
Competing interests
The authors declare no competing interests.
Reviewer information
Nature Reviews Immunology thanks R. Casellas, J. Chaudhuri and M. Clark for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fugmann SD, Lee AI, Shockett PE, Villey IJ & Schatz DG The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol. 18, 495–527 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Chaudhuri J et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv. Immunol. 94, 157–214 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Fong YW, Cattoglio C & Tjian R The intertwined roles of transcription and repair proteins. Mol. Cell 52, 291–302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciccia A & Elledge SJ The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan C & Shiloh MU Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl Acad. Sci. USA 97, 8841–8848 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang HHY, Pannunzio NR, Adachi N & Lieber MR Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moynahan ME & Jasin M Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackford AN & Jackson SP ATM. ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Stracker TH & Petrini JH The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuoka S et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 (2007). This study demonstrates that DDR kinases phosphorylate hundreds of proteins in response to DNA DSBs, with most functioning in networks that have no known role in the DDR. [DOI] [PubMed] [Google Scholar]
- 11.Bassing CH, Swat W & Alt FW The mechanism and regulation of chromosomal V(D)J recombination. Cell 109, S45–S55 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Helmink BA & Sleckman BP The response to and repair of RAG-mediated DNA double-strand breaks. Annu. Rev. Immunol. 30, 175–202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gellert M V(D)J recombination: rag proteins, repair factors, and regulation. Annu. Rev. Biochem. 71, 101–132 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Desiderio S, Lin WC & Li Z The cell cycle and V(D)J recombination. Curr. Top. Microbiol. Immunol. 217, 45–59 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Ma Y, Pannicke U, Schwarz K & Lieber MR Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108, 781–794 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Bredemeyer AL et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442, 466–470 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Melchers F Checkpoints that control B cell development. J. Clin. Invest. 125, 2203–2210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark MR, Mandal M, Ochiai K & Singh H Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 14, 69–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemazee D & Weigert M Revising B cell receptors. J. Exp. Med. 191, 1813–1817 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhuri J & Alt FW Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 4, 541–552 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Bredemeyer AL et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature 456, 819–823 (2008).This report shows that RAG DSBs activate an ncDDR including a genetic programme with many genes encoding proteins that function in diverse lymphocyte developmental processes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandal M et al. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 12, 1212–1220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sleckman BP, Gorman JR & Alt FW Accessibility control of antigen-receptor variable-region gene assembly: role of cis-acting elements. Annu. Rev. Immunol. 14, 459–481 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Amin RH & Schlissel MS Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat. Immunol. 9, 613–622 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ochiai K et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat. Immunol. 13, 300–307 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goetz CA, Harmon IR, O’Neil JJ, Burchill MA & Farrar MA STAT5 activation underlies IL7 receptor-dependent B cell development. J. Immunol. 172, 4770–4778 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Bednarski JJ et al. RAG-induced DNA double-strand breaks signal through Pim2 to promote pre-B cell survival and limit proliferation. J. Exp. Med. 209, 11–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domen J et al. Pim-1 levels determine the size of early B lymphoid compartments in bone marrow. J. Exp. Med. 178, 1665–1673 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson K et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity 28, 335–345 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Casellas R et al. Contribution of receptor editing to the antibody repertoire. Science 291, 1541–1544 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Fistonich C et al. Cell circuits between B cell progenitors and IL-7+ mesenchymal progenitor cells control B cell development. J. Exp. Med. 215, 2586–2599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tokoyoda K, Egawa T, Sugiyama T, Choi BI & Nagasawa T Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20, 707–718 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Bednarski JJ et al. RAG-mediated DNA double-strand breaks activate a cell type-specific checkpoint to inhibit pre-B cell receptor signals. J. Exp. Med. 213, 209–223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guidos CJ et al. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 10, 2038–2054 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Wu ZH, Shi Y, Tibbetts RS & Miyamoto S Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science 311, 1141–1146 (2006).. [DOI] [PubMed] [Google Scholar]
- 36.Huang TT, Wuerzberger-Davis SM, Wu ZH & Miyamoto S Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell 115, 565–576 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Rosen SD Ligands for L-selectin: homing, inflammation, and beyond. Annu. Rev. Immunol. 22, 129–156 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Shiow LR et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Pearce G et al. Signaling protein SWAP-70 is required for efficient B cell homing to lymphoid organs. Nat. Immunol. 7, 827–834 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Fox CJ et al. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 17, 1841–1854 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JL, Limnander A & Rothman PB Pim-1 and Pim-2 kinases are required for efficient pre-B cell transformation by v-Abl oncogene. Blood 111, 1677–1685 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Derudder E et al. Development of immunoglobulin lambda-chain-positive B cells, but not editing of immunoglobulin kappa-chain, depends on NF-κB signals. Nat. Immunol. 10, 647–654 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Y et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404, 897–900 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Zhu C et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 109, 811–821 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Hu J, Tepsuporn S, Meyers RM, Gostissa M & Alt FW Developmental propagation of V(D)J recombination-associated DNA breaks and translocations in mature B cells via dicentric chromosomes. Proc. Natl Acad. Sci. USA 111, 10269–10274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirao A et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287, 1824–1827 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Wossning T et al. Deregulated Syk inhibits differentiation and induces growth factor-independent proliferation of pre-B cells. J. Exp. Med. 203, 2829–2840 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rolink A, Kudo A, Karasuyama H, Kikuchi Y & Melchers F Long-term proliferating early pre B cell lines and clones with the potential to develop to surface Ig-positive, mitogen reactive B cells in vitro and in vivo. EMBO J. 10, 327–336 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bemark M, Martensson A, Liberg D & Leanderson T Spi-C, a novel Ets protein that is temporally regulated during B lymphocyte development. J. Biol. Chem. 274, 10259–10267 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Hashimoto S et al. Prf, a novel Ets family protein that binds to the PU.1 binding motif, is specifically expressed in restricted stages of B cell development. Int. Immunol. 11, 1423–1429 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Sharrocks AD The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2, 827–837 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Carlsson R, Persson C & Leanderson T SPI-C, a PU-box binding ETS protein expressed temporarily during B cell development and in macrophages, contains an acidic transactivation domain located to the N-terminus. Mol. Immunol. 39, 1035–1043 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Schweitzer BL et al. Spi-C has opposing effects to PU.1 on gene expression in progenitor B cells. J. Immunol. 177, 2195–2207 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Xu LS et al. Regulation of B cell linker protein transcription by PU.1 and Spi-B in murine B cell acute lymphoblastic leukemia. J. Immunol. 189, 3347–3354 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Thornton TM et al. Inactivation of nuclear GSK3β by Ser389 phosphorylation promotes lymphocyte fitness during DNA double-strand break response. Nat. Commun. 7, 10553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeMicco A et al. Lymphocyte lineage-specific and developmental stage specific mechanisms suppress cyclin D3 expression in response to DNA double strand breaks. Cell Cycle 15, 2882–2894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vettermann C & Schlissel MS Allelic exclusion of immunoglobulin genes: models and mechanisms. Immunol. Rev. 237, 22–42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mostoslavsky R et al. Kappa chain monoallelic demethylation and the establishment of allelic exclusion. Genes Dev. 12, 1801–1811 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bergman Y, Fisher A & Cedar H Epigenetic mechanisms that regulate antigen receptor gene expression. Curr. Opin. Immunol. 15, 176–181 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Karki S et al. Regulated capture of Vκ gene topologically associating domains by transcription factories. Cell Rep. 24, 2443–2456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hewitt SL et al. RAG-1 and ATM coordinate monoallelic recombination and nuclear positioning of immunoglobulin loci. Nat. Immunol. 10, 655–664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steinel NC et al. The Ataxia Telangiectasia mutated kinase controls Igκ allelic exclusion by inhibiting secondary Vκ-to-Jκ rearrangements. J. Exp. Med. 210, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinel NC, Fisher MR, Yang-Iott KS & Bassing CH The ataxia telangiectasia mutated and cyclin D3 proteins cooperate to help enforce TCRβ and IgH allelic exclusion. J. Immunol. 193, 2881–2890 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo J et al. Regulation of the TCRa repertoire by the survival window of CD4+CD8+ thymocytes. Nat. Immunol. 3, 469–476 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Yamagami T, ten Boekel E, Andersson J, Rolink A & Melchers F Frequencies of multiple IgL chain gene rearrangements in single normal or κL chain-deficient B lineage cells. Immunity 11, 317–327 (1999). [DOI] [PubMed] [Google Scholar]
- 66.Huang CY, Sleckman BP & Kanagawa O Revision of T cell receptor a chain genes is required for normal T lymphocyte development. Proc. Natl Acad. Sci. USA 102, 14356–14361 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Villey I, Caillol D, Selz F, Ferrier P & de Villartay JP Defect in rearrangement of the most 5’ TCR-Jα following targeted deletion of T early α (TEA): implications for TCR α locus accessibility. Immunity 5, 331–342 (1996). [DOI] [PubMed] [Google Scholar]
- 68.Hawwari A, Bock C & Krangel MS Regulation of T cell receptor alpha gene assembly by a complex hierarchy of germline Jα promoters. Nat. Immunol. 6, 481–489 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matei IR et al. ATM deficiency disrupts TCRα locus integrity and the maturation of CD4+CD8+thymocytes. Blood 109, 1887–1896 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Batista CR, Li SK, Xu LS, Solomon LA & DeKoter RP PU.1 regulates Ig light chain transcription and rearrangement in pre-B cells during B cell development. J. Immunol. 198, 1565–1574 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Schwarzenbach H, Newell JW & Matthias P Involvement of the Ets family factor PU.1 in the activation of immunoglobulin promoters. J. Biol. Chem. 270, 898–907 (1995). [DOI] [PubMed] [Google Scholar]
- 72.Fisher MR, Rivera-Reyes A, Bloch NB Schatz DG. & Bassing CH. Immature lymphocytes inhibit Rag1 and Rag2 transcription and V(D)J recombination in response to DNA double-strand breaks. J. Immunol. 198, 2943–2956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ochodnicka-Mackovicova K et al. The DNA damage response regulates RAG1/2 expression in pre-B cells through ATM-FOXO1 signaling. J. Immunol. 197, 2918–2929 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Hewitt SL et al. The conserved ATM kinase RAG2-S365 phosphorylation site limits cleavage events in individual cells independent of any repair defect. Cell Rep. 21, 979–993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basu U et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 144, 353–363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boboila C, Alt FW & Schwer B Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116, 1–49 (2012). [DOI] [PubMed] [Google Scholar]
- 77.Ranuncolo SM et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat. Immunol. 8, 705–714 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Phan RT & Dalla-Favera R The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639(2004). [DOI] [PubMed] [Google Scholar]
- 79.Phan RT, Saito M, Basso K, Niu H & Dalla-Favera R BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 6, 1054–1060 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Sherman MH et al. AID-induced genotoxic stress promotes B cell differentiation in the germinal center via ATM and LKB1 signaling. Mol. Cell 39, 873–885 (2010). In this study, the authors show that DNA DSBs made during CSR in mature B cells activate an ncDDR that includes a genetic programme that functions in plasma cell differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vuong BQ et al. A DNA break-and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat. Immunol. 14, 1183–1189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reina-San-Martin B, Chen HT, Nussenzweig A & Nussenzweig MC ATM is required for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 200, 1103–1110 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dudley DD et al. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc. Natl Acad. Sci. USA 99, 9984–9989 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gu H, Zou YR & Rajewsky K Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73, 1155–1164 (1993). [DOI] [PubMed] [Google Scholar]
- 85.Wuerffel R et al. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity 27, 711–722 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feldman S et al. 53BP1 contributes to Igh locus chromatin topology during class switch recombination. J. Immunol. 198, 2434–2444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khair L et al. ATM increases activation-induced cytidine deaminase activity at downstream S regions during class-switch recombination. J. Immunol. 192, 4887–4896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morales AJ et al. A type I IFN-dependent DNA damage response regulates the genetic program and inflammasome activation in macrophages. eLife 6, e24655 (2017). This paper shows that reactive nitrogen compounds made by activated macrophages generate DNA DSBs that activate an ncDDR, which regulates macrophage functions in innate immune responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo Z, Kozlov S, Lavin MF, Person MD & Paull TT ATM activation by oxidative stress. Science 330, 517–521 (2010). [DOI] [PubMed] [Google Scholar]
- 90.MacMicking J, Xie QW & Nathan C Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 (1997). [DOI] [PubMed] [Google Scholar]
- 91.Purbey PK et al. Defined sensing mechanisms and signaling pathways contribute to the global inflammatory gene expression output elicited by ionizing radiation. Immunity 47, 421–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Teresa Pinto A et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 6, 18765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brzostek-Racine S, Gordon C, Van Scoy S & Reich NC The DNA damage response induces IFN. J. Immunol. 187, 5336–5345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Broz P & Dixit VM Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Erttmann SF et al. Loss of the DNA damage repair kinase ATM impairs inflammasome-dependent anti-bacterial innate immunity. Immunity 45, 106–118 (2016). [DOI] [PubMed] [Google Scholar]
- 96.Pereira-Lopes S et al. NBS1 is required for macrophage homeostasis and functional activity in mice. Blood 126, 2502–2510 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Herrtwich L et al. DNA damage signaling instructs polyploid macrophage fate in granulomas. Cell 167, 1264–1280 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Borghesi L et al. B lineage-specific regulation of V(D)J recombinase activity is established in common lymphoid progenitors. J. Exp. Med. 199, 491–502 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Welner RS et al. Asynchronous RAG-1 expression during B lymphopoiesis. J. Immunol. 183, 7768–7777 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fronkova E et al. Lymphoid differentiation pathways can be traced by TCR delta rearrangements. J. Immunol. 175, 2495–2500 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Pilbeam K et al. The ontogeny and fate of NK cells marked by permanent DNA rearrangements. J. Immunol. 180, 1432–1441 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karo JM, Schatz DG & Sun JC The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell 159, 94–107 (2014). This paper describes NK cells derived from progenitors that experienced RAG DSBs, and presumably DDR signalling, exhibit greater fitness during activation and immune responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Andrews DM & Smyth MJ A potential role for RAG-1 in NK cell development revealed by analysis of NK cells during ontogeny. Immunol. Cell Biol. 88, 107–116 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Innes CL et al. DNA damage activates a complex transcriptional response in murine lymphocytes that includes both physiological and cancer-predisposition programs. BMC Genomics 14, 163 (2013).This reference shows that many of the genes activated in lymphocytes by RAG DSBs are also activated by genotoxic DSBs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Liu H et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 (2018). [DOI] [PubMed] [Google Scholar]